
 Hiroshi Kogo
Hiroshi Kogo- Division of Molecular Genetics, Institute for Comprehensive Medical Science, Fujita Health University, Toyoake, Aichi, Japan
The prophase of meiosis I ensures the correct segregation of chromosomes to each daughter cell. This includes the pairing, synapsis, and recombination of homologous chromosomes. A subset of chromosomal abnormalities, including translocation and inversion, disturbs these processes, resulting in the failure to complete synapsis. This activates the meiotic pachytene checkpoint, and the gametes are fated to undergo cell cycle arrest and subsequent apoptosis. Spermatogenic cells appear to be more vulnerable to the pachytene checkpoint, and male carriers of chromosomal abnormalities are more susceptible to infertility. In contrast, oocytes tend to bypass the checkpoint and instead generate other problems, such as chromosome imbalance that often leads to recurrent pregnancy loss in female carriers. Recent advances in genetic manipulation technologies have increased our knowledge about the pachytene checkpoint and surveillance systems that detect chromosomal synapsis. This review focuses on the consequences of synapsis failure in humans and provides an overview of the mechanisms involved. We also discuss the sexual dimorphism of the involved pathways that leads to the differences in reproductive outcomes between males and females.
Introduction
Down syndrome is the most common chromosomal abnormality that manifests in newborns. The karyotype of 90% of Down syndrome cases is standard trisomy 21, whereas in approximately 4% of cases an extra chromosome 21 is associated with the Robertsonian translocation of chromosome 21 (Gardner and Sutherland, 2004). About three-quarters of Down syndrome cases with Robertsonian translocation occur de novo, while the remaining one-quarter are familial cases in which either parent is a balanced carrier of the same translocation. In these familial cases, a distinct bias in the transmission of the translocation occurs: the balanced carrier of the translocation is almost always the mother (Gardner and Sutherland, 2004). Likewise, reciprocal translocation between autosomes shows similar transmission bias. Constitutional t(11;22)(q23;q11.2) translocation is a well-known recurrent reciprocal translocation. The only viable unbalanced offspring of this translocation manifest Emanuel syndrome (Kurahashi et al., 2010). Either of the parents of a patient with Emanuel syndrome is almost always a balanced carrier of the translocation. Similar to the Robertsonian translocation, there is a distinct transmission bias to the t(11;22) translocation: in more than 90% of cases with Emanuel syndrome, the mother is found to be the balanced carrier, whereas de novo production of t(11;22) is, conversely, always of paternal origin (Kurahashi and Emanuel, 2001; Carter et al., 2009; Ohye et al., 2010).
Robertsonian translocations and other autosomal translocations are often associated with recurrent pregnancy loss (RPL), when one of the couple is a balanced carrier of the translocation. This is due to the transmission of the unbalanced translocation to the gametes via adjacent segregation (or 3:1 segregation) during meiotic division. The resulting zygote with the unbalanced chromosomes will mostly develop into a fetus but subsequently die in utero. In a survey of RPL cases, 3% of the couples carried an autosomal translocation and 2% carried a Robertsonian translocation (Fryns et al., 1984). In these cases, the female carries the translocation at a frequency twice as high as that of the male.
These female biases in translocation transmission are occasionally confused with the maternal bias of the extra chromosome of standard trisomy 21 in Down syndrome. It is formally accepted that this maternal bias is most likely due to differences in the timing of meiotic division between males and females. In males, meiosis starts at adolescence and the cell division of germ cells continues almost throughout a lifetime. In contrast, oocytes enter meiosis during the fetal period, but their development is arrested during the middle of prophase during meiosis I (dictyate stage), and they restart maturation just prior to ovulation in females of reproductive age. In these days, there is mounting evidence that the degradation of meiosis-specific cohesin that occurs during the long meiotic arrest of oogenesis induces the nondisjunction or predivision of homologous chromosomes, thereby leading to age-dependent aneuploidy in females (Hunt and Hassold, 2010; Kurahashi et al., 2012). Obviously, this does not explain the female bias in the transmission of unbalanced chromosomes to offspring and the reproductive outcome of the translocation carrier.
It is accepted that sex bias results from the nature of two sex-specific reproduction mechanisms and the process of meiosis. The first is that a part of the male translocation carrier population does not transmit the translocation to his offspring because these carriers are infertile due to azoospermia. Females and men without azoospermia might suffer from RPL but can also produce offspring with either of normal chromosome, balanced translocation or unbalanced. Indeed, a family with autosomal translocation and male-only sterility has been reported, in which the female carrier had two consecutive spontaneous abortions (Paoloni-Giacobino et al., 2000). This might exert a large impact on the female bias of transmission. Another factor that affects bias is transmission distortion of the translocation chromosomes. In general, female translocation carriers are more frequently transmit the translocation to the offspring than male carriers without azoospermia (Gardner and Sutherland, 2004). This latter phenomenon might be independent of the former, but the nature of this phenomenon is less understood.
In this review, we focus on chromosomal translocation and male-only sterility. Recently, mutations in the SYCP3 gene were reported in RPL women experiencing more than three spontaneous abortions during early gestation (Bolor et al., 2009). The mutant genes consistently encode C-terminally mutated proteins that might inhibit the normal function of the SYCP3 protein via dominant negative effects and have been predicted to generate aneuploid oocytes. Interestingly, a mutation in a similar region in the SYCP3 gene was also reported in azoospermia patients with meiotic arrest (Miyamoto et al., 2003). These data indicate that the quality controls of meiotic chromosome segregation are more stringent in males than in females (Hunt and Hassold, 2002). “Checkpoint” is defined as a cellular surveillance system that detects abnormalities and is dispensable when everything is normal. It has been speculated that the sexual dimorphism of these reproductive outcomes might be due to differences in checkpoint robustness between males and females (Kurahashi et al., 2009).
Meiotic Arrest of Spermatogenesis in the Male Translocation Carrier
There is an emerging consensus that the events that occur during the prophase of meiosis I are essential for the proper segregation of homologous chromosomes (Hassold et al., 2000; Handel and Schimenti, 2010). Homologous chromosomes that behave independently during mitotic division have to segregate into two different daughter cells during meiosis I (Figure 1A). To accomplish this process, homologous chromosomes interact with each other, utilizing a specialized pathway known as homologous recombination (HR; Figure 1B). Initially, programmed double-strand-breaks (DSBs) manifest in chromosomal DNA by the action of SPO11 endonuclease at more than 100 sites throughout the entire genome. Activation of the DSB sites is controlled by PRDM9, which encodes a H3K4 methylase. PRDM9 binds to the 13 bp recombination hotspot consensus sequence and facilitates DSB formation via methylation of surrounding histones (Borde et al., 2009; Buard et al., 2009; Baudat et al., 2010; Myers et al., 2010; Parvanov et al., 2010). To correctly repair these DSBs, a subsequent HR pathway is activated and the broken DNA ends begin to look for homologous regions. As the consequence, two homologous chromosomes are brought together in close association, a process known as homolog-pairing. A proteinaceous structure known as the synaptonemal complex (SC) is subsequently formed between the paired homologous chromosomes. This step is called synapsis. The DNA lesions are subsequently repaired via HR with the aid of recombination proteins RAD51 and DMC1. During the final step of HR, a four-stranded DNA structure, the Holliday junction that physically connects the two chromosomes is resolved in one of two ways, crossover or non-crossover. Crossover maintains the physical linkage of the chromosomes (chiasmata) and produces the appropriate bi-oriented tension at the opposite spindle poles during metaphase of meiosis I. Thus, the number and location of the crossover events is strictly regulated (crossover assurance and interference). Meiotic recombination, which is well known as a mechanism that shuffles genetic material in order to produce variation among individuals, is also indispensable for the proper segregation of homologous chromosomes (Kurahashi et al., 2009). Failure of each of these processes triggers cell cycle arrest and the subsequent apoptosis of meiotic cells (Hochwagen and Amon, 2006).
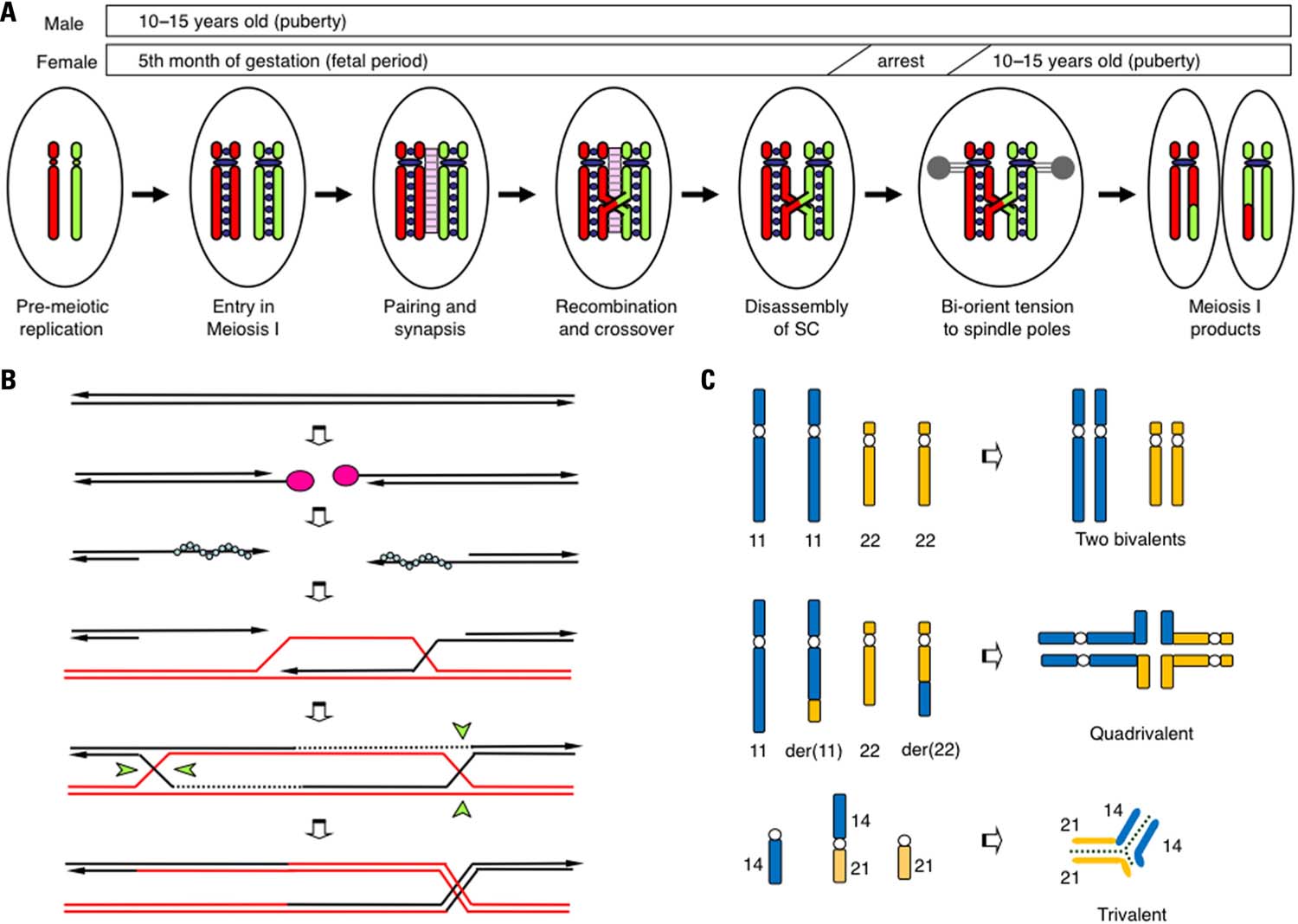
FIGURE 1. Schematic representation of the prophase of meiosis I. (A) The critical steps that affect chromosomal segregation in meiosis I. During the pre-meiotic S-phase, both maternal (red) and paternal (green) chromosomes are replicated and tightly connected to cohesin complexes (purple circles). Then, two homologous chromosomes undergo pairing, and a synaptonemal complex (pink ladder) is established between them. HR results in the production of at least one obligatory chiasma via crossover. After the disassembly of SC, two homologous chromosomes are pulled in opposite directions to the two spindle poles (gray). (B) Schematic representation of meiotic recombination. Initially, programmed DSBs are induced by SPO11 endonuclease (pink circles). The 5′-ends of the DSBs are then resected and the 3′-protruding single-stranded ends are generated. With the aid of RAD51 or DMC1, the DNA ends produce nucleofilament complexes (blue circles) that facilitate genome-wide homology scanning in order to find homologous chromosomes (red lines). Next, a single-stranded DNA end invades the homologous duplex DNA, forming a D-loop structure. DNA synthesis seals the DSBs and the double Holliday junctions emerge. These Holliday junctions are resolved in one of two ways, crossover (right) or non-crossover (left), with the Holliday junction resolvase (green wedges). (C) Pachytene configuration in balanced carriers of chromosomal translocation. In t(11;22) reciprocal translocation, two translocated chromosomes in concert with two normal counterparts form a quadrivalent to complete synapsis. In a t(14;21) Robertsonian translocation, the translocated chromosome and the two normal counterparts form a trivalent.
The developmental stage during which the SC between the homologous chromosomes is established is called the pachytene. In testicular specimens from infertile men with chromosomal translocations, meiotic arrest and massive cell death at pachytene have been consistently observed (Solari, 1999; Egozcue et al., 2000). The spermatocytes appear to be eliminated by a quality control system, known as the pachytene checkpoint (Roeder and Bailis, 2000). Obviously, the pachytene configuration of the homologous chromosomes is considerably affected in the presence of translocated chromosomes in the translocation carrier. In the case of reciprocal translocation, homologous pairing can be theoretically achieved by the formation of a quadrivalent (the trivalent of the Robertsonian translocation; Figure 1C). However, the pachytene configuration of translocated chromosomes is variable and partly dependent on the locations of the translocation breakpoints. The most likely configuration is the quadrivalent, while asynapsis, partial synapsis, and heterosynapsis between non-homologous chromosomes have also been observed. Even in the quadrivalent, full synapsis is not always achieved and small but substantial portions around breakpoints are often unsynapsed. The degree of synapsis failure most likely affects the consequences of the gametes (Martin, 2008; Burgoyne et al., 2009).
DSB-Dependent and -Independent Pachytene Checkpoints
The mechanism of the pachytene checkpoint has been long studied using yeast, but recent advances in the genetic manipulation of mice have also revealed the presence of the pachytene checkpoint in mammals. Over the past decade, a number of knockout mice that were deficient in genes involved in the meiotic HR pathway have been created. Because some of the HR proteins are commonly utilized in the DSB repair machinery of mitotic cells, the mutants of these genes manifest as extragonadal phenotypes such as increased cancer susceptibility (Matzuk and Lamb, 2002; Cohen et al., 2006). However, regardless of whether the gene is meiosis-specific or not, infertility is a common phenotype in both male and female mutants. Defects in HR, e.g., defects in the DNA repair system, leave unrepaired DSBs that most likely trigger the checkpoint system (Hochwagen and Amon, 2006). Hence, most of meiotic recombination mutants manifest as meiotic arrest due to the DSB-dependent pachytene checkpoint, leading to azoospermia and oocyte loss by massive apoptosis. In the testicular specimens from Dmc1-deficient mice, extensive apoptosis is consistently observed in stage IV of the seminiferous tubules that mostly includes pachytene spermatocytes (Pittman et al., 1998; Yoshida et al., 1998; de Rooij and de Boer, 2003). In the ovaries of mutants, no follicles have been detected, suggesting that oocytes cannot develop into the subsequent dictyate stage due to extensive apoptosis at the pachytene checkpoint. These data suggest that the DSB-dependent pachytene checkpoint operates similarly both in males and females.
Genetic defects related to SC also manifest as reproductive failure as their only phenotype. These genes do not directly take part in the HR pathway, but defects in SC formation generally impair DSB repair efficiency, which may also activate the DSB-dependent pachytene checkpoint, leading to meiotic arrest (Handel and Schimenti, 2010). However, in such mutants, phenotype severity often differs between males and females but is consistently less severe in females. For instances, knockout mice for genes that encodes the components of SC, such as Sycp2 and Sycp3, manifest as male infertility by meiotic arrest, whereas female mice demonstrate subfertility and a variable degree of fetal lethality in utero (Yuan et al., 2000, 2002; Yang et al., 2006). It is still possible that reproductive failure is not simply due to the activation of the DSB-dependent pachytene checkpoint and that some different pathways might be able to cope with SC defects.
On the other hand, several lines of evidence suggest that in a subset of mutants, the meiotic phenotype appears to be driven by a DSB-independent pachytene checkpoint. Spo11 initiates meiotic recombination by catalyzing the programmed DSBs. Both male and female Spo11-deficient mice are made sterile by meiotic arrest during the pachytene stage (Baudat et al., 2000; Romanienko and Camerini-Otero, 2000). Mutants demonstrate defects in homologous pairing due to the absence of DSBs and the subsequent development of extensive synapsis failure with some degree of heterosynapsis. Given the lack of DSBs, checkpoints for unrepaired DSBs do not contribute to meiotic arrest. Thus, the pachytene checkpoint in mutants is most likely activated in a DSB-independent manner. Thorough examinations have revealed that putative DSB-independent meiotic arrest is a bit later and less severe than the DSB-dependent (Barchi et al., 2005; Di Giacomo et al., 2005). Similar observations have also been obtained in other DSB-deficient mutant mice (Reinholdt and Schimenti, 2005). These data suggest the possibility of DSB-independent synapsis checkpoints in mammals.
Next, we set out to determine which mechanism is involved in the massive cell death that occurs during pachytene in human males with translocation, the DSB-dependent or -independent pachytene checkpoint. Because synapsis failure can induce meiotic recombination defects, partial synapsis failure due to translocation might instigate enough recombination efficiency to activate the DSB-dependent checkpoint, leading to cell death during the pachytene as well. Indeed, a reduced number of MLH1 foci, a marker of crossover sites at the pachytene, was reported in the spermatocytes of infertile male translocation carrier (Sun et al., 2005; Ferguson et al., 2008; Leng et al., 2009). However, this might not have much of an effect on the fate of the gametes because only the cells harboring DSBs around the breakpoints might have difficulty initiating DSB repair due to functional SC defects. Further, DSB-dependent checkpoints, which appear to operate similarly in males and females, cannot account for the sexual dimorphism of the reproductive outcomes. Thus, the DSB-dependent checkpoint is unlikely to exert a profound effect on the partial synapsis of pachytene chromosomes in translocation carriers.
Etiology Underlying Sex Differences: Involvement of the Sex Body
How are the chromosomes that undergo partial synapsis subsequently processed? A good example can help imagine this process: the behavior of sex chromosomes during male meiosis. In general, because male sex chromosomes are heteromorphic with small homologous regions known as pseudoautosomal regions at both chromosomal ends, they can make a pair and undergo HR to form the obligatory chiasma required for correct segregation during meiosis I. However, male sex chromosomes cannot be fully synapsed throughout the X or Y chromosome-specific regions and, instead, form a specialized nuclear territory known as the sex body or XY body (Figure 2A; Handel, 2004). The sex body is typically found at the periphery of the nucleus with synapsed chromosomal ends that are anchored to the nuclear lamina. In females, the meiotic stage progress from the pachytene to the subsequent diplotene stage after almost all of the chromosomes are fully synapsed and all of the DSBs are repaired. In contrast, because sex chromosomes cannot be fully synapsed and the DSB repair is delayed in males, the sex body is likely to mask the unsynapsed and unrepaired regions of the male sex chromosomes and facilitate cell cycle progression to the subsequent stage despite the presence of unsynapsed and unrepaired chromosomes.
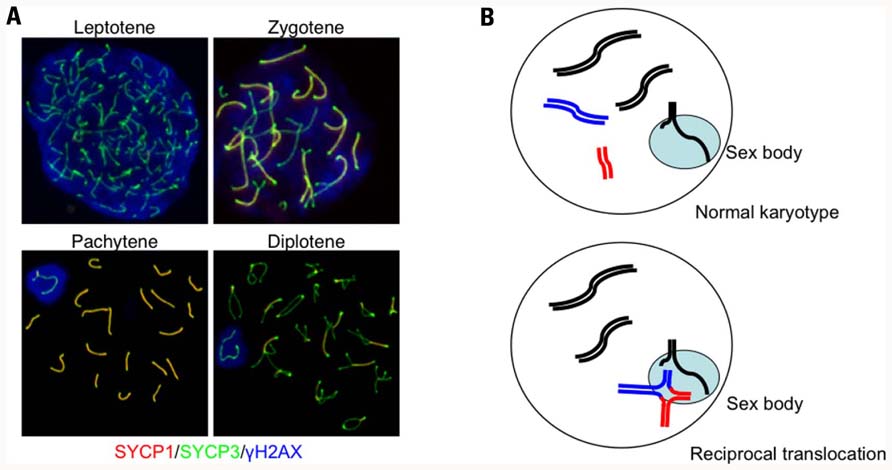
FIGURE 2. Autosome–sex body association in a translocation carrier. (A) Progression of prophase in meiosis I and the formation of the sex body in male mice. Prophase I begins during the leptotene stage when each chromosome begins condensation. The subsequent stage is the zygotene, which occurs when homologous chromosomes are paired and initiate SC formation. Because the chromosomal axes are stained with anti-SYCP3 (green) and the central elements of the SC are indicated with anti-SYCP1 staining (red), the regions that complete synapsis appear as yellow. During pachytene, all of the autosomes finish SC formation, while the male sex chromosomes cannot complete synapsis and remain green. The unsynapsed X and Y chromosomes are confined within the γH2AX-stained nuclear domain (blue). (B) Diagrams illustrating the autosome–sex body association. The upper panel shows sex body formation during normal spermatogenesis. Unsynapsed regions of the X and Y chromosomes are confined within the sex body (light blue). In male translocation carriers (lower panel), translocated chromosomes form quadrivalents (red and blue lines), which often have unsynapsed regions around the breakpoints, that become associated with the sex body.
Within the sex body, unsynapsed regions of the male sex chromosomes are transcriptionally inactivated and a number of heterochromatin-associated proteins are assembled into this specialized chromatin domain. This phenomenon is called meiotic sex chromosome inactivation (MSCI; Turner, 2007; Yan and McCarrey, 2009). Similar to X chromosome inactivation in terms of dosage compensation in the female, MSCI is characterized by several epigenetic modifications. For example, the phosphorylated variant of histone H2AX (γH2AX) is detected at sites of MSCI. Other possible suppressive modifications detected include substitution of H3 by variant H3.3 and enrichment of H2A variant, macro-H2A1 (Baarends and Grootegoed, 2003; Hoyer-Fender, 2003). MSCI also involves acquisition of other histone modifications, including H2A and H2B ubiquitination, with localization of small ubiquitin-like modifier proteins to the sex body (Kota and Feil, 2010). Although this inactivation is transient during prophase I, and the transcription of some of the genes on sex chromosomes restarts after the completion of meiosis, most sex chromosome genes remain inactivated through late spermatogenesis within a similar nuclear domain called the post-meiotic sex chromatin (Namekawa et al., 2006; Turner et al., 2006). This process might serve to mark the parental origin of the paternal X chromosome in the zygote. It is still unclear why MSCI occurs. One possible explanation is to prevent the transcription of template chromosomal DNA with unrepaired DSBs, because DSB repair is delayed in unsynapsed sex chromosomes (Cloutier and Turner, 2010; Inagaki et al., 2010). Indeed, mediator of DNA damage checkpoint 1 (MDC1), which binds to the DSB-induced γH2AX, recruits ATR kinase and induces chromosome-wide spreading of the γH2AX leading to the MSCI (Ichijima et al., 2011).
To date, the meiotic behavior of translocated chromosomes has been extensively studied by light microscopic and electron microscopic examinations of the SC in the testicular tissues of infertile men with balanced translocation. A large body of data has been accumulated, demonstrating that translocated chromosomes are often located near the sex chromosomes and occasionally interacted with them (Figure 2B; Luciani et al., 1984; Rosenmann et al., 1985; Saadallah and Hulten, 1985; Chandley et al., 1986; Gabriel-Robez et al., 1986; Johannisson et al., 1987). In the pachytene spermatocytes of these translocation carriers, not only the asynapsed univalents, but the multivalents are also associated with sex chromosomes and the sex body, supporting the observation that at least small regions around the breakpoints are unsynapsed in multivalents. Later, interactions between the sex chromosome and translocated chromosome were clearly confirmed by immunohistochemistry with antibodies against the SC component in combination with fluorescence in situ hybridization technique (Oliver-Bonet et al., 2005a). Notably, these autosome–sex body associations have been observed in translocation carriers with azoospermia, but have never been seen in normozoospermic carriers (Oliver-Bonet et al., 2005b). Thus, the autosome–sex body association is most likely involved with the etiology of male infertility in translocation carriers.
Meiotic Silencing of the Unsynapsed Chromosome
Turner et al. (2005) performed elegant experiments using mice with various types of sex chromosome aneuploidy, demonstrating that MSCI is essentially the transcriptional silencing of genes located in the unsynapsed region, which is called meiotic silencing of unsynapsed chromosomes (MSUC; Schimenti, 2005; Turner et al., 2005). This phenomenon had been reported earlier in yeast as a mechanism for protecting the genome against invasion from transposable elements, but it also appears to play a vital role in the surveillance of stage progression during meiosis (Shiu et al., 2001; Kelly and Aramayo, 2007). It is not unreasonable to hypothesize that MSUC, because it operates in indispensable autosomal genes, might possibly function as surveillance against synapsis failure. Interestingly, Spo11-deficient mice, having no meiotic DSBs and, thereby, manifesting extensive asynapsis, demonstrate the formation of a nuclear domain that is similar to the sex body in both males and females, independent of the sex chromosomes, which is called the pseudo-sex body (Barchi et al., 2005; Bellani et al., 2005). Some of the proteins that participate in heterochromatin formation in MSCI, such as BRCA1, γH2AX, and ATR, are also localized within the pseudo-sex body, suggesting that transcription is silenced in this chromatin domain. This implicates that cell death via MSUC might contribute to the DSB-independent surveillance mechanism against synapsis defects.
The evidence of an autosome–sex body association raises a reasonable hypothesis that MSUC operating in unsynapsed regions of the translocation chromosomes contributes to the infertility of the male translocation carrier. Indeed, translocated chromosomes are consistently observed within the transcriptionally inactivated chromatin domain of sex body marked with γH2AX staining (Sciurano et al., 2007). Inactivation of the autosome most likely results in male infertility, at least according to data indicating that most X; autosome translocation results in male infertility regardless of the location of the breakpoint (Madan, 1983). In the case of X; autosome translocation, many of the indispensable genes around the breakpoint of the autosome are involved in the sex body and silenced leading to cell death. In the case of reciprocal translocation between autosomes, the autosome–sex body association most likely inactivates genes near the breakpoints (Homolka et al., 2007; Ferguson et al., 2008). According to this hypothesis, the reproductive outcome of the translocation depends on the breakpoints location and the number and/or nature of the genes that are inactivated. Regardless, inactivation of autosomal genes might possibly render a subset of male translocation carriers infertile.
This hypothesis, in the context of the autosome–sex body association, appears to illustrate the differences in reproductive outcomes between males and females. However, this hypothesis contains an intrinsic paradox regarding the involvement of MSUC/MSCI as the mechanism that eliminates cells with synapsis failure. Given that MSCI is a simple manifestation of MSUC that operates in the heteromorphic sex chromosomes of males, the MSUC should also operate in females. Indeed, mouse experiments have demonstrated the presence of MSUC in both males and females (Turner et al., 2005). Several lines of evidence also suggest that human females also possess a kind of surveillance system for synapsis. Female infertility is a common symptom in females with Turner syndrome and the 45, X karyotype, where X chromosome often remains unsynapsed. Examination of the ovaries of females with Turner syndrome reveals the depletion of pachytene oocytes (Speed, 1986). In Turner females with various structural abnormalities of the X chromosome, the size of the unsynapsed region appears to correlate with the severity of the sexual phenotype, suggesting that synapsis failure triggers apoptosis in oocytes (Ogata and Matsuo, 1995). These data suggest that the unsynapsed chromosome is detected by a quality control system even in oogenesis. According to this hypothesis, the surveillance system should respond to regional unsynapsis around the translocation breakpoints of the quadrivalent that leads to meiotic arrest both in males and females. This does not appear to reconcile with male-only infertility.
Here, there are two lines of conceptual thinking about the involvement of MSUC in synapsis failure that subsequently leads to the cell death of spermatocytes. One is that MSUC itself inactivates the indispensable autosomal genes, thereby leading to cell death. The inactivation of the involved autosomal segments might be enhanced by the presence of the sex body in pachytene spermatocytes, which can potentially explain the sex-specific difference in stringency of the system leading to male-only infertility. The alternative is MSCI failure, which means the derepression of transcription in sex chromosomal genes that leads to the subsequent apoptosis (Mahadevaiah et al., 2008). The unsynapsed region of the translocated chromosomes might inhibit the assembly of MSUC proteins in the sex body, although it is unclear whether a partially unsynapsed region is enough to impact this inhibitory effect. In addition, heterosynapsis between autosomes and sex chromosomes, which has been occasionally observed, might inhibit the silencing of the sex chromosomes. MSCI failure only happens in males, which might account for the sexual differences in the robustness of the checkpoints for synapsis in the presence of translocation.
Identification of Synapsis Checkpoint Protein in Mammals
To further investigate the mechanisms of the pachytene checkpoints at the molecular level, we searched for candidate genes that are essential to synapsis by examining the expression profiles of mouse genes expressed during the prophase of meiosis I (Kogo et al., 2010). Among the hundreds of candidate genes, we focused on Hormad1, the mammalian ortholog of yeast Hop1, by its potential function in the synapsis surveillance system. Hormad1 is a HORMA domain protein that is located on the SC in the unsynapsed region (Wojtasz et al., 2009; Fukuda et al., 2010). Recently, we and others have reported detailed analyses of Hormad1-deficient mice. Hormad1-deficient mice are infertile and demonstrate extensive failure of homologous pairing and synapsis in both males and females (Shin et al., 2010; Daniel et al., 2011; Kogo et al., 2012). In males, spermatogenesis arrests during the pachytene due to the severe synapsis failure, and all spermatogenic cells undergo stage IV apoptosis. Unexpectedly, Hormad1-deficient ovaries contain a normal number of oocytes, despite the extensive asynapsis, and consequently produce aneuploid oocytes that lead to subsequent fetal death in utero (Figures 3A,B). The failure to eliminate oocytes with extensive synapsis failure in the Hormad1-deficient ovary prompted the hypothesis that Hormad1 itself might be required for the mammalian pachytene checkpoint mechanism.
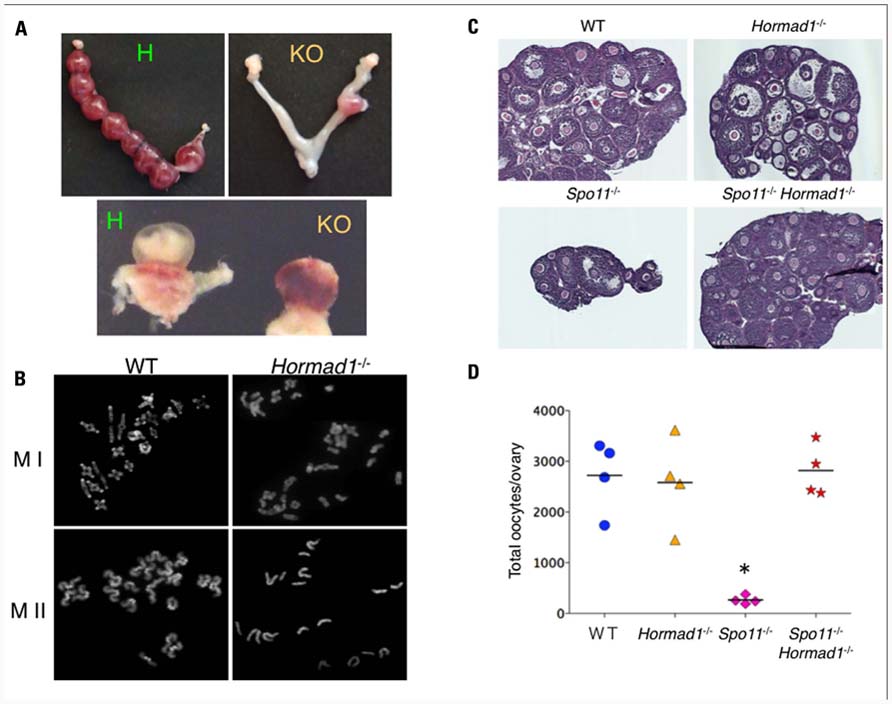
FIGURE 3. Hormad1 deficiency abrogates massive apoptosis in Spo11 deficiency.(A) Representative image of a uterus. Hormad1-deficient females carry only a small number of implantation sites (right) compared with the control (left). The lower panel shows a representative image of a conceptus from a Hormad1-deficient female with a non-developing fetus (right). (B) Representative data of the first meiotic metaphase (MI) of wild-type (WT) and Hormad1-deficient oocytes with only a small number of bivalents (right). The lower panel shows a representative photograph of the second meiotic metaphase (MII) of Hormad1-deficient oocytes with some monovalents (right). (C) Hematoxylin and eosin staining of ovary sections from 20-day-old female mice. The ovaries from Spo11-deficient mice are small with only a small number of follicles, while Spo11/Hormad1 double-knockout mice demonstrate the same size and oocyte number as the wild-type levels. (D) The number of oocytes in 20-day-old mice ovaries. The c-Kit positive oocytes are almost absent in the Spo11-deficient ovary, but are abundant in the Spo11/Hormad1 double-knockout ovaries (*P <0.05).
To exclude the effect of the DSB-dependent pachytene checkpoint and to independently analyze the checkpoint for synapsis, we utilized the Spo11-deficient mice that do not demonstrate meiotic DSB and thus do not demonstrate an active DSB-dependent pathway. Analysis of Hormad1/Spo11 double-knockout mice demonstrated that Hormad1 deficiency abrogates the massive oocyte loss that takes place in Spo11-deficient ovaries (Figures 3C,D). The double-mutant spermatocytes still undergo stage IV apoptosis, but they evade arrest at the zygotene/pachytene transition and the timing of cell death is slightly delayed (Kogo et al., unpublished data). These results raise speculation that the Hormad1 plays an important role in the DSB-independent pachytene checkpoint of both males and females, and possibly the surveillance for synapsis. Our present data thus highlight a novel DNA damage-independent synapsis checkpoint of mammalian meiosis.
Because spermatocytes still undergo apoptosis in male double-mutants, these mutants allowed us to analyze the male-specific backup pathway that is available for when the Hormad1-dependent pachytene checkpoint does not work. In Hormad1/Spo11 double-knockout males, derepression of sex chromosome genes was observed (Kogo et al., 2012). In the absence of Hormad1, spermatocytes fail to form the sex body. This might be due in part to the proteins required for heterochromatin formation, such as BRCA1, γH2AX, and ATR, that cannot be recruited to form the specialized chromatin domain if extensive asynapsis occurs, causing them to remain localized at DSB sites in unsynapsed regions (Mahadevaiah et al., 2008). Furthermore, in the absence of DSB, these components form the pseudo-sex body, as mentioned above, and do not assemble around the sex chromosomes. Consequently, transcriptional repression of the sex chromosome genes is impaired in male double-mutants, and this MSCI failure leads to spermatocyte apoptosis. Our data clearly demonstrate that one of these pathways acts as the male-specific surveillance system for synapsis failure.
Next, we set out to determine what mechanism triggers apoptosis at the Hormad1-dependent pachytene checkpoint for synapsis that operates both in males and females. One plausible idea is the transcriptional silencing of autosomes via MSUC. As mentioned above, Spo11 deficiency induces the formation of the pseudo-sex body, within which the transcription of autosomes might be silenced, possibly leading to cell death. Hormad1 deficiency abrogates formation of the pseudo-sex body in Hormad1/Spo11 double-knockout females (Daniel et al., 2011; Kogo et al., 2012). Therefore, MSUC within the pseudo-sex body might be the cause of cell death in Spo11-deficient mice and, furthermore, it appears to be Hormad1-dependent. On the other hand, another pathway that is not involved in MSUC also underlies the etiology of Hormad1-dependent cell death in synapsis failure (Kouznetsova et al., 2009). We found that Hormad1 is abundant at the unsynapsed chromosomes axes and highly phosphorylated in Spo11-deficient mice (Kogo et al., 2012). We propose the hypothesis that extensive asynapsis activates the pachytene checkpoint signaling pathway via the phosphorylation of Hormad1, thereby leading to stage IV apoptosis in both males and females. A possible mechanism is illustrated in Figure 4A.
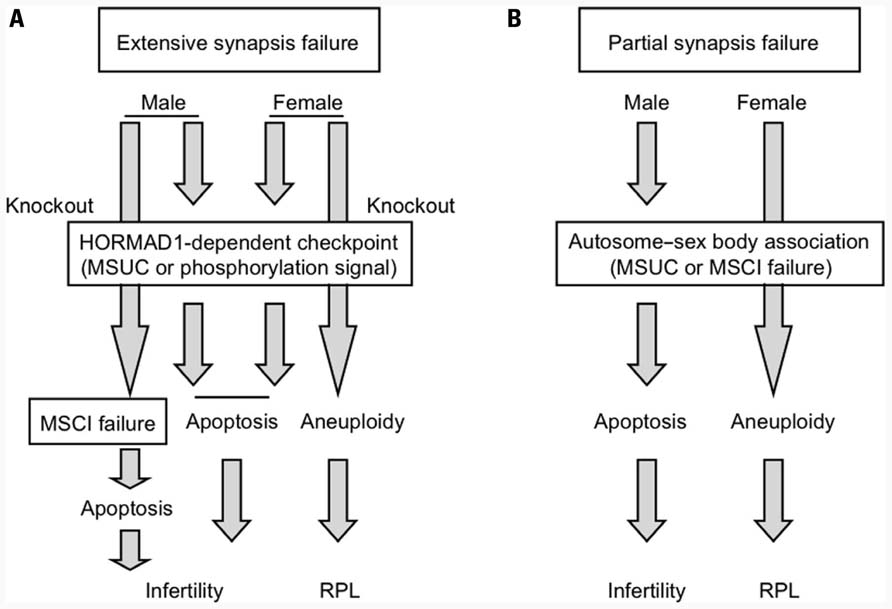
FIGURE 4. Putative mechanism of the DSB-independent surveillance system for synapsis during meiosis I. (A) Surveillance system for detecting extensive synapsis failure that is typically observed in Spo11 deficiency. (B) Surveillance system for detecting partial synapsis failure that is typically observed in translocation carriers.
Future Perspectives
To analyze the DSB-independent synapsis surveillance system independent of the DSB-dependent pachytene checkpoint, we had to utilize Spo11-knockout mice. However, Spo11 deficiency is a specific situation where most of the chromosomes are unsynapsed. It is still unknown whether the Hormad1-dependent pathway contributes to the consequence of partial unsynapsis due to translocation or other chromosomal abnormalities during normal meiotic prophase. Given the sexual dimorphism of the reproductive outcomes between male and female translocation carriers, the autosome–sex body association should provide useful clues regarding the molecular nature of the checkpoint mechanism for partial unsynapsis in spermatogenesis, regardless if this mechanism involves MSUC or MSCI failure (Figure 4B). If MSUC is the mechanism, Hormad1 might be an important part of the surveillance system, even during partial synapsis failure, because formation of the sex body is dependent on Hormad1, whereas in MSCI failure Hormad1 might not be essential. To date, there are a couple of studies on the etiology of infertile male mice with reciprocal translocation that have attempted to determine which mechanism is essential, but this is still an open question (Jaafar et al., 1993; Homolka et al., 2007). To evaluate these possibilities, investigations on spermatogenesis in male infertile mice with reciprocal translocation under conditions of deficiency in MSUC component are needed.
On the other hand, it may also be possible that such autosome–sex body interactions do not necessarily lead to cell death. Infertile males with the Robertsonian translocation, whose spermatocytes show autosome–sex body association, mostly manifest as oligozoospermia, not azoospermia, suggesting that spermatocytes with partial unsynapsis could develop beyond the pachytene (Navarro et al., 1991). Indeed, mouse spermatocytes with the Robertsonian translocation manifest as partial synapsis failure with some evidence of MSUC, but can develop beyond the diplotene stage to undergo cell death at metaphase, possibly via the spindle assembly checkpoint (SAC; Eaker et al., 2001; Manterola et al., 2009). In addition to the reduced stringency of the oocyte SAC, attachment of a quadrivalent to the sex body might generate a problem and trigger male-specific effects at SAC during meiosis I (Oliver-Bonet et al., 2005a; Nagaoka et al., 2011). The involvement of SAC in the reproductive failure of translocation carriers warrants further investigations.
Another unsolved question is why some translocation carriers suffer from male infertility and others do not. The breakpoint location should have a considerable impact on MSUC and the reproductive outcome. Accordingly, Robertsonian translocations are expected to be less severe than reciprocal translocations because the breakpoints are located at the centromeric repeats that constitutes heterochromatin without any important genes, but in reality this is not true (Page et al., 1996). In non-Robertsonian reciprocal translocations, acrocentric chromosomes are frequently involved in cases of male infertility, but a relatively low rate of autosome–sex body association has been observed in reciprocal translocations not involved in acrocentric chromosomes (Chandley et al., 1986; Guichaoua et al., 1990). The close proximity of the sex body and nucleolus during pachytene stage might affect the susceptibility of MSUC to Robertsonian translocations (Knibiehler et al., 1981; Tsutsumi et al., 2011).
In addition, there is one intriguing example that demonstrates whether infertility is dependent or independent of the location of the translocation breakpoints. In males with recurrent t(11;22)(q23;q11)s, only a subset of male translocation carriers are infertile, although they all possess almost identical breakpoints (Kurahashi et al., 2000). It can be hypothesized that the etiology behind synapsis failure-related infertility due to is multifactorial and that genetic factors govern the robustness of the synapsis checkpoint, thereby determining one’s susceptibility to infertility. Whole genome genotyping analysis of patients with reproductive failure and fertile controls might allow us to identify the risk factors and protective variants that might be located within the genes participating in the synapsis checkpoint (Aston and Carrell, 2009; Hu et al., 2011).
Together, MSUC and MSCI failure via autosome–sex body association might possibly render a subset of male translocation carriers infertile. Thus, this male-only infertility most likely contributes to the female predominance of the transmission of translocated chromosomes. The identification of Hormad1, which is the driver of the checkpoint system for extensive synapsis failure under conditions of Spo11 deficiency, might allow us to elucidate the mechanism that governs the surveillance system for partial unsynapsis that has actually observed in cases of human reproductive failure caused by translocation. In the future, the identification of putative variants that add susceptibility to the checkpoint robustness will allow evidence-based risk assessments of reproductive failure, infertility or RPL, due to chromosomal abnormalities.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Takaya Abe (RIKEN CDB, Japan) for the generation of Hormad1-deficient mice, and Drs. Maria Jasin and Scott Keeney (Memorial Sloan-Kettering Cancer Center, NY, USA) for providing Spo11-deficient mice. These studies were supported by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
Aston, K. I., and Carrell, D. T. (2009). Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J. Androl. 30, 711–725.
Baarends, W. M., and Grootegoed, J. A. (2003). Chromatin dynamics in the male meiotic prophase. Cytogenet. Genome Res.103, 225–234.
Barchi, M., Mahadevaiah, S., Di Giacomo, M., Baudat, F., de Rooij, D. G., Burgoyne, P. S., Jasin, M., and Keeney, S. (2005). Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol. Cell Biol. 25, 7203–7215.
Baudat, F., Buard, J., Grey, C., Fledel-Alon, A., Ober, C., Przeworski, M., Coop, G., and de Massy, B. (2010). PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840.
Baudat, F., Manova, K., Yuen, J. P., Jasin, M., and Keeney, S. (2000). Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol. Cell 6, 989–998.
Bellani, M. A., Romanienko, P. J., Cairatti, D. A., and Camerini-Otero, R. D. (2005). SPO11 is required for sex-body formation, and Spo11 heterozygosity rescues the prophase arrest of Atm-/- spermatocytes. J. Cell Sci. 118, 3233–3245.
Bolor, H., Mori, T., Nishiyama, S., Ito, Y., Hosoba, E., Inagaki, H., Kogo, H., Ohye, T., Tsutsumi, M., Kato, T., Tong, M., Nishizawa, H., Pryor-Koishi, K., Kitaoka, E., Sawada, T., Nishiyama, Y., Udagawa, Y., and Kurahashi, H. (2009). Mutations of the SYCP3 gene in women with recurrent pregnancy loss. Am. J. Hum. Genet. 84, 14–20.
Borde, V., Robine, N., Lin, W., Bonfils, S., Géli, V., and Nicolas, A. (2009). Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J. 28, 99–111.
Buard, J., Barthès, P., Grey, C., and de Massy, B. (2009). Distinct histone modifications define initiation and repair of meiotic recombination in the mouse. EMBO J. 28, 2616–2624.
Burgoyne, P. S., Mahadevaiah, S. K., and Turner, J. M. (2009). The consequences of asynapsis for mammalian meiosis. Nat. Rev. Genet. 10, 207–216.
Carter, M. T., St. Pierre, S. A., Zackai, E. H., Emanuel, B. S., and Boycott, K. M. (2009). Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): clinical features of 63 individuals. Am. J. Med. Genet. A 149A, 1712–1721.
Chandley, A. C., Speed, R. M., McBeath, S., and Hargreave, T. B. (1986). A human 9;20 reciprocal translocation associated with male infertility analyzed at prophase and metaphase I of meiosis. Cytogenet. Cell Genet. 41, 145–153.
Cloutier, J. M., and Turner, J. M. (2010). Meiotic sex chromosome inactivation. Curr. Biol. 20, R962–R963.
Cohen, P. E., Pollack, S. E., and Pollard, J. W. (2006). Genetic analysis of chromosome pairing, recombination, and cell cycle control during first meiotic prophase in mammals. Endocr. Rev. 27, 398–426.
Daniel, K., Lange, J., Hached, K., Fu, J., Anastassiadis, K., Roig, I., Cooke, H. J., Stewart, A. F., Wassmann, K., Jasin, M., Keeney, S., and Tóth, A. (2011). Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat. Cell Biol. 13, 599–610.
de Rooij, D. G., and de Boer, P. (2003). Specific arrests of spermatogenesis in genetically modified and mutant mice. Cytogenet. Genome Res. 103, 267–276.
Di Giacomo, M., Barchi, M., Baudat, F., Edelmann, W., Keeney, S., and Jasin, M. (2005). Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc. Natl. Acad. Sci. U.S.A. 102, 737–742.
Eaker, S., Pyle, A., Cobb, J., and Handel, M. A. (2001). Evidence for meiotic spindle checkpoint from analysis of spermatocytes from Robertsonian-chromosome heterozygous mice. J. Cell Sci. 114, 2953–2965.
Egozcue, S., Blanco, J., Vendrell, J. M., García, F., Veiga, A., Aran, B., Barri, P. N., Vidal, F., and Egozcue, J. (2000). Human male infertility: chromosome anomalies, meiotic disorders, abnormal spermatozoa and recurrent abortion. Hum. Reprod. Update 6, 93–105.
Ferguson, K. A., Chow, V., and Ma, S. (2008). Silencing of unpaired meiotic chromosomes and altered recombination patterns in an azoospermic carrier of a t(8;13) reciprocal translocation. Hum. Reprod. 23, 988–995.
Fryns, J. P., Kleczkowska, A., Kubień, E., Petit, P., and Van den Berghe, H. (1984). Cytogenetic survey in couples with recurrent fetal wastage. Hum. Genet. 65, 336–354.
Fukuda, T., Daniel, K., Wojtasz, L., Toth, A., and Höög, C. (2010). A novel mammalian HORMA domain-containing protein, HORMAD1, preferentially associates with unsynapsed meiotic chromosomes. Exp. Cell Res. 316, 158–171.
Gabriel-Robez, O., Ratomponirina, C., Dutrillaux, B., Carré-Pigeon, F., and Rumpler, Y. (1986). Meiotic association between the XY chromosomes and the autosomal quadrivalent of a reciprocal translocation in two infertile men, 46,XY,t(19;22) and 46,XY,t(17;21). Cytogenet. Cell Genet. 43, 154–160.
Gardner, R. J., and Sutherland, G. R. (2004). Chromosome Abnormalities and Genetic Counseling, 3rd Edn. New York: Oxford University Press.
Guichaoua, M. R., Quack, B., Speed, R. M., Noel, B., Chandley, A. C., and Luciani, J. M. (1990). Infertility in human males with autosomal translocations: meiotic study of a 14;22 Robertsonian translocation. Hum. Genet. 86, 162–166.
Handel, M. A. (2004). The XY body: a specialized meiotic chromatin domain. Exp. Cell Res. 296, 57–63.
Handel, M. A., and Schimenti, J. C. (2010). Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 11, 124–136.
Hassold, T., Sherman, S., and Hunt, P. (2000). Counting cross-overs: characterizing meiotic recombination in mammals. Hum. Mol. Genet. 9, 2409–2419.
Hochwagen, A., and Amon, A. (2006). Checking your breaks: surveillance mechanisms of meiotic recombination. Curr. Biol. 16, R217–R228.
Homolka, D., Ivanek, R., Capkova, J., Jansa, P., and Forejt, J. (2007). Chromosomal rearrangement interferes with meiotic X chromosome inactivation. Genome Res. 17, 1431–1437.
Hoyer-Fender, S. (2003). Molecular aspects of XY body formation. Cytogenet. Genome Res. 103, 245–255.
Hu, Z., Xia, Y., Guo, X., Dai, J., Li, H., Hu, H., Jiang, Y., Lu, F., Wu, Y., Yang, X., Li, H., Yao, B., Lu, C., Xiong, C., Li, Z., Gui, Y., Liu, J., Zhou, Z., Shen, H., Wang, X., and Sha, J. (2011). A genome-wide association study in Chinese men identifies three risk loci for non-obstructive azoospermia. Nat. Genet. 44, 183–186.
Hunt, P., and Hassold, T. (2010). Female meiosis: coming unglued with age. Curr. Biol. 20, R699–R702.
Ichijima, Y., Ichijima, M., Lou, Z., Nussenzweig, A., Camerini-Otero, R. D., Chen, J., Andreassen, P. R., and Namekawa, S. H. (2011). MDC1 directs chromosome-wide silencing of the sex chromosomes in male germ cells. Genes Dev. 25, 959–971.
Inagaki, A., Schoenmakers, S., and Baarends, W. M. (2010). DNA double strand break repair, chromosome synapsis and transcriptional silencing in meiosis. Epigenetics 5, 255–266.
Jaafar, H., Gabriel-Robez, O., and Rumpler, Y. (1993). Chromosomal anomalies and disturbance of transcriptional activity at the pachytene stage of meiosis: relationship to male sterility. Cytogenet. Cell Genet. 64, 273–280.
Johannisson, R., Löhrs, U., Wolff, H. H., and Schwinger, E. (1987). Two different XY-quadrivalent associations and impairment of fertility in men. Cytogenet. Cell Genet. 45, 222–230.
Kelly, W. G., and Aramayo, R. (2007). Meiotic silencing and the epigenetics of sex. Chromosome Res. 15, 633–651.
Knibiehler, B., Mirre, C., Hartung, M., Jean, P., and Stahl, A. (1981). Sex vesicle-associated nucleolar organizers in mouse spermatocytes: localization, structure, and function. Cytogenet. Cell Genet. 31, 47–57.
Kogo, H., Kowa-Sugiyama, H., Yamada, K., Bolor, H., Tsutsumi, M., Ohye, T., Inagaki, H., Taniguchi, M., Toda, T., and Kurahashi, H. (2010). Screening of genes involved in chromosome segregation during meiosis I: toward the identification of genes responsible for infertility in humans. J. Hum. Genet. 55, 293–299.
Kogo, H., Tsutsumi, M., Ohye, T., Inagaki, H., Abe, T., and Kurahashi, H. (2012). HORMAD1-dependent checkpoint/surveillance mechanism eliminates asynaptic oocytes. Genes Cells 17, 439–454.
Kota, S. K., and Feil, R. (2010). Epigenetic transitions in germ cell development and meiosis. Dev. Cell 19, 675–686.
Kouznetsova, A., Wang, H., Bellani, M., Camerini-Otero, R. D., Jessberger, R., and Höög, C. (2009). BRCA1-mediated chromatin silencing is limited to oocytes with a small number of asynapsed chromosomes. J. Cell Sci. 122, 2446–2452.
Kurahashi, H., Shaikh, T. H., Zackai, E. H., Celle, L., Driscoll, D. A., Budarf, M. L., and Emanuel, B. S. (2000). Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22). Am. J. Hum. Genet. 67, 763–768.
Kurahashi, H., and Emanuel, B. S. (2001). Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat. Genet. 29, 139–140.
Kurahashi, H., Bolor, H., Kato, T., Kogo, H., Tsutsumi, M., Inagaki, H., and Ohye, T. (2009). Recent advance in our understanding of the molecular nature of chromosomal abnormalities. J. Hum. Genet. 54, 253–260.
Kurahashi, H., Inagaki, H., Ohye, T., Kogo, H., Tsutsumi, M., Kato, T., Tong, M., and Emanuel, B. S. (2010). The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin. Genet. 78, 299–309.
Kurahashi, H., Tsutsumi, M., Nishiyama, S., Kogo, H., Inagaki, H., and Ohye, T. (2012). Molecular basis of maternal age-related increase in oocyte aneuploidy. Congenit. Anom. (Kyoto) 52, 8–15.
Leng, M., Li, G., Zhong, L., Hou, H., Yu, D., and Shi, Q. (2009). Abnormal synapses and recombination in an azoospermic male carrier of a reciprocal translocation t(1;21). Fertil. Steril. 91, 1293.e17-22.
Luciani, J. M., Guichaoua, M. R., Mattei, A., and Morazzani, M. R. (1984). Pachytene analysis of a man with a 13q;14q translocation and infertility. Behavior of the trivalent and nonrandom association with the sex vesicle. Cytogenet. Cell Genet. 38, 14–22.
Madan, K. (1983). Balanced structural changes involving the human X: effect on sexual phenotype. Hum. Genet. 63, 216–221.
Mahadevaiah, S. K., Bourc’his, D., de Rooij, D. G., Bestor, T. H., Turner, J. M., and Burgoyne, P. S. (2008). Extensive meiotic asynapsis in mice antagonises meiotic silencing of unsynapsed chromatin and consequently disrupts meiotic sex chromosome inactivation. J. Cell Biol. 182, 263–276.
Manterola, M., Page, J., Vasco, C., Berríos, S., Parra, M. T., Viera, A., Rufas, J. S., Zuccotti, M., Garagna, S., and Fernández-Donoso, R. (2009). A high incidence of meiotic silencing of unsynapsed chromatin is not associated with substantial pachytene loss in heterozygous male mice carrying multiple simple Robertsonian translocations. PLoS Genet. 5, e1000625. doi: 10.1371/journal.pgen.1000625
Matzuk, M. M., and Lamb, D. J. (2002). Genetic dissection of mammalian fertility pathways. Nat. Cell Biol. 4(Suppl.), S41–S49.
Miyamoto, T., Hasuike, S., Yogev, L., Maduro, M. R., Ishikawa, M., Westphal, H. and Lamb, D. J. (2003). Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet 362, 1714–1719.
Myers, S., Bowden, R., Tumian, A., Bontrop, R. E., Freeman, C., MacFie, T. S., McVean, G., and Donnelly, P. (2010). Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879.
Nagaoka, S. I., Hodges, C. A., Albertini, D. F., and Hunt, P. A. (2011). Oocyte-specific differences in cell-cycle control create an innate susceptibility to meiotic errors. Curr. Biol. 21, 651–657.
Namekawa, S. H., Park, P. J., Zhang, L. F., Shima, J. E., McCarrey, J. R., Griswold, M. D., and Lee, J. T. (2006). Postmeiotic sex chromatin in the male germline of mice. Curr. Biol. 16, 660–667.
Navarro, J., Vidal, F., Benet, J., Templado, C., Marina, S., and Egozcue, J. (1991). XY-trivalent association and synaptic anomalies in a male carrier of a Robertsonian t(13;14) translocation. Hum. Reprod. 6, 376–381.
Ogata, T., and Matsuo, N. (1995). Turner syndrome and female sex chromosome aberrations: deduction of the principal factors involved in the development of clinical features. Hum. Genet. 95, 607–629.
Ohye, T., Inagaki, H., Kogo, H., Tsutsumi, M., Kato, T., Tong, M., Macville, M. V., Medne, L., Zackai, E. H., Emanuel, B. S., and Kurahashi, H. (2010). Paternal origin of the de novo constitutional t(11;22)(q23;q11). Eur. J. Hum. Genet. 18, 783–787.
Oliver-Bonet, M., Ko, E., and Martin, R. H. (2005a). Male infertility in reciprocal translocation carriers: the sex body affair. Cytogenet. Genome Res. 111, 343–346.
Oliver-Bonet, M., Benet, J., Sun, F., Navarro, J., Abad, C., Liehr, T., Starke, H., Greene, C., Ko, E., and Martin, R. H. (2005b). Meiotic studies in two human reciprocal translocations and their association with spermatogenic failure. Hum. Reprod. 20, 683–688.
Page, S. L., Shin, J. C., Han, J. Y., Choo, K. H., and Shaffer, L. G. (1996). Breakpoint diversity illustrates distinct mechanisms for Robertsonian translocation formation. Hum. Mol. Genet. 5, 1279–1288.
Paoloni-Giacobino, A., Kern, I., Rumpler, Y., Djlelati, R., Morris, M. A., and Dahoun, S. P. (2000). Familial t(6;21)(p21.1;p13) translocation associated with male-only sterility. Clin. Genet. 58, 324–328.
Parvanov, E. D., Petkov, P. M., and Paigen, K. (2010). Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835.
Pittman, D. L., Cobb, J., Schimenti, K. J., Wilson, L. A., Cooper, D. M., Brignull, E., Handel, M. A., and Schimenti, J. C. (1998). Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol. Cell 1, 697–705.
Reinholdt, L. G., and Schimenti, J. C. (2005). Mei1 is epistatic to Dmc1 during mouse meiosis. Chromosoma 114, 127–134.
Romanienko, P. J., and Camerini-Otero, R. D. (2000). The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 6, 975–987.
Rosenmann, A., Wahrman, J., Richler, C., Voss, R., Persitz, A., and Goldman, B. (1985). Meiotic association between the XY chromosomes and unpaired autosomal elements as a cause of human male sterility. Cytogenet. Cell Genet. 39, 19–29.
Saadallah, N., and Hulten, M. (1985). A complex three breakpoint translocation involving chromosomes 2, 4, and 9 identified by meiotic investigations of a human male ascertained for subfertility. Hum. Genet. 71, 312–320.
Sciurano, R., Rahn, M., Rey-Valzacchi, G., and Solari, A. J. (2007). The asynaptic chromatin in spermatocytes of translocation carriers contains the histone variant gamma-H2AX and associates with the XY body. Hum. Reprod. 22, 142–150.
Shin, Y. H., Choi, Y., Erdin, S. U., Yatsenko, S. A., Kloc, M., Yang, F., Wang, P. J., Meistrich, M. L., and Rajkovic, A. (2010). Hormad1 mutation disrupts synaptonemal complex formation, recombination, and chromosome segregation in mammalian meiosis. PLoS Genet. 6, e1001190. doi: 10.1371/journal.pgen.1001190
Shiu, P. K., Raju, N. B., Zickler, D., and Metzenberg, R. L. (2001). Meiotic silencing by unpaired DNA. Cell 107, 905–916.
Solari, A. J. (1999). Synaptonemal complex analysis in human male infertility. Eur. J. Histochem. 43, 265–276.
Speed, R. M. (1986). Oocyte development in XO foetuses of man and mouse: the possible role of heterologous X-chromosome pairing in germ cell survival. Chromosoma 94, 115–124.
Sun, F., Oliver-Bonet, M., Turek, P. J., Ko, E., and Martin, R. H. (2005). Meiotic studies in an azoospermic human translocation (Y;1) carrier. Mol. Hum. Reprod. 11, 361–364.
Tsutsumi, M., Kogo, H., Kowa-Sugiyama, H., Inagaki, H., Ohye, T., and Kurahashi, H. (2011). Characterization of a novel mouse gene encoding an SYCP3-like protein that relocalizes from the XY body to the nucleolus during prophase of male meiosis I. Biol. Reprod. 85, 165–171.
Turner, J. M., Mahadevaiah, S. K., Ellis, P. J., Mitchell, M. J., and Burgoyne, P. S. (2006). Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev. Cell 10, 521–529.
Turner, J. M., Mahadevaiah, S. K., Fernandez-Capetillo, O., Nussenzweig, A., Xu, X., Deng, C. X., and Burgoyne, P. S. (2005). Silencing of unsynapsed meiotic chromosomes in the mouse. Nat. Genet. 37, 41–47.
Wojtasz, L., Daniel, K., Roig, I., Bolcun-Filas, E., Xu, H., Boonsanay, V., Eckmann, C. R., Cooke, H. J., Jasin, M., Keeney, S., McKay, M. J., and Toth, A. (2009). Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 5, e1000702. doi: 10.1371/journal.pgen.1000702
Yan, W., and McCarrey, J. R. (2009). Sex chromosome inactivation in the male. Epigenetics 4, 452–456.
Yang, F., De La Fuente, R., Leu, N. A., Baumann, C., McLaughlin, K. J., and Wang, P. J. (2006). Mouse SYCP2 is required for synaptonemal complex assembly and chromosomal synapsis during male meiosis. J. Cell Biol. 173, 497–507.
Yoshida, K., Kondoh, G., Matsuda, Y., Habu, T., Nishimune, Y., and Morita, T. (1998). The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol. Cell 1, 707–718.
Yuan, L., Liu, J. G., Hoja, M. R., Wilbertz, J., Nordqvist, K., and Hoog, C. (2002). Female germ cell aneuploidy and embryo death in mice lacking the meiosis-specific protein SCP3. Science 296, 1115–1118.
Keywords: synapsis, pachytene checkpoint, translocation, sex body, meiotic sex chromosome silencing, meiotic silencing of unsynapsed chromosomes, Hormad1
Citation: Kurahashi H, Kogo H, Tsutsumi M, Inagaki H and Ohye T (2012) Failure of homologous synapsis and sex-specific reproduction problems. Front. Gene. 3:112. doi: 10.3389/fgene.2012.00112
Received: 27 March 2012; Accepted: 30 May 2012;
Published online: 18 June 2012.
Edited by:
Ryan Yuen, The Hospital for Sick Children, CanadaReviewed by:
Amy C. Lossie, Purdue University, USAGirdhari Lal, National Centre for Cell Science, India
Copyright: © 2012 Kurahashi, Kogo, Tsutsumi, Inagaki and Ohye. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hiroki Kurahashi, Division of Molecular Genetics, Institute for Comprehensive Medical Science, Fujita Health University, 1-98 Dengakugakubo, Kutsukake-cho, Toyoake, Aichi 470-1192, Japan. e-mail:a3VyYUBmdWppdGEtaHUuYWMuanA=