

- 1Department of Chemical Physiology, The Scripps Research Institute, La Jolla, CA, USA
- 2Department of Molecular and Integrative Neurosciences, The Scripps Research Institute, La Jolla, CA, USA
Core body temperature (CBT) and calorie intake are main components of energy homeostasis and two important regulators of health, longevity, and aging. In homeotherms, CBT can be influenced by calorie intake as food deprivation or calorie restriction (CR) lowers CBT whereas feeding has hyperthermic effects. The finding that in mice CBT prolonged lifespan independently of CR, suggested that the mechanisms modulating CBT may represent important regulators of aging. Here we summarize the current knowledge on the signaling molecules and their receptors that participate in the regulation of CBT responses to calorie intake. These include hypothalamic neuropeptides regulating feeding but also energy expenditure via modulation of thermogenesis. We also report studies indicating that nutrient signals can contribute to regulation of CBT by direct action on hypothalamic preoptic warm-sensitive neurons that in turn regulate adaptive thermogenesis and hence CBT. Finally, we show the role played by two orphans G protein-coupled receptor: GPR50 and GPR83, that were recently demonstrated to regulate temperature-dependent energy expenditure.
Introduction
Experimental work on calorie restriction (CR), core body temperature (CBT), and the insulin-like growth factor 1 (IGF1)/Insulin pathway point at energy homeostasis as an important regulator of health, longevity, and aging. The two main components of energy homeostasis are nutrient and temperature homeostasis. Each contributes to energy intake and energy expenditure, respectively, and in homeotherms, they are regulated primarily in the hypothalamus. Although nutrient and temperature homeostasis are typically investigated independently, there is a distinct relationship between them. Calorie intake can affect CBT, with feeding producing acute hyperthermic effects, whereas food deprivation as well as the controlled reduction of nutrient intake in CR, can induce longer lasting hypothermia (Rampone and Shirasu, 1964; Walford and Spindler, 1997; Smirnov and Kiyatkin, 2008). CBT response to calorie intake reduction is regarded as an adaptive mechanism, decreasing energy expenditure when nutrient availability is limited. This mechanism may have evolved to prolong survival until food became available, and at least under controlled experimental conditions of CR, it contributes to increased lifespan. With some strain and diet specific differences, CR reduced CBT across species including mice, rats, primates, and humans (Duffy et al., 1989; Lane et al., 1996; Rikke et al., 2003; Soare et al., 2011). Work on transgenic mice with lowered CBT showed that even a modest (~0.5°C) but prolonged CBT reduction increased median lifespan of up to 20%. This was observed in animals on ad libitum diet and with a calorie intake similar to that of wild type (wt) littermates, demonstrating that the effects of CBT on longevity were independent from those of CR (Conti et al., 2006). These findings also suggested that the reduction of CBT occurring during CR may contribute to the effects of CR on longevity. Thus, the molecules and the pathways regulating CBT responses to calorie intake may be important regulators of aging.
Adaptive thermogenesis is controlled via the sympathetic nervous system (SNS), which influences heat production in the brown adipose tissue (BAT). BAT is a specialized tissue responsible for producing heat for adaptive thermogenesis by dissipating the mitochondrial proton gradient via the uncoupling protein 1 (UCP1). In rodents and human infants, BAT has been shown to be the major source of induced heat (Cannon and Nedergaard, 2004). In addition to BAT, the SNS can also influence CBT by affecting heat production in the skeletal muscles and the liver, as well as by restricting heat dissipation via regulation of peripheral vasoconstriction.
This review will focus on signaling molecules demonstrated in mouse or in rat to be produced by and/or to act on two hypothalamic regions pivotal in the regulation of temperature or nutrient homeostasis, and that are in polysynaptic contact with the BAT (Elmquist et al., 2005). One such region includes the paraventricular (PVN), the arcuate (ARC), and the lateral hypothalamic (LH) nuclei (Figure 1); another is the preoptic area (POA; Figure 2).
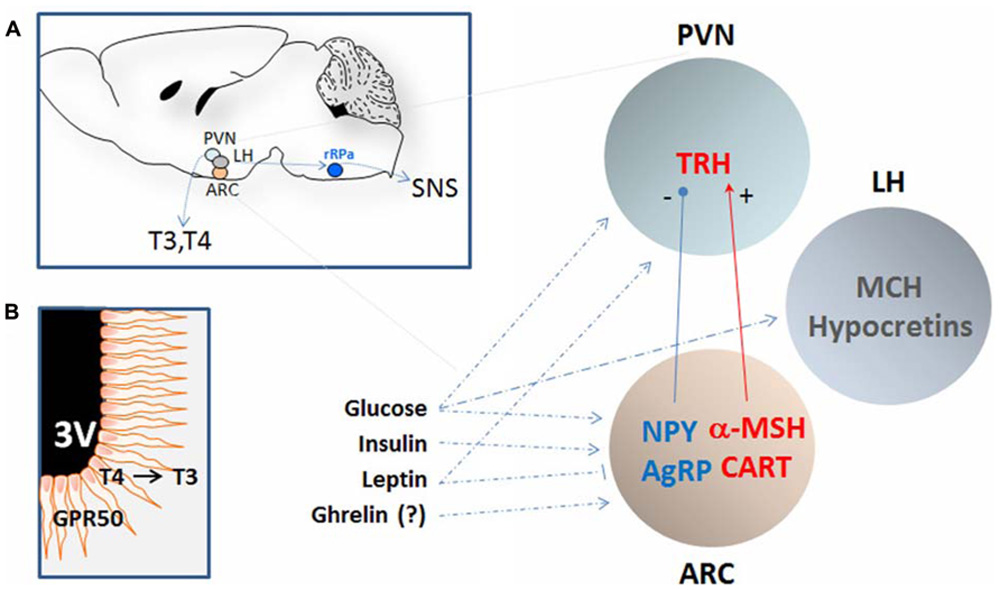
FIGURE 1. Schematic representation of the hypothalamic nuclei and the neuropeptides that regulates calorie intake and energy expenditure by affecting core body temperature. (A) The approximate location of the periventricular nucleus (PVN), the arcuate nucleus (ARC), and the lateral hypothalamus (LH) are shown in a sagittal section of the mouse brain (left). These regions are in polysynaptic contact with the BAT (not shown) via the raphe pallidus (RPa) and can regulate CBT by affecting the sympathetic nervous system (SNS). In addition, CBT can also be affected by thyrotropin-releasing hormones (TRH) that determines the levels of circulating thyroid hormones T3 and T4. The PVN and the ARC contain neurons that can respond to nutritional state by modulating the level of specific orexigenic or anorexigenic neuropeptides. The orexigenic neuropeptide Y (NPY) and agouti-related protein (AgRP) are elevated during calorie restriction or food deprivation and are strong stimulator of appetite and reduce CBT by acting on the SNS as well as by reducing the level of TRH in the PVN. In contrast, the anorexigenic peptide α-melanocyte-stimulating hormone (α-MSH), and possibly the cocaine-and amphetamine-regulated transcript (CART), stimulates thermogenesis. The LH is also an area important in the regulation of energy homeostasis and the melanin-concentrating hormone (MCH) and the hypocretins (Hcrt) can contribute to CBT elevation at least indirectly by increasing the locomotor activity associated with food seeking behavior and possibly via BAT activation. Neurons in these nuclei are sensitive to changes in the level of glucose, leptin, insulin, and possibly ghrelin all positively correlated to CBT. (B) Schematic representation of tanycytes cells lining the third ventricle (3V) and projecting into the hypothalamus. These cells can contribute to the regulation of CBT in response to nutrient signals in at least two ways: (1) by producing of T3 from T4 thus increasing its local concentration and in doing so inhibiting the synthesis of TRH even when circulating T3 levels are low; (2) by expressing GPR50, an orphan G-coupled protein receptor proposed to serve as a regulator of adaptive thermogenesis in response to nutrient signals. See text for details and references.
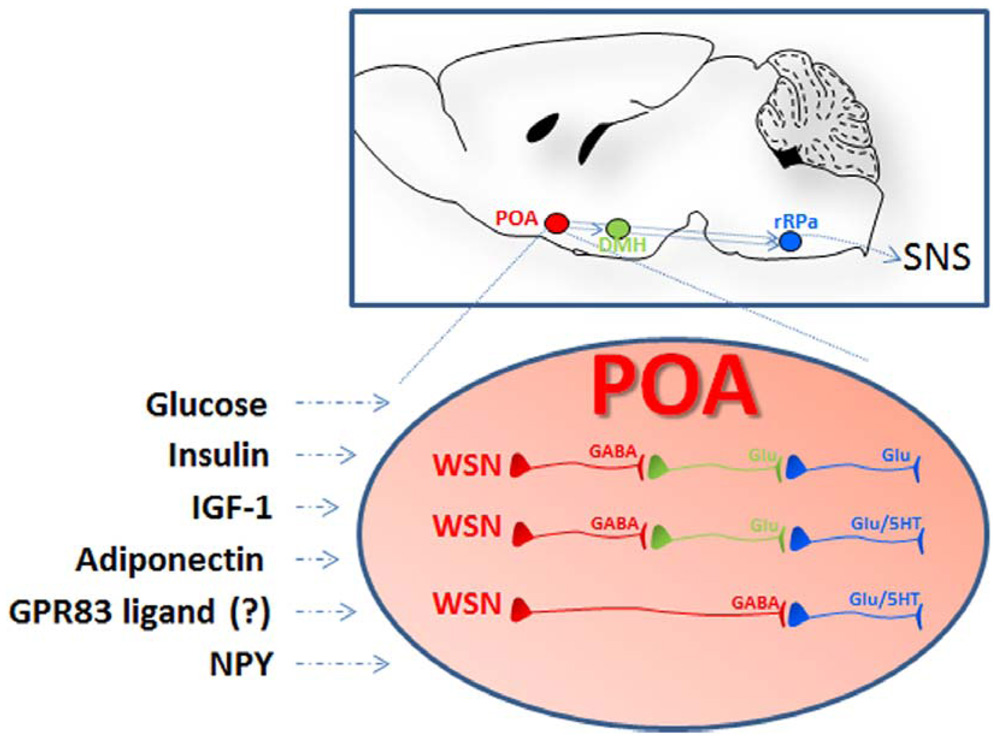
FIGURE 2. Schematic representation of the localization and the organization of the nuclei and the cells participating to central thermoregulation. Lesions studies indicate that the hypothalamic preoptic area (POA) exerts the function of a bona fide thermostat allowing sensing and proper thermoregulatory responses to local as well as peripheral temperature changes (afferent pathways are not shown). In addition to respond to changes in temperature the POA can also sense nutrient signals: POA injection of insulin, IGF-1, and adiponectin were followed by hyperthermia via BAT activation while treatment with NPY or downregulation of the G-protein coupled receptor 83 (GPR83) induced hypothermia. The receptors for some of these ligands, as well as GPR83, were demonstrated in the POA warm-sensitive neurons (WSN). These specialized GABAergic cells exert a tonic inhibition on the raphe pallidus (aRP) either directly or through neurons in the dorsomedial hypothalamus (DMH) and control thermogenesis by activation of brown adipose tissue (BAT), muscular shivering, or the regulation of vasodilation (scheme adapted from Morrison and Nakamura, 2011). WSN are one component of the POA thermoregulatory neurocircuitry that comprises also temperature insensitive and cold-sensitive neurons that might also participate in the POA responses to nutrients (not shown in this scheme).
The PVN, ARC, and LH express neuropeptides and their receptors, which together regulate feeding in addition to influencing CBT. These peptides include the neuropeptide Y (NPY), the cocaine- and amphetamine-regulated transcript (CART), the agouti-related protein (AgRP), the α-melanocyte-stimulating hormone (α-MSH), the melanin-concentrating hormone (MCH) and the thyrotropin-releasing hormone (TRH). These hypothalamic regions and neuropeptides are only reviewed here for their role in temperature homeostasis, and we refer to comprehensive reviews for their role in feeding (Elmquist et al., 2005; Morton et al., 2006; Gao and Horvath, 2007; Sanchez-Lasheras et al., 2010).
The POA contains temperature sensitive neurons that are pivotal in the sensing and the regulation of CBT (Hammel et al., 1960; Nakayama et al., 1963; Boulant, 2000). Among them are the warm-sensitive neurons (WSN), which are GABAergic neurons that exert a tonic inhibition on the dorsomedial hypothalamus (DMH) and the raphe pallidus (rRPa), both of which can activate spinal sympathetic and somatic motor circuits to drive adaptive thermogenesis in BAT (Morrison and Nakamura, 2011). WSN are typically investigated for their role in regulating fever or response to peripheral (skin) and local changes in temperature. However, electrophysiological studies and more recent molecular characterization have demonstrated that these specialized cells also respond to nutrient signals including glucose, insulin, and adiponectin (Silva and Boulant, 1984; Sanchez-Alavez et al., 2010; Eberwine and Bartfai, 2011; Klein et al., 2011). Evidence that these and other peripheral nutrient signals may contribute to CBT regulation via their action in the PVN, the ARC and the LH will also be summarized. Finally, we will discuss the role of the two orphan G protein-coupled receptors (GPCRs) GPR50 and GPR83 that were recently proposed to mediate effects of yet unidentified endogenous signals on energy expenditure via CBT regulation.
Hypothalamic Orexigenic and Anorexigenic Peptides
Neuropeptide Y
Neuropeptide Y is a 36 aa, C terminally amidated neuropeptide. NPY acts at five different GPCR type of NPY-receptors (Y1–Y5), and is found in the autonomic nervous system and the brain, where its expression is highest in the ARC (Figure 1). NPY is a strong stimulator of feeding: its expression and synaptic level in the ARC is associated with hunger and is elevated during food deprivation or CR. Importantly, central administration of NPY not only increased food intake but also caused hypothermia (-1 to -3°C), reducing metabolic rate (Stanley et al., 1986). Such effect is at least in part due to decreased SNS-mediated thermogenesis, resulting from the NPY-mediated presynaptic (auto) inhibition of noradrenaline release from neurons that contain both norepinephrine (NE) and NPY. This also leads to a lower level of thermogenesis in BAT. In addition, Y1 and Y5 postsynaptic receptors on brown adipocytes also counteract the effect of NE at beta 3 adrenoreceptors (Billington et al., 1991; Egawa et al., 1991; Walker and Romsos, 1993; Bouali et al., 1995; Currie and Coscina, 1995; Pedrazzini et al., 1998; Williams et al., 2001). The Y5 agonists increased feeding, reduced oxygen consumption and energy expenditure in rats, probably by acting on ARC and BAT. Furthermore, the Y5 subtype selective antagonist increased CBT and the transcription of UCP1 in the BAT of mice (Hwa et al., 1999; Mashiko et al., 2007). Working with cold-acclimated Siberian hamsters, (Pelz and Dark (2007) and Dark and Pelz (2008)) found that activation of Y1 induced a prolonged reduction in CBT similar to that observed during natural torpor. Finally, inhibition of Y1 (albeit not of Y5) in hamster or its downregulation (knock-down) with antisense oligodeoxynucleotides in rats produced a transient hyperthermia (Lopez-Valpuesta et al., 1996; Pelz and Dark, 2007; Dark and Pelz, 2008).
Since the hypothermic action of NPY was observed not only after its administration into the ARC or the PVN, but also the POA, it was proposed that NPY influenced the activity of thermoregulatory neurons (Currie and Coscina, 1995; Jolicoeur et al., 1995; Dark and Pelz, 2008). Molecular profiling showed that POA WSN express Y2 as well as the GPR83, an orphan receptor sharing homology to Y2 and found by one group to interact with NPY in vitro (Sah et al., 2007; Eberwine and Bartfai, 2011; Dubins et al., 2012). Interestingly, downregulation of GPR83 expression in the POA by shRNA was recently shown to reduce CBT (discussed below; Dubins et al., 2012).
Agouti-Related Protein and α-Melanocyte-Stimulating Hormone
The neuropeptides AgRP and α-MSH are the endogenous antagonist and agonist, respectively, of the melanocortin receptors and the main ligands of the central melanocortin system (Figure 1). In the hypothalamus they are produced in the ARC, where AgRP co-localizes with NPY, and where the precursor of α-MSH, the pro-opiomelanocortin (POMC), is co-expressed with CART (discussed below). AgRP and α-MSH stimulate and inhibit appetite, respectively, to modulate nutrient intake, but they can also contribute to the regulation of energy expenditure and can influence CBT (reviewed in Cone, 1999; Robinson et al., 2000; Schwartz et al., 2000; Spiegelman and Flier, 2001; Fan et al., 2005). This action is mediated by the melanocortin 4 receptor (MC4R) subtype. Mice null for MC4R, or treated with MC4R antagonists, including AgRP, have reduced thermogenesis and fail to upregulate UCP1 in the intercapsular BAT when fed high fat diet or when exposed to cold (Ste Marie et al., 2000; Butler et al., 2001; Voss-Andreae et al., 2007). Conversely, administration of MC3/4R agonist into the ventricle or in the RPa increased temperature via BAT activation (Yasuda et al., 2004).
Double labeling with retrograde tracing experiments demonstrated a connection between BAT and MC4R neurons in the RPa, the PVN, and the DMH MC4R mRNA was also demonstrated in LH, VMH, as well as in the POA, suggesting that the α-MSH and AgRP regulate BAT thermogenesis by acting on several different neuronal circuits (Kishi et al., 2003; Liu et al., 2003).
The melanocortin system can also influence the circulating levels of thyroid hormones (TRH and T3/T4), which were increased or reduced by intracerebroventricular (i.c.v.) injection of α-MSH or AgRP, respectively (Fekete et al., 2000a, 2002a; Kim et al., 2000). This is achieved by direct innervation of both α-MSH and AgRP fibers in the ARC to the TRH neurons in the PVN (Legradi and Lechan, 1999; Fekete et al., 2004; see below for the thermoregulatory action of TRH and thyroid hormones).
Interestingly, the melanocortin system was also recently demonstrated to be modulated by the NAD+-dependent deacetylase Sirt1, the mammalian homolog of Sir2 which was implicated in mediating some of the lifespan prolonging effects of CR (Guarente and Picard, 2005; Coppari, 2012). Sirt1 is expressed in the hypothalamus and its pharmacological inhibition decreased AgRP neuronal activity, producing a reduced inhibition of POMC neurons that was MCR4-dependent. Similar results were observed in animals with ablation of Sirt1 in AgRP neurons: these mice displayed decreased sensitivity to ghrelin, food intake, and body weight (Dietrich et al., 2010).
Cocaine- and Amphetamine-Regulated Transcript
The CART is a neuropeptide co-expressed with POMC in neurons in the ARC (Figure 1). CART primarily affects energy homeostasis through its anorexigenic action, and can possibly contribute to energy expenditure by influencing CBT. In the rat, i.c.v. injection of CART was followed by hypothermic effects that were reduced by exendin-9-39, an antagonist of the glucagon like peptide 1 receptor also demonstrated to mediate the hypophagic of CART (Skibicka et al., 2009). However, a different study showed that CART injected in the PVN induced the expression of UCP1 in the BAT, suggesting that CART may be capable of stimulating adaptive thermogenesis by mitochondrial uncoupling (Wang et al., 2000).
Melanin-Concentrating Hormone and Hypocretins
The LH is another region involved in the regulation of feeding and energy expenditure, and lesions to this nucleus caused hypophagia as well as hyperthermia (Teitelbaum and Epstein, 1962; Stevenson and Montemurro, 1963). Two distinct families of signaling molecules are recognized as main modulators of nutrient homeostasis and energy expenditure in the LH: the MCH and hypocretins (also known as Orexins; Figure 1). MCH is a powerful stimulator of feeding and its ablation results in a lean phenotype (Shimada et al., 1998). Hypocretins are neuropeptides involved in the regulation of sleep, wakefulness, and reward, and they are also able to increase appetite and CBT (de Lecea et al., 1998; Sakurai et al., 1998; Yoshimichi et al., 2001; Adamantidis and de Lecea, 2009; Bonnavion and de Lecea, 2010).
Retrograde labeling studies demonstrated a link between MCH neurons and hypocretin neurons and BAT innervation and activity (Oldfield et al., 2002; Cerri and Morrison, 2005). The level of UCP1 transcripts in brown adipocytes was increased in mice null for MCH, and this mutation normalized CBT in leptin-deficient animals (Segal-Lieberman et al., 2003). Consistently, UCP1 mRNA levels in BAT were reduced by MCH infusion (Ito et al., 2003). However, MCH and hypocretins have profound effects on locomotion, rendering the contribution of muscular activity to energy expenditure difficult to evaluate. In our experience, for instance, a deletion of up to 90% of hypocretin neurons reduced locomotion without affecting CBT (Conti et al., 2006).
Thyrotropin-Releasing Hormone
The TRH is recognized as an important regulator of energy metabolism (reviewed in Lechan and Fekete, 2006). TRH exerts this action mainly via the modulation of the hypothalamic–pituitary–thyroid (HPT) axis, regulating the level of the thyroid hormones thyroxine (T4) and triiodothyroxine (T3). TRH neurons in the PVN regulate the release of the pituitary thyroid-stimulating hormones (TSH) into the circulation that, in turn, act on the thyroid gland to release T3 and T4 (Figure 1). Importantly, thyroid hormones inhibit TRH secretion, providing a negative regulatory feedback onto the axis. Thyroid hormones have long been recognized as mediators of thyroid thermogenesis, a phenomenon mainly investigated as a peripheral event evoked via direct thyroid action on muscle cells, involving altered muscle cell Ca2+ homeostasis, and possibly UCP3 (reviewed in Silva, 2006). More recently, it was proposed that the mechanisms of T3-induced thermogenesis are central and involve the sympathetic activation of BAT, requiring the activation of the lipogenic pathway in the ventromedial hypothalamus (Cannon and Nedergaard, 2010; Lopez et al., 2010).
Thyrotropin-releasing hormone, as well as T3, are also important regulators of feeding, and experimental work indicated that both hormones can regulate thermogenesis in response to calorie intake. Fasting induced a fall in T3 and T4 levels and a reduction of TRH expression in the PVN, an effect at least in part due to decreased level of anorexigenic peptides (Spencer et al., 1983; Connors et al., 1985; Blake et al., 1991, 1992; Ahima et al., 1996; Legradi et al., 1998). These actions are mediated in two manners: by leptin acting directly on TRH neurons in the PVN, or by leptin acting indirectly by exerting opposite actions on α-MSH/CART and NPY/AgRP neurons localized in the ARC and projecting to the PVN (Ahima et al., 1996, 2000; Ahima, 2000; Nillni et al., 2000; Harris et al., 2001; Bjorbaek and Hollenberg, 2002; Perello et al., 2010). Fasting-induced reduction of TRH can be restored by i.c.v. injection of α-MSH or CART, which activate TRH neurons and stimulate hormone release (Fekete et al., 2000a,b; Kim et al., 2000; Nillni et al., 2000). Both AgRP and NPY can inhibit TRH neurons, reducing TRH transcript and circulating thyroid hormone levels. The NPY action is also mediated via Y1 and Y5 receptors (Fekete et al., 2001, 2002a,b; Costa-e-Sousa et al., 2011; Vella et al., 2011). Recently, it was reported that MC4R and NPY are both required for hepatic metabolism of T4 during fasting (Vella et al., 2011).
A distinct hypothalamic mechanism for the downregulation of the HPT axis is represented by local increase of T3 via fast-induced elevation of type 2 iodothyronine deiodinase in tanycytes, a group of ependymal cells that are located at the base of the third ventricle and extend into the hypothalamus (Figure 1B). During fasting, the type 2 deiodinase, D2 can convert T4 into the more potent T3 whose feedback inhibits the HPT axis, lowering the level of circulating thyroid hormones (Diano et al., 1998). Interestingly, these cells produce high level of GPR50, an orphan receptor also expressed in several hypothalamic nuclei and in pituitary neurons. GPR50 was recently shown to be a strong regulator of energy expenditure and thermogenesis in the context of the state of torpor (Bechtold et al., 2012; and see below).
Finally, TRH may also influence thermogenesis by direct action on POA temperature sensitive neurons and without affecting the HPT axis. Central injection of TRH was, in fact, capable of decreasing the activity of a fraction of WSN and increasing that of cold-sensitive neurons in the POA (Hori et al., 1988). This finding is consistent with the central hyperthermic effect of TRH, although its possible role in influencing CBT in response to nutrient intake remains to be investigated.
Peripheral Nutrient Signals
Glucose
A central role of glucose in influencing CBT was first revealed by experiments in which i.c.v. injection of the glucose analog 2-DG was followed by reduced sympathetic activation of BAT and hypothermia (Freinkel et al., 1972; Egawa et al., 1989). Glucose-sensing neurons are found in most hypothalamic nuclei (Ritter and Dinh, 1994; Dunn-Meynell et al., 1998; Silver and Erecinska, 1998; Briski and Sylvester, 2001; Fioramonti et al., 2004; Wang et al., 2004; Yang et al., 2004) as well as at the brain stem level in the area postrema (AP), the nucleus of the solitary tract (NTS), the dorsal motor nucleus of the vagus (DMNX), and the basolateral medulla (BLM) (Adachi et al., 1984; Mizuno and Oomura, 1984; Ritter and Dinh, 1994; Yettefti et al., 1995; Dunn-Meynell et al., 1998; Dallaporta et al., 1999; Briski and Sylvester, 2001). It is possible to distinguish two categories of neurons depending on whether the elevation of extracellular glucose level has excitatory or inhibitory action on their activity (Anand et al., 1964; Adachi et al., 1984; Routh, 2002; Yang et al., 2004). Using mice with inactivation of the glucose transporter type 2 (Glut2), Mounien et al. (2010) demonstrated that the effects of glucose on thermogenesis are at least in part mediated via decreased leptin sensitivity of NPY and POMC expressing neurons in the ARC. These actions on the ARC may not be direct, but mediated by glucose-sensing neurons located in the LH, the dorsal vagal complex, and the basal medulla.
Interestingly, Glut2 neurons were also found in the DMH, an area that receives projections from POA WSN, suggesting the possibility that glucose may also influence adaptive thermogenesis via this neuronal circuitry. Finally, electrophysiological studies revealed that POA neurons, including a fraction of warm and cold-sensitive neurons, are sensitive to glucose (Silva and Boulant, 1984; Figure 2).
Leptin
Leptin is a small protein produced by adipose tissue that acts peripherally as well as centrally to regulate appetite and energy expenditure (Campfield et al., 1995; Halaas et al., 1995; Pelleymounter et al., 1995). Mice null for leptin receptor or for the transcription factor STAT3, which is involved in leptin receptor signaling, are obese and have reduced CBT and oxygen consumption (Gao et al., 2004). Leptin-deficient mice spontaneously enter into torpor when deprived of food, a response that is prevented by leptin administration (Gavrilova et al., 1999). Conversely, in wt mice leptin reduced food intake, elevated CBT, and increased the sympathetic activation of BAT (Pelleymounter et al., 1995; Haynes et al., 1997).
The effects of leptin on energy expenditure and thermogenesis have mostly been investigated for leptin’s ability to regulate TRH either by direct action on PVN neurons, or by indirect action via inhibition of NPY/AgRP and stimulation of POMC/CART neurons in the ARC (Ahima et al., 1996, 2000; Ahima, 2000; Nillni et al., 2000; Schwartz et al., 2000; Harris et al., 2001; Bjorbaek and Hollenberg, 2002; Perello et al., 2010; see also Thyrotropin-Releasing Hormone).
A distinct mechanism of action for leptin-induced thermogenesis was also proposed to occur via stimulation of the release of the endogenous pyrogen interleukin-1β and prostaglandins (Luheshi et al., 1999) acting on POA and MPO neurons.
Ghrelin
The gastrointestinal peptide ghrelin is a hunger-stimulating hormone produced mainly by specialized cells in the fundus of the stomach and the pancreas. Ghrelin promotes an increase in food intake and a reduction in energy expenditure, resulting in a positive energy balance and an increase in body weight (Tschop et al., 2000; Lawrence et al., 2002; Theander-Carrillo et al., 2006).
Definitive proof for a role of ghrelin in regulating CBT is still lacking since findings remain few and contrasting. Central i.c.v. injection of ghrelin was reported to not only to be able to provoke a transient reduction of CBT associated with decreased spontaneous activity, but also to promote a small but significant reduction of BAT temperature, which indicates that ghrelin may be capable of reducing energy expenditure by affecting temperature homeostasis (Lawrence et al., 2002; Yasuda et al., 2003; Tang-Christensen et al., 2004). A single case of severe hypothermia in humans subject to prolonged treatment with ghrelin was also reported (Wiedmer et al., 2011). When the same group further investigated the hypothermic effect of ghrelin in rodents, they found evidence that ghrelin could bind to axon terminals in the POA, but they did not see any effects on CBT when the peptide was injected i.c.v. or subcutaneously.
Findings that CBT reduction may not be one of the mechanisms by which ghrelin regulates energy expenditure also came from experiments using mice null for ghrelin O-acyltransferase (GOAT), the enzyme that catalyzes the octanoylation of ghrelin, that is a post-translational modification necessary for the biological activity of this peptide. CBT profiles in GOAT null mice were similar to that of their wt littermates in different nutritional states, including fasting, or when exposed to different ambient temperatures (Heppner et al., 2012).
Instead, two distinct studies suggest that ghrelin may have a role in fasting-induced torpor. One found that the torpor induced by food deprivation was more severe if animals were treated with ghrelin peripherally. These effects were lost in animals with chemical ablation of the ARC, or in mice null for NPY, but not in mice blocked in α-MSH pathway (Gluck et al., 2006). Another study found that mice null for pre-pro-ghrelin had increased sensitivity to fasting and lowered ambient temperature, resulting in a precipitous drop of CBT, impaired sleep pattern, and decreased survival (Szentirmai et al., 2009). However, such a phenotype was not observed in mice lacking ghrelin receptor, suggesting that additional ghrelin receptor subtypes may exist. In addition, some of the differences in these studies may be due to the distinct ambient temperature at which experiments were carried out, with the hypothermic effects of ghrelin reported only at 17–18°C, but not at 25°C, a value closer to thermo-neutrality.
Insulin/IGF-1
The pancreatic hormone insulin is the main regulator of peripheral glucose homeostasis and has been also investigated for its role as regulator of energy homeostasis in the central nervous system (Woods et al., 1979; Baskin et al., 1987; for recent reviews, see Plum et al., 2006; Belgardt and Bruning, 2010). Indeed, the insulin receptor (IR) is expressed in several brain regions, including the hypothalamus where it is abundant in the ARC (Havrankova et al., 1978; Werther et al., 1987; Marks et al., 1990). Pharmacological studies with central insulin injection, as well as elegant transgenic models of selective IR-ablation, showed that insulin can act centrally to cause reduced food intake, increased weight loss, and helped to regulate peripheral glucose homeostasis (Woods et al., 1979; McGowan et al., 1992a; Chavez et al., 1995; Bruning et al., 2000; Obici et al., 2002; Brown et al., 2006; Koch et al., 2008).
A role of insulin in regulating thermogenesis in response to feeding was proposed when it was observed that pharmacological inhibition of its secretion effectively attenuated diet-induced thermogenesis (Rothwell and Stock, 1981, 1986, 1988). Since either peripheral or central administration of insulin activated the SNS, the involvement of BAT in this response was promptly hypothesized (McCormack, 1982; Rothwell and Stock, 1986; Muntzel et al., 1995).
Injection of insulin into the hypothalamus had hyperthermic effects, increasing CBT and energy expenditure (Menendez and Atrens, 1991; McGowan et al., 1992a,b). This was proposed to occur via the insulin-mediated inhibition of the NPY/AgRP neurons expressing IR (Porte et al., 2002, 2005; Fekete et al., 2006; Mayer and Belsham, 2009).
The presence of IRs in the POA raised the possibility that insulin may influence thermogenesis by also acting on neurons in this region (Unger et al., 1989; Cardona-Gomez et al., 2000; Plum et al., 2005; van Baak, 2008). Central i.c.v. injection of insulin reduced the unit activity of POA neurons sensitive to peripheral changes in scrotum temperature, indicating that this hormone may modulate thermoregulatory responses by affecting these specialized cells (Wang and Lin, 1985). Recently, IR was demonstrated on at least a fraction of POA WSN, and electrophysiological studies on hypothalamic slices demonstrated that insulin acted directly on intrinsically WSN, inducing hyperpolarization and reducing their firing rate (Sanchez-Alavez et al., 2010). Retrograde transport and double labeling studies also demonstrated that the IR-positive WSN are GABAergic and project to the RPa (thus a likely synaptic connection to BAT was established). Finally, POA injection of insulin induced a specific, PI3K-involving and dose-dependent elevation of CBT mediated by stimulation of BAT (Figure 2).
A similar finding was reported for the IGF-1 (Sanchez-Alavez et al., 2011). Its receptor can be expressed on WSN and POA, and an injection of IGF-1 elicited a dose-dependent increase of CBT and activated BAT. Although the effects of IGF-1 on WSN activity remain to be demonstrated, the CBT effects of central IGF-1 were reduced in mice lacking neuronal IR. Since IGF-1 can also activate IR, the IR homodimers or the IGF-1R/IR heterodimers may contribute to the thermogenic action of IGF-1 (Sanchez-Alavez et al., 2011).
Adiponectin
Adiponectin is a protein hormone secreted by adipose tissue. It has insulin-sensitizing effects, and is an important regulator of metabolism in peripheral tissues, enhancing fatty acid oxidation and glucose uptake in muscle, and reducing hepatic glucose production (Berg et al., 2001, 2002; Fruebis et al., 2001; Yamauchi et al., 2001; Tomas et al., 2002; Shklyaev et al., 2003; Qi et al., 2004). The adiponectin receptors AdipoR1 and AdipoR2 are expressed in different brain regions such as the hypothalamus, where adiponectin is beginning to be investigated for its possible central effects (Yamauchi et al., 2003; Fry et al., 2006; Kos et al., 2007; Kubota et al., 2007; Coope et al., 2008; Guillod-Maximin et al., 2009; Psilopanagioti et al., 2009; Hoyda and Ferguson, 2010; Thundyil et al., 2011).
So far, only a limited number of studies have measured the effects of adiponectin on CBT and energy expenditure and these have reported contrasting findings. For instance, one group found i.c.v. injection of adiponectin recapitulated its peripheral effects and increased energy expenditure via BAT-induced thermogenesis (Qi et al., 2004). Another showed increased BAT UCP1 and rectal temperature following peripheral, but not central injection with adiponectin (Masaki et al., 2003). A third group instead reported that intravenous injection of adiponectin reduced BAT UCP1 mRNA and energy expenditure while exerting central orexigenic effects via direct action on the ARC (Kubota et al., 2007). The effects of central injections of adiponectin on calorie intake are also contrasting, reporting either pro-anorexigenic or pro-orexigenic effects, as well as no effects at all (Masaki et al., 2003; Kubota et al., 2007; Coope et al., 2008).
Since both AdipoR1 or AdipoR2 were recently found in a fraction of POA WSN we tested the effects of adiponectin on thermogenesis in mice null for either one of the adiponectin receptors (Eberwine and Bartfai, 2011; Klein et al., 2011). When injected locally into the POA of wt mice, adiponectin had thermogenic effects elevating CBT and fatty acid oxidation (measured as decreased respiratory exchange ratio). These effects were nearly abolished in mice lacking AdipoR1, and were only diminished in animals null for AdipoR2. It is possible that some of the contrasting findings may be due to differences in the experimental conditions used, or to the putative opposite roles that AdipoR1 and AdipoR2 were found to have on energy metabolism (Bjursell et al., 2007). Another confounding factor may be that the oligomer form of adiponectin used as the adiponectin monomer can oligomerize to form 3-mers that can further aggregate into 6-, 12-, or 18-mers (Pajvani et al., 2003). Kubota et al. (2007) reported that in mice, only 3- and 6-mers can enter the CSF from the circulation.
Two Interesting Orphan GPCRs with Hypothalamic Expression
The GPCRs are the favorite drug target class of the pharmaceutical industry and many of the most used and safest drugs are ligands to this class of receptors, including beta blockers, the antihistamines, and the D2 receptor antagonist antipsychotics to mention a few. The relative ease by which ligands to GPCRs are developed is the reason for excitement in the discovery of orphan GPCRs with physiologically and pharmacologically interesting and robust effects. Thus we examine into the effects mediated by GPR83 and GPR50, because it is likely that the validation of their role in integration of nutrient and energy homeostasis will lead to the development of useful drugs that affect feeding body weight and life span.
GPR83
Profiling of WSN revealed that these cells express several orphan GPCRs (Eberwine and Bartfai, 2011). Among these is GPR83 (also known as GIR, GPR72, or JP05), originally identified as a stress–response element from a murine thymoma cDNA library treated with glucocorticoids and forskolin (Harrigan et al., 1989, 1991; Baughman et al., 1991), and subsequently shown to be highly expressed in several brain regions including the hypothalamus, the cortex, the thalamus, the hippocampus, and the amygdala (Pesini et al., 1998; Brezillon et al., 2001; Wang et al., 2001; Adams et al., 2003; Sah et al., 2005). GPR83 shares some homology with a variety of known peptide receptors, including the neuropeptide Y2 receptor. One study reported that NPY C-terminus fragments can bind and activate rat GPR83 with moderate affinity suggesting that GPR83 might participate in the regulation of nutrient intake (Sah et al., 2007).
Local downregulation of GPR83 in the hypothalamic POA, by injection of lentiviral vectors expressing a pool of shRNAs directed against all known isoforms of mouse GPR83 recently demonstrated its role in temperature homeostasis (Dubins et al., 2012). Reduction of POA GPR83 in the range of 30–50% caused a modest (0.15°C) but significant reduction of CBT, starting at day 4 post-treatment, that lasted at least until recording was stopped at day 18. CBT reduction was observed only in the dark period of the day, when the mice are active, and was not significant during the light-inactive phase. The downregulation of the expression GPR83 did not alter calorie intake, and animals treated with silencing GPR83 shRNA ate similarly to those treated with the non-silencing counterpart. However, the silencing shRNA treated group showed an increase in body weight gain that became significant 3 weeks after treatment and was associated with reduced hypothalamic receptor expression. This phenotype was similar to that observed in the long-lived transgenic mice, with reduced CBT achieved by producing heat through uncoupling neuronal mitochondria in the vicinity of WSN cells in the POA (Conti et al., 2006).
GPR50
GPR50 is a GPCR recently demonstrated to play an important role in adaptive thermogenesis in response to calorie intake (Bechtold et al., 2012). It was originally cloned from human pituitary gland and termed melatonin-related receptor (MRR) for its homology with the melatonin receptors (Reppert et al., 1996; Dufourny et al., 2008, 2012). GPR50 does not bind to melatonin, and although it may dimerize with melatonin receptors (possibly influencing melatonin action) to date it remains an orphan receptor (Levoye et al., 2006). Expression of GPR50 is high in the hypothalamus, where it localized in the medial POA, the LH neurons of the dorsomedial nucleus, and in tanycytes (Reppert et al., 1996; Drew et al., 1998, 2001; Hamouda et al., 2007; Sidibe et al., 2010; Batailler et al., 2011; Bechtold et al., 2012).
When fed ad libitum, mice null for GPR50 (Gpr50-/-) showed a modest (~0.5°C) reduction of CBT, that like in the Hcrt-UCP2 mice and the GPR83 shRNA mice, respectively, was observed only during the dark-active part of the day. In response to 24 h food deprivation, CBT of Gpr50-/- mice dropped up to 10°C. O2 consumption and CO2 production were also reduced, and mice entered a torpor-like state. The exact mechanisms by which GPR50 may affect thermogenesis remain to be elucidated. The experimental evidence collected thus far suggest that GPR50 can affect thermal responses to energy signals by directly reducing the responses to leptin and melanocortin during fasting in the ARC, and indirectly by suppressing TRH in the PVN, possibly normally inhibiting entry into a hypometabolic state (Bechtold et al., 2012).
Summary
Lowered CBT increased lifespan and its value in homeotherms can be affected by calorie intake. Here we reviewed the current knowledge on the molecules and signals that mediate CBT responses to calorie intake as these may influence longevity and aging.
At least two hypothalamic regions are involved in mediating these responses. One is the region containing the ARC, the PVN, and the LH nuclei, which synthesize neuropeptides to regulate feeding, but are also able to affect CBT. The second is the POA, recognized for integrating and regulating peripheral as well as central temperature information, and containing temperature sensitive neurons that can also respond to nutrient signals. Both regions can activate the SNS and are in polysynaptic contact with BAT, a tissue specialized in heat production, and that in small animals, such as mice and rats, is the main contributor to thermogenesis. Within the ARC and the LH, the anabolic neuropeptides NPY, AgRP, and MCH are elevated during food deprivation, stimulating appetite and concomitantly reducing thermogenesis. In contrast, the catabolic neuropeptide α-MSH has opposite effects. In addition, AgRP and α-MSH were also proposed to regulate thermogenesis by inhibiting or stimulating the release of TRH from the PVN, thus influencing the level of circulating thyroid hormone.
Nutrient signals can influence thermoregulation by affecting the level of the aforementioned neuropeptides or by inhibiting the activity of WSN POA neurons. In particular, glucose, insulin, and leptin can act on neurons in the PVN and the ARC, stimulating the release of TRH and α-MSH, and reducing that of NPY and AgRP, promoting CBT increase. The POA was demonstrated to be responsive to glucose, insulin, IGF-1, NPY, and adiponectin, and some of these signals were demonstrated to modulate WSN activity. Two orphan GPCRs, GPR83 and GPR50, were also recently proposed to be instrumental in influencing CBT in response to calorie intake. GPR83 is elevated in the hypothalamus during CR, is expressed on WSN, and its local downregulation in the POA reduced CBT. GPR50, primarily expressed in tanycytes lining the third ventricle, appears to be an important regulator of the hypothermic response to fasting.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Supported by NIH grant AG028040. The authors thank Nikki Bortell for editing the manuscript.
Abbreviations
α-MSH, α-melanocyte-stimulating hormone; AgRP, agouti-related protein; ARC, arcuate nucleus; BAT, brown adipose tissue; CART, cocaine- and amphetamine-regulated transcript; CBT, core body temperature; CR, calorie restriction; DMH, dorsomedial hypothalamus nucleus; DMNX, dorsal motor nucleus of the vagus; Glut2, glucose transporter type 2; GOAT, ghrelin O-acyltransferase; GPR83, G protein-coupled receptor; IGF-1, insulin-like growth factor 1; IR, insulin receptor; LH, lateral hypothalamus; MCH, melanin-concentrating hormone; MC4R, melanocortin 4 receptor; MRR, melatonin-related receptor; NE, norepinephrine; NPY, neuropeptide Y; NTS, nucleus of the solitary tract; POA, preoptic area; POMC, pro-opiomelanocortin; PVN, paraventricular nucleus; rRPa, raphe pallidus; SNS, sympathetic nervous system; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormones; UCP1, uncoupling protein 1; WSN, warm-sensitive neurons.
References
Adachi, A., Shimizu, N., Oomura, Y., and Kobashi, M. (1984). Convergence of hepatoportal glucose-sensitive afferent signals to glucose-sensitive units within the nucleus of the solitary tract. Neurosci. Lett. 46, 215–218.
Adamantidis, A., and de Lecea, L. (2009). The hypocretins as sensors for metabolism and arousal. J. Physiol. 587, 33–40.
Adams, F., Grassie, M., Shahid, M., Hill, D. R., and Henry, B. (2003). Acute oral dexamethasone administration reduces levels of orphan GPCR glucocorticoid-induced receptor (GIR) mRNA in rodent brain: potential role in HPA-axis function. Brain Res. Mol. Brain Res. 117, 39–46.
Ahima, R. S., Prabakaran, D., Mantzoros, C., Qu, D., Lowell, B., Maratos-Flier, E., et al. (1996). Role of leptin in the neuroendocrine response to fasting. Nature 382, 250–252.
Ahima, R. S., Saper, C. B., Flier, J. S., and Elmquist, J. K. (2000). Leptin regulation of neuroendocrine systems. Front. Neuroendocrinol. 21, 263–307.
Anand, B. K., Chhina, G. S., Sharma, K. N., Dua, S., and Singh, B. (1964). Activity of single neurons in the hypothalamic feeding centers: effect of glucose. Am. J. Physiol. 207, 1146–1154.
Baskin, D. G., Figlewicz, D. P., Woods, S. C., Porte, D. Jr., and Dorsa, D. M. (1987). Insulin in the brain. Annu. Rev. Physiol. 49, 335–347.
Batailler, M., Mullier, A., Sidibe, A., Delagrange, P., Prevot, V., Jockers, R., et al. (2011). Neuroanataomical distribution of the orphan GPR50 receptor in adult sheep and rodent brains. J. Neuroendocrinol. doi: 10.1111/j.1365-2826.2011.02274.x [Epub ahead of print].
Baughman, G., Harrigan, M. T., Campbell, N. F., Nurrish, S. J., and Bourgeois, S. (1991). Genes newly identified as regulated by glucocorticoids in murine thymocytes. Mol. Endocrinol. 5, 637–644.
Bechtold, D. A., Sidibe, A., Saer, B. R., Li, J., Hand, L. E., Ivanova, E. A., et al. (2012). A role for the melatonin-related receptor GPR50 in leptin signaling, adaptive thermogenesis, and torpor. Curr. Biol. 22, 70–77.
Belgardt, B. F., and Bruning, J. C. (2010). CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 1212, 97–113.
Berg, A. H., Combs, T. P., Du, X., Brownlee, M., and Scherer, P. E. (2001). The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 7, 947–953.
Berg, A. H., Combs, T. P., and Scherer, P. E. (2002). ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 13, 84–89.
Billington, C. J., Briggs, J. E., Grace, M., and Levine, A. S. (1991). Effects of intracerebroventricular injection of neuropeptide Y on energy metabolism. Am. J. Physiol. 260, 21–27.
Bjorbaek, C., and Hollenberg, A. N. (2002). Leptin and melanocortin signaling in the hypothalamus. Vitam. Horm. 65, 281–311.
Bjursell, M., Ahnmark, A., Bohlooly, Y. M., William-Olsson, L., Rhedin, M., Peng, X. R., et al. (2007). Opposing effects of adiponectin receptors 1 and 2 on energy metabolism. Diabetes 56, 583–593.
Blake, N. G., Eckland, D. J., Foster, O. J., and Lightman, S. L. (1991). Inhibition of hypothalamic thyrotropin-releasing hormone messenger ribonucleic acid during food deprivation. Endocrinology 129, 2714–2718.
Blake, N. G., Johnson, M. R., Eckland, D. J., Foster, O. J., and Lightman, S. L. (1992). Effect of food deprivation and altered thyroid status on the hypothalamic–pituitary–thyroid axis in the rat. J. Endocrinol. 133, 183–188.
Bonnavion, P., and de Lecea, L. (2010). Hypocretins in the control of sleep and wakefulness. Curr. Neurol. Neurosci. Rep. 10, 174–179.
Bouali, S. M., Fournier, A., St-Pierre, S., and Jolicoeur, F. B. (1995). Effects of NPY and NPY2-36 on body temperature and food intake following administration into hypothalamic nuclei. Brain Res. Bull. 36, 31–35.
Boulant, J. A. (2000). Role of the preoptic-anterior hypothalamus in thermoregulation and fever. Clin. Infect. Dis. 31(Suppl. 5) S157–S161.
Brezillon, S., Detheux, M., Parmentier, M., Hokfelt, T., and Hurd, Y. L. (2001). Distribution of an orphan G-protein coupled receptor (JP05) mRNA in the human brain. Brain Res. 921, 21–30.
Briski, K. P., and Sylvester, P. W. (2001). Co-distribution of Fos- and mu opioid receptor immunoreactivity within the rat septopreoptic area and hypothalamus during acute glucose deprivation: effects of the mu receptor antagonist CTOP. Neurosci. Lett. 306, 141–144.
Brown, L. M., Clegg, D. J., Benoit, S. C., and Woods, S. C. (2006). Intraventricular insulin and leptin reduce food intake and body weight in C57BL/6J mice. Physiol. Behav. 89, 687–691.
Bruning, J. C., Gautam, D., Burks, D. J., Gillette, J., Schubert, M., Orban, P. C., et al. (2000). Role of brain insulin receptor in control of body weight and reproduction. Science 289, 2122–2125.
Butler, A. A., Marks, D. L., Fan, W. Kuhn, C. M., Bartolome, M., and Cone, R. D. (2001). Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat. Neurosci. 4, 605–611.
Campfield, L. A., Smith, F. J., Guisez, Y., Devos, R., and Burn, P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546–549.
Cannon, B., and Nedergaard, J. (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359.
Cannon, B., and Nedergaard, J. (2010). Thyroid hormones: igniting brown fat via the brain. Nat. Med. 16, 965–967.
Cardona-Gomez, G. P., DonCarlos, L., and Garcia-Segura, L. M. (2000). Insulin-like growth factor I receptors and estrogen receptors colocalize in female rat brain. Neuroscience 99, 751–760.
Cerri, M., and Morrison, S. F. (2005). Activation of lateral hypothalamic neurons stimulates brown adipose tissue thermogenesis. Neuroscience 135, 627–638.
Chavez, M., Kaiyala, K., Madden, L. J., Schwartz, M. W., and Woods, S. C. (1995). Intraventricular insulin and the level of maintained body weight in rats. Behav. Neurosci. 109, 528–531.
Cone, R. D. (1999). The central melanocortin system and energy homeostasis. Trends Endocrinol. Metab. 10, 211–216.
Connors, J. M., DeVito, W. J., and Hedge, G. A. (1985). Effects of food deprivation on the feedback regulation of the hypothalamic–pituitary–thyroid axis of the rat. Endocrinology 117, 900–906.
Conti, B., Sanchez-Alavez, M., Winsky-Sommerer, R., Morale, M. C., Lucero, J., Brownell, S., et al. (2006). Transgenic mice with a reduced core body temperature have an increased life span. Science 314, 825–828.
Coope, A., Milanski, M., Araujo, E. P., Tambascia, M., Saad, M. J., Geloneze, B., et al. (2008). AdipoR1 mediates the anorexigenic and insulin/leptin-like actions of adiponectin in the hypothalamus. FEBS Lett. 582, 1471–1476.
Costa-e-Sousa, R. H., Souza, L. L., Calvino, C., Cabanelas, A., Almeida, N. A., Oliveira, K. J., et al. (2011). Central NPY-Y5 receptors activation plays a major role in fasting-induced pituitary–thyroid axis suppression in adult rat. Regul. Pept. 171, 43–47.
Currie, P. J., and Coscina, D. V. (1995). Dissociated feeding and hypothermic effects of neuropeptide Y in the paraventricular and perifornical hypothalamus. Peptides 16, 599–604.
Dallaporta, M., Himmi, T., Perrin, J., and Orsini, J. C. (1999). Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport 10, 2657–2660.
Dark, J., and Pelz, K. M. (2008). NPY Y1 receptor antagonist prevents NPY-induced torpor-like hypothermia in cold-acclimated Siberian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R236–R245.
de Lecea, L., Kilduff, T. S., Peyron, C., Gao, X., Foye, P. E., Danielson, P. E., et al. (1998). The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U.S.A. 95, 322–327.
Diano, S., Naftolin, F., Goglia, F., and Horvath, T. L. (1998). Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology 139, 2879–2884.
Dietrich, M. O., Antunes, C., Geliang, G., Liu, Z. W., Borok, E., Nie, Y., et al. (2010). Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci. 30, 11815–11825.
Drew, J. E., Barrett, P., Mercer, J. G., Moar, K. M., Canet, E., Delagrange, P., et al. (2001). Localization of the melatonin-related receptor in the rodent brain and peripheral tissues. J. Neuroendocrinol. 13, 453–458.
Drew, J. E., Barrett, P., Williams, L. M., Conway, S., and Morgan, P. J. (1998). The ovine melatonin-related receptor: cloning and preliminary distribution and binding studies. J. Neuroendocrinol. 10, 651–661.
Dubins, J. S., Sanchez-Alavez, M., Zhukov, V., Sanchez-Gonzalez, A., Moroncini, G., Carvajal-Gonzalez, S., et al. (2012). Downregulation of GPR83 in the hypothalamic preoptic area reduces core body temperature and elevates circulating levels of adiponectin. Metabolism. doi: 10.1016/j.metabol.2012.03.015 [Epub ahead of print].
Duffy, P. H., Feuers, R. J., Leakey, J. A., Nakamura, K., Turturro, A., and Hart, R. W. (1989). Effect of chronic caloric restriction on physiological variables related to energy metabolism in the male Fischer 344 rat. Mech. Ageing Dev. 48, 117–l33.
Dufourny, L., Levasseur, A., Migaud, M., Callebaut, I., Pontarotti, P., Malpaux, B., et al. (2008). GPR50 is the mammalian ortholog of Mel1c: evidence of rapid evolution in mammals. BMC Evol. Biol. 8, 105. doi: 10.1186/1471-2148-8-105
Dufourny, L., Levasseur, A., Migaud, M., Callebaut, I., Pontarotti, P., Malpaux, B., et al. (2012). Correction: GPR50 is the mammalian ortholog of Mel1c: evidence of rapid evolution in mammals. BMC Evol. Biol. 12, 28. doi: 10.1186/1471-2148-12-28
Dunn-Meynell, A. A., Rawson, N. E., and Levin, B. E. (1998). Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. 814, 41–54.
Eberwine, J., and Bartfai, T. (2011). Single cell transcriptomics of hypothalamic warm sensitive neurons that control core body temperature and fever response signaling asymmetry and an extension of chemical neuroanatomy. Pharmacol. Ther. 129, 241–259.
Egawa, M., Yoshimatsu, H., and Bray, G. A. (1989). Lateral hypothalamic injection of 2-deoxy-D-glucose suppresses sympathetic activity. Am. J. Physiol. 257, R1386–R1392.
Egawa, M., Yoshimatsu, H., and Bray, G. A. (1991). Neuropeptide Y suppresses sympathetic activity to interscapular brown adipose tissue in rats. Am. J. Physiol. 260, R328–R334.
Elmquist, J. K., Coppari, R., Balthasar, N., Ichinose, M., and Lowell, B. B. (2005). Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J. Comp. Neurol. 493, 63–71.
Fan, W., Voss-Andreae, A., Cao, W. H., and Morrison, S. F. (2005). Regulation of thermogenesis by the central melanocortin system. Peptides 26, 1800–1813.
Fekete, C., Kelly, J., Mihaly, E., Sarkar, S., Rand, W. M., Legradi, G., et al. (2001). Neuropeptide Y has a central inhibitory action on the hypothalamic–pituitary–thyroid axis. Endocrinology 142, 2606–2613.
Fekete, C., Legradi, G., Mihaly, E., Huang, Q. H., Tatro, J. B., Rand, W. M., et al. (2000a). alpha-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J. Neurosci. 20, 1550–1558.
Fekete, C., Mihaly, E., Luo, L. G., Kelly, J., Clausen, J. T., Mao, Q., et al. (2000b). Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamic–pituitary–thyroid axis during fasting. J. Neurosci. 20, 9224–9234.
Fekete, C., Marks, D. L., Sarkar, S., Emerson, C. H., Rand, W. M., Cone, R.D., et al. (2004). Effect of agouti-related protein in regulation of the hypothalamic–pituitary–thyroid axis in the melanocortin 4 receptor knockout mouse. Endocrinology 145, 4816–4821.
Fekete, C., Sarkar, S., Rand, W. M., Harney, J. W., Emerson, C. H., Bianco, A. C., and Lechan, R. M. (2002a). Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic–pituitary–thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology 143, 3846–3853.
Fekete, C., Sarkar, S., Rand, W. M., Harney, J. W., Emerson, C. H., Bianco, A. C., et al. (2002b). Neuropeptide Y1 and Y5 receptors mediate the effects of neuropeptide Y on the hypothalamic–pituitary–thyroid axis. Endocrinology 143, 4513–4519.
Fekete, C., Singru, P. S., Sanchez, E. S., Sarkar, S., Christoffolete, M. A., Riberio, R. S., et al. (2006). Differential effects of central leptin, insulin, or glucose administration during fasting on the hypothalamic–pituitary–thyroid axis and feeding-related neurons in the arcuate nucleus. Endocrinology 147, 520–529.
Fioramonti, X., Lorsignol, A., Taupignon, A., and Penicaud, L. (2004). A new ATP-sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes 53, 2767–2775.
Freinkel, N., Metzger, B. E., Harris, E., Robinson, S., and Mager, M. (1972). The hypothermia of hypoglycemia. Studies with 2-deoxy-D-glucose in normal human subjects and mice. N. Engl. J. Med. 287, 841–845.
Fruebis, J., Tsao, T. S., Javorschi, S., Ebbets-Reed, D., Erickson, M. R., Yen, F. T., et al. (2001). Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. U.S.A. 98, 2005–2010.
Fry, M., Smith, P. M., Hoyda, T. D., Duncan, M., Ahima, R. S., Sharkey, K. A., et al. (2006). Area postrema neurons are modulated by the adipocyte hormone adiponectin. J. Neurosci. 26, 9695–9702.
Gao, Q., and Horvath, T. L. (2007). Neurobiology of feeding and energy expenditure. Annu. Rev. Neurosci. 30, 367–398.
Gao, Q., Wolfgang, M. J., Neschen, S., Morino, K., Horvath, T. L., Shulman, G. I., et al. (2004). Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc. Natl. Acad. Sci. U.S.A. 101, 4661–4666.
Gavrilova, O., Leon, L. R., Marcus-Samuels, B., Mason, M. M., Castle, A. L. Refetoff, S., et al. (1999). Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 96, 14623–14628.
Gluck, E. F., Stephens, N., and Swoap, S. J. (2006). Peripheral ghrelin deepens torpor bouts in mice through the arcuate nucleus neuropeptide Y signaling pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1303–R1309.
Guillod-Maximin, E., Roy, A. F., Vacher, C. M., Aubourg, A., Bailleux, V., Lorsignol, A., et al. (2009). Adiponectin receptors are expressed in hypothalamus and colocalized with proopiomelanocortin and neuropeptide Y in rodent arcuate neurons. J. Endocrinol. 200, 93–105.
Halaas, J. L., Gajiwala, K. S., Maffei, M., Cohen, S. L., Chait, B. T., Rabinowitz, D., et al. (1995). Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269, 543–546.
Hammel, H., Hardy, J. D., and Fusco, M. M. (1960). Thermoregulatory responses to hypothalamic cooling in unanesthetized dogs. Am. J. Physiol. 198, 481–486.
Hamouda, H. O., Chen, P., Levoye, A., Sozer-Topcular, N., Daulat, A. M., Guillaume, J. L., et al. (2007). Detection of the human GPR50 orphan seven transmembrane protein by polyclonal antibodies mapping different epitopes. J. Pineal Res. 43, 10–15.
Harrigan, M. T., Baughman, G., Campbell, N. F., and Bourgeois, S. (1989). Isolation and characterization of glucocorticoid- and cyclic AMP-induced genes in T lymphocytes. Mol. Cell. Biol. 9, 3438–3446.
Harrigan, M. T., Campbell, N. F., and Bourgeois, S. (1991). Identification of a gene induced by glucocorticoids in murine T-cells: a potential G protein-coupled receptor. Mol. Endocrinol. 5, 1331–1338.
Harris, M., Aschkenasi, C., Elias, C. F., Chandrankunnel, A., Nillni, E. A., Bjoorbaek, C., et al. (2001). Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J. Clin. Invest. 107, 111–120.
Havrankova, J., Roth, J., and Brownstein, M. (1978). Insulin receptors are widely distributed in the central nervous system of the rat. Nature 272, 827–829.
Haynes, W. G., Morgan, D. A., Walsh, S. A., Mark, A. L., and Sivitz, W. I. (1997). Receptor-mediated regional sympathetic nerve activation by leptin. J. Clin. Invest. 100, 270–278.
Heppner, K. M., Muller, T. D., Kirchner, H., Perez-Tilve, D., Pfluger, P. T., Tschop, M. H., et al. (2012). The role of ghrelin-octanoyl-acyl-transferase (GOAT) in thermoregulation. J. Endocrinol. Invest. doi: 10.3275/8388 [Epub ahead of print].
Hori, T., Yamasaki, M., Asami, T., Koga, H., and Kiyohara, T. (1988). Responses of anterior hypothalamic-preoptic thermosensitive neurons to thyrotropin releasing hormone and cyclo(His-Pro). Neuropharmacology 27, 895–901.
Hoyda, T. D., and Ferguson, A. V. (2010). Adiponectin modulates excitability of rat paraventricular nucleus neurons by differential modulation of potassium currents. Endocrinology 151, 3154–3162.
Hwa, J. J., Witten, M. B., Williams, P., Ghibaudi, L., Gao, J., Salisbury, B. G., et al. (1999). Activation of the NPY Y5 receptor regulates both feeding and energy expenditure. Am. J. Physiol. 277, R1428–R1434.
Ito, M., Gomori, A., Ishihara, A., Oda, Z., Mashiko, S., Matsushita, H., et al. (2003). Characterization of MCH-mediated obesity in mice. Am. J. Physiol. Endocrinol. Metab. 284, E940–E945.
Jolicoeur, F. B., Bouali, S. M., Fournier, A., and St-Pierre, S. (1995). Mapping of hypothalamic sites involved in the effects of NPY on body temperature and food intake. Brain Res. Bull. 36, 125–129.
Kim, M. S., Small, C. J., Stanley, S. A., Morgan, D. G., Seal, L. J., Kong, W. M., et al. (2000). The central melanocortin system affects the hypothalamo-pituitary thyroid axis and may mediate the effect of leptin. J. Clin. Invest. 105, 1005–1011.
Kishi, T., Aschkenasi, C. J., Lee, C. E., Mountjoy, K. G., Saper, C. B., and Elmquist, J. K. (2003). Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J. Comp. Neurol. 457, 213–235.
Klein, I., Sanchez-Alavez, M., Tabarean, I., Schaefer, J., Holmberg, K. H., Klaus, J., et al. (2011). AdipoR1 and 2 are expressed on warm sensitive neurons of the hypothalamic preoptic area and contribute to central hyperthermic effects of adiponectin. Brain Res. 1423, 1–9.
Koch, L., Wunderlich, F. T., Seibler, J., Konner, A. C., Hampel, B., Irlenbusch, S., et al. (2008). Central insulin action regulates peripheral glucose and fat metabolism in mice. J. Clin. Invest. 118, 2132–2147.
Kos, K., Harte, A. L., da Silva, N. F., Tonchev, A., Chaldakov, G., James, S., et al. (2007). Adiponectin and resistin in human cerebrospinal fluid and expression of adiponectin receptors in the human hypothalamus. J. Clin. Endocrinol. Metab. 92, 1129–1136.
Kubota, N., Yano, W., Kubota, T., Yamauchi, T., Itoh, S., Kumagai, H., et al. (2007). Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 6, 55–68.
Lane, M. A., Baer, D. J., Rumpler, W. V., Weindruch, R., Ingram, D. K., Tilmont, E. M., et al. (1996). Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc. Natl. Acad. Sci. U.S.A. 93, 4159–4164.
Lawrence, C. B., Snape, A. C., Baudoin, F. M., and Luckman, S. M. (2002). Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology 143, 155–162.
Lechan, R. M., and Fekete, C. (2006). The TRH neuron: a hypothalamic integrator of energy metabolism. Prog. Brain Res. 153, 209–235.
Legradi, G., Emerson, C. H., Ahima, R. S., Rand, W. M., Flier, J. S., and Lechan, R. M. (1998). Arcuate nucleus ablation prevents fasting-induced suppression of ProTRH mRNA in the hypothalamic paraventricular nucleus. Neuroendocrinology 68, 89–97.
Legradi, G., and Lechan, R. M. (1999). Agouti-related protein containing nerve terminals innervate thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology 140, 3643–3652.
Levoye, A., Dam, J., Ayoub, M. A., Guillaume, J. L., Couturier, C., Delagrange, P., et al. (2006). The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J. 25, 3012–3023.
Liu, H., Kishi, T., Roseberry, A. G., Cai, X., Lee, C. E., Montez, J. M., et al. (2003). Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J. Neurosci. 23, 7143–7154.
Lopez, M., Varela, L., Vazquez, M. J., Rodriguez-Cuenca, S., Gonzalez, C. R., Velagapudi, V. R., et al. (2010). Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 16, 1001–1008.
Lopez-Valpuesta, F. J., Nyce, J. W., and Myers, R. D. (1996). NPY-Y1 receptor antisense injected centrally in rats causes hyperthermia and feeding. Neuroreport 7, 2781–2784.
Luheshi, G. N., Gardner, J. D., Rushforth, D. A., Loudon, A. S., and Rothwell, N. J. (1999). Leptin actions on food intake and body temperature are mediated by IL-1. Proc. Natl. Acad. Sci. U.S.A. 96, 7047–7052.
Marks, J. L., Porte, D. Jr., Stahl, W. L., and Baskin, D. G. (1990). Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 127, 3234–3236.
Masaki, T., Chiba, S., Yasuda, T., Tsubone, T., Kakuma, T., Shimomura, I., et al. (2003). Peripheral, but not central, administration of adiponectin reduces visceral adiposity and upregulates the expression of uncoupling protein in agouti yellow (Ay/a) obese mice. Diabetes 52, 2266–2273.
Mashiko, S., Ishihara, A., Iwaasa, H., Sano, H., Ito, J., Gomori, A., et al. (2007). A pair-feeding study reveals that a Y5 antagonist causes weight loss in diet-induced obese mice by modulating food intake and energy expenditure. Mol. Pharmacol. 71, 602–608.
Mayer, C. M., and Belsham, D. D. (2009). Insulin directly regulates NPY and AgRP gene expression via the MAPK MEK/ERK signal transduction pathway in mHypoE-46 hypothalamic neurons. Mol. Cell. Endocrinol. 307, 99–108.
McCormack, J. G. (1982). The regulation of fatty acid synthesis in brown adipose tissue by insulin. Prog. Lipid. Res. 21, 195–223.
McGowan, M. K., Andrews, K. M., and Grossman, S. P. (1992a). Chronic intrahypothalamic infusions of insulin or insulin antibodies alter body weight and food intake in the rat. Physiol. Behav. 51, 753–766.
McGowan, M. K., Andrews, K. M., and Grossman, S. P. (1992b). Role of intrahypothalamic insulin in circadian patterns of food intake, activity, and body temperature. Behav. Neurosci. 106, 380–385.
Menendez, J. A., and Atrens, D. M. (1991). Insulin and the paraventricular hypothalamus: modulation of energy balance. Brain Res. 555, 193–201.
Mizuno, Y., and Oomura, Y. (1984). Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res. 307, 109–116.
Morrison, S. F., and Nakamura, K. (2011). Central neural pathways for thermoregulation. Front. Biosci. 16, 74–104.
Morton, G. J., Cummings, D. E., Baskin, D. G., Barsh, G. S., and Schwartz, M. W. (2006). Central nervous system control of food intake and body weight. Nature 443, 289–295.
Mounien, L., Marty, N., Tarussio, D., Metref, S., Genoux, D., Preitner, F., et al. (2010). Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J. 24, 1747–1758.
Muntzel, M. S., Anderson, E. A., Johnson, A. K., and Mark, A. L. (1995). Mechanisms of insulin action on sympathetic nerve activity. Clin. Exp. Hypertens. 17, 39–50.
Nakayama, T., Hammel, H. T., Hardy, J. D., and Eisenman, J. S. (1963). Thermal stimulation of electrical activity of single units of the preoptic region. Am. J. Physiol. 204, 1122–1126.
Nillni, E. A., Vaslet, C., Harris, M. A., Hollenberg, A., Bjorbak, C., and Flier, J. S. (2000). Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J. Biol. Chem. 275, 36124–36133.
Obici, S., Feng, Z., Karkanias, G., Baskin, D. G., and Rossetti, L. (2002). Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 5, 566–572.
Oldfield, B. J., Giles, M. E., Watson, A., Anderson, C., Colvill, L. M., and McKinley, M. J. (2002). The neurochemical characterisation of hypothalamic pathways projecting polysynaptically to brown adipose tissue in the rat. Neuroscience 110, 515–526.
Pajvani, U. B., Du, X., Combs, T. P., Berg, A. H., Rajala, M. W., Schulthess, T., et al. (2003). Structure–function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J. Biol. Chem. 278, 9073–9085.
Pedrazzini, T., Seydoux, J., Kunstner, P., Aubert, J. F., Grouzmann, E., Beermann, F., et al. (1998). Cardiovascular response, feeding behavior and locomotor activity in mice lacking the NPY Y1 receptor. Nat. Med. 4, 722–726.
Pelleymounter, M. A., Cullen, M. J., Baker, M. B., Hecht, R., Winters, D., Boone, T., et al. (1995). Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269, 540–543.
Pelz, K. M., and Dark, J. (2007). ICV NPY Y1 receptor agonist but not Y5 agonist induces torpor-like hypothermia in cold-acclimated Siberian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R2299–R2311.
Perello, M., Cakir, I., Cyr, N. E., Romero, A., Stuart, R. C., Chiappini, F., et al. (2010). Maintenance of the thyroid axis during diet-induced obesity in rodents is controlled at the central level. Am. J. Physiol. Endocrinol. Metab. 299, E976–E989.
Pesini, P., Detheux, M., Parmentier, M., and Hokfelt, T. (1998). Distribution of a glucocorticoid-induced orphan receptor (JP05) mRNA in the central nervous system of the mouse. Brain Res. Mol. Brain Res. 57, 281–300.
Plum, L., Belgardt, B. F., and Bruning, J. C. (2006). Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 116, 1761–1766.
Plum, L., Schubert, M., and Bruning, J. C. (2005). The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 16, 59–65.
Porte, D. Jr., Baskin, D. G., and Schwartz, M. W. (2002). Leptin and insulin action in the central nervous system. Nutr. Rev. 60, S20–S29; discussion S68–S84, 85–87.
Porte, D. Jr., Baskin, D. G., and Schwartz, M. W. (2005). Insulin signaling in the central nervous system: a critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes 54, 1264–1276.
Psilopanagioti, A., Papadaki, H., Kranioti, E. F., Alexandrides, T. K., and Varakis, J. N. (2009). Expression of adiponectin and adiponectin receptors in human pituitary gland and brain. Neuroendocrinology 89, 38–47.
Qi, Y., Takahashi, N., Hileman, S. M., Patel, H. R., Berg, A. H., Pajvani, U. B., et al. (2004). Adiponectin acts in the brain to decrease body weight. Nat. Med. 10, 524–529.
Rampone, A. J., and Shirasu, M. E. (1964). Temperature changes in the rat in response to feeding. Science 144, 317–319.
Reppert, S. M., Weaver, D. R., Ebisawa, T., Mahle, C. D., and Kolakowski, L. F. Jr. (1996). Cloning of a melatonin-related receptor from human pituitary. FEBS Lett. 386, 219–224.
Rikke, B. A., Yerg, J. E. III, Battaglia, M. E., Nagy, T. R., Allison, D. B., and Johnson, T. E. (2003). Strain variation in the response of body temperature to dietary restriction. Mech. Ageing Dev. 124, 663–678.
Ritter, S., and Dinh, T. T. (1994). 2-Mercaptoacetate and 2-deoxy-D-glucose induce Fos-like immunoreactivity in rat brain. Brain Res. 641, 111–1120.
Robinson, S. W., Dinulescu, D. M., and Cone, R. D. (2000). Genetic models of obesity and energy balance in the mouse. Annu. Rev. Genet. 34, 687–745.
Rothwell, N. J., and Stock, M. J. (1981). A role for insulin in the diet-induced thermogenesis of cafeteria-fed rats. Metabolism 30, 673–678.
Rothwell, N. J., and Stock, M. J. (1986). “Brown adipose tissue and diet-induced thermogenesis,” in Brown Adipose Tissue, eds P. Trayhurn and D. G. Nicholls (New York: Edward Arnold), 269–298.
Routh, V. H. (2002). Glucose-sensing neurons: are they physiologically relevant? Physiol. Behav. 76, 403–413.
Sah, R., Parker, S. L., Sheriff, S., Eaton, S., Balasubramaniam, A., and Sallee, F. R. (2007). Interaction of NPY compounds with the rat glucocorticoid-induced receptor (GIR) reveals similarity to the NPY-Y2 receptor. Peptides 28, 302–309.
Sah, R., Pritchard, L. M., Richtand, N. M., Ahlbrand, R., Eaton, K., Sallee, F. R., et al. (2005). Expression of the glucocorticoid-induced receptor mRNA in rat brain. Neuroscience 133, 281–292.
Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., et al. (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585.
Sanchez-Alavez, M., Osborn, O., Tabarean, I. V., Holmberg, K. H., Eberwine, J., Kahn, C. R., et al. (2011). Insulin-like growth factor 1-mediated hyperthermia involves anterior hypothalamic insulin receptors. J. Biol. Chem. 286, 14983–14990.
Sanchez-Alavez, M., Tabarean, I. V., Osborn, O., Mitsukawa, K., Schaefer, J., Dubins, J., et al. (2010). Insulin causes hyperthermia by direct inhibition of warm-sensitive neurons. Diabetes 59, 43–50.
Sanchez-Lasheras, C., Konner, A. C., and Bruning, J. C. (2010). Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front. Neuroendocrinol. 31, 4–15.
Schwartz, M. W., Woods, S. C., Porte, D. Jr., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671.
Segal-Lieberman, G., Bradley, R. L., Kokkotou, E., Carlson, M., Trombly, D. J., Wang, X., et al. (2003). Melanin-concentrating hormone is a critical mediator of the leptin-deficient phenotype. Proc. Natl. Acad. Sci. U.S.A. 100, 10085–10090.
Shimada, M., Tritos, N. A., Lowell, B. B., Flier, J. S., and Maratos-Flier, E. (1998). Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396, 670–674.
Shklyaev, S., Aslanidi, G., Tennant, M., Prima, V., Kohlbrenner, E., Kroutov, V., et al. (2003). Sustained peripheral expression of transgene adiponectin offsets the development of diet-induced obesity in rats. Proc. Natl. Acad. Sci. U.S.A. 100, 14217–14222.
Sidibe, A., Mullier, A., Chen, P., Baroncini, M., Boutin, J. A., Delagrange, P., et al. (2010). Expression of the orphan GPR50 protein in rodent and human dorsomedial hypothalamus, tanycytes and median eminence. J. Pineal Res. 48, 263–269.
Silva, J. E. (2006). Thermogenic mechanisms and their hormonal regulation. Physiol. Rev. 86, 435–464.
Silva, N. L., and Boulant, J. A. (1984). Effects of osmotic pressure, glucose, and temperature on neurons in preoptic tissue slices. Am. J. Physiol. 247, R335–R345.
Silver, I. A., and Erecinska, M. (1998). Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J. Neurophysiol. 79, 1733–1745.
Skibicka, K. P., Alhadeff, A. L., and Grill, H. J. (2009). Hindbrain cocaine- and amphetamine-regulated transcript induces hypothermia mediated by GLP-1 receptors. J. Neurosci. 29, 6973–6981.
Smirnov, M. S., and Kiyatkin, E. A. (2008). Fluctuations in central and peripheral temperatures associated with feeding behavior in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1415–R1424.
Soare, A., Cangemi, R., Omodei, D., Holloszy, J. O., and Fontana, L. (2011). Long-term calorie restriction, but not endurance exercise, lowers core body temperature in humans. Aging (Albany NY) 3, 374–379.
Spencer, C. A., Lum, S. M., Wilber, J. F., Kaptein, E. M., and Nicoloff, J. T. (1983). Dynamics of serum thyrotropin and thyroid hormone changes in fasting. J. Clin. Endocrinol. Metab. 56, 883–888.
Spiegelman, B. M., and Flier, J. S. (2001). Obesity and the regulation of energy balance. Cell 104, 531–543.
Stanley, B. G., Kyrkouli, S. E., Lampert, S., and Leibowitz, S. F. (1986). Neuropeptide-Y chronically injected into the hypothalamus – a powerful neurochemical inducer of hyperphagia and obesity. Peptides 7, 1189–1192.
Ste Marie, L., Miura, G. I., Marsh, D. J., Yagaloff, K., and Palmiter, R. D. (2000). A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 12339–12344.
Stevenson, J. A., and Montemurro, D. G. (1963). Loss of weight and metabolic rate of rats with lesions in the medial and lateral hypothalamus. Nature 198, 92.
Szentirmai, E., Kapas, L., Sun, Y., Smith, R. G., and Krueger, J. M. (2009). The preproghrelin gene is required for the normal integration of thermoregulation and sleep in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 14069–14074.
Tang-Christensen, M., Vrang, N., Ortmann, S., Bidlingmaier, M., Horvath, T. L., and Tschop, M. (2004). Central administration of ghrelin and agouti-related protein (83–132) increases food intake and decreases spontaneous locomotor activity in rats. Endocrinology 145, 4645–4652.
Teitelbaum, P., and Epstein, A. N. (1962). The lateral hypothalamic syndrome: recovery of feeding and drinking after lateral hypothalamic lesions. Psychol. Rev. 69, 74–90.
Theander-Carrillo, C., Wiedmer, P., Cettour-Rose, P., Nogueiras, R., Perez-Tilve, D., Pfluger, P., et al. (2006). Ghrelin action in the brain controls adipocyte metabolism. J. Clin. Invest. 116, 1983–1993.
Thundyil, J., Pavlovski, D., Sobey, C. G., and Arumugam, T. V. (2011). Adiponectin receptor signalling in the brain. Br. J. Pharmacol. 165, 313–327.
Tomas, E., Tsao, T. S., Saha, A. K., Murrey, H. E., Zhang Cc, C., Itani, S. I., et al. (2002). Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. U.S.A. 99, 16309–16313.
Tschop, M., Smiley, D. L., and Heiman, M. L. (2000). Ghrelin induces adiposity in rodents. Nature 407, 908–913.
Unger, J., McNeill, T. H., Moxley, R. T. III, White, M., Moss, A., and Livingston, J. N. (1989). Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 31, 143–157.
van Baak, M. A. (2008). Meal-induced activation of the sympathetic nervous system and its cardiovascular and thermogenic effects in man. Physiol. Behav. 94, 178–186.
Vella, K. R., Ramadoss, P., Lam, F. S., Harris, J. C., Ye, F. D., Same, P. D., et al. (2011). NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab. 14, 780–790.
Voss-Andreae, A., Murphy, J. G., Ellacott, K. L., Stuart, R. C., Nillni, E. A., Cone, R. D., et al. (2007). Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology 148, 1550–1560.
Walford, R. L., and Spindler, S. R. (1997). The response to calorie restriction in mammals shows features also common to hibernation: a cross-adaptation hypothesis. J. Gerontol. A Biol. Sci. Med. Sci. 52, B179–B183.
Walker, H. C., and Romsos, D. R. (1993). Similar effects of NPY on energy metabolism and on plasma insulin in adrenalectomized ob/ob and lean mice. Am. J. Physiol. 264, E226–E230.
Wang, C., Billington, C. J., Levine, A. S., and Kotz, C. M. (2000). Effect of CART in the hypothalamic paraventricular nucleus on feeding and uncoupling protein gene expression. Neuroreport 11, 3251–3255.
Wang, D., Herman, J. P., Pritchard, L. M., Spitzer, R. H., Ahlbrand, R. L., Kramer, G. L., et al. (2001). Cloning, expression, and regulation of a glucocorticoid-induced receptor in rat brain: effect of repetitive amphetamine. J. Neurosci. 21, 9027–9035.
Wang, H. S., and Lin, M. T. (1985). Effects of insulin on thermoregulatory responses and hypothalamic neuronal activity. Pharmacology 30, 86–94.
Wang, R., Liu, X., Hentges, S. T., Dunn-Meynell, A. A., Levin, B. E., Wang, W., et al. (2004). The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53, 1959–1965.
Werther, G. A., Hogg, A., Oldfield, B. J., McKinley, M. J., Figdor, R., Allen, A. M., et al. (1987). Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 121, 1562–1570.
Wiedmer, P., Strasser, F., Horvath, T. L., Blum, D., Dimarchi, R., Lutz, T., et al. (2011). Ghrelin-induced hypothermia: a physiological basis but no clinical risk. Physiol. Behav. 105, 43–51.
Williams, G., Bing, C., Cai, X. J., Harrold, J. A., King, P. J., and Liu, X. H. (2001). The hypothalamus and the control of energy homeostasis: different circuits, different purposes. Physiol. Behav. 74, 683–701.
Woods, S. C., Lotter, E. C., McKay, L. D., and Porte, D. Jr. (1979). Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282, 503–505.
Yamauchi, T., Kamon, J., Ito, Y., Tsuchida, A., Yokomizo, T., Kita, S., et al. (2003). Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769.
Yamauchi, T., Kamon, J., Waki, H., Terauchi, Y., Kubota, N., Hara, K., et al. (2001). The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 7, 941–946.
Yang, X. J., Kow, L. M., Pfaff, D. W., and Mobbs, C. V. (2004). Metabolic pathways that mediate inhibition of hypothalamic neurons by glucose. Diabetes 53, 67–73.
Yasuda, T., Masaki, T., Kakuma, T., and Yoshimatsu, H. (2003). Centrally administered ghrelin suppresses sympathetic nerve activity in brown adipose tissue of rats. Neurosci. Lett. 349, 75–78.
Yasuda, T., Masaki, T., Kakuma, T., and Yoshimatsu, H. (2004). Hypothalamic melanocortin system regulates sympathetic nerve activity in brown adipose tissue. Exp. Biol. Med. (Maywood) 229, 235–239.
Yettefti, K., Orsini, J. C., el Ouazzani, T., Himmi, T., Boyer, A., and Perrin, J. (1995). Sensitivity of nucleus tractus solitarius neurons to induced moderate hyperglycemia, with special reference to catecholaminergic regions. J. Auton. Nerv. Syst. 51, 191–197.
Keywords: core body temperature, calorie restriction, hypothalamus, neuropeptides, GPCR, homeostasis, warm-sensitive neurons
Citation: Bartfai T and Conti B (2012) Molecules affecting hypothalamic control of core body temperature in response to calorie intake. Front. Gene. 3:184. doi: 10.3389/fgene.2012.00184
Received: 09 July 2012; Accepted: 31 August 2012;
Published online: 05 October 2012.
Edited by:
Elena G. Pasyukova, Russian Academy of Sciences, RussiaReviewed by:
Rozalyn Anderson, University of Wisconsin Madison, USAWilliam K. Scott, University of Miami, USA
William Mair, Harvard School of Public Health, USA
Mei-Jie Jou, Chang Gung University, Taiwan
Copyright: © 2012 Bartfai and Conti. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Bruno Conti, Department of Molecular and Integrative Neurosciences, The Scripps Research Institute, 10550 North Torrey Pines Road, SR307, La Jolla, CA 92037, USA. e-mail:YmNvbnRpQHNjcmlwcHMuZWR1