
- Mirna Therapeutics, Inc., Austin, TX, USA
MicroRNA-34 (miR-34) is a master regulator of tumor suppression. It is downregulated in numerous cancers and inhibits malignant growth by repressing genes involved in various oncogenic signaling pathways. Consequently, miR-34 antagonizes processes that are necessary for basic cancer cell viability as well as cancer stemness, metastasis, and chemoresistance. This broad anti-oncogenic activity holds the prospect of creating a new remedy that is effective against tumor heterogeneity. This review focuses on the molecular mechanisms of miR-34-mediated tumor suppression, pharmacologies in animal models of cancer, and a status update of a miR-34 therapy that may be among the first miRNA mimics to reach the clinic.
Introduction
Results generated in recent years from genomic and proteomic approaches have changed our view on cancer. From a disease previously assumed to be manifested in the alteration of merely several genes, cancer is in fact genetically very complex. Tumor cells frequently harbor an array of mutated genes, and each tumor mass can comprise hundreds of cancer cells with distinct cancer genotypes (Jones et al., 2008; Parsons et al., 2008; Gerlinger et al., 2012). As a result, approaches to cancer therapies are shifting to accommodate these new findings and to devise more effective and safer remedies. Conventional therapies, still the prevailing form of therapy, are frequently deemed too toxic or inadequate due to chemoresistance. These treatments are followed or replaced by targeted therapies that intercept with a known oncogene. At an increasing rate, these targeted therapies are not solely prescribed based on the histotype but, rather, on the genetic cancer profile. Thus, those originally developed for a specific tumor type can also have activity in another, yet histologically unrelated cancer type with the same underlying mutation (Welch and Moore, 2007). However, many targeted therapies alone are insufficiently effective and will have to be used in combination. This confirms that cancer is the product of various genetic and epigenetic changes and requires interference with multiple oncogenic pathways for successful intervention.
The therapeutic use of miRNAs, a class of non-coding RNAs, has gained much attention because their mechanisms of action are in line with our current thinking of cancer as a pathway disease. miRNAs function as master regulators of the genome by modulating the expression of tens to hundreds of genes and by controlling several cellular pathways at once. Certain miRNAs are frequently deregulated in cancer cells and, consequently, so are pathways downstream. As a result, a cellular proto-oncogene can escape miRNA-mediated repression and, thereby, acquire a gain of function that is independent of mutations to the proto-oncogene itself (Johnson et al., 2005). A modest number of miRNAs shows a loss of function in a very broad range of cancer types, indicating that they play a central role in the regulation of tumor suppression. Among these are let-7, miR-34, and miR-200, all of which are evolutionarily conserved from plant to man and presumably control basic programs during cellular proliferation and differentiation. Correcting these deficiencies by miRNA replacement results in an activation of anti-proliferative and pro-apoptotic pathways, and cancer cell death (Bader et al., 2010; Wiggins et al., 2010). An obvious patient eligible for miRNA replacement would be one with miRNA levels that are lower in the tumor than the normal tissue. However, there is increasing evidence demonstrating that cancer cells with normal miRNA expression levels are also susceptible to miRNA treatment (Wiggins et al., 2010). A plausible explanation for this observation is the threshold phenomenon – it is likely that oncogenic pathways induced by mutations in other genes are controlled, albeit insufficiently counteracted, by the endogenous miRNA. Increasing the expression of the miRNA may enhance its antagonizing function beyond the threshold at which it becomes dominantly tumor suppressive.
Master Tumor Suppressor miR-34
One of the challenges that drug developers are faced with is picking the right target. This also applies to miRNAs. There are many miRNAs that are deregulated in cancerous tissues; however, only few of these induce robust phenotypes in cancer cells, and even fewer are potent enough to be used as therapeutics. Thus, similar to other cancer genes, it is possible that miRNAs play distinct roles in which only a subset function as “drivers” of cancer progression. In addition, several other miRNAs have been associated with both tumor suppressive and oncogenic functions, or assume roles in diseases outside oncology which makes their practical application as cancer therapeutics difficult to predict (Valastyan et al., 2009; Liu et al., 2010).
A miRNA for which the tumor suppressor role is well-defined is miR-34. Human miR-34 comprises three family members: miR-34a, miR-34b, and miR-34c. The mature miR-34a sequence consists of 22 nucleotides and shares 86% homology (19/22 nt) and 82% homology (18/22 nt) with miR-34b and miR-34c, respectively. Both miR-34b and miR-34c are 23 nucleotides in length and 83% homologous (19/23 nt). Residues identical between all three family members span across the entire miRNA sequence and include those in the “seed region,” a stretch of 8 nucleotides from position 2–9 adjacent to the 5′ terminus that guides the selection of target mRNAs. Due to this homology, the miR-34 family members control a similar set of target genes and appear to be functionally redundant (He et al., 2007). In normal human tissues, miR-34a is the prevailing family member (Hsu et al., 2008). In contrast, endogenous miR-34b/c levels are low in most tissues except lung, ovary, testes, and trachea (Hsu et al., 2008). The miR-34a gene is located on chromosome 1p36.22, and miR-34b and miR-34c are expressed from a polycistronic transcript encoded on chromosome 11q23.1. Both gene loci are in regions that have been associated with fragile sites of the genome that are frequently altered in cancer (Calin et al., 2004). Hypermethylation of the miR-34 promoters is another mechanism that can lead to reduced levels of endogenous miR-34 family members (Lodygin et al., 2008; Lujambio et al., 2008; Vogt et al., 2011). Thus, genetic and epigenetic mechanisms contribute to a loss of miR-34a expression that has been noted in a wide range of solid and hematological malignancies, including cancers of the lung (Bommer et al., 2007; Lodygin et al., 2008; Gallardo et al., 2009; Wiggins et al., 2010), prostate (Fujita et al., 2008; Lodygin et al., 2008), breast (Lodygin et al., 2008; Vogt et al., 2011), pancreas (Chang et al., 2007; Lodygin et al., 2008; Vogt et al., 2011), colon (Tazawa et al., 2007; Lodygin et al., 2008; Vogt et al., 2011), kidney (Lodygin et al., 2008; Vogt et al., 2011), liver (Li et al., 2009; Tryndyak et al., 2009), bladder (Lodygin et al., 2008), skin (Lodygin et al., 2008), esophagus (Chen et al., 2011), brain (Welch et al., 2007; Cole et al., 2008; Wei et al., 2008; Feinberg-Gorenshtein et al., 2009), cervix (Wang et al., 2009; Li et al., 2010), ovary (Kuo et al., 2009; Corney et al., 2010; Vogt et al., 2011), urothelium (Vogt et al., 2011), and the lymphoid system (Mraz et al., 2009; Chim et al., 2010; Craig et al., 2011). Consistent with this extensive aberrant expression profile, reintroducing mimics of miR-34a inhibits numerous cancer cell types and suggests that miR-34a functions at the core of tumorigenic processes shared among cancer cells. This anti-oncogenic activity of miR-34a has been demonstrated in cancer cell types of lung (Bommer et al., 2007; He et al., 2007; Raver-Shapira et al., 2007; Tarasov et al., 2007; Sun et al., 2008; Wiggins et al., 2010), liver (Li et al., 2009), pancreas (Lodygin et al., 2008; Ji et al., 2009), colon (Chang et al., 2007; He et al., 2007; Tazawa et al., 2007; Lize et al., 2009), brain (Welch et al., 2007; Cole et al., 2008; Wei et al., 2008; Luan et al., 2010; Li et al., 2011), skin (Yan et al., 2009; Greenberg et al., 2011), prostate (Lodygin et al., 2008; Liu et al., 2011), bone (Tarasov et al., 2007; Yan et al., 2012), ovary (Corney et al., 2010), as well as lymphoma and leukemia (Asslaber et al., 2010; Craig et al., 2011).
Molecular Mechanisms of miR-34 Tumor Suppression
How does miR-34a work? The study of miR-34a-induced phenotypes is beginning to outline a picture in which miR-34a acts as a master of tumor suppression (Figure 1). miR-34a can antagonize many different oncogenic processes by regulating genes that function in various cellular pathways (Table 1). A majority of these genes encode well-known proto-oncoproteins that represent attractive drug targets themselves. It is possible that the repression of only a few select targets is responsible for the miR-34a phenotype. Indeed, some of the miR-34a-regulated transcripts have directly been implicated during miR-34a-dependent tumor suppression (Craig et al., 2011). However, none were able to fully recapitulate the miR-34a phenotype, suggesting that the miR-34a function depends on multiple and perhaps the majority of all targets. The miR-34a-induced change on any of the targeted gene products is moderate and less than 1.23-fold for most proteins (Kaller et al., 2011). And yet, the collective repression of a wide range of target genes is likely to a have a significant impact on cellular phenotypes (Selbach et al., 2008). Interestingly, miR-34a targets downregulated on the protein level were also affected on the mRNA level which suggests that miR-34a simultaneously affects protein synthesis and mRNA abundance (Kaller et al., 2011).

FIGURE 1. Cancer processes counteracted by miR-34.
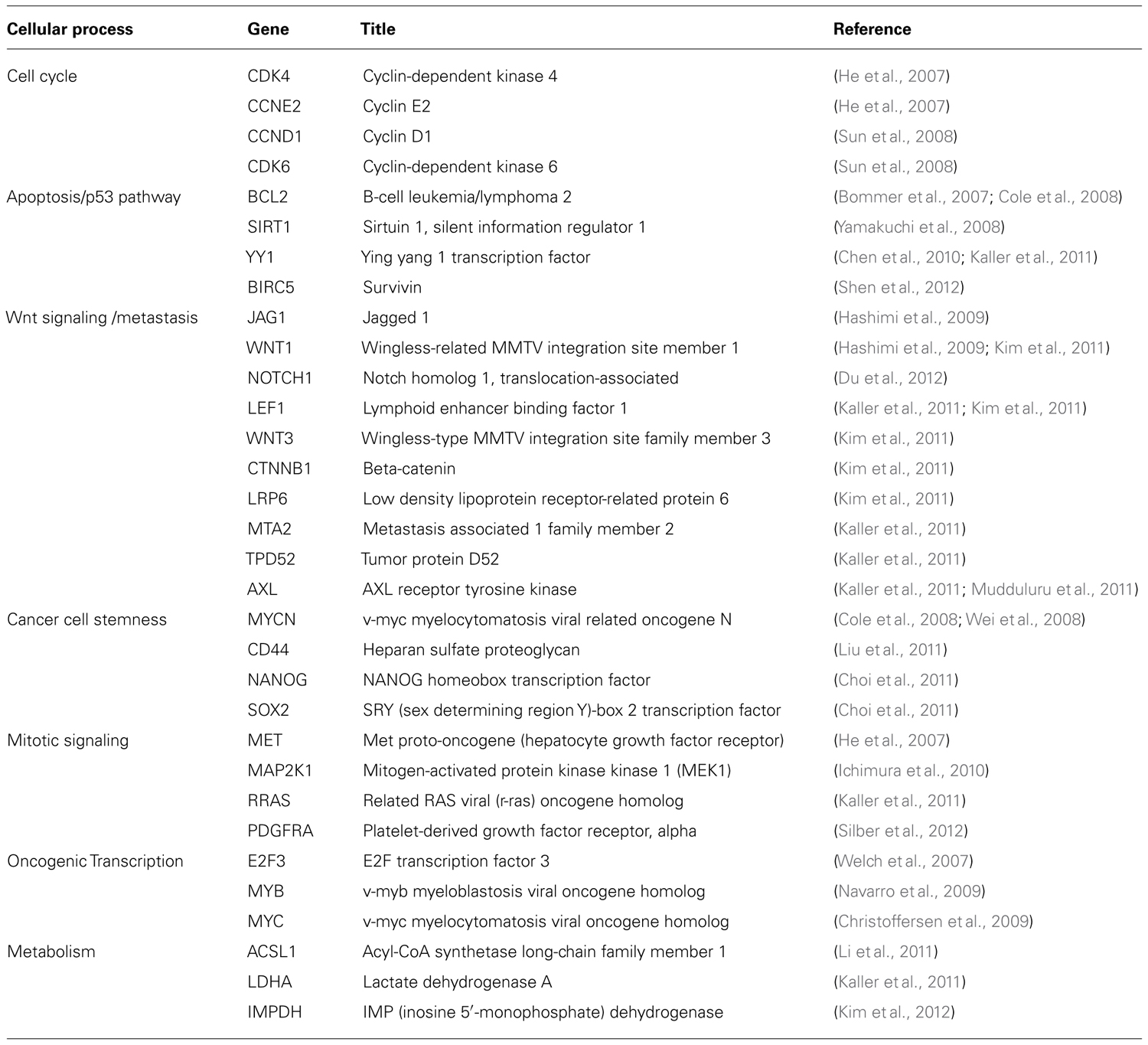
TABLE 1. Gene products directly repressed by miR-34a.
A major function attributed to miR-34a is the control of cellular proliferation, cell cycle, and senescence. In cancer cells, ectopic expression of miR-34a decreases cell doubling rates (Lodygin et al., 2008; Wiggins et al., 2010), leads to G1/G2 arrest (He et al., 2007; Lodygin et al., 2008), and transforms cells into large, flat bodies that stain positive for senescence-associated beta-galactosidase (He et al., 2007; Tazawa et al., 2007; Lodygin et al., 2008). miR-34a is upregulated during various forms of senescence, including quiescence, replicative senescence, H2O2-induced premature senescence, and senescence induced by an oncogene (Christoffersen et al., 2009; Maes et al., 2009). In agreement with these phenotypes are transcriptome and proteome profiles generated in miR-34a overexpressing cells. These results show that genes differentially expressed function predominantly in the regulation of the cell cycle (Bommer et al., 2007; Chang et al., 2007; He et al., 2007). Genes that are downregulated are enriched for those containing putative miR-34a binding sites, indicating that several interactions are direct. Transcripts for which the interactions with miR-34a have experimentally been validated include cyclins D1 and E2, as well as cyclin-dependent kinases 4 and 6. In addition, miR-34a directly represses a series of signaling molecules that drive cellular proliferation and play roles in the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways. These are mitogen-activated protein kinase kinase 1 (MEK1, MAP2K1), R-Ras (RRAS), platelet-derived growth factor receptor (PDGFRA), and hepatocyte growth factor receptor (MET).
The functions of miR-34a in the control of the cell cycle are complemented by those that regulate apoptosis. All miR-34 family members are transcriptionally induced by p53 and are important effector molecules in the execution of molecular programs directed by p53 (Bommer et al., 2007; Chang et al., 2007; He et al., 2007; Raver-Shapira et al., 2007; Tarasov et al., 2007). miR-34a represses BCL2 and survivin; however, it is possible that other mechanisms contribute to the pro-apoptotic functions of miR-34a (Bommer et al., 2007; Shen et al., 2012). Indeed, miR-34a can stimulate endogenous p53 activity in a positive feedback-loop by targeting SIRT1 (silent information regulator 1), a NAD-dependent deacetylase that deactivates TP53 (tumor protein 53), and YY1 (yin yang 1), a protein that binds to p53 and promotes p53 ubiquitination and degradation (Sui et al., 2004; Yamakuchi et al., 2008; Chen et al., 2010). Thus, it appears that miR-34a takes advantage of upstream signaling to maintain its own expression levels and to activate tumor suppressor pathways exclusively regulated by p53. However, the requirement for p53 in miR-34a-induced tumor suppression remains unknown. Emerging evidence suggests that endogenous p53 is not a prerequisite for miR-34a tumor suppression – miR-34a transcription can occur independently of p53, and miR-34a is capable of inhibiting cancer cells that lack endogenous p53 (Christoffersen et al., 2009; Wiggins et al., 2010).
A valuable aspect of miR-34a is its ability to inhibit cancer stem cells. Cancer stem cells, also known as tumor-initiating cells, are defined as a small fraction of cancer cells that have the ability to self-renew and to give rise to identical daughter cells (Reya et al., 2001). As such, cancer stem cells act as the “seed” of the tumor that has an enhanced propensity to form tumors in the animal, metastasize and withstand conventional cancer therapy. Thus, chemoresistance and recurrence are likely due to the presence of these tumor-initiating cells that ought to be eliminated for successful patient outcome. For instance, miR-34a levels are lower in prostate cancer stem cells that express either the CD44 or the CD133 epitope, and in those enriched in the side population when compared to the bulk of prostate tumor cells (Liu et al., 2011). Overexpression of miR-34a, either by transient transfection or stable genomic integration via a retrovirus, interferes with sphere formation in soft agar and tumorigenicity in vivo (Liu et al., 2011). Both are abilities that have specifically been associated with the presence of prostate cancer stem cells. miR-34a also inhibited the propagation properties of tumor-initiating cells derived from pancreatic carcinoma and medulloblastoma (Ji et al., 2009; de Antonellis et al., 2011). Thus, the relative amounts of endogenous miR-34a proportionally decline as cells transform from normal to cancer and to cancer stem-like cells. This progression appears to be functionally linked to tumor viability and abolished by miR-34 replacement.
The notion that the modulation of miR-34a abundance has a direct impact on pluripotency is supported by observations that implicate miR-34a in somatic cell reprogramming (Choi et al., 2011). miR-34a prevents cell reprogramming by directly inhibiting pluripotency genes, including NANOG, SOX2, and MYCN (Choi et al., 2011). Similarly, the ability to inhibit prostate cancer stem cells was causally linked to miR-34a-dependent repression of CD44 which may further define its specific effects on cancer stem cells (Liu et al., 2011). Hence, the repression of these pluripotency genes provides a molecular explanation for miR-34a functions that govern stemness and differentiation.
Other functions ascribed to miR-34a are during inhibition of metastasis and chemoresistance. Both may be a consequence of miR-34a-induced cancer stem cell elimination but may also be an independent result of miR-34a regulation in non-stem cancer cells. The Wnt signaling pathway is intimately involved in the transition from an epithelial to a mesenchymal state (EMT) and metastasis. It is subject to miR-34-dependent regulation at multiple levels. miR-34a and miR-34b/c directly repress the translation of WNT1 and WNT3, as well as LRP6, a co-receptor of Frizzled that binds to Wnt ligands (Kim et al., 2011). The miR-34 family members also represses LEF1 and beta-catenin (CTNNB1) that function as the primary transcriptional mediators downstream of Wnt. Accordingly, miR-34 interferes with phenotypes attributed to Wnt signaling such as a block of Wnt-induced axis duplication during Xenopus development, a decrease in the tissue-invasive activity of colorectal cancer cells and inhibition of TCF/LEF target genes (Kim et al., 2011). Interestingly, several of the genes transcriptionally induced by TCF/LEF are CCND1, MYC, and CD44 that are also independently repressed by miR-34a. The miRNA also downregulates other pro-metastatic genes not necessarily linked to the Wnt signaling pathway (MET, AXL, MTA2; He et al., 2007; Kaller et al., 2011). This corroborates the concept that the regulatory mechanisms of miRNAs involve multiple components within a given pathway and across several others.
The ability to impact multiple cellular pathways may also suggest that miR-34a can act synergistically with conventional, cytotoxic therapies. The modes of action of both drug classes are likely sufficiently distinct for the combination to provide an added benefit. This has experimentally been tested in cancer cell models of prostate, pancreas, colon, bladder, stomach, and brain. As shown in Table 2, miR-34a mitigates resistance to taxol- and platinum-based agents, as well as resistance to a few others. The effects of the combination in normal tissues are currently unknown. The mechanisms through which miR-34a exerts its chemo-sensitizing functions remain ill-defined and require further study; and yet, the preliminary data derived from these cell models may indicate that the utility of a future miR-34a therapeutic can be substantially expanded.
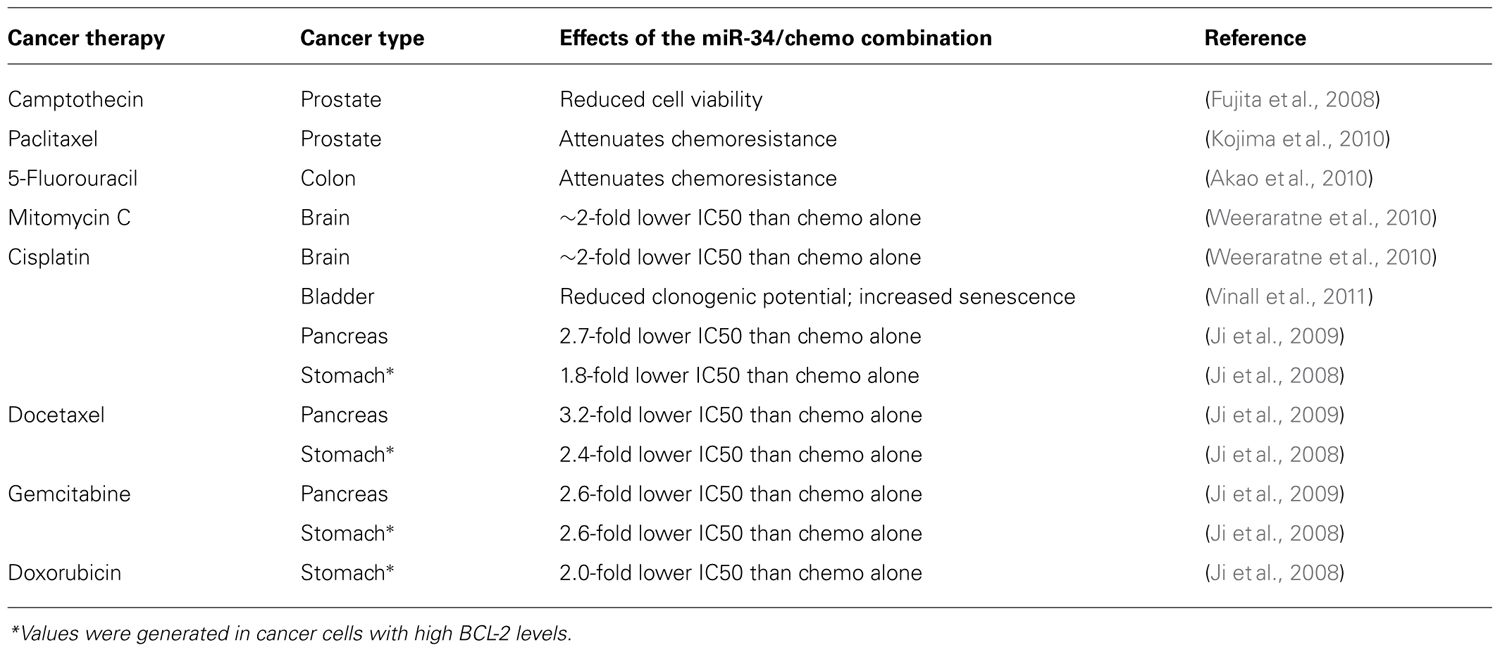
TABLE 2. Effects of miR-34a in combination with conventional therapies.
Pharmacology of miR-34 in Animal Models of Cancer
The biological activity of miR-34a identified in cell models suggests that it can also suppress tumor growth in vivo. Although a thorough assessment of miR-34a activity in animals primarily hinges on the performance of in vivo delivery systems, there are a few examples that illustrate its therapeutic potential (Table 3). A common approach to achieve miRNA expression in vivo is based on vectoral systems that function similarly to those used for traditional gene therapy (Esquela-Kerscher et al., 2008; Kumar et al., 2008; Kota et al., 2009; Qiu et al., 2011). These can be used either on xenografts before transplantation or systemically as nanoparticles by parenteral administration. Another approach is in form of nanoparticles that contain ~19–23-nt double-stranded miRNA mimics and can be administered by intravenous tail vein injections (Chen et al., 2010; Wiggins et al., 2010). Here, examples of the systemic delivery mode are discussed in further detail.
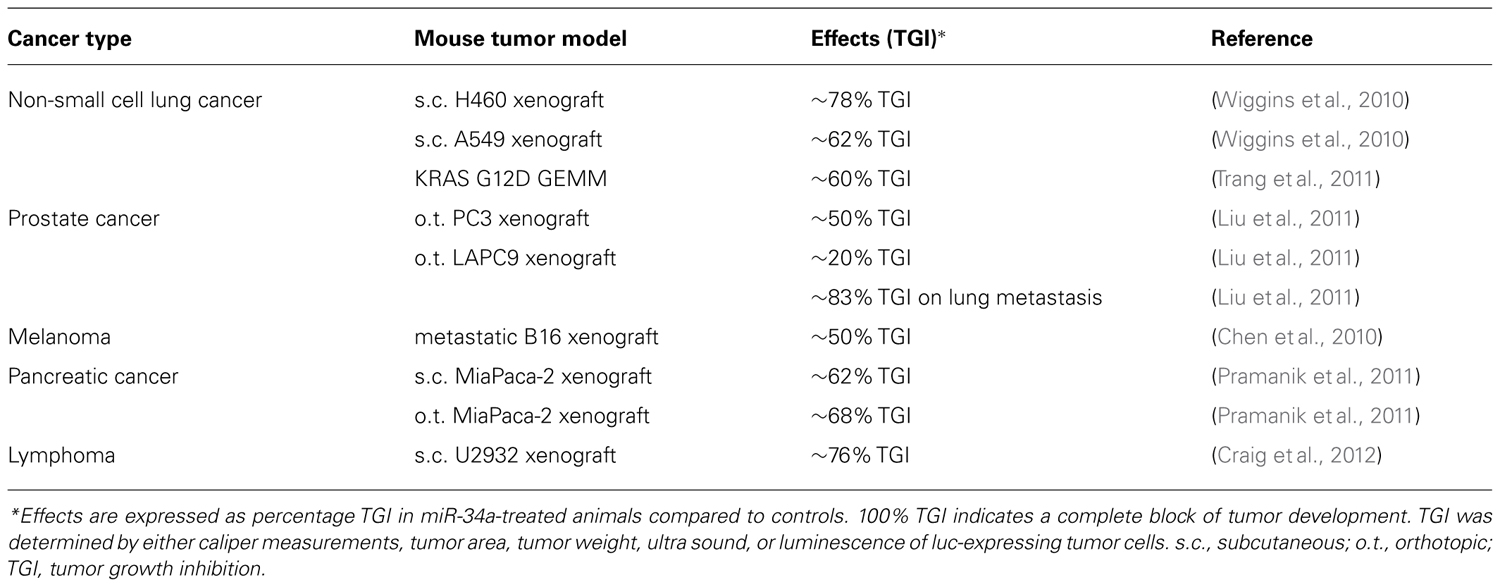
TABLE 3. Therapeutic activity of miR-34a in animal models of cancer.
One example of systemic delivery is provided by the intravenous delivery of miR-34a in polycationic liposome-hyaluronic acid nanoparticles that are modified with a GC4 single-chain antibody fragment (scFv; Chen et al., 2010). The antibody moiety facilitates tumor targeting to human glioma tumor sphere cells as well as murine B16 melanoma cells. To test the therapeutic utility, the formulation was administered on two consecutive days (0.3 mg/kg) to mice that had lung metastases induced by the B16F10 melanoma xenograft. Eleven days after the first treatment, the tumor burden was reduced by approximately 50% as assessed by luminescence imaging of metastatic lesions. In a separate experiment, miR-34a activity was confirmed by an increase of miR-34a-induced apoptosis and the downregulation of survivin protein levels in lung metastases.
Pramanik et al. (2011) investigated the therapeutic potential of miR-34a in subcutaneous and orthotopic pancreas MiaPaca-2 xenografts. miR-34a was delivered systemically using a 100 nm-lipid-based nanoparticle that contains a miR-34a-encoding vector. Animals with confirmed tumors were treated three times a week for three consecutive weeks, and tumor growth was determined by either caliper measurements or ultra sound. The data showed that tumors from mice treated with miR-34a were significantly smaller and showed signs of necrosis and apoptosis. Orthotopic tumors also displayed reduced levels of Ki67. In addition, SIRT1 protein as well as CD44 and ALDH mRNA levels were reduced in subcutaneous tumor tissues of miR-34a-treated animals.
A series of experiments were performed with a neutral lipid emulsion (NLE) that forms particles in the nanometer range and carry a net charge close to zero. Proof of concept for the therapeutic utility of the miR-34a/NLE formulation was generated in subcutaneous H460 and A549 non-small cell lung cancer xenografts (Wiggins et al., 2010). Mice carrying palpable H460 tumors received a dose of 3 mg/kg via intravenous tail vein injections every 3 days over a period of 9 days. A549-carrying animals were dosed at 1 mg/kg every other day for a total of 15 days. In comparison to controls, tumor growth of miR-34a-treated groups was inhibited by 78 and 62%, respectively. Both xenografts displayed an increase in apoptosis and a reduction in proliferation. The anti-oncogenic effects were accompanied by an accumulation of the miR-34a mimic in tumor tissues and a downregulation of MET, CDK4, and BCL2 protein levels (Wiggins et al., 2010).
Similar results were obtained in a genetically engineered mouse model of lung cancer that is based on the constitutively active KRAS mutant G12D (Trang et al., 2011). Ten weeks after the activation of the KRAS-G12D mutant and the initiation of orthotopic lung tumors, miR-34a mimics conjugated with NLE were systemically introduced every other day for a total of eight injections at a concentration of 1 mg/kg each time. At sacrifice, mice given a NLE formulation that contained a scrambled negative control oligonucleotide showed extensive diffuse hyperplasia and adenomas in lungs. In contrast, the lesions observed in miR-34a-treated animals were few and significantly smaller. The remaining tumor area was only 40% of the overall tumor area that was noted in controls. This result correlated with a significant elevation of miR-34a levels in lung tissue, reduced expression of Ki67 and an increase in TUNEL-positive cells.
In models of prostate cancer, the efficacy of miR-34a complexed in NLE was demonstrated by Liu et al. (2011). Orthotopic PC-3 tumors were generated by the surgical implantation of tumor cells into the dorsal prostate of male NOD/SCID mice and allowed to grow for 3 weeks. Then, mice were treated every other day by intravenous tail injections using a dose of 1 mg/kg per administration. After the fifth dose, mice were sacrificed to determine tumor weights. Tumors were half the weight of those from control animals (Liu et al., 2011). A similar experimental set-up was used to evaluate miR-34a activity in the orthotopic LAPC9 tumor xenograft. In these animals, the effect on the primary tumor was only moderate (20% tumor inhibition). However, animals in the miR-34a study group greatly benefitted from significantly enhanced survival, most likely due to a substantial decrease in LAPC9 metastasis to lung and other organs (Liu et al., 2011).
During the transition from low-grade to high-grade cancers, diffuse large B-cell lymphomas frequently show a gain of MYC function and a concurrent loss of miR-34a. Reintroduction of miR-34a mimics into cultured lymphoma cells proved to be more effective in inhibiting lymphoma cells than siRNAs directed against MYC or FOXP1, another key oncogene in this type of cancer. This suggests that miR-34a replacement is the preferred therapeutic modality in this model (Craig et al., 2011). To explore this experimentally, mice with existing subcutaneous U2932 lymphoma xenografts were given intravenous injections of the NLE/miR-34a formulation every other day. Each dose was equivalent to 1 mg/kg and was repeated eight times over 13 days. Caliper measurements indicated that miR-34a was able to reduce tumor growth by 76% which was also reflected in tumor weights. Thus, the data indicate that the therapeutic utility of miR-34a is not limited to solid tumor types and can also be applied to hematological malignancies (Craig et al., 2012).
The studies featuring NLE were carried out using double-stranded miR-34a mimics in which the active strand is identical to the endogenous miRNA. Therefore, non-specific off-target effects are unlikely because the mimic is expected to repress the same set of genes that is also regulated by the natural counterpart. However, the pleiotropism of miRNAs raises concerns about miR-34a-induced toxicity in normal tissues. It is possible that the induction of miRNA-regulated pathways that drives the elimination of cancer cells is also detrimental to normal cells. In discord with this view, however, the transient transfection of miR-34a had no noticeable effects in a series of non-cancerous cells (Wiggins et al., 2010). Likewise, animal studies evaluating the therapeutic effects of miR-34a formulations failed to detect unwanted side effects associated with miR-34a. These observations were based on mouse gross behavior, body weights, pathologic examination of organ morphologies, and blood chemistries indicative of injuries to liver, kidney, and muscle (Chen et al., 2010; Wiggins et al., 2010; Pramanik et al., 2011; Craig et al., 2012). In addition, formulated miR-34a mimics did not induce a non-specific immune response as assessed by serum cytokine levels in immunocompetent mice (Chen et al., 2010; Wiggins et al., 2010). The average level of endogenous miR-34a retrieved from most tissues ranges around 50,000 copies/ng RNA (Hsu et al., 2008). This is equivalent to approximately 500–1,000 copies/cell, given an overall RNA content of 10–20 pg/cell. Thus, the abundance of endogenous miR-34a in normal tissues suggests that pathways affected by exogenous miR-34a are already activated or deactivated by the endogenous miRNA. Therapeutically increasing the miRNA level may not sufficiently change the levels of target genes to impact the direction of these pathways and normal cell activities. Experimental evidence to support this hypothesis, however, is missing.
Development Progress of a miR-34 Therapy
The transition from bench to bedside of a miR-34a-based cancer therapy depends on the availability of a clinically relevant delivery system. Many technologies that were used to establish proof of concept in the animal are promising candidates but are not advanced far enough to demonstrate the robustness, scalable manufacture, and quality controls necessary to support clinical trials.
To accelerate the path to the clinic, Mirna Therapeutics screened multiple existing delivery systems that are in pre-clinical development or have already entered clinical testing with other oligonucleotide therapeutics. This evaluation encompassed an assessment of (i) efficacy in appropriate mouse models of cancer, (ii) miRNA biodistribution, and (iii) preliminary safety. The delivery technology with the best combination of efficacy, biodistribution, and safety was the NOV340 technology (SMARTICLES®, Marina Biotech, Bothell, WA; Mirna Therapeutics Inc., 2011), an ionizable liposome that forms a particle with a diameter of ~120 nm. The liposome contains amphoteric lipids that are cationic at low pH and neutral or anionic at neutral and higher pH. The lipids and miRNA mimics are mixed under acidic conditions to facilitate efficient miRNA encapsulation and liposome formation. In biofluids with a pH of 7–7.5, the nanoparticles assume a slightly anionic character that may prevent unwanted interactions with the negative charge of cellular membranes in the endothelium and other tissues. Since the pH tends to be lower in tumor areas, the NOV340 particles may become cationic in these areas and adhere to tumor cells.
The pharmacology of the NOV340/miR-34a formulation was tested in an orthotopic model of hepatocellular carcinoma. A model of liver cancer was chosen because (i) the majority of hepatic malignancies present mutations in pathways that can be antagonized by miR-34a (Catalogue of Somatic Mutations in Cancer), (ii) miR-34a inhibits human hepatocellular carcinoma cells in culture (Li et al., 2009), and (iii) efficient NOV340 liposome delivery to liver. The data showed that treatment of mice carrying existing tumors led to a significant tumor regression and prolonged survival (Daige et al., 2011; Bader et al., 2012). A histologic examination of livers revealed that several of the mice appeared to be tumor-free. This was in stark contrast to mice dosed with NOV340 particles that were loaded with a scrambled control. The NOV340/miR-34a-treated animals lacked notable drug-related side effects as determined by normal organ morphology, blood chemistries, and serum cytokine levels.
In summary, the data demonstrate the therapeutic utility of the clinically relevant NOV340/miR-34 formulation in a model of hepatocellular carcinoma. Mirna Therapeutics has initiated a preclinical development program to support the manufacture of cGMP materials and the conduction of IND-enabling studies. The company anticipates initiating clinical trials in 2013 for miR-34 that may be one of the first miRNA mimics to reach the clinic.
Closing Remarks
The replacement of miRNAs provides a new therapeutic concept. It aims to restore a loss of function that has been a largely inaccessible approach for drug developers. In addition, it builds on the regulation of multiple cellular pathways associated with human disease which appears to be a requirement for successful cancer therapy. Data generated in the animal suggest that the pharmacological delivery of miRNA mimics effectively inhibits tumor growth and is well-tolerated by normal tissues. This has cleared the path for a miR-34 therapy to the clinic. The emphasis is now on safety in higher species and man.
Conflict of Interest Statement
The author is an employee of Mirna Therapeutics and declares potential conflicts of interest relevant to the subject matter or materials discussed in this article.
Acknowledgments
The author thanks Drs Annette Schlageter, Paul Lammers, and David Brown for critical reading of the manuscript. This work was supported by a grant from the National Institutes of Health (1R43CA137939) and a commercialization grant from the Cancer Prevention and Research Institute of Texas (CPRIT).
References
Akao, Y., Noguchi, S., Iio, A., Kojima, K., Takagi, T., and Naoe, T. (2010). Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett. 300, 197–204.
Asslaber, D., Pinon, J. D., Seyfried, I., Desch, P., Stocher, M., Tinhofer, I., Egle, A., Merkel, O., and Greil, R. (2010). microRNA-34a expression correlates with MDM2 SNP309 polymorphism and treatment-free survival in chronic lymphocytic leukemia. Blood 115, 4191–4197.
Bader, A. G., Brown, D., and Winkler, M. (2010). The promise of microRNA replacement therapy. Cancer Res. 70, 7027–7030.
Bader, A. G., Daige, C. L., Kelnar, K., Priddy, L., Dysart, S., Wiggins, J., Zhao, J., Leatherbury, N., Omotola, M., Stoudemire, J., Lammers, P., and Brown, D. (2012). Preclinical data of a microRNA-based therapy for hepatocellular carcinoma. Annual AACR Conference Presentation, March 31–April 4, 2012, Chicago, IL.
Bommer, G. T., Gerin, I., Feng, Y., Kaczorowski, A. J., Kuick, R., Love, R. E., Zhai, Y., Giordano, T. J., Qin, Z. S., Moore, B. B., MacDougald, O. A., Cho, K. R., and Fearon, E. R. (2007). p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 17, 1298–1307.
Calin, G. A., Sevignani, C., Dumitru, C. D., Hyslop, T., Noch, E., Yendamuri, S., Shimizu, M., Rattan, S., Bullrich, F., Negrini, M., and Croce, C. M. (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. U.S.A. 101, 2999–3004.
Catalogue of Somatic Mutations in Cancer. Wellcome Sanger Trust Institute, WSAU. Available at: http://www.sanger.ac.uk
Chang, T. C., Wentzel, E. A., Kent, O. A., Ramachandran, K., Mullendore, M., Lee, K. H., Feldmann, G., Yamakuchi, M., Ferlito, M., Lowenstein, C. J., Arking, D. E., Beer, M. A., Maitra, A., and Mendell, J. T. (2007). Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 26, 745–752.
Chen, Q. R., Yu, L. R., Tsang, P., Wei, J. S., Song, Y. K., Cheuk, A., Chung, J. Y., Hewitt, S. M., Veenstra, T. D., and Khan, J. (2010). Systematic proteome analysis identifies transcription factor YY1 as a direct target of miR-34a. J. Proteome Res. 10, 479–487.
Chen, X., Hu, H., Guan, X., Xiong, G., Wang, Y., Wang, K., Li, J., Xu, X., Yang, K., and Bai, Y. (2011). CpG island methylation status of miRNAs in esophageal squamous cell carcinoma. Int. J. Cancer 130, 1607–1613.
Chen, Y., Zhu, X., Zhang, X., Liu, B., and Huang, L. (2010). Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol. Ther. 18, 1650–1656.
Chim, C. S., Wong, K. Y., Qi, Y., Loong, F., Lam, W. L., Wong, L. G., Jin, D. Y., Costello, J. F., and Liang, R. (2010). Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 31, 745–750.
Choi, Y. J., Lin, C. P., Ho, J. J., He, X., Okada, N., Bu, P., Zhong, Y., Kim, S. Y., Bennett, M. J., Chen, C., Ozturk, A., Hicks, G. G., Hannon, G. J., and He, L. (2011). miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 13, 1353–1360.
Christoffersen, N. R., Shalgi, R., Frankel, L. B., Leucci, E., Lees, M., Klausen, M., Pilpel, Y., Nielsen, F. C., Oren, M., and Lund, A. H. (2009). p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 17, 236–245.
Cole, K. A., Attiyeh, E. F., Mosse, Y. P., Laquaglia, M. J., Diskin, S. J., Brodeur, G. M., and Maris, J. M. (2008). A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 6, 735–742.
Corney, D. C., Hwang, C. I., Matoso, A., Vogt, M., Flesken-Nikitin, A., Godwin, A. K., Kamat, A. A., Sood, A. K., Ellenson, L. H., Hermeking, H., and Nikitin, A. Y. (2010). Frequent downregulation of miR-34 family in human ovarian cancers. Clin. Cancer Res. 16, 1119–1128.
Craig, V. J., Cogliatti, S. B., Imig, J., Renner, C., Neuenschwander, S., Rehrauer, H., Schlapbach, R., Dirnhofer, S., Tzankov, A., and Muller, A. (2011). Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 117, 6227–6236.
Craig, V. J., Tzankov, A., Flori, M., Schmid, C. A., Bader, A. G., and Muller, A. (2012). Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia. doi: 10.1038/leu.2012.110 [Epub ahead of print].
Daige, C., Priddy, L., Kelnar, K., Zhao, J., Dysart, S., Bader, A., and Brown, D. (2011). The development of a miRNA-based therapeutic candidate for hepatocellular carcinoma. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, November 12–16, 2011, San Francisco, CA. Abstract no. C142.
de Antonellis, P., Medaglia, C., Cusanelli, E., Andolfo, I., Liguori, L., De Vita, G., Carotenuto, M., Bello, A., Formiggini, F., Galeone, A., De Rosa, G., Virgilio, A., Scognamiglio, I., Sciro, M., Basso, G., Schulte, J. H., Cinalli, G., Iolascon, A., and Zollo, M. (2011). MiR-34a targeting of Notch ligand delta-like 1 impairs CD15+/CD133+ tumor-propagating cells and supports neural differentiation in medulloblastoma. PLoS ONE 6, e24584. doi: 10.1371/journal.pone.0024584
Du, R., Sun, W., Xia, L., Zhao, A., Yu, Y., Zhao, L., Wang, H., Huang, C., and Sun, S. (2012). Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS ONE 7, e30771. doi: 10.1371/journal.pone.0030771
Esquela-Kerscher, A., Trang, P., Wiggins, J. F., Patrawala, L., Cheng, A., Ford, L., Weidhaas, J. B., Brown, D., Bader, A. G., and Slack, F. J. (2008). The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 7, 759–764.
Feinberg-Gorenshtein, G., Avigad, S., Jeison, M., Halevy-Berco, G., Mardoukh, J., Luria, D., Ash, S., Steinberg, R., Weizman, A., and Yaniv, I. (2009). Reduced levels of miR-34a in neuroblastoma are not caused by mutations in the TP53 binding site. Genes Chromosomes Cancer 48, 539–543.
Fujita, Y., Kojima, K., Hamada, N., Ohhashi, R., Akao, Y., Nozawa, Y., Deguchi, T., and Ito, M. (2008). Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem. Biophys. Res. Commun. 377, 114–119.
Gallardo, E., Navarro, A., Vinolas, N., Marrades, R. M., Diaz, T., Gel, B., Quera, A., Bandres, E., Garcia-Foncillas, J., Ramirez, J., and Monzo, M. (2009). miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis 30, 1903–1909.
Gerlinger, M., Rowan, A. J., Horswell, S., Larkin, J., Endesfelder, D., Gronroos, E., Martinez, P., Matthews, N., Stewart, A., Tarpey, P., Varela, I., Phillimore, B., Begum, S., McDonald, N. Q., Butler, A., Jones, D., Raine, K., Latimer, C., Santos, C. R., Nohadani, M., Eklund, A. C., Spencer-Dene, B., Clark, G., Pickering, L., Stamp, G., Gore, M., Szallasi, Z., Downward, J., Futreal, P. A., and Swanton, C. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892.
Greenberg, E., Hershkovitz, L., Itzhaki, O., Hajdu, S., Nemlich, Y., Ortenberg, R., Gefen, N., Edry, L., Modai, S., Keisari, Y., Besser, M. J., Schachter, J., Shomron, N., and Markel, G. (2011). Regulation of cancer aggressive features in melanoma cells by microRNAs. PLoS ONE 6, e18936. doi: 10.1371/journal.pone.0018936
Hashimi, S. T., Fulcher, J. A., Chang, M. H., Gov, L., Wang, S., and Lee, B. (2009). MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood 114, 404–414.
He, L., He, X., Lim, L. P., de Stanchina, E., Xuan, Z., Liang, Y., Xue, W., Zender, L., Magnus, J., Ridzon, D., Jackson, A. L., Linsley, P. S., Chen, C., Lowe, S. W., Cleary, M. A., and Hannon, G. J. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134.
Hsu, S. D., Chu, C. H., Tsou, A. P., Chen, S. J., Chen, H. C., Hsu, P. W., Wong, Y. H., Chen, Y. H., Chen, G. H., and Huang, H. D. (2008). miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res. 36, D165–D169.
Ichimura, A., Ruike, Y., Terasawa, K., Shimizu, K., and Tsujimoto, G. (2010). MicroRNA-34a inhibits cell proliferation by repressing mitogen-activated protein kinase kinase 1 during megakaryocytic differentiation of K562 cells. Mol. Pharmacol. 77, 1016–1024.
Ji, Q., Hao, X., Meng, Y., Zhang, M., Desano, J., Fan, D., and Xu, L. (2008). Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 8, 266. doi: 10.1186/1471-2407-8-266
Ji, Q., Hao, X., Zhang, M., Tang, W., Yang, M., Li, L., Xiang, D., Desano, J. T., Bommer, G. T., Fan, D., Fearon, E. R., Lawrence, T. S., and Xu, L. (2009). MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE 4, e6816. doi: 10.1371/journal.pone.0006816
Johnson, S. M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K. L., Brown, D., and Slack, F. J. (2005). RAS is regulated by the let-7 microRNA family. Cell 120, 635–647.
Jones, S., Zhang, X., Parsons, D. W., Lin, J. C., Leary, R. J., Angenendt, P., Mankoo, P., Carter, H., Kamiyama, H., Jimeno, A., Hong, S. M., Fu, B., Lin, M. T., Calhoun, E. S., Kamiyama, M., Walter, K., Nikolskaya, T., Nikolsky, Y., Hartigan, J., Smith, D. R., Hidalgo, M., Leach, S. D., Klein, A. P., Jaffee, E. M., Goggins, M., Maitra, A., Iacobuzio-Donahue, C., Eshleman, J. R., Kern, S. E., Hruban, R. H., Karchin, R., Papadopoulos, N., Parmigiani, G., Vogelstein, B., Velculescu, V. E., and Kinzler, K. W. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806.
Kaller, M., Liffers, S. T., Oeljeklaus, S., Kuhlmann, K., Roh, S., Hoffmann, R., Warscheid, B., and Hermeking, H. (2011). Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis. Mol. Cell. Proteomics 10, M111.010462.
Kim, H. R., Roe, J. S., Lee, J. E., Hwang, I. Y., Cho, E. J., and Youn, H. D. (2012). A p53-inducible microRNA-34a downregulates Ras signaling by targeting IMPDH. Biochem. Biophys. Res. Commun. 418, 682–688.
Kim, N. H., Kim, H. S., Kim, N. G., Lee, I., Choi, H. S., Li, X. Y., Kang, S. E., Cha, S. Y., Ryu, J. K., Na, J. M., Park, C., Kim, K., Lee, S., Gumbiner, B. M., Yook, J. I., and Weiss, S. J. (2011). p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 4, ra71.
Kojima, K., Fujita, Y., Nozawa, Y., Deguchi, T., and Ito, M. (2010). MiR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. Prostate 70, 1501–1512.
Kota, J., Chivukula, R. R., O ’Donnell, K. A., Wentzel, E. A., Montgomery, C. L., Hwang, H. W., Chang, T. C., Vivekanandan, P., Torbenson, M., Clark, K. R., Mendell, J. R., and Mendell, J. T. (2009). Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137, 1005–1017.
Kumar, M. S., Erkeland, S. J., Pester, R. E., Chen, C. Y., Ebert, M. S., Sharp, P. A., and Jacks, T. (2008). Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. U.S.A. 105, 3903–3908.
Kuo, K. T., Guan, B., Feng, Y., Mao, T. L., Chen, X., Jinawath, N., Wang, Y., Kurman, R. J., Shih Ie, M., and Wang, T. L. (2009). Analysis of DNA copy number alterations in ovarian serous tumors identifies new molecular genetic changes in low-grade and high-grade carcinomas. Cancer Res. 69, 4036–4042.
Li, B., Hu, Y., Ye, F., Li, Y., Lv, W., and Xie, X. (2010). Reduced miR-34a expression in normal cervical tissues and cervical lesions with high-risk human papillomavirus infection. Int. J. Gynecol. Cancer 20, 597–604.
Li, N., Fu, H., Tie, Y., Hu, Z., Kong, W., Wu, Y., and Zheng, X. (2009). miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 275, 44–53.
Li, W. B., Ma, M. W., Dong, L. J., Wang, F., Chen, L. X., and Li, X. R. (2011). MicroRNA-34a targets notch1 and inhibits cell proliferation in glioblastoma multiforme. Cancer Biol. Ther. 12, 477–483.
Li, W. Q., Chen, C., Xu, M. D., Guo, J., Li, Y. M., Xia, Q. M., Liu, H. M., He, J., Yu, H. Y., and Zhu, L. (2011). The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J. 278, 1522–1532.
Liu, C., Kelnar, K., Liu, B., Chen, X., Calhoun-Davis, T., Li, H., Patrawala, L., Yan, H., Jeter, C., Honorio, S., Wiggins, J. F., Bader, A. G., Fagin, R., Brown, D., and Tang, D. G. (2011). The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 17, 211–215.
Liu, X., Sempere, L. F., Ouyang, H., Memoli, V. A., Andrew, A. S., Luo, Y., Demidenko, E., Korc, M., Shi, W., Preis, M., Dragnev, K. H., Li, H., Direnzo, J., Bak, M., Freemantle, S. J., Kauppinen, S., and Dmitrovsky, E. (2010). MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J. Clin. Invest. 120, 1298–1309.
Lize, M., Pilarski, S., and Dobbelstein, M. (2009). E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death Differ. 17, 452–458.
Lodygin, D., Tarasov, V., Epanchintsev, A., Berking, C., Knyazeva, T., Korner, H., Knyazev, P., Diebold, J., and Hermeking, H. (2008). Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7, 2591–2600.
Luan, S., Sun, L., and Huang, F. (2010). MicroRNA-34a: a novel tumor suppressor in p53-mutant glioma cell line U251. Arch. Med. Res. 41, 67–74.
Lujambio, A., Calin, G. A., Villanueva, A., Ropero, S., Sanchez-Cespedes, M., Blanco, D., Montuenga, L. M., Rossi, S., Nicoloso, M. S., Faller, W. J., Gallagher, W. M., Eccles, S. A., Croce, C. M., and Esteller, M. (2008). A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 105, 13556–13561.
Maes, O. C., Sarojini, H., and Wang, E. (2009). Stepwise up-regulation of microRNA expression levels from replicating to reversible and irreversible growth arrest states in WI-38 human fibroblasts. J. Cell. Physiol. 221, 109–119.
Mirna Therapeutics, Inc. (2011). Marina Biotech and Mirna Therapeutics announce license agreement for the development of microRNA-based therapeutics. Available at: www.mirnarx.com.
Mraz, M., Malinova, K., Kotaskova, J., Pavlova, S., Tichy, B., Malcikova, J., Stano Kozubik, K., Smardova, J., Brychtova, Y., Doubek, M., Trbusek, M., Mayer, J., and Pospisilova, S. (2009). miR-34a, miR-29c and miR-17-5p are downregulated in CLL patients with TP53 abnormalities. Leukemia 23, 1159–1163.
Mudduluru, G., Ceppi, P., Kumarswamy, R., Scagliotti, G. V., Papotti, M., and Allgayer, H. (2011). Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 30, 2888–2899.
Navarro, F., Gutman, D., Meire, E., Caceres, M., Rigoutsos, I., Bentwich, Z., and Lieberman, J. (2009). miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood 114, 2181–2192.
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C., Leary, R. J., Angenendt, P., Mankoo, P., Carter, H., Siu, I. M., Gallia, G. L., Olivi, A., McLendon, R., Rasheed, B. A., Keir, S., Nikolskaya, T., Nikolsky, Y., Busam, D. A., Tekleab, H., Diaz, L. A., Jr., Hartigan, J., Smith, D. R., Strausberg, R. L., Marie, S. K., Shinjo, S. M., Yan, H., Riggins, G. J., Bigner, D. D., Karchin, R., Papadopoulos, N., Parmigiani, G., Vogelstein, B., Velculescu, V. E., and Kinzler, K. W. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812.
Pramanik, D., Campbell, N. R., Karikari, C., Chivukula, R., Kent, O. A., Mendell, J. T., and Maitra, A. (2011). Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 10, 1470–1480.
Qiu, X., Friedman, J. M., and Liang, G. (2011). Creating a flexible multiple microRNA expression vector by linking precursor microRNAs. Biochem. Biophys. Res. Commun. 411, 276–280.
Raver-Shapira, N., Marciano, E., Meiri, E., Spector, Y., Rosenfeld, N., Moskovits, N., Bentwich, Z., and Oren, M. (2007). Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 26, 731–743.
Reya, T., Morrison, S. J., Clarke, M. F., and Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111.
Selbach, M., Schwanhausser, B., Thierfelder, N., Fang, Z., Khanin, R., and Rajewsky, N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63.
Shen, Z., Zhan, G., Ye, D., Ren, Y., Cheng, L., Wu, Z., and Guo, J. (2012). MicroRNA-34a affects the occurrence of laryngeal squamous cell carcinoma by targeting the antiapoptotic gene survivin. Med. Oncol. doi: 10.1007/s12032-011-0156-x.
Silber, J., Jacobsen, A., Ozawa, T., Harinath, G., Pedraza, A., Sander, C., Holland, E. C., and Huse, J. T. (2012). miR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis. PLoS ONE 7, e33844. doi: 10.1371/journal.pone.0033844
Sui, G., Affar el, B., Shi, Y., Brignone, C., Wall, N. R., Yin, P., Donohoe, M., Luke, M. P., Calvo, D., Grossman, S. R., and Shi, Y. (2004). Yin Yang 1 is a negative regulator of p53. Cell 117, 859–872.
Sun, F., Fu, H., Liu, Q., Tie, Y., Zhu, J., Xing, R., Sun, Z., and Zheng, X. (2008). Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 582, 1564–1568.
Tarasov, V., Jung, P., Verdoodt, B., Lodygin, D., Epanchintsev, A., Menssen, A., Meister, G., and Hermeking, H. (2007). Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 6, 1586–1593.
Tazawa, H., Tsuchiya, N., Izumiya, M., and Nakagama, H. (2007). Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 104, 15472–15477.
Trang, P., Wiggins, J. F., Daige, C. L., Cho, C., Omotola, M., Brown, D., Weidhaas, J. B., Bader, A. G., and Slack, F. J. (2011). Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 19, 1116–1122.
Tryndyak, V. P., Ross, S. A., Beland, F. A., and Pogribny, I. P. (2009). Down-regulation of the microRNAs miR-34a, miR-127, and miR-200b in rat liver during hepatocarcinogenesis induced by a methyl-deficient diet. Mol. Carcinog. 48, 479–487.
Valastyan, S., Reinhardt, F., Benaich, N., Calogrias, D., Szasz, A. M., Wang, Z. C., Brock, J. E., Richardson, A. L., and Weinberg, R. A. (2009). A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell 137, 1032–1046.
Vinall, R. L., Ripoll, A. Z., Wang, S., Pan, C. X., and Devere White, R. W. (2011). MiR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int. J. Cancer 130, 2526–2538.
Vogt, M., Munding, J., Gruner, M., Liffers, S. T., Verdoodt, B., Hauk, J., Steinstraesser, L., Tannapfel, A., and Hermeking, H. (2011). Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 458, 313–322.
Wang, X., Wang, H. K., McCoy, J. P., Banerjee, N. S., Rader, J. S., Broker, T. R., Meyers, C., Chow, L. T., and Zheng, Z. M. (2009). Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 15, 637–647.
Weeraratne, S. D., Amani, V., Neiss, A., Teider, N., Scott, D. K., Pomeroy, S. L., and Cho, Y. J. (2010). miR-34a confers chemosensitivity through modulation of MAGE-A and p53 in medulloblastoma. Neuro Oncol. 13, 165–175.
Wei, J. S., Song, Y. K., Durinck, S., Chen, Q. R., Cheuk, A. T., Tsang, P., Zhang, Q., Thiele, C. J., Slack, A., Shohet, J., and Khan, J. (2008). The MYCN oncogene is a direct target of miR-34a. Oncogene 27, 5204–5213.
Welch, C., Chen, Y., and Stallings, R. L. (2007). MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 26, 5017–5022.
Welch, S. A. and Moore, M. J. (2007). Erlotinib: success of a molecularly targeted agent for the treatment of advanced pancreatic cancer. Future Oncol. 3, 247–254.
Wiggins, J. F., Ruffino, L., Kelnar, K., Omotola, M., Patrawala, L., Brown, D., and Bader, A. G. (2010). Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 70, 5923–5930.
Yamakuchi, M., Ferlito, M., and Lowenstein, C. J. (2008). miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 13421–13426.
Yan, D., Zhou, X., Chen, X., Hu, D. N., Dong, X. D., Wang, J., Lu, F., Tu, L., and Qu, J. (2009). MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Invest. Ophthalmol. Vis. Sci. 50, 1559–1565.
Keywords: miR-34, miR-34a, miRNA, miRNA replacement, miRNA therapeutics
Citation: Bader AG (2012) miR-34 – A microRNA replacement therapy is headed to the clinic. Front. Gene. 3:120. doi:10.3389/fgene.2012.00120
Received: 30 April 2012; Accepted: 14 June 2012;
Published online: 02 July 2012.
Edited by:
Michael Rossbach, Genome Institute of Singapore, SingaporeReviewed by:
Navjotsingh Pabla, California Institute of Technology, USAMelanie Carless, Texas Biomedical Research Institute, USA
Copyright: © 2012 Bader. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andreas G. Bader, Mirna Therapeutics, Inc., 2150 Woodward Street, Suite 100, Austin, TX 78744, USA. e-mail:YWJhZGVyQG1pcm5hcnguY29t
†Andreas G. Bader is an employee of Mirna Therapeutics which develops miRNA-based therapies.