


- 1 Cancer Epigenetics Group, Research Center of the Portuguese Oncology Institute – Porto, Rua Dr. António Bernardino de Almeida, Porto, Portugal
- 2 Department of Pathology, Portuguese Oncology Institute – Porto, Rua Dr. António Bernardino de Almeida, Porto, Portugal
- 3 Department of Pathology and Molecular Immunology, Institute of Biomedical Sciences Abel Salazar, University of Porto, Porto, Portugal
- 4 Department of Genetics, Portuguese Oncology Institute – Porto, Rua Dr. António Bernardino de Almeida, Porto, Portugal
Renal cell tumors (RCT) collectively constitute the third most common type of genitourinary neoplasms, only surpassed by prostate and bladder cancer. They comprise a heterogeneous group of neoplasms with distinctive clinical, morphological, and genetic features. Epigenetic alterations are a hallmark of cancer cells and their role in renal tumorigenesis is starting to emerge. Aberrant DNA methylation, altered chromatin remodeling/histone onco-modifications and deregulated microRNA expression not only contribute to the emergence and progression of RCTs, but owing to their ubiquity, they also constitute a promising class of biomarkers tailored for disease detection, diagnosis, assessment of prognosis, and prediction of response to therapy. Moreover, due to their dynamic and reversible properties, those alterations represent a target for epigenetic-directed therapies. In this review, the current knowledge about epigenetic mechanisms and their altered status in RCT is summarized and their envisaged use in a clinical setting is also provided.
Introduction
Renal cell tumors (RCT) are the most common neoplasms of the kidney in adults, representing 2–3% of all non-cutaneous malignant neoplasms, ranking 14th in incidence for both genders, with a mortality rate of 1.6/100,000, worldwide (Ferlay et al., 2010). RCTs are a very heterogeneous group of neoplasms, ranging from benign to malignant behavior, displaying variable clinical aggressiveness and considerable histopathological diversity (Lopez-Beltran et al., 2009). The most prevalent benign RCT is oncocytoma, whereas among malignant RCTs the most common subtypes are clear cell renal cell carcinoma (ccRCC, 70–75% of cases), papillary renal cell carcinoma (pRCC, 10–15% of cases), and chromophobe renal cell carcinoma (chRCC, 5–10% of cases). Owing to the fact that RCT are mostly asymptomatic at their earliest stages, when curative treatment is more likely to be effective, a significant proportion of cases (up to a third of all RCCs) are diagnosed at a late stage, when the neoplasm has already spread locally or systemically (Lam et al., 2005). On the other hand, an increasing number of RCT is being identified through routine ultrasonography, posing new diagnostic challenges at a pre-operative stage, such as the discrimination between benign and malignant tumors.
Presently, there are no effective screening or early diagnostic tools for RCT that might be used in at-risk groups or in a wider population setting (Rini et al., 2009). However, the development of such tools might have a relevant clinical impact, mainly if they would be based on non-invasive approaches (Scelo and Brennan, 2007). In this regard, cancer-related genetic and/or epigenetic alterations might be used as biomarkers enabling the detection of cancerous cells in clinical samples, thus increasing the ability to identify tumors at their earliest stages (Esteller, 2008). Eventually, those alterations might also prove useful for assessing prognosis, response to therapy, and also for patient monitoring (Esteller, 2008). RCTs display characteristic and often discriminative chromosomal alterations (Baldewijns et al., 2008). These, however, are not easily amenable for detection in clinical samples because it requires tissue culture for karyotyping or FISH analysis using tumor biopsies. On the contrary, epigenetic alterations carry an enormous potential as specific cancer biomarkers (Mulero-Navarro and Esteller, 2008). Furthermore, owing to the reversible nature of epigenetic alterations, these might constitute attractive therapeutic targets, as demonstrated for some hemato-lymphoid neoplasms (Rodriguez-Paredes and Esteller, 2011). Thus, in this review, we address not only the role of epigenetic alterations in renal carcinogenesis, but also their clinical potential in RCT management.
Epigenetic Mechanisms and their Deregulation in Carcinogenesis
Epigenetics might be defined as modifications of the DNA or associated proteins, other than DNA sequence variation itself, that carry information content during cell division (Feinberg and Tycko, 2004). Presently, three main epigenetic mechanisms are recognized: DNA methylation, chromatin remodeling/post-translational histone modifications, and microRNA (miRNA) regulation. DNA methylation and chromatin remodeling/post-translational histone modifications cause changes in chromatin structure that modulate the accessibility of the nuclear transcriptional machinery to specific DNA sequences. On the other hand, miRNAs interact with mRNAs regulating translation to protein.
DNA Methylation
The most extensively studied epigenetic mechanism is DNA methylation which, in mammals, consists on the addition of a methyl group to cytosines preceding guanines (so-called “CpG dinucleotides”), a chemical reaction which is catalyzed by DNA methyltransferases (DNMTs; Goldberg et al., 2007; Lopez-Serra and Esteller, 2008). These CpG dinucleotides are usually clustered in DNA stretches called “CpG Islands,” preferentially found at the regulatory regions of genes, i.e., promoter, untranslated regions, and exon 1. In a normal cell, CpG methylation is present mainly in repetitive sequences, retrotransposons, and parasitic sequences, thus contributing to genetic stability, whereas most of the CpG islands within promoters remain unmethylated during development and even after differentiation, with the notable exception of some imprinted genes, genes on the silenced copy of X chromosome in females, and tissue-specific genes (Vaissiere et al., 2008). Gene silencing associated with methylation of promoter regions containing CpG islands may be due to an obstruction of the access of transcription factors or through the promotion of binding of methylcytosine-binding proteins (MBD), which cooperate with DNMTs and histone deacetylases (HDACs; Fraga et al., 2005; Sharma et al., 2010). Recently, an important role in gene expression regulation has been also credited to lower density CpG regions located in the vicinity of CpG islands, the so-called “CpG island shores” (Doi et al., 2009). These are sequences up to 2 kb distant from CpG islands, which are associated with gene expression (Doi et al., 2009; Irizarry et al., 2009). Remarkably, the methylation pattern at CpG island shores is mostly tissue-specific and cancer-associated alterations in those patterns occur at sites that vary normally in tissue differentiation (Irizarry et al., 2009). These observations are in line with the so-called “epigenetic progenitor model of cancer” (Feinberg et al., 2006).
Aberrant DNA methylation is probably the best characterized cancer-related epigenetic alteration and it is considered by some as one of the earliest events in the process of tumorigenesis (Feinberg et al., 2006). Those aberrations include both global and gene-specific hypomethylation as well as gene-specific CpG island promoter hypermethylation (Mulero-Navarro and Esteller, 2008; Sharma et al., 2010). The impact of gene-specific alterations in DNA methylation depends on the function of the affected gene and the type of alteration. Whereas promoter hypomethylation may cause activation of proto-oncogenes, hypermethylation induces silencing of cancer-related genes with tumor suppressive properties (Feinberg et al., 2006; Sharma et al., 2010). On the other hand, decrease of global genome methylation (genome-wide hypomethylation) may lead to genomic instability in repetitive sequences, especially at pericentromeric regions, predisposing to abnormal recombination, facilitating translocations, deletions, and chromosomal rearrangements (Ehrlich, 2005; Mulero-Navarro and Esteller, 2008; Sharma et al., 2010). It was also demonstrated that aberrant methylation may affect large extensions of DNA in cancer cells, resulting in extensive epigenetic reprogramming of entire chromosomal regions (Frigola et al., 2006).
Chromatin Remodeling and Histone Modifications
Chromatin remodeling refers to covalent and non-covalent modifications of histones, the proteins that form the core of nucleosomes which are the basic unit of chromatin. The N-terminal tails of histones protrude from the nucleosome and may be subjected to a wide range of post-translational covalent changes (methylation, acetylation, ubiquitylation, sumoylation, and phosphorylation) of specific amino acid residues and these are involved in regulation of transcription (Kouzarides, 2007a). This set of histone modifications – the so-called “histone code” – determines the configuration of chromatin, adjusting the accessibility to effector proteins (Kouzarides, 2007a). For instance, whereas lysine acetylation is associated with active transcription, lysine methylation may lead to transcription activation or repression depending on the residue affected and the degree of modification (Kouzarides, 2007a). For example, trimethylation of lysine 4 on histone H3 (H3K4me3) is associated with active transcription, whilst H3K27me3 and H3K9me3 are the two chief repressive marks (Kouzarides, 2007a). There is a large number of enzymes involved in the addition or removal of the covalent modifications, including histone acetyltransferases (HATs), deacetylases (HDACs), methyltransferases (HMTs), and demethylases (HDMs; Kouzarides, 2007a,b; Shi, 2007). Besides their important role in regulation of gene expression in somatic cells, covalent histone modifications are also critical for embryonic stem cell (ESC) development and differentiation (Kouzarides, 2007a,b; Shi, 2007). Indeed, ESCs display bivalent chromatin domains which provide epigenomic plasticity during normal development. Those bivalent chromatin marks consist on the simultaneous occupancy of trimethylated histone H3 lysine 27 (H3K27me3, a repressive mark) and of di- and trimethylated histone 3 lysine 4 (H3K4me2 and H3K4me3, which are both active marks) at the promoters of genes encoding transcription factors that regulate developmentally important genes (Mikkelsen et al., 2007). Thus, neoplastic transformation would be associated with an alteration of these marks, leading to silencing of tumor suppressor genes (TSG; through the loss of H3K4me2 and H3K4me3) with associated promoter hypermethylation and/or activation of proto-oncogenes with concomitant loss of methylation (through loss of H3K27me3; Bloushtain-Qimron et al., 2008; Kondo et al., 2008). On the other hand, non-covalent mechanism involved in chromatin remodeling include alterations in nucleosome positioning and incorporation of histone variants (Henikoff, 2008). It should also be emphasized that DNA methylation and histone modifications work in concert to achieve stable patterns of gene expression (Ballestar and Esteller, 2005; Vaissiere et al., 2008). Indeed, nucleosome remodeling may be involved in inappropriate gene silencing through the cooperation with aberrant DNA methylation and repressive histone covalent modifications (Lin et al., 2007). Concerning histone modifications, global loss of both acetylation of lysine 16 (H4K16ac) and of trimethylation of lysine 20 of histone H4 (H4K20me3) are commonly found in cancer cells, usually in association with DNA hypomethylation at repetitive DNA sequences (Fraga et al., 2005). Loss of H4K16ac correlates with gene silencing (Kapoor-Vazirani et al., 2008), but H4K20me3 is normally found in constitutive heterochromatin regions and other regions that contain silenced genes (Fullgrabe et al., 2011). Thus, it seems paradoxical that, in cancer development, the presence of H4K20me3 is also associated with local silencing of genes (Fullgrabe et al., 2011). In addition, altered methylation patterns of H3K9 and H3K27 have also been found in cancer cells (Nguyen et al., 2002; Valk-Lingbeek et al., 2004). Those modifications, which result in inappropriate gene silencing, are accomplished by histone-modifying enzymes. The expression of some of those enzymes, most notably of HDAC1 and EZH2, is upregulated in several cancers and this is thought to contribute decisively for cancer initiation and progression (Halkidou et al., 2004; Valk-Lingbeek et al., 2004; Song et al., 2005). In this regard, HDMs, like LSD1 which targets methylated H3K4 and H3K9, seem also to play an important role in some cancer models (Metzger et al., 2005; Schulte et al., 2009).
MicroRNAs
MicroRNAs constitute a class of small (∼18–25 nt in length), non-coding RNAs, which are synthesized (pri-miR) and processed in the nucleus (pre-miR), and subsequently in the cytoplasm (mature miR), to finally incorporate the RISC (RNA-induced silencing complex) protein complex (Garzon et al., 2009; Guil and Esteller, 2009). MicroRNAs interact with the 3′UTR of target mRNAs, inducing translational repression or degradation of the latter (Guil and Esteller, 2009). Interestingly, a specific miRNA may regulate multiple mRNAs in the same manner that a single mRNA may be targeted by multiple miRNAs (Shenouda and Alahari, 2009). Moreover, a large proportion of human gene transcripts (estimated between 30 and 70%) are known to be regulated by miRNAs, and these display temporal and tissue-specific patterns. Thus, miRNAs are one of the main classes of regulatory human genes, involved in multiple important cell processes, including cell proliferation, apoptosis, differentiation, development, and metabolism (Guil and Esteller, 2009).
The altered expression of miRNAs in cancer has been more recently documented (Lu et al., 2005). Owing to their role as regulators of mRNA expression, they may act as oncogenes (overexpressed miRNAs that target TSG) or TSG (downregulated miRNAs targeting proto-oncogenes; Guil and Esteller, 2009). Examples of the former include miR-21, targeting PTEN (Chan et al., 2005), whereas miR-15 and miR-16, which target BCL2, and let-7, targeting RAS, typify the latter (Zhang et al., 2007; Ventura and Jacks, 2009). The mechanisms that lead to altered miRNA expression are similar to those of regular genes and include amplification, deletion, mutation, and promoter hypermethylation (Guil and Esteller, 2009; Dudziec et al., 2011).
Epigenetic Aberrations in Renal Cell Tumors and Clinical Applications
As mentioned in the previous section, the delicate epigenetic homeostasis that characterizes normal cells is frequently disturbed in cancer. Indeed, when compared to genetic alterations such as point mutations, deletions, and amplifications, epigenetic events seem to occur much more frequently and at earlier stages (Sharma et al., 2010; Berdasco and Esteller, 2011). These characteristics not only endow epigenetic alterations a critical role in tumorigenesis, but they also set the basis for their use as cancer biomarkers for early detection, diagnosis, assessment of prognosis, patient monitoring, and prediction of response to therapy, as previously stated (Sharma et al., 2010; Berdasco and Esteller, 2011). In the next sections, the role of each epigenetic mechanism in renal cell tumorigenesis, as well as its potential use as biomarker and therapeutic target, will be discussed.
DNA Methylation
Most studies that have investigated promoter hypermethylation in RCT used either a candidate-gene approach (i.e., cancer-related genes deregulated through this mechanism in other malignancies and/or genes known to be mutated in familial RCC) or a functional epigenomic approach, based on genome-wide CpG methylation analysis platforms (e.g., CpG island arrays). Table 1 provides an overview of the most commonly methylated gene promoters in RCTs according to the cellular pathway in which they are involved. Studies using the candidate-gene strategy disclosed promoters with a high frequency (>70% of cases) of hypermethylation, which include APAF1 (Christoph et al., 2006a,b), MDR1 (Costa et al., 2007), and PTGS2 (Costa et al., 2007), those with intermediate frequency (70–20%), like RASSF1A (Battagli et al., 2003; Dulaimi et al., 2004; Gonzalgo et al., 2004; Hoque et al., 2004; Costa et al., 2007; Peters et al., 2007), CDH1 (Esteller et al., 2001; Morris et al., 2003; Hoque et al., 2004; Costa et al., 2007), DAPK1 (Morris et al., 2003; Christoph et al., 2006a), KRT19 (Paiva et al., 2011), TIMP3 (Esteller et al., 2001; Battagli et al., 2003; Dulaimi et al., 2004; Hoque et al., 2004; Costa et al., 2007), UCHL1 (Kagara et al., 2008; Seliger et al., 2009), PCDH17 (Costa et al., 2011), and TCF21 (Costa et al., 2011), and infrequently methylated (<20%) such as APC (Esteller et al., 2001; Dulaimi et al., 2004; Gonzalgo et al., 2004; Hoque et al., 2004; Costa et al., 2007), CASP-8 (Morris et al., 2003; Christoph et al., 2006b), CDH13 (Morris et al., 2003), GSTP1 (Esteller et al., 2001; Dulaimi et al., 2004; Hoque et al., 2004; Costa et al., 2007), JUP (Breault et al., 2005), MGMT (Esteller et al., 2001; Morris et al., 2003; Dulaimi et al., 2004; Hoque et al., 2004; Costa et al., 2007), p14ARF (Esteller et al., 2001; Dulaimi et al., 2004; Hoque et al., 2004; Costa et al., 2007), p16INK4a (Herman et al., 1995; Esteller et al., 2001; Morris et al., 2003; Dulaimi et al., 2004; Hoque et al., 2004; Costa et al., 2007), RARβ2 (Battagli et al., 2003; Morris et al., 2003; Dulaimi et al., 2004; Hoque et al., 2004; Costa et al., 2007), and VHL (Herman et al., 1994; Clifford et al., 1998; Dulaimi et al., 2004; Banks et al., 2006).
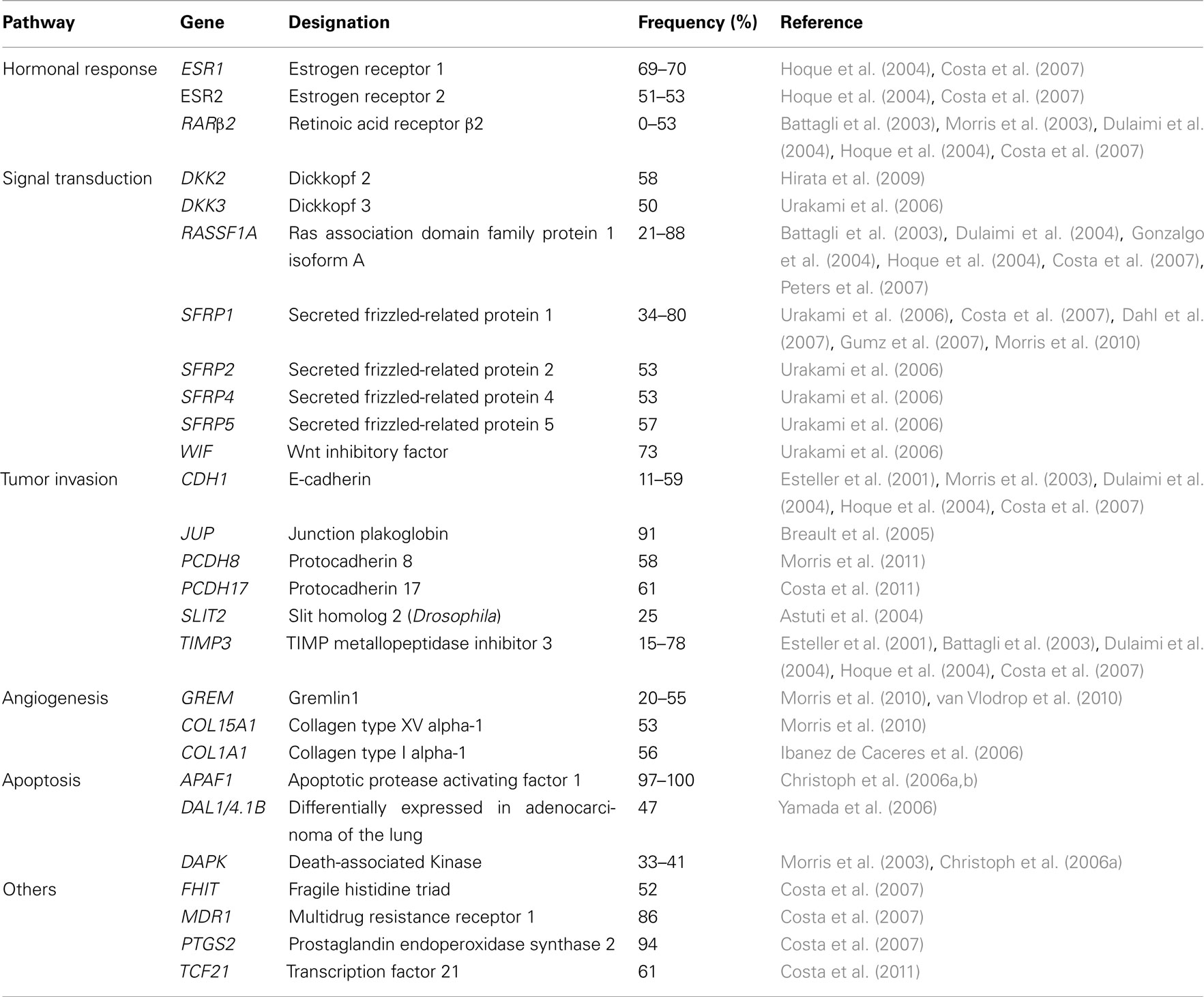
Table 1. Genes frequently methylated in renal cell tumors, according to their function/pathway.
Moreover, because the frequency of point mutations affecting genes involved in familial RCC is relatively low in the sporadic cases, except for VHL, the relevance of aberrant promoter methylation in RCC carcinogenesis has been highlighted (McRonald et al., 2009). Indeed, several genes, like RASSF1A, SFRP1, DAPK1, and SPINT2, were consistently found to be silenced by promoter hypermethylation but rarely mutated in sporadic RCC (Morris et al., 2011).
In this setting, functional epigenomic approaches emerged as efficient strategies to identify genes whose expression was silenced by promoter methylation in RCC, through the identification of genes re-expressed after treatment with demethylating drugs in RCC cell lines and further validated in primary tumor samples. This strategy allowed for the discovery of more than 10 candidate TSG in RCC: SPINT2 (Morris et al., 2005), IGFBP1, IGFBP3, COL1A1 (Ibanez de Caceres et al., 2006), UCHL1 (Kagara et al., 2008), CXCL16, KTN19 (Morris et al., 2008), BNC1, COL14A1, SFRP1, CST6, and PDLIM4 (Morris et al., 2010). Furthermore, gene promoters found to be methylated in cell lines but not in primary tumors (e.g., SST, PTGS1, ISG15, and THY1) might be important for cancer progression, whereas upregulated genes following treatment of cell lines with demethylating drugs but without promoter methylation (e.g., BAP, IGSF4, RRM2, PMAIP1, Claudin1, and ICAM), might be reactivated due to changes in promoter methylation status at upstream regulators (Morris et al., 2008). The identification of new candidate TSG may also be accomplished using high-throughput CpG methylation analysis platforms (McRonald et al., 2009) or by isolating methylated DNA by immunoprecipitation (methylated DNA immunoprecipitation, MeDIP) which is then used to perform whole-genome microarray analysis (Morris et al., 2011). These strategies enabled the identification of several new candidate TSG in RCC, including HTR1B, CALCA, IGFBP2, SOX17, COL1A2, BMP4, HS3ST2, FRZB, TAL1, MCM2, KCNK4, HOXC6, PITX2, SEPT5, IRF7, CCNA1, HOXA11, TERT, TMEFF2, EPHA3, PGF, MYOD1, MMP2, TNFRSF10C, PENK, EYA4, MYLK, IRAK3, ZNF215, SMARCB1, TWIST1, SCGB3A1, and IGFBP7 (McRonald et al., 2009), as well as ATP5G2, PCDH8, CORO6, KLHL35, QPCT, SCUBE3, ZSCAN18, CCDC8, and FBN2 (Morris et al., 2011). Deriving from data collected in several studies, a CpG island methylator phenotype (CIMP) for RCC has been suggested (Dulaimi et al., 2004; McRonald et al., 2009), but this topic warrants further investigation in larger series of cases.
Loss of promoter methylation has been seldom reported in RCC, although several gene promoters less frequently methylated in RCC tumor samples compared to normal kidney tissue have been found, including CARD15 (methylated in 18% of tumors), HLA-DRA (24%), SPARC (34%), IL8 (35%), SEPT9 (39%), HLA-DPB1 (45%), TNFSF10 (47%), VAMP8 (50%), PRKCDBP (55%), HLA-DPA1 (56%), HDAC1 (58%), BTK (58%), S100A2 (60%), MPO (61%), CRK (61%), CAPG (61%), NEU1 (69%), ELL (71%; McRonald et al., 2009), and CA 9 (Cho et al., 2001).
The accumulating data on gene promoter hypermethylation in RCC supports the development of clinically relevant biomarkers. Interestingly, different RCC subtypes seem to display different gene sets deregulated by promoter hypermethylation (Dulaimi et al., 2004), and a gene panel (CDH1, PTGS2, and RASSF2) intended for the discrimination among the most frequent RCT subtypes in tissue samples has been evaluated (Costa et al., 2007). However, the application of this approach in an early detection setting requires testing of clinical samples obtained by minimally invasive or (ideally) non-invasive means, of which urine and serum stand as the most likely candidates. This has been attempted using a three-gene panel (APC, RARβ2, RASSF1A) which detected RCC with high specificity and sensitivity (Hoque et al., 2004). Moreover, RASSF1A promoter methylation might also prove useful for tumor surveillance/monitoring of RCC cancer patients (Peters et al., 2007).
Promoter hypermethylation of some genes has been associated with clinical and pathological features of tumor aggressiveness and also to have prognostic relevance. Thus, aberrant promoter methylation of APAF1 and DAPK1 (Christoph et al., 2006a), as well as of GREM1 (van Vlodrop et al., 2010) was has been associated with aggressive forms of RCC. Moreover, APAF1, DAPK1 (Christoph et al., 2006a), JUP (Breault et al., 2005), PTEN (Kim et al., 2005), UCHL1 (Kagara et al., 2008), DAL1-4.1B/EPB41L3 (Yamada et al., 2006), BNC1, COL14A1, and SFRP1 (Morris et al., 2010) promoter methylation have been associated with poorer survival, and most of them (JUP, APAF1, DAPK1, PTEN, DAL1-4.1B, BNC1, and COL14A1) retained independent prognostic value in multivariate analysis (Breault et al., 2005; Christoph et al., 2006a; Morris et al., 2010). In addition, a genome-wide methylation profile of ccRCC using bacterial artificial chromosome array-based methylated CpG island amplification (BAMCA) and unsupervised hierarchical clustering analysis, found that the clusters obtained for ccRCC tumor tissues predicted recurrence and these were clinicopathological valid since tumors with vascular invasion, renal vein neoplastic thrombi and higher pathological TNM stage clustered together (Arai et al., 2009). Interestingly, DNA methylation status of non-cancerous tissues was similar to that of respective RCC samples, and they were also associated with patient outcome, suggesting an association of RCC prognosis with precancerous molecular alterations (Arai et al., 2009). Finally, it has been also reported that ABCG2/MDR1 gene promoter was methylated in RCC cell lines, and because this gene is associated with resistance of cancer cells to chemotherapeutic agents, this biomarker might become clinically useful for prediction of response to therapy (To et al., 2008).
Due to the association between promoter methylation and gene silencing, inhibition of DMNTs has been proposed as a therapeutic strategy to reactivate dormant genes. The nucleoside analog 5-aza-2′-deoxycytidine (DAC), a DNMTs inhibitor (DNMTi) has been tested in combination with conventional chemotherapeutic agents (e.g., vinblastine or paclitaxel; Takano et al., 2010; Shang et al., 2011) in RCC cell lines. A synergistic effect was observed in both settings, but clinical studies are required to conclusively demonstrate the therapeutic usefulness of DAC in RCC.
Chromatin Remodeling and Histone Onco-Modifications
Most studies on chromatin remodeling and histone onco-modifications in RCT have dealt with the relationship with hypoxia, as well as with the prognostic relevance of those alterations and the potential therapeutic use of histone deacetylase inhibitors.
It is widely acknowledged that VHL dysfunction is a pivotal event in ccRCC carcinogenesis (Baldewijns et al., 2008), leading to constitutive expression of genes that mediate cellular adaptation to hypoxia. This gene encodes for the VHL protein (pVHL), a substrate recognition component of an E3 ubiquitin ligase complex that targets hypoxia-inducible factors (HIF; e.g., HIF1α and HIF2α) for ubiquitination and proteasome degradation. HIFs are constitutively expressed and under normoxic conditions they are hydroxylated, allowing for their recognition by pVHL and subsequent targeting for degradation. Lack of pVHL leads to increased levels of HIFs and increased expression of genes involved in cellular response to hypoxia, promoting angiogenesis, invasion and metastasis, evasion of cell death, and cellular metabolism (Rathmell and Chen, 2008; Linehan et al., 2010).
Recent evidence unraveled a strong link between hypoxic cellular response and epigenetic regulation, especially histone modifications, albeit in non-RCC cancer cell lines (Figure 1; Johnson et al., 2008). Indeed, hypoxia was reported to be associated with widespread repression of total RNA and mRNA synthesis and to induce global histone modifications typically associated with transcriptional repression (loss of H3K9ac, increase in H3K9me2, H3K27me2, H3K9me3, H3K27me3, H3K4me1), but also unpredictably with gene activation (increase in H3K14ac, H4R3me2 which may facilitate acetylation of histones associated with activation of transcription, H3K4me2, H3K4me3, and H3K7me2; Chen et al., 2006; Johnson et al., 2008). Gene-specific histone modifications included a decrease in H3K9ac and an increase in H3K9/27me2 at hypoxia-repressed genes, an increase in H3K9ac and a decrease in H3K9/27me2 at hypoxia-induced genes, and an increase in H3K4me3 and a decrease in H3K27me3 in all hypoxia-responsive genes (activated and repressed), suggesting that repressed chromatin H3K4me3 enriched and H3K27me3 deprived might be rapidly activated when hypoxia is reversed (Chen et al., 2006; Johnson et al., 2008).
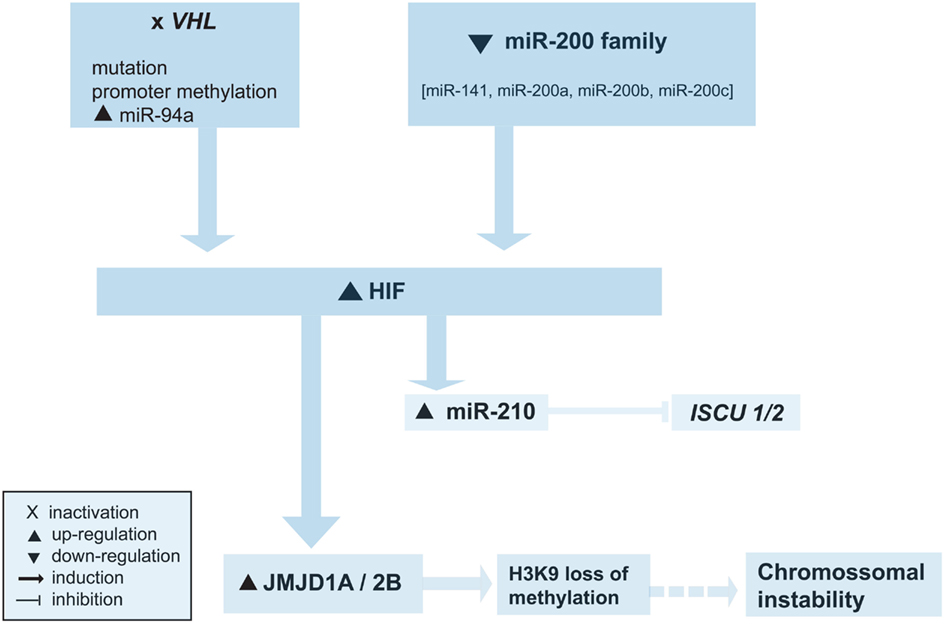
Figure 1. Overview of the epigenetic alterations involved in abnormal hypoxic cellular response. See text for details.
One of the mechanisms that might be involved in altered epigenetic landscape due to hypoxic effects is the regulation of Jumonji domain containing histone demethylases by HIF1α, a critical mediator of hypoxic response (Beyer et al., 2008; Pollard et al., 2008; Figure 1). Genes encoding Jumonji domain containing 2-oxoglutarate-dependent oxygenases, characterized by a catalytic Jumonji C (JmjC) domain, were defined as a class of HIF-responsive hypoxia-inducible genes (Pollard et al., 2008). Histone demethylases Jumonji domain containing 1A (JMJD1A), Jumonji domain containing 2B (JMJD2B), and Jumonji domain containing 2C (JMJD2C) showed consistent patterns of regulation by hypoxia, especially JMJD1A and JMJD2B, which were found to be induced by HIF1α but not by HIF2α (Pollard et al., 2008). Since HIF upregulation is a feature of ccRCC, the regulation of histone methylation status, and thus of genetic expression, by JmjC histone demethylases might be another mechanism leading to epigenetic changes in RCC carcinogenesis. Indeed, both JMJD1A and JMJD2B were found to be elevated in a RCC cell line with VHL loss of function (Beyer et al., 2008), and the expression of JMJD1A was reported to be higher in RCC cancer tissue than in adjacent normal renal tissue, mainly in cancer cells surrounding blood vessels, suggesting that JMJD1A is involved in tumor angiogenesis (Guo et al., 2011).
In addition, genes encoding for histone-modifying enzymes have been also reported to be mutated in ccRCC (van Haaften et al., 2009; Dalgliesh et al., 2010; Table 2). Specifically, inactivating mutations were described for SETD2 (H3K36 methyltransferase), JARID1C/KDM5C (H3K4 demethylase), UTX/KMD6A (H3K27 demethylase), and MLL2 (an H3K4 methyltransferase; van Haaften et al., 2009; Dalgliesh et al., 2010). More recently, mutations in the SWI/SNF chromatin remodeling complex gene PBRM1 has been identified in 41% of ccRCCs (Varela et al., 2011). Interestingly, the induction of the H3K9 specific demethylases JMJD1A and JMJD2B, which target H3K9me1/me2 and H3K9me2/me3 respectively, might contribute to an increased mutation rate in tumors with upregulation of HIF, as H3K9 loss of methylation promotes chromosomal instability (Beyer et al., 2008).

Table 2. Histone onco-modifications and deregulated modifiers in renal cell tumors.
The diagnostic potential of histone modifications and/or histone modifiers in RCC has not been explored thus far. They have been, however, proposed as molecular biomarkers of prognosis, easily translated to routine pathology because they may be assessed by immunohistochemistry in formalin-fixed, paraffin-embedded tissue sections. Several histone marks have been associated with poor prognosis in RCC, including low H3K4me2, H3K18ac, and H3K9me2 (Seligson et al., 2009). H3K4me1–3 levels were also found to be inversely correlated with Fuhrman grade, pT stage, lymph node involvement and distant metastases, and an H3K4me score (combining staining levels of H3K4me) was an independent factor for RCC progression-free survival (Ellinger et al., 2010). Similar observations were made for global H3Ac and H4Ac levels, as well as for H3K9Ac levels in RCCs treated with partial nephrectomy (Minardi et al., 2009), whereas H3K18Ac levels were an independent predictor of RCC progression after surgery (Mosashvilli et al., 2010). Concerning histone modifiers, EZH2 was found to be upregulated in ccRCC, but this alteration was unexpectedly associated with a favorable prognosis (Hinz et al., 2009).
Histone onco-modifications might also carry therapeutic implications, as patients with marks of poor prognosis including low levels of H3K4me2, H3K18ac, and H3K9me2 could benefit from innovative treatments with histone deacetylase inhibitors (HDACi) Histone onco-modifications might also carry therapeutic implications, as patients with marks of poor prognosis including low levels of H3K4me2, H3K18ac, and H3K9me2 could benefit from innovative treatments with histone deacetylase inhibitors (HDACi; Seligson et al., 2009). Some preclinical studies on RCC cell lines using the HDACi Vorinostat demonstrated an increase in the anticancer activity of temsirolimus, a mammalian target of rapamycin (mTOR) inhibitor, through survivin downregulation, leading to increased apoptosis, and enhanced inhibition of angiogenesis (Mahalingam et al., 2010). Moreover, the proteasome mediated degradation of Aurora A and Aurora B kinases through inhibition of HDAC3 and HDAC6 by the HDACi LBH589, was found to induce G2-M arrest and apoptosis in RCC, highlighting its potential therapeutic use (Cha et al., 2009). Valproic acid is also an HDACi which causes growth arrest, preventing tumor cell attachment to endothelium and matrix proteins, and blocking integrin-dependent signaling (Jones et al., 2009a). The combination of valproic acid with interferon-alpha enhanced the effects of the former (Jones et al., 2009a,b) and similar synergism was found when combined with AEE788, a multiple receptor tyrosine kinase inhibitor (Juengel et al., 2010). The combination of HDACi and retinoids might also provide an alternative therapeutic strategy because RARβ2 expression is reduced in RCC, in part owing to gene-specific histone hypoacetylation, and its re-expression is associated with anti-neoplastic effects through the abrogation of retinoid-resistance (Touma et al., 2005; Wang et al., 2005). Interestingly, this synergistic activity with retinoids has been demonstrated for both HDACi MS-275 (Wang et al., 2005) and trichostatin A (TSA; Touma et al., 2005) in RCC cell lines.
MicroRNAs
Deregulation of miRNA expression seems to be pivotal for RCC development and progression (Valera et al., 2011). Indeed, several miRNA have been found to be deregulated in RCTs, although most studies have focused mainly on ccRCC. Upregulation of miR-16 (Jung et al., 2009; Zhou et al., 2010), miR-18a (Neal et al., 2010), miR-20a (Neal et al., 2010), miR-21 (Liu et al., 2010a; Neal et al., 2010; Zhou et al., 2010), miR-34a (Liu et al., 2010a; Zhou et al., 2010), miR-34b (Liu et al., 2010a; Zhou et al., 2010), miR-92a (Valera et al., 2011), miR-155 (Jung et al., 2009; Liu et al., 2010a; Neal et al., 2010; Zhou et al., 2010), miR-185 (Liu et al., 2010a; Zhou et al., 2010), miR-210 (Jung et al., 2009; Liu et al., 2010a; Neal et al., 2010; Zhou et al., 2010), miR-224 (Jung et al., 2009; Liu et al., 2010a) and let-7 i (Neal et al., 2010; Zhou et al., 2010), and downregulation of miR-125b (Liu et al., 2010a; Zhou et al., 2010), miR-141 (Nakada et al., 2008; Jung et al., 2009; Liu et al., 2010a), miR-133b (Liu et al., 2010a; Zhou et al., 2010), miR-200b (Jung et al., 2009; Liu et al., 2010a), miR-200c (Nakada et al., 2008; Jung et al., 2009; Liu et al., 2010a), miR-429 (Jung et al., 2009; Liu et al., 2010a; Zhou et al., 2010), miR-506 (Zhou et al., 2010; Li et al., 2011), miR-508-3p (Zhou et al., 2010; Li et al., 2011), miR-509-5p (Zhou et al., 2010; Li et al., 2011), miR-509-3-5p (Zhou et al., 2010; Li et al., 2011), miR-510 (Liu et al., 2010a; Zhou et al., 2010; Li et al., 2011), and miR-514 (Jung et al., 2009; Liu et al., 2010a; Zhou et al., 2010; Li et al., 2011) are the most consistently reported alterations.
Renal cell tumors display distinct cytogenetic alterations and these might cause miRNA deregulation as matching patterns between deregulated miRNAs and chromosomal aberrations have been reported in ccRCC (Chow et al., 2010a,b). On the other hand, miRNA deregulation might serve as an alternative mechanism for gene expression alterations due to chromosomal aberrations. This is well illustrated by the miR-204/211 family. Whereas in pRCC gain of 3q is a common finding, leading to upregulation of several genes including C3orf58, CCDC50, DTX3L, PLD1, TRIM59, ECT2, RAP2B, and SERP1, targeted by miR-204/211 (Liu et al., 2010a), in ccRCC miR-204/211 downregulation might be the mechanism causing upregulation of the aforementioned genes, since 3q gain is rare (Liu et al., 2010a). Moreover, it has been postulated that most miRNAs are tandemly clustered (Lee et al., 2002; Seitz et al., 2003), and, accordingly, a co-expression pattern for miRNA families miR-8 (or miR-200), miR-199, and miR-506 has been found in ccRCC (Li et al., 2011). Another interesting example is provided by the miR-506 family members miR-506, miR-508-3p, miR-509-5p, miR-509-3-5p, miR-510, and miR-514, which are downregulated in ccRCC (Zhou et al., 2010). The corresponding genes are tandemly clustered in the fragile site Xq27.3 and their predicted targets are upregulated, including some genes involved in key signaling pathways like LDHA, HK1, VEGFB, and PSMA1 (Zhou et al., 2010).
Bioinformatics, anti-correlation analysis of miRNA/mRNA levels and functional studies in paired tumorous and normal tissues are also revealing interesting data on cell function alterations due to deregulated miRNA in RCTs. Those have showed that deregulated microRNAs target genes are commonly involved in metabolic (71 target genes of 13 deregulated microRNAs), focal adhesion, cell adhesion molecules and ECM receptor interactions (30 target genes of 25 deregulated microRNAs), cell cycle regulation (24 target genes of 22 deregulated microRNAs), and apoptosis (14 target genes of 11 deregulated microRNAs) pathways in ccRCC (Zhou et al., 2010; Table 3).
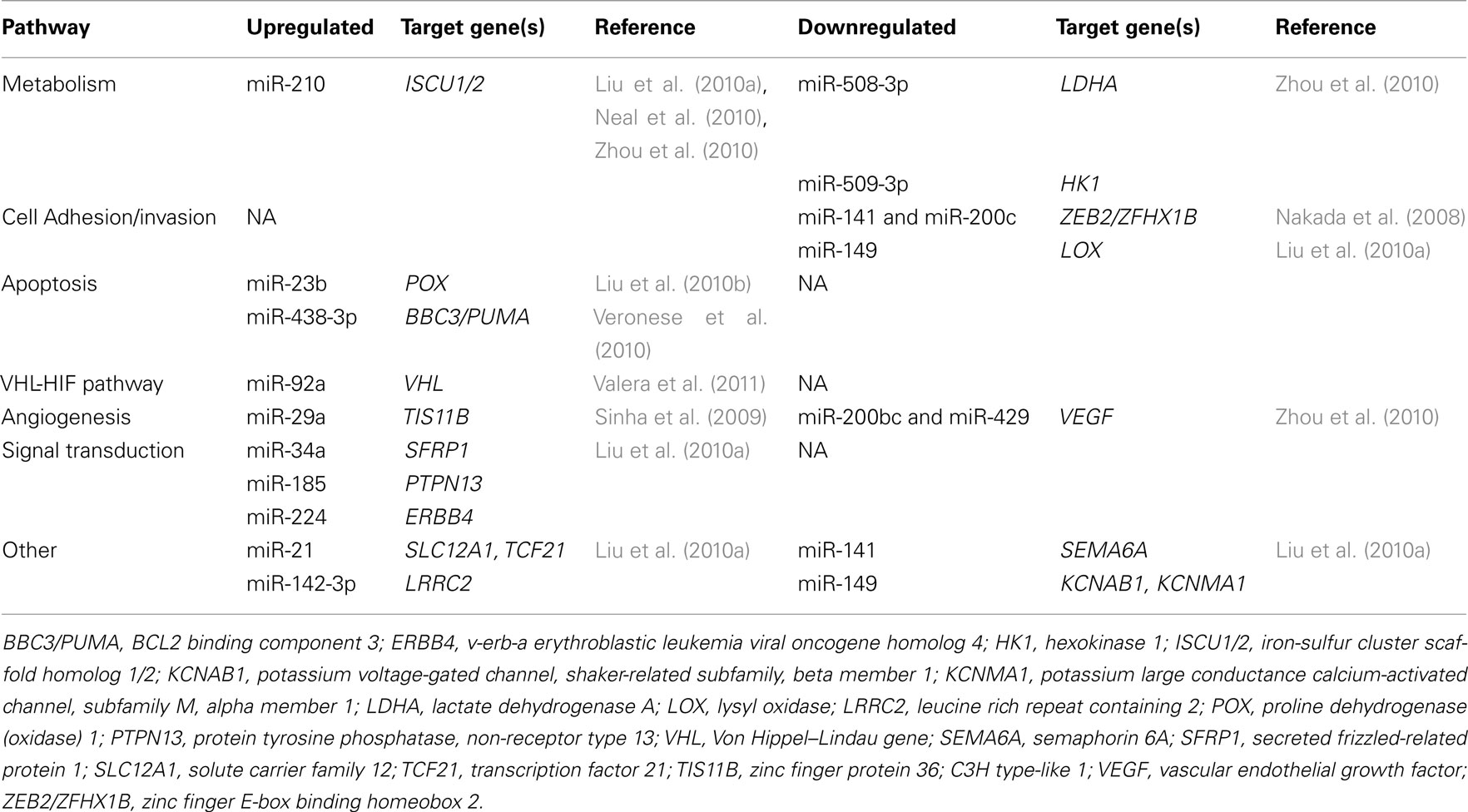
Table 3. MicroRNA deregulation in renal cell tumors, according to their function/pathway and target genes’.
Furthermore, the miR-200 family is also known to be involved in epithelial-to-mesenchymal transition, and its downregulation might contribute to tumor invasion and metastasis (Liu et al., 2010a; Zhou et al., 2010; Li et al., 2011).
Downregulation of TSG in RCC has been also correlated with upregulation of oncogenic miRNAs. Indeed, elevated miR-185 was correlated with downregulation of PTEN (with subsequent activation of PI3K-AKT-mTOR signaling pathway), PTPN13 (a Fas-associated tyrosine phosphatase that can inhibit PI3K/AKT signaling, induce apoptosis and suppress the cell survival effects of IGF-I), and KCNJ16 (a cell growth-related membrane protein). In addition, upregulation of miR-34a was correlated with downregulation of SFRP1 (a Wnt signaling pathway regulator) whereas miR-224 upregulation was associated with downregulation of ERBB4 (an EGFR family member and putative TSG; Liu et al., 2010a).
Hypoxic regulation of miRNAs is also emerging as an important mechanism implicated in RCC tumorigenesis (Figure 1). The deregulation of miRNAs in cell lines lacking VHL was been shown to be either mediated largely via HIF induction (miR-210 and miR-155) or by HIF independent VHL actions (miR-31, miR-21, miR-18a, miR-17, let-7i, miR-20a; Neal et al., 2010). Furthermore, the expression levels of HIF2α and of its downstream targets (VEGFα, TGFβ) seems to be regulated by several members of the miR-200 family (miR-141, miR-200a*, miR-200b, miR-200c), and downregulation of miR-200 (or miR-8) family and VHL loss activate the HIF pathway (Zhou et al., 2010). On the other hand, HIF1α can induce miR-210 in many solid tumors, and miR-210 has been consistently found to be overexpressed in ccRCC (Jung et al., 2009; Liu et al., 2010a; Neal et al., 2010; Zhou et al., 2010). Upregulation of miR-210 was associated with a reduced expression of its target gene ISCU1/2, which encodes assembly proteins involved in the biogenesis of [4Fe-4S] and [2Fe-2S] iron-sulfur clusters (Neal et al., 2010). These are implicated in electron transport and mitochondrial oxidation-reduction reactions, and, thus, downregulation of ISCU1/2 might contribute to the repression of mitochondrial proteins and to the anaerobic metabolism in ccRCC (Neal et al., 2010). Interestingly, hypoxia induced microRNAs have been also correlated with SLC12A1 and TCF21 downregulation (encoding cell adhesion proteins) in ccRCC, whose mRNAs were identified as direct targets of hypoxia induced miR-21 (Liu et al., 2010a).
Only a few studies addressed the potential use of miRNAs as RCC biomarkers for detection. In this regard, differential miRNA expression patterns between neoplastic and non-neoplastic renal tissues, as well as among different renal tumor subtypes have been described. The discrimination between ccRCC and normal kidney tissue might be accomplished by a panel of nine miRs (miR-21, miR-34a, miR-142-3p, miR-155, miR-185, miR-200c, miR-210, miR-224, and miR-592; Juan et al., 2010), a combination of miR-141 and miR-155 (Jung et al., 2009) or through the differential expression of miR-92a, miR-210, and miR-200c (Valera et al., 2011).
Concerning distinctive miRNA signatures for each of the main RCT subtypes, unsupervised hierarchical cluster analysis of miRNA microarray data showed that tumors derived from the proximal nephron (ccRCC and pRCC type I) and tumors derived from the distal nephron (oncocytomas and chRCC) can be distinguished by their miRNA profile (Valera et al., 2011), extending previous observations for ccRCC and chRCC (Nakada et al., 2008). These differential expression patterns of microRNAs might be also used to subclassify RCT (Petillo et al., 2009; Fridman et al., 2010; Youssef et al., 2011). In ccRCC 23 miRNA are differentially expressed (let-7e, let-7f, let-7g, miR10b, miR-124, miR-126, miR-138, miR-140-5p, miR-142-5p, miR-144, miR-184, miR-200c, miR-203, miR-206, miR-210, miR-218, miR-27a, miR-27b, miR-335, miR-373, miR-378, miR-92a, miR-98; Valera et al., 2011). However, some miRNAs are characteristic of sporadic ccRCC (let-7c, let-7d, miR-1, miR-100, miR-10a, miR-148b, miR-191, miR-199a-3p, miR-19a, miR-215, miR-29b, miR-30c, miR-363, miR-9) and others of hereditary (Von Hippel–Lindau syndrome-related) RCC (let-7a, miR-125a-5p, miR-125b, miR-143, miR-146b-5p, miR-15b, miR-17, miR-193a-5p, miR-193b, miR-196a, miR-20b, miR-214, miR-23b, miR-32, miR-372; Valera et al., 2011). Furthermore, ccRCC and pRCC differentially express 27 miRNAs, with miR-203 and miR-424 being more expressed in ccRCC than in pRCC, and miR-31 and mir-504 being more expressed in pRCC than in ccRCC (Petillo et al., 2009). Remarkably, a similar miRNA expression pattern was reported for chRCC and oncocytoma. These tumors share 60 miRNAs, with miR-143, miR-19a, miR-21, miR-29a, miR-181a, and miR-378 unique to chRCC and mir-146a being preferentially expressed in oncocytoma (Valera et al., 2011). Moreover, chRCC displayed higher levels of miR-203, mir-200b, miR-197, and miR-320 than oncocytoma, whereas miR-186 is more highly expressed in oncocytoma than in chRCC (Petillo et al., 2009). Finally, a recent study which investigated miRNAs expression levels in sera of RCC patients and healthy controls, identified miR-1233 as promising biomarker for RCC detection and monitoring (Wulfken et al., 2011).
Altered levels of miRNA might also provide prognostic information. Whereas miR-155 and miR-21 expression in ccRCC tumors has been found to correlate with tumor size (Neal et al., 2010), higher miR-210 levels were found in tumors displaying higher Fuhrman grade (Valera et al., 2011). In addition, in ccRCC, overexpression of miR-32 as well as of miR-210, miR-21, let-7i, and miR-18a correlated with poor survival (Petillo et al., 2009; Neal et al., 2010) and lower miR-106b levels were associated with metastatic disease and poorer relapse-free survival (Slaby et al., 2010). High miR-210 expression was also found in tumors with lymph node metastasis (Valera et al., 2011), suggesting unique miRNA signatures in RCC metastasis, distinct from those of primary tumors (White et al., 2011).
Conclusion
The ubiquity of epigenetic alterations in RCT supports their fundamental role in renal carcinogenesis. Those alterations not only provide further insight into the complex mechanisms underlying the genesis and progression of RCT, but they also grant the opportunity for the development of innovative biomarkers which might aid in disease detection, diagnosis, assessment of prognosis, and prediction of response to therapy (Table 4). Finally, owing to the reversible and plastic nature of epigenetic alterations, these constitute an attractive target for novel therapeutic approaches that might tackle one of the most chemoresistant types of human cancer.
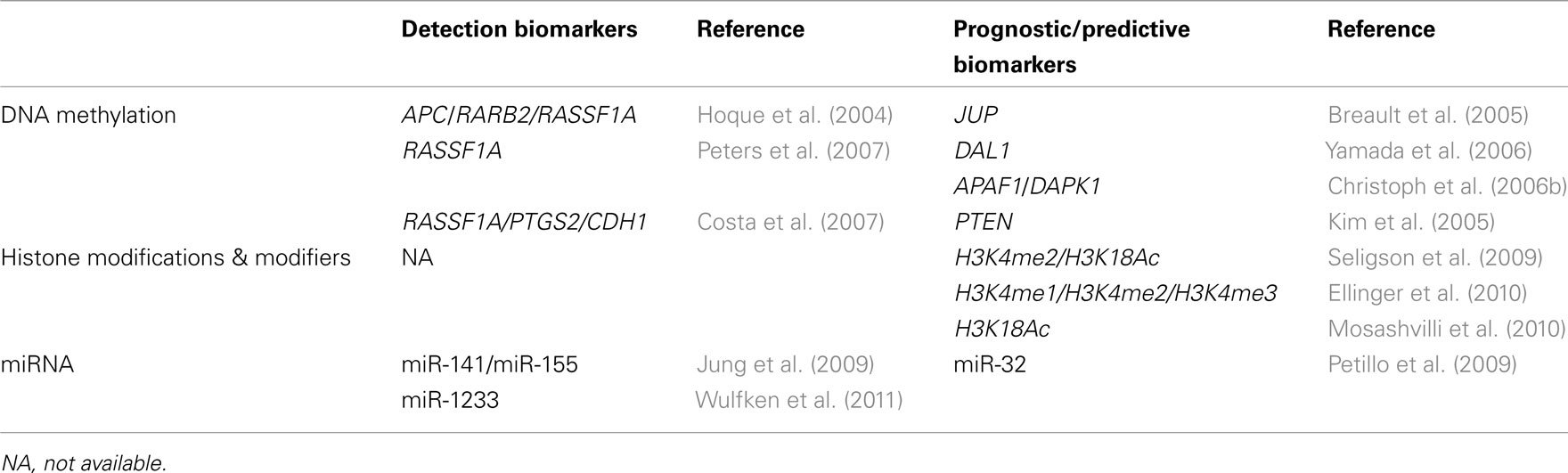
Table 4. Diagnostic and prognostic information in renal cell tumors provided by epigenetic biomarkers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants to from the European Community’s Seventh Framework Program – Grant number FP7-HEALTH-F5-2009-241783 (Carmen Jerónimo), Liga Portuguesa Contra o Cancro – Núcleo Regional do Norte (Carmen Jerónimo, Rui Henrique), Research Centre of the Portuguese Oncology Institute Porto – CI-IPOP-4-2008 (Carmen Jerónimo), and Associação Portuguesa de Urologia (Rui Henrique, Ana Sílvia Luís, Carmen Jerónimo).
References
Arai, E., Ushijima, S., Fujimoto, H., Hosoda, F., Shibata, T., Kondo, T., Yokoi, S., Imoto, I., Inazawa, J., Hirohashi, S., and Kanai, Y. (2009). Genome-wide DNA methylation profiles in both precancerous conditions and clear cell renal cell carcinomas are correlated with malignant potential and patient outcome. Carcinogenesis 30, 214–221.
Astuti, D., Da Silva, N. F., Dallol, A., Gentle, D., Martinsson, T., Kogner, P., Grundy, R., Kishida, T., Yao, M., Latif, F., and Maher, E. R. (2004). SLIT2 promoter methylation analysis in neuroblastoma, Wilms’ tumour and renal cell carcinoma. Br. J. Cancer 90, 515–521.
Baldewijns, M. M., Van Vlodrop, I. J., Schouten, L. J., Soetekouw, P. M., De Bruine, A. P., and Van Engeland, M. (2008). Genetics and epigenetics of renal cell cancer. Biochim. Biophys. Acta 1785, 133–155.
Ballestar, E., and Esteller, M. (2005). Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem. Cell Biol. 83, 374–384.
Banks, R. E., Tirukonda, P., Taylor, C., Hornigold, N., Astuti, D., Cohen, D., Maher, E. R., Stanley, A. J., Harnden, P., Joyce, A., Knowles, M., and Selby, P. J. (2006). Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 66, 2000–2011.
Battagli, C., Uzzo, R. G., Dulaimi, E., Ibanez De Caceres, I., Krassenstein, R., Al-Saleem, T., Greenberg, R. E., and Cairns, P. (2003). Promoter hypermethylation of tumor suppressor genes in urine from kidney cancer patients. Cancer Res. 63, 8695–8699.
Berdasco, M., and Esteller, M. (2011). DNA methylation in stem cell renewal and multipotency. Stem Cell Res. Ther. 2, 42.
Beyer, S., Kristensen, M. M., Jensen, K. S., Johansen, J. V., and Staller, P. (2008). The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 283, 36542–36552.
Bloushtain-Qimron, N., Yao, J., Snyder, E. L., Shipitsin, M., Campbell, L. L., Mani, S. A., Hu, M., Chen, H., Ustyansky, V., Antosiewicz, J. E., Argani, P., Halushka, M. K., Thomson, J. A., Pharoah, P., Porgador, A., Sukumar, S., Parsons, R., Richardson, A. L., Stampfer, M. R., Gelman, R. S., Nikolskaya, T., Nikolsky, Y., and Polyak, K. (2008). Cell type-specific DNA methylation patterns in the human breast. Proc. Natl. Acad. Sci. U.S.A. 105, 14076–14081.
Breault, J. E., Shiina, H., Igawa, M., Ribeiro-Filho, L. A., Deguchi, M., Enokida, H., Urakami, S., Terashima, M., Nakagawa, M., Kane, C. J., Carroll, P. R., and Dahiya, R. (2005). Methylation of the gamma-catenin gene is associated with poor prognosis of renal cell carcinoma. Clin. Cancer Res. 11, 557–564.
Cha, T. L., Chuang, M. J., Wu, S. T., Sun, G. H., Chang, S. Y., Yu, D. S., Huang, S. M., Huan, S. K., Cheng, T. C., Chen, T. T., Fan, P. L., and Hsiao, P. W. (2009). Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin. Cancer Res. 15, 840–850.
Chan, J. A., Krichevsky, A. M., and Kosik, K. S. (2005). MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65, 6029–6033.
Chen, H., Yan, Y., Davidson, T. L., Shinkai, Y., and Costa, M. (2006). Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 66, 9009–9016.
Cho, M., Uemura, H., Kim, S. C., Kawada, Y., Yoshida, K., Hirao, Y., Konishi, N., Saga, S., and Yoshikawa, K. (2001). Hypomethylation of the MN/CA9 promoter and upregulated MN/CA9 expression in human renal cell carcinoma. Br. J. Cancer 85, 563–567.
Chow, T. F., Mankaruos, M., Scorilas, A., Youssef, Y., Girgis, A., Mossad, S., Metias, S., Rofael, Y., Honey, R. J., Stewart, R., Pace, K. T., and Yousef, G. M. (2010a). The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J. Urol. 183, 743–751.
Chow, T. F., Youssef, Y. M., Lianidou, E., Romaschin, A. D., Honey, R. J., Stewart, R., Pace, K. T., and Yousef, G. M. (2010b). Differential expression profiling of microRNAs and their potential involvement in renal cell carcinoma pathogenesis. Clin. Biochem. 43, 150–158.
Christoph, F., Kempkensteffen, C., Weikert, S., Kollermann, J., Krause, H., Miller, K., Schostak, M., and Schrader, M. (2006a). Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br. J. Cancer 95, 1701–1707.
Christoph, F., Weikert, S., Kempkensteffen, C., Krause, H., Schostak, M., Kollermann, J., Miller, K., and Schrader, M. (2006b). Promoter hypermethylation profile of kidney cancer with new proapoptotic p53 target genes and clinical implications. Clin. Cancer Res. 12, 5040–5046.
Clifford, S. C., Prowse, A. H., Affara, N. A., Buys, C. H., and Maher, E. R. (1998). Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosomes Cancer 22, 200–209.
Costa, V. L., Henrique, R., Danielsen, S. A., Eknaes, M., Patricio, P., Morais, A., Oliveira, J., Lothe, R. A., Teixeira, M. R., Lind, G. E., and Jeronimo, C. (2011). TCF21 and PCDH17 methylation: an innovative panel of biomarkers for a simultaneous detection of urological cancers. Epigenetics 6, 1120–1130.
Costa, V. L., Henrique, R., Ribeiro, F. R., Pinto, M., Oliveira, J., Lobo, F., Teixeira, M. R., and Jeronimo, C. (2007). Quantitative promoter methylation analysis of multiple cancer-related genes in renal cell tumors. BMC Cancer 7, 133. doi:10.1186/1471-2407-7-133
Dahl, E., Wiesmann, F., Woenckhaus, M., Stoehr, R., Wild, P. J., Veeck, J., Knuchel, R., Klopocki, E., Sauter, G., Simon, R., Wieland, W. F., Walter, B., Denzinger, S., Hartmann, A., and Hammerschmied, C. G. (2007). Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene 26, 5680–5691.
Dalgliesh, G. L., Furge, K., Greenman, C., Chen, L., Bignell, G., Butler, A., Davies, H., Edkins, S., Hardy, C., Latimer, C., Teague, J., Andrews, J., Barthorpe, S., Beare, D., Buck, G., Campbell, P. J., Forbes, S., Jia, M., Jones, D., Knott, H., Kok, C. Y., Lau, K. W., Leroy, C., Lin, M. L., Mcbride, D. J., Maddison, M., Maguire, S., Mclay, K., Menzies, A., Mironenko, T., Mulderrig, L., Mudie, L., O’Meara, S., Pleasance, E., Rajasingham, A., Shepherd, R., Smith, R., Stebbings, L., Stephens, P., Tang, G., Tarpey, P. S., Turrell, K., Dykema, K. J., Khoo, S. K., Petillo, D., Wondergem, B., Anema, J., Kahnoski, R. J., Teh, B. T., Stratton, M. R., and Futreal, P. A. (2010). Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463, 360–363.
Doi, A., Park, I. H., Wen, B., Murakami, P., Aryee, M. J., Irizarry, R., Herb, B., Ladd-Acosta, C., Rho, J., Loewer, S., Miller, J., Schlaeger, T., Daley, G. Q., and Feinberg, A. P. (2009). Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 41, 1350–1353.
Dudziec, E., Miah, S., Choudhry, H. M., Owen, H. C., Blizard, S., Glover, M., Hamdy, F. C., and Catto, J. W. (2011). Hypermethylation of CpG islands and shores around specific microRNAs and mirtrons is associated with the phenotype and presence of bladder cancer. Clin. Cancer Res. 17, 1287–1296.
Dulaimi, E., Ibanez De Caceres, I., Uzzo, R. G., Al-Saleem, T., Greenberg, R. E., Polascik, T. J., Babb, J. S., Grizzle, W. E., and Cairns, P. (2004). Promoter hypermethylation profile of kidney cancer. Clin. Cancer Res. 10, 3972–3979.
Ehrlich, M. (2005). DNA methylation and cancer-associated genetic instability. Adv. Exp. Med. Biol. 570, 363–392.
Ellinger, J., Kahl, P., Mertens, C., Rogenhofer, S., Hauser, S., Hartmann, W., Bastian, P. J., Buttner, R., Muller, S. C., and Von Ruecker, A. (2010). Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int. J. Cancer 127, 2360–2366.
Esteller, M., Corn, P. G., Baylin, S. B., and Herman, J. G. (2001). A gene hypermethylation profile of human cancer. Cancer Res. 61, 3225–3229.
Feinberg, A. P., Ohlsson, R., and Henikoff, S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33.
Feinberg, A. P., and Tycko, B. (2004). The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153.
Ferlay, J., Shin, H. R., Bray, F., Forman, D., Mathers, C., and Parkin, D. M. (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917.
Fraga, M. F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., Bonaldi, T., Haydon, C., Ropero, S., Petrie, K., Iyer, N. G., Perez-Rosado, A., Calvo, E., Lopez, J. A., Cano, A., Calasanz, M. J., Colomer, D., Piris, M. A., Ahn, N., Imhof, A., Caldas, C., Jenuwein, T., and Esteller, M. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400.
Fridman, E., Dotan, Z., Barshack, I., David, M. B., Dov, A., Tabak, S., Zion, O., Benjamin, S., Benjamin, H., Kuker, H., Avivi, C., Rosenblatt, K., Polak-Charcon, S., Ramon, J., Rosenfeld, N., and Spector, Y. (2010). Accurate molecular classification of renal tumors using microRNA expression. J. Mol. Diagn. 12, 687–696.
Frigola, J., Song, J., Stirzaker, C., Hinshelwood, R. A., Peinado, M. A., and Clark, S. J. (2006). Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 38, 540–549.
Fullgrabe, J., Kavanagh, E., and Joseph, B. (2011). Histone onco-modifications. Oncogene 30, 3391–3403.
Garzon, R., Calin, G. A., and Croce, C. M. (2009). MicroRNAs in cancer. Annu. Rev. Med. 60, 167–179.
Goldberg, A. D., Allis, C. D., and Bernstein, E. (2007). Epigenetics: a landscape takes shape. Cell 128, 635–638.
Gonzalgo, M. L., Yegnasubramanian, S., Yan, G., Rogers, C. G., Nicol, T. L., Nelson, W. G., and Pavlovich, C. P. (2004). Molecular profiling and classification of sporadic renal cell carcinoma by quantitative methylation analysis. Clin. Cancer Res. 10, 7276–7283.
Guil, S., and Esteller, M. (2009). DNA methylomes, histone codes and miRNAs: tying it all together. Int. J. Biochem. Cell Biol. 41, 87–95.
Gumz, M. L., Zou, H., Kreinest, P. A., Childs, A. C., Belmonte, L. S., Legrand, S. N., Wu, K. J., Luxon, B. A., Sinha, M., Parker, A. S., Sun, L. Z., Ahlquist, D. A., Wood, C. G., and Copland, J. A. (2007). Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin. Cancer Res. 13, 4740–4749.
Guo, X., Shi, M., Sun, L., Wang, Y., Gui, Y., Cai, Z., and Duan, X. (2011). The expression of histone demethylase JMJD1A in renal cell carcinoma. Neoplasma 58, 153–157.
Halkidou, K., Gaughan, L., Cook, S., Leung, H. Y., Neal, D. E., and Robson, C. N. (2004). Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59, 177–189.
Henikoff, S. (2008). Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 9, 15–26.
Herman, J. G., Latif, F., Weng, Y., Lerman, M. I., Zbar, B., Liu, S., Samid, D., Duan, D. S., Gnarra, J. R., Linehan, W. M., and Baylin, S. B. (1994). Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. U.S.A. 91, 9700–9704.
Herman, J. G., Merlo, A., Mao, L., Lapidus, R. G., Issa, J. P., Davidson, N. E., Sidransky, D., and Baylin, S. B. (1995). Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 55, 4525–4530.
Hinz, S., Weikert, S., Magheli, A., Hoffmann, M., Engers, R., Miller, K., and Kempkensteffen, C. (2009). Expression profile of the polycomb group protein enhancer of Zeste homologue 2 and its prognostic relevance in renal cell carcinoma. J. Urol. 182, 2920–2925.
Hirata, H., Hinoda, Y., Nakajima, K., Kawamoto, K., Kikuno, N., Kawakami, K., Yamamura, S., Ueno, K., Majid, S., Saini, S., Ishii, N., and Dahiya, R. (2009). Wnt antagonist gene DKK2 is epigenetically silenced and inhibits renal cancer progression through apoptotic and cell cycle pathways. Clin. Cancer Res. 15, 5678–5687.
Hoque, M. O., Begum, S., Topaloglu, O., Jeronimo, C., Mambo, E., Westra, W. H., Califano, J. A., and Sidransky, D. (2004). Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer Res. 64, 5511–5517.
Ibanez de Caceres, I., Dulaimi, E., Hoffman, A. M., Al-Saleem, T., Uzzo, R. G., and Cairns, P. (2006). Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 66, 5021–5028.
Irizarry, R. A., Ladd-Acosta, C., Wen, B., Wu, Z., Montano, C., Onyango, P., Cui, H., Gabo, K., Rongione, M., Webster, M., Ji, H., Potash, J. B., Sabunciyan, S., and Feinberg, A. P. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 41, 178–186.
Johnson, A. B., Denko, N., and Barton, M. C. (2008). Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. 640, 174–179.
Jones, J., Juengel, E., Mickuckyte, A., Hudak, L., Wedel, S., Jonas, D., and Blaheta, R. A. (2009a). The histone deacetylase inhibitor valproic acid alters growth properties of renal cell carcinoma in vitro and in vivo. J. Cell. Mol. Med. 13, 2376–2385.
Jones, J., Juengel, E., Mickuckyte, A., Hudak, L., Wedel, S., Jonas, D., Hintereder, G., and Blaheta, R. A. (2009b). Valproic acid blocks adhesion of renal cell carcinoma cells to endothelium and extracellular matrix. J. Cell. Mol. Med. 13, 2342–2352.
Juan, D., Alexe, G., Antes, T., Liu, H., Madabhushi, A., Delisi, C., Ganesan, S., Bhanot, G., and Liou, L. S. (2010). Identification of a microRNA panel for clear-cell kidney cancer. Urology 75, 835–841.
Juengel, E., Engler, J., Mickuckyte, A., Jones, J., Hudak, L., Jonas, D., and Blaheta, R. A. (2010). Effects of combined valproic acid and the epidermal growth factor/vascular endothelial growth factor receptor tyrosine kinase inhibitor AEE788 on renal cell carcinoma cell lines in vitro. BJU Int. 105, 549–557.
Jung, M., Mollenkopf, H. J., Grimm, C., Wagner, I., Albrecht, M., Waller, T., Pilarsky, C., Johannsen, M., Stephan, C., Lehrach, H., Nietfeld, W., Rudel, T., Jung, K., and Kristiansen, G. (2009). MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J. Cell. Mol. Med. 13, 3918–3928.
Kagara, I., Enokida, H., Kawakami, K., Matsuda, R., Toki, K., Nishimura, H., Chiyomaru, T., Tatarano, S., Itesako, T., Kawamoto, K., Nishiyama, K., Seki, N., and Nakagawa, M. (2008). CpG hypermethylation of the UCHL1 gene promoter is associated with pathogenesis and poor prognosis in renal cell carcinoma. J. Urol. 180, 343–351.
Kapoor-Vazirani, P., Kagey, J. D., Powell, D. R., and Vertino, P. M. (2008). Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 68, 6810–6821.
Kim, H. L., Seligson, D., Liu, X., Janzen, N., Bui, M. H., Yu, H., Shi, T., Belldegrun, A. S., Horvath, S., and Figlin, R. A. (2005). Using tumor markers to predict the survival of patients with metastatic renal cell carcinoma. J. Urol. 173, 1496–1501.
Kondo, Y., Shen, L., Cheng, A. S., Ahmed, S., Boumber, Y., Charo, C., Yamochi, T., Urano, T., Furukawa, K., Kwabi-Addo, B., Gold, D. L., Sekido, Y., Huang, T. H., and Issa, J. P. (2008). Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 40, 741–750.
Lam, J. S., Leppert, J. T., Figlin, R. A., and Belldegrun, A. S. (2005). Role of molecular markers in the diagnosis and therapy of renal cell carcinoma. Urology 66, 1–9.
Lee, Y., Jeon, K., Lee, J. T., Kim, S., and Kim, V. N. (2002). MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 21, 4663–4670.
Li, X., Chen, J., Hu, X., Huang, Y., Li, Z., Zhou, L., Tian, Z., Ma, H., Wu, Z., Chen, M., Han, Z., Peng, Z., Zhao, X., Liang, C., Wang, Y., Sun, L., Zhao, J., Jiang, B., Yang, H., Gui, Y., Cai, Z., and Zhang, X. (2011). Comparative mRNA and microRNA expression profiling of three genitourinary cancers reveals common hallmarks and cancer-specific molecular events. PLoS ONE 6, e22570. doi:10.1371/journal.pone.0022570
Lin, J. C., Jeong, S., Liang, G., Takai, D., Fatemi, M., Tsai, Y. C., Egger, G., Gal-Yam, E. N., and Jones, P. A. (2007). Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell 12, 432–444.
Linehan, W. M., Srinivasan, R., and Schmidt, L. S. (2010). The genetic basis of kidney cancer: a metabolic disease. Nat. Rev. Urol. 7, 277–285.
Liu, H., Brannon, A. R., Reddy, A. R., Alexe, G., Seiler, M. W., Arreola, A., Oza, J. H., Yao, M., Juan, D., Liou, L. S., Ganesan, S., Levine, A. J., Rathmell, W. K., and Bhanot, G. V. (2010a). Identifying mRNA targets of microRNA dysregulated in cancer: with application to clear cell renal cell carcinoma. BMC Syst. Biol. 4, 51. doi:10.1186/1752-0509-4-51
Liu, W., Zabirnyk, O., Wang, H., Shiao, Y. H., Nickerson, M. L., Khalil, S., Anderson, L. M., Perantoni, A. O., and Phang, J. M. (2010b). miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 29, 4914–4924.
Lopez-Beltran, A., Carrasco, J. C., Cheng, L., Scarpelli, M., Kirkali, Z., and Montironi, R. (2009). 2009 Update on the classification of renal epithelial tumors in adults. Int. J. Urol. 16, 432–443.
Lopez-Serra, L., and Esteller, M. (2008). Proteins that bind methylated DNA and human cancer: reading the wrong words. Br. J. Cancer 98, 1881–1885.
Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B. L., Mak, R. H., Ferrando, A. A., Downing, J. R., Jacks, T., Horvitz, H. R., and Golub, T. R. (2005). MicroRNA expression profiles classify human cancers. Nature 435, 834–838.
Mahalingam, D., Medina, E. C., Esquivel, J. A. II, Espitia, C. M., Smith, S., Oberheu, K., Swords, R., Kelly, K. R., Mita, M. M., Mita, A. C., Carew, J. S., Giles, F. J., and Nawrocki, S. T. (2010). Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin. Cancer Res. 16, 141–153.
McRonald, F. E., Morris, M. R., Gentle, D., Winchester, L., Baban, D., Ragoussis, J., Clarke, N. W., Brown, M. D., Kishida, T., Yao, M., Latif, F., and Maher, E. R. (2009). CpG methylation profiling in VHL related and VHL unrelated renal cell carcinoma. Mol. Cancer 8, 31.
Metzger, E., Wissmann, M., Yin, N., Muller, J. M., Schneider, R., Peters, A. H., Gunther, T., Buettner, R., and Schule, R. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439.
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., Alvarez, P., Brockman, W., Kim, T. K., Koche, R. P., Lee, W., Mendenhall, E., O’Donovan, A., Presser, A., Russ, C., Xie, X., Meissner, A., Wernig, M., Jaenisch, R., Nusbaum, C., Lander, E. S., and Bernstein, B. E. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560.
Minardi, D., Lucarini, G., Filosa, A., Milanese, G., Zizzi, A., Di Primio, R., Montironi, R., and Muzzonigro, G. (2009). Prognostic role of global DNA-methylation and histone acetylation in pT1a clear cell renal carcinoma in partial nephrectomy specimens. J. Cell. Mol. Med. 13, 2115–2121.
Morris, M. R., Gentle, D., Abdulrahman, M., Clarke, N., Brown, M., Kishida, T., Yao, M., Teh, B. T., Latif, F., and Maher, E. R. (2008). Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br. J. Cancer 98, 496–501.
Morris, M. R., Gentle, D., Abdulrahman, M., Maina, E. N., Gupta, K., Banks, R. E., Wiesener, M. S., Kishida, T., Yao, M., Teh, B., Latif, F., and Maher, E. R. (2005). Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 65, 4598–4606.
Morris, M. R., Hesson, L. B., Wagner, K. J., Morgan, N. V., Astuti, D., Lees, R. D., Cooper, W. N., Lee, J., Gentle, D., Macdonald, F., Kishida, T., Grundy, R., Yao, M., Latif, F., and Maher, E. R. (2003). Multigene methylation analysis of Wilms’ tumour and adult renal cell carcinoma. Oncogene 22, 6794–6801.
Morris, M. R., Ricketts, C., Gentle, D., Abdulrahman, M., Clarke, N., Brown, M., Kishida, T., Yao, M., Latif, F., and Maher, E. R. (2010). Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene 29, 2104–2117.
Morris, M. R., Ricketts, C. J., Gentle, D., Mcronald, F., Carli, N., Khalili, H., Brown, M., Kishida, T., Yao, M., Banks, R. E., Clarke, N., Latif, F., and Maher, E. R. (2011). Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene 30, 1390–1401.
Mosashvilli, D., Kahl, P., Mertens, C., Holzapfel, S., Rogenhofer, S., Hauser, S., Buttner, R., Von Ruecker, A., Muller, S. C., and Ellinger, J. (2010). Global histone acetylation levels: prognostic relevance in patients with renal cell carcinoma. Cancer Sci. 101, 2664–2669.
Mulero-Navarro, S., and Esteller, M. (2008). Epigenetic biomarkers for human cancer: the time is now. Crit. Rev. Oncol. Hematol. 68, 1–11.
Nakada, C., Matsuura, K., Tsukamoto, Y., Tanigawa, M., Yoshimoto, T., Narimatsu, T., Nguyen, L. T., Hijiya, N., Uchida, T., Sato, F., Mimata, H., Seto, M., and Moriyama, M. (2008). Genome-wide microRNA expression profiling in renal cell carcinoma: significant down-regulation of miR-141 and miR-200c. J. Pathol. 216, 418–427.
Neal, C. S., Michael, M. Z., Rawlings, L. H., Van Der Hoek, M. B., and Gleadle, J. M. (2010). The VHL-dependent regulation of microRNAs in renal cancer. BMC Med. 8, 64. doi:10.1186/1741-7015-8-64
Nguyen, C. T., Weisenberger, D. J., Velicescu, M., Gonzales, F. A., Lin, J. C., Liang, G., and Jones, P. A. (2002). Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2’-deoxycytidine. Cancer Res. 62, 6456–6461.
Paiva, F., Duarte-Pereira, S., Costa, V. L., Ramalho-Carvalho, J., Patricio, P., Ribeiro, F. R., Lobo, F., Oliveira, J., Jeronimo, C., and Henrique, R. (2011). Functional and epigenetic characterization of the KRT19 gene in renal cell neoplasms. DNA Cell Biol. 30, 85–90.
Peters, I., Rehmet, K., Wilke, N., Kuczyk, M. A., Hennenlotter, J., Eilers, T., Machtens, S., Jonas, U., and Serth, J. (2007). RASSF1A promoter methylation and expression analysis in normal and neoplastic kidney indicates a role in early tumorigenesis. Mol. Cancer 6, 49.
Petillo, D., Kort, E. J., Anema, J., Furge, K. A., Yang, X. J., and Teh, B. T. (2009). MicroRNA profiling of human kidney cancer subtypes. Int. J. Oncol. 35, 109–114.
Pollard, P. J., Loenarz, C., Mole, D. R., Mcdonough, M. A., Gleadle, J. M., Schofield, C. J., and Ratcliffe, P. J. (2008). Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 416, 387–394.
Rathmell, W. K., and Chen, S. (2008). VHL inactivation in renal cell carcinoma: implications for diagnosis, prognosis and treatment. Expert Rev. Anticancer Ther. 8, 63–73.
Rodriguez-Paredes, M., and Esteller, M. (2011). Cancer epigenetics reaches mainstream oncology. Nat. Med. 17, 330–339.
Scelo, G., and Brennan, P. (2007). The epidemiology of bladder and kidney cancer. Nat. Clin. Pract. Urol. 4, 205–217.
Schulte, J. H., Lim, S., Schramm, A., Friedrichs, N., Koster, J., Versteeg, R., Ora, I., Pajtler, K., Klein-Hitpass, L., Kuhfittig-Kulle, S., Metzger, E., Schule, R., Eggert, A., Buettner, R., and Kirfel, J. (2009). Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 69, 2065–2071.
Seitz, H., Youngson, N., Lin, S. P., Dalbert, S., Paulsen, M., Bachellerie, J. P., Ferguson-Smith, A. C., and Cavaille, J. (2003). Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat. Genet. 34, 261–262.
Seliger, B., Handke, D., Schabel, E., Bukur, J., Lichtenfels, R., and Dammann, R. (2009). Epigenetic control of the ubiquitin carboxyl terminal hydrolase 1 in renal cell carcinoma. J. Transl. Med. 7, 90.
Seligson, D. B., Horvath, S., Mcbrian, M. A., Mah, V., Yu, H., Tze, S., Wang, Q., Chia, D., Goodglick, L., and Kurdistani, S. K. (2009). Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 174, 1619–1628.
Shang, D., Liu, Y., Xu, X., Han, T., and Tian, Y. (2011). 5-Aza-2’-deoxycytidine enhances susceptibility of renal cell carcinoma to paclitaxel by decreasing LEF1/phospho-beta-catenin expression. Cancer Lett. 311, 230–236.
Shenouda, S. K., and Alahari, S. K. (2009). MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev. 28, 369–378.
Shi, Y. (2007). Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 8, 829–833.
Sinha, S., Dutta, S., Datta, K., Ghosh, A. K., and Mukhopadhyay, D. (2009). Von Hippel-Lindau gene product modulates TIS11B expression in renal cell carcinoma: impact on vascular endothelial growth factor expression in hypoxia. J. Biol. Chem. 284, 32610–32618.
Slaby, O., Jancovicova, J., Lakomy, R., Svoboda, M., Poprach, A., Fabian, P., Kren, L., Michalek, J., and Vyzula, R. (2010). Expression of miRNA-106b in conventional renal cell carcinoma is a potential marker for prediction of early metastasis after nephrectomy. J. Exp. Clin. Cancer Res. 29, 90.
Song, J., Noh, J. H., Lee, J. H., Eun, J. W., Ahn, Y. M., Kim, S. Y., Lee, S. H., Park, W. S., Yoo, N. J., Lee, J. Y., and Nam, S. W. (2005). Increased expression of histone deacetylase 2 is found in human gastric cancer. Acta Pathol. Microbiol. Immunol. Scand. 113, 264–268.
Takano, Y., Iwata, H., Yano, Y., Miyazawa, M., Virgona, N., Sato, H., Ueno, K., and Yano, T. (2010). Up-regulation of connexin 32 gene by 5-aza-2’-deoxycytidine enhances vinblastine-induced cytotoxicity in human renal carcinoma cells via the activation of JNK signalling. Biochem. Pharmacol. 80, 463–470.
To, K. K., Polgar, O., Huff, L. M., Morisaki, K., and Bates, S. E. (2008). Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol. Cancer Res. 6, 151–164.
Touma, S. E., Goldberg, J. S., Moench, P., Guo, X., Tickoo, S. K., Gudas, L. J., and Nanus, D. M. (2005). Retinoic acid and the histone deacetylase inhibitor trichostatin a inhibit the proliferation of human renal cell carcinoma in a xenograft tumor model. Clin. Cancer Res. 11, 3558–3566.
Urakami, S., Shiina, H., Enokida, H., Hirata, H., Kawamoto, K., Kawakami, T., Kikuno, N., Tanaka, Y., Majid, S., Nakagawa, M., Igawa, M., and Dahiya, R. (2006). Wnt antagonist family genes as biomarkers for diagnosis, staging, and prognosis of renal cell carcinoma using tumor and serum DNA. Clin. Cancer Res. 12, 6989–6997.
Vaissiere, T., Sawan, C., and Herceg, Z. (2008). Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 659, 40–48.
Valera, V. A., Walter, B. A., Linehan, W. M., and Merino, M. J. (2011). Regulatory effects of microRNA-92 (miR-92) on VHL gene expression and the hypoxic activation of miR-210 in clear cell renal cell carcinoma. J. Cancer 2, 515–526.
Valk-Lingbeek, M. E., Bruggeman, S. W., and Van Lohuizen, M. (2004). Stem cells and cancer; the polycomb connection. Cell 118, 409–418.
van Haaften, G., Dalgliesh, G. L., Davies, H., Chen, L., Bignell, G., Greenman, C., Edkins, S., Hardy, C., O’Meara, S., Teague, J., Butler, A., Hinton, J., Latimer, C., Andrews, J., Barthorpe, S., Beare, D., Buck, G., Campbell, P. J., Cole, J., Forbes, S., Jia, M., Jones, D., Kok, C. Y., Leroy, C., Lin, M. L., Mcbride, D. J., Maddison, M., Maquire, S., Mclay, K., Menzies, A., Mironenko, T., Mulderrig, L., Mudie, L., Pleasance, E., Shepherd, R., Smith, R., Stebbings, L., Stephens, P., Tang, G., Tarpey, P. S., Turner, R., Turrell, K., Varian, J., West, S., Widaa, S., Wray, P., Collins, V. P., Ichimura, K., Law, S., Wong, J., Yuen, S. T., Leung, S. Y., Tonon, G., Depinho, R. A., Tai, Y. T., Anderson, K. C., Kahnoski, R. J., Massie, A., Khoo, S. K., Teh, B. T., Stratton, M. R., and Futreal, P. A. (2009). Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 41, 521–523.
van Vlodrop, I. J., Baldewijns, M. M., Smits, K. M., Schouten, L. J., Van Neste, L., Van Criekinge, W., Van Poppel, H., Lerut, E., Schuebel, K. E., Ahuja, N., Herman, J. G., De Bruine, A. P., and Van Engeland, M. (2010). Prognostic significance of Gremlin1 (GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am. J. Pathol. 176, 575–584.
Varela, I., Tarpey, P., Raine, K., Huang, D., Ong, C. K., Stephens, P., Davies, H., Jones, D., Lin, M. L., Teague, J., Bignell, G., Butler, A., Cho, J., Dalgliesh, G. L., Galappaththige, D., Greenman, C., Hardy, C., Jia, M., Latimer, C., Lau, K. W., Marshall, J., Mclaren, S., Menzies, A., Mudie, L., Stebbings, L., Largaespada, D. A., Wessels, L. F., Richard, S., Kahnoski, R. J., Anema, J., Tuveson, D. A., Perez-Mancera, P. A., Mustonen, V., Fischer, A., Adams, D. J., Rust, A., Chan-On, W., Subimerb, C., Dykema, K., Furge, K., Campbell, P. J., Teh, B. T., Stratton, M. R., and Futreal, P. A. (2011). Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542.
Ventura, A., and Jacks, T. (2009). MicroRNAs and cancer: short RNAs go a long way. Cell 136, 586–591.
Veronese, A., Lupini, L., Consiglio, J., Visone, R., Ferracin, M., Fornari, F., Zanesi, N., Alder, H., D’Elia, G., Gramantieri, L., Bolondi, L., Lanza, G., Querzoli, P., Angioni, A., Croce, C. M., and Negrini, M. (2010). Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 70, 3140–3149.
Wang, X. F., Qian, D. Z., Ren, M., Kato, Y., Wei, Y., Zhang, L., Fansler, Z., Clark, D., Nakanishi, O., and Pili, R. (2005). Epigenetic modulation of retinoic acid receptor beta2 by the histone deacetylase inhibitor MS-275 in human renal cell carcinoma. Clin. Cancer Res. 11, 3535–3542.
White, N. M., Khella, H. W., Grigull, J., Adzovic, S., Youssef, Y. M., Honey, R. J., Stewart, R., Pace, K. T., Bjarnason, G. A., Jewett, M. A., Evans, A. J., Gabril, M., and Yousef, G. M. (2011). miRNA profiling in metastatic renal cell carcinoma reveals a tumour-suppressor effect for miR-215. Br. J. Cancer 105, 1741–1749.
Wulfken, L. M., Moritz, R., Ohlmann, C., Holdenrieder, S., Jung, V., Becker, F., Herrmann, E., Walgenbach-Brunagel, G., Von Ruecker, A., Muller, S. C., and Ellinger, J. (2011). MicroRNAs in renal cell carcinoma: diagnostic implications of serum miR-1233 levels. PLoS ONE 6, e25787. doi:10.1371/journal.pone.0025787
Yamada, D., Kikuchi, S., Williams, Y. N., Sakurai-Yageta, M., Masuda, M., Maruyama, T., Tomita, K., Gutmann, D. H., Kakizoe, T., Kitamura, T., Kanai, Y., and Murakami, Y. (2006). Promoter hypermethylation of the potential tumor suppressor DAL-1/4.1B gene in renal clear cell carcinoma. Int. J. Cancer 118, 916–923.
Youssef, Y. M., White, N. M., Grigull, J., Krizova, A., Samy, C., Mejia-Guerrero, S., Evans, A., and Yousef, G. M. (2011). Accurate molecular classification of kidney cancer subtypes using microRNA signature. Eur. Urol. 59, 721–730.
Zhang, B., Pan, X., Cobb, G. P., and Anderson, T. A. (2007). MicroRNAs as oncogenes and tumor suppressors. Dev. Biol. 302, 1–12.
Zhou, L., Chen, J., Li, Z., Li, X., Hu, X., Huang, Y., Zhao, X., Liang, C., Wang, Y., Sun, L., Shi, M., Xu, X., Shen, F., Chen, M., Han, Z., Peng, Z., Zhai, Q., Zhang, Z., Yang, R., Ye, J., Guan, Z., Yang, H., Gui, Y., Wang, J., Cai, Z., and Zhang, X. (2010). Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 5, e15224. doi:10.1371/journal.pone.0015224
Keywords: epigenetics, kidney, renal cell tumors, DNA methylation, chromatin remodeling, histone post-translational modifications, microRNAs, epigenetic-based therapeutics
Citation: Henrique R, Luís AS and Jerónimo C (2012) The epigenetics of renal cell tumors: from biology to biomarkers. Front. Gene. 3:94. doi: 10.3389/fgene.2012.00094
Received: 25 January 2012; Paper pending published: 22 February 2012;
Accepted: 09 May 2012; Published online: 30 May 2012.
Edited by:
Trygve Tollefsbol, University of Alabama at Birmingham, USAReviewed by:
Tomas J. Ekstrom, Karolinska Institutet, SwedenBradford Coffee, Emory University, USA
Jorg Tost, Commissariat a l’Energie Atomique, France
Copyright: © 2012 Henrique, Luís and Jerónimo. This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Rui Henrique, Department of Pathology, Portuguese Oncology Institute – Porto, Rua Dr. António Bernardino de Almeida, 4200-072 Porto, Portugal. e-mail:cm1oZW5yaXF1ZUBpY2Jhcy51cC5wdA==