Abstract
The nervous system becomes increasingly vulnerable to insults and prone to dysfunction during aging. Age-related decline of neuronal function is manifested by the late onset of many neurodegenerative disorders, as well as by reduced signaling and processing capacity of individual neuron populations. Recent findings indicate that impairment of Ca2+ homeostasis underlies the increased susceptibility of neurons to damage, associated with the aging process. However, the impact of aging on Ca2+ homeostasis in neurons remains largely unknown. Here, we survey the molecular mechanisms that mediate neuronal Ca2+ homeostasis and discuss the impact of aging on their efficacy. To address the question of how aging impinges on Ca2+ homeostasis, we consider potential nodes through which mechanisms regulating Ca2+ levels interface with molecular pathways known to influence the process of aging and senescent decline. Delineation of this crosstalk would facilitate the development of interventions aiming to fortify neurons against age-associated functional deterioration and death by augmenting Ca2+ homeostasis.
INTRODUCTION
Fluctuations in intracellular calcium concentration act as signals for a variety of processes in neurons. Most notably, Ca2+ is the major trigger of neurotransmitter release, a process that has been thoroughly investigated over the past decades (Neher and Sakaba, 2008). Moreover, it has also become clear that Ca2+ is essential for a variety of other neuronal functions, including neuronal excitability (Marty and Zimmerberg, 1989), integration of electrical signals (Llinas, 1988; Marty and Zimmerberg, 1989), synaptic plasticity (Malenka et al., 1989), gene expression (Szekely et al., 1990), metabolism (McCormack and Denton, 1990), and programmed cell death (Chalfie and Wolinsky, 1990). Given its central role in processes that are fundamental to the excitable nature of neurons, Ca2+ homeostasis is tightly regulated in these cells (see Table 1 for a summary of the key effectors of Ca2+ homeostasis, in neurons). Here, we briefly overview the main mechanisms neurons use in order to achieve an intricate regulation of the intracellular concentration of Ca2+. In addition, we discuss the accumulating evidence on the potential role of deregulated Ca2+ homeostasis in aging and disease of the nervous system.
Table 1
| Sub-cellular localization | Function | |
|---|---|---|
| Channels | ||
| Voltage-gated Ca2+ channels NMDA receptor | Plasma membrane | Influx of Ca2+ into the cell |
| PMCA, ATP driven ca2+ pump NCX, “Na+/ca2+ exchanger” | Efflux of ca2+ from the cell | |
| SERCA 1, 2a, 2b, 3 | ER and Golgi | Influx of ca2+ into the ER or Golgi |
| Inositol 3-phosphate (InsP3) receptors | ER | Efflux of ca2+ from the ER |
| Ryanodine receptors (RyRs) | ||
| NAADP receptors | ||
| polycystin-2 channels | ||
| presenilin 1 and 2 | ||
| SPCA 1a, 1b, 1c, 1d, 2 | Golgi | Influx of ca2+ into the GolgiX |
| ca2+ uniporter | Mitochondria | Influx of ca2+ into mitochondria |
| NCX mitochondrial Na+/ca2+ exchanger mPTP | Efflux of ca2+ from mitochondria | |
| Buffers | ||
| Calreticulin | ER | Reversible sequestering of ca2+ |
| Calsequestrin | ||
| Endoplasmin | ||
| BiP/grp78 | ||
| Reticulocalbin | ||
| CREC family proteins | ||
| Calretinin | Cytosol, mainly CNS GABAergic interneurons | |
| Calbindin | ||
| Parvalbumin | ||
| Nucleo-calbindin | Golgi | |
| Glycerophosphate dehydrogenase Aralar ARE | Mitochondrial | |
| Sensors | ||
| Calmodulin | Cytosol | Translation of graded ca2+ concentration changes into graded signaling responses via interaction with ca2+ sensitive enzymes |
| Recoverins | Cytosol, photoreceptors | |
| Guanylyl cyclase activating protein 1 (GCAP1) Frequenins | Cytosol, CNS neurons | Kv channel interacting proteins (KChIPs) |
Summary of different Ca2+ channels, buffers and sensors, their subcellular localization and function.
MECHANISMS OF NEURONAL CALCIUM HOMEOSTASIS RELEVANT TO AGING AND DEGENERATION
Ca2+ INFLUX THROUGH THE PLASMA MEMBRANE
Plasma membrane Ca2+ channels allow the passive influx of calcium ions down their electrochemical gradient. These channels are categorized into two major groups depending on the mechanism controlling their transition between the open and closed conformations: channels gated by voltage (also known as voltage-operated Ca2+ channels, VOCC), and channels gated by ligand binding, in neurons usually L-glutamate (Figure 1; Table 1).
FIGURE 1
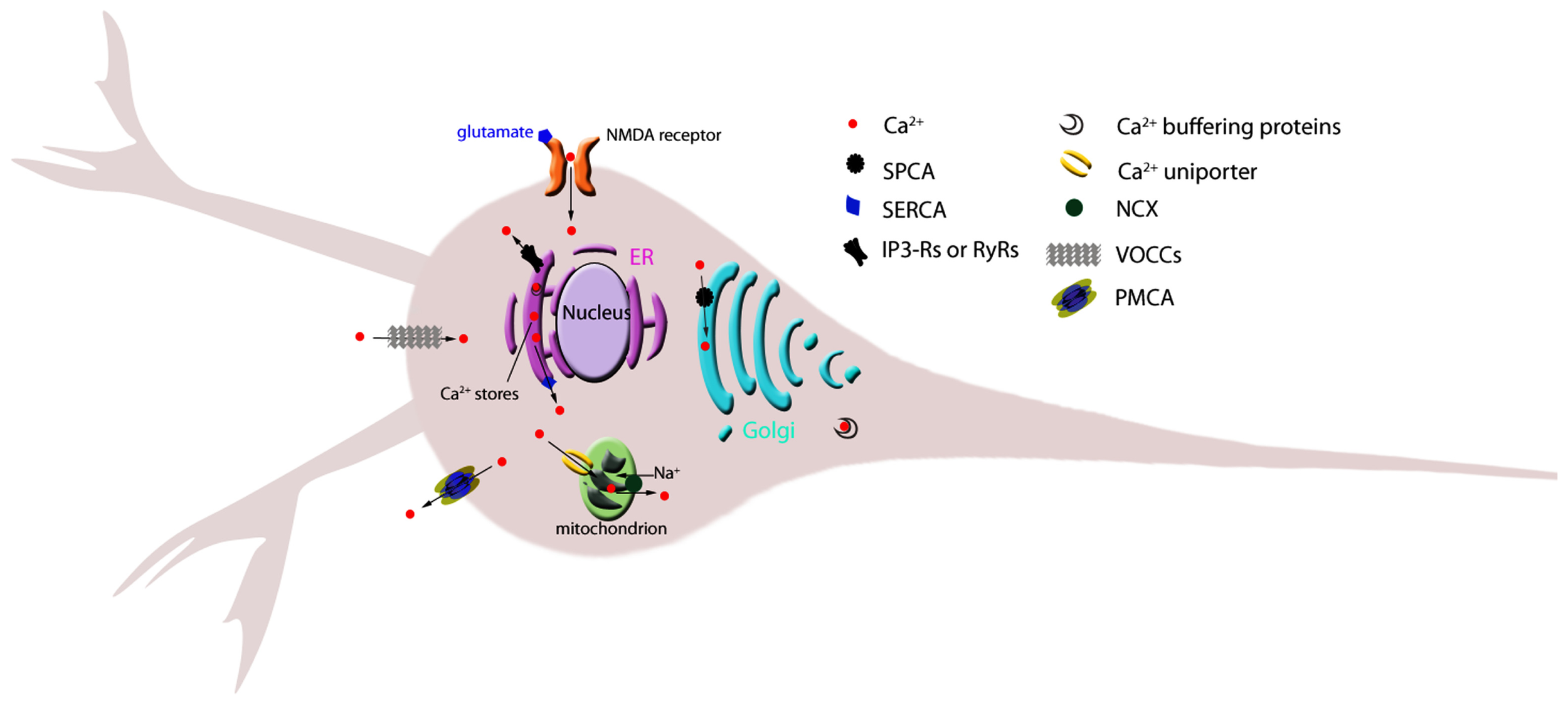
Schematic representation of the main Ca2+ homeostatic machinery components in neurons. Individual, key components of calcium homeostatic mechanisms discussed in the text are shown. Arrows indicate direction of ion flux. ER, endoplasmic reticulum; IP3-R, inositol 3-phosphate receptor; NCX, sodium calcium exchanger; NMDA, N-methyl-D-aspartate; PMCA, plasma membrane Ca2+ ATPase; RyR, ryanodine receptor; SERCA, sarco(endo)plasmic reticulum Ca2+ ATPase; SPCA, secretory-pathway Ca2+-ATPase; VOCC, voltage-operated calcium channel.
Voltage-gated Ca2+ channels are multi-protein complexes comprising several different subunits: α1, α2δ, β1-4, and γ (Takahashi and Catterall, 1987; Catterall et al., 1990). The α1 subunit is the largest and it contains the conduction pore, the voltage sensors, and gating apparatus, and most of the known sites of channel regulation by second messengers, drugs, and toxins. The α1 subunits are associated with distinct auxiliary protein subunits (Catterall et al., 1990): the intracellular β subunit, the transmembrane, disulfide-linked α2δ subunit complex, and the γ subunit, a component of skeletal muscle Ca2+ channels also expressed in heart and brain having four transmembrane segments. Although these auxiliary subunits modulate the functional properties of the Ca2+ channel complex, the pharmacological and physiological diversity of Ca2+ channels arises primarily from the existence of multiple α1 subunits. These are encoded by 10 distinct genes in mammals, further divided into three subfamilies based on sequence similarity (Catterall et al., 1990; Snutch and Reiner, 1992; Ertel et al., 2000). Division of Ca2+ channels into these three subfamilies is phylogenetically ancient, as single representatives of each are found in the Caenorhabditis elegans genome. Recently, calcium homeostasis modulator 1 (CALHM1), a glycosylated membrane protein expressed throughout the brain, was identified as the pore-forming subunit of a unique plasma membrane Ca2+-permeable voltage-gated ion channel (Ma et al., 2012).
Based on the characteristics of channel composition, distinct classes of Ca2+ currents have been described (Tsien et al., 1988). In summary, N-type, P/Q-type, and R-type Ca2+ currents are induced upon strong depolarization (Tsien et al., 1991) and are pharmacologically blocked by specific toxins derived from snail and spider venoms (Miljanich and Ramachandran, 1995). N-type and P/Q-type Ca2+ currents are observed primarily in neurons where they initiate neurotransmission at most fast conventional synapses (Catterall et al., 1990; Olivera et al., 1994; Dunlap et al., 1995). More specifically, the CaV2 subfamily members (CaV2.1, CaV2.2, and CaV2.3) conduct P/Q-type, N-type, and R-type Ca2+ currents, respectively (Catterall et al., 1990; Snutch and Reiner, 1992; Olivera et al., 1994; Ertel et al., 2000). Ca2+ entering neurons through the CaV2.1 and CaV2.2 channels is primarily responsible for initiating synaptic transmission at conventional fast synapses (Olivera et al., 1994; Dunlap et al., 1995). CaV2.2 channels are most prevalent at synapses formed by neurons of the peripheral nervous system. In contrast, CaV2.1 channels play a major role at most synapses formed by neurons of the mammalian central nervous system. However, in some central synapses, including a subset of inhibitory interneurons of the hippocampus (Poncer et al., 1997), CaV2.2 channels are predominant in neurotransmitter release.
Ca2+ entry through a voltage-gated Ca2+ channel initiates neurotransmission by triggering vesicular release (Stanley, 1993). Ca2+-triggered synaptic vesicle exocytosis depends on the assembly of the SNARE complex, in which the vesicle-associated v-SNARE protein synaptobrevin (VAMP) interacts with two plasma membrane-associated t-SNARE proteins, SNAP-25 and syntaxin-1 (Sollner et al., 1993; Bajjalieh and Scheller, 1995; Sudhof, 1995,2004). Maturation into a release-ready SNARE complex requires synaptotagmin, an integral Ca2+-binding protein of the synaptic vesicle membrane that provides Ca2+-dependent regulation of the fusion machinery. Ca2+ influx into the presynaptic terminal binds to the Ca2+ sensor, synaptotagmin, and the SNARE complex changes conformation from a trans to a cis state, resulting in the fusion of apposing membranes and the release of neurotransmitter. Neurotransmitter release occurs in two phases: a fast synchronous (phasic) component and a slow asynchronous (tonic) component (Hubbard, 1963; Barrett and Stevens, 1972; Rahamimoff and Yaari, 1973; Goda and Stevens, 1994; Atluri and Regehr, 1998). Both forms of transmission are Ca2+ dependent. Synchronous release driven by the precisely timed presynaptic Ca2+ current results in a large, fast postsynaptic response (Llinas et al., 1981; Sabatini and Regehr, 1996), whereas the slower asynchronous component, resulting from residual Ca2+ remaining in the terminal after an action potential, provides a basal or tonic level of neurotransmitter release at many synapses (Atluri and Regehr, 1998; Lu and Trussell, 2000; Hagler and Goda, 2001).
In addition to voltage-gated channels, a number of Ca2+ channels on the plasma membrane of neurons are activated by the interaction of ligands with their own plasma membrane receptors. The most prominent such ligand in the nervous system is L-glutamate, by far the most widespread excitatory transmitter in the vertebrate central nervous system. L-glutamate activates two general classes of receptors, the “ionotropic” receptors, which are ionic channels, and the G-protein coupled “metabotropic” receptors. Of these, the ionotropic receptors mediate the direct penetration of Ca2+ into the cell. Three forms of ionotropic receptors have been characterized and named after their most widely used agonists. These are the kainate (KA) receptors, the α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors, and the N-methyl-D-aspartate (NMDA) receptors. The channels formed by AMPA and KA receptors are primarily permeable to Na+ and K+ and exhibit a rather low conductance to Ca2+ (Mayer and Westbrook, 1987). By contrast, the NMDA receptors have a considerably higher conductance and are permeable to Na+ and Ca2+ (MacDermott et al., 1986). These receptors do not mediate rapid synaptic transmission, their contribution being primarily to the slow component of excitatory postsynaptic currents. At the resting plasma membrane potential they are powerfully inhibited by Mg2+, whose block is reversed by plasma membrane depolarization (Nowak et al., 1984). Thus, the rapid increase of membrane depolarization following the activation of KA/AMPA receptors by glutamate released into the synaptic cleft reduces the inhibition of NMDA receptors by Mg2+. Therefore, the excitatory postsynaptic potential produced by activation of an NMDA receptor highly increases the concentration of Ca2+ in the cell. The Ca2+ in turn functions as a key second messenger in various signaling pathways. The ability of the NMDA receptor to act as a “coincidence receptor,” requiring the concomitant presence of its ligand and membrane depolarization in order to be activated, explains many aspects of its functional involvement in long-term potentiation (LTP) and synaptic plasticity, a process associated with memory and learning as discussed later.
EFFLUX OF CALCIUM THROUGH THE PLASMA MEMBRANE
Two major plasma membrane mechanisms are responsible for the extrusion of Ca2+ from cells (Figure 1; Table 1). One is the ATP-driven plasma membrane Ca2+ pump (PMCA) and the other is the Na+/Ca2+ exchanger (NCX), a complex similar to that discussed later for the removal of Ca2+ from the mitochondrial matrix into the cytoplasm (Baker and Allen, 1984; Carafoli and Longoni, 1987; Blaustein, 1988). Unlike in mitochondria, plasma membrane NCX has the inherent ability to move Ca2+ into or out of the cell depending on the prevailing conditions. When the system is acting to remove Ca2+, energy is supplied by the electrochemical gradient that ultimately results from the activity of the plasma membrane Na+/K+ ATPase (Na+ pump).
Plasma membrane Ca2+ pump has a higher affinity for Ca2+ (Kd = 100 nM) but a very slow turnover, whereas NCX has a much lower affinity (Kd = 1000 nM) but a higher turnover. Both types of transporters are co-expressed in neurons and in astrocytes (DiPolo and Beauge, 1983; Juhaszova et al., 2000). However, the precise role that each plays in removing excess Ca2+ loads under different physiological and pathophysiological conditions remains rather unclear. A major difference is the fact that they exhibit distinct subcellular localization patterns. In particular, some if not all of PMCA found in neurons seems to be localized very close to the neurotransmitter release sites (active zone) of the presynaptic terminals, whereas NCX is excluded from these sites and present in a more dispersed fashion on the rest of the neuron (Juhaszova et al., 2000; Blaustein et al., 2002). Therefore, the PMCA may help keep active zone Ca2+ very low, and function to re-prime the neurotransmitter release mechanism following activity. NCX, on the other hand, is believed to efflux Ca2+ that has diffused away from the active zone and perhaps been temporarily sequestered by the endoplasmic reticulum (ER). Moreover, the discovery of a multitude of PMCA isoforms and alternative splice variants (Strehler and Treiman, 2004; Strehler et al., 2007), as well as recent results on PMCA “knockout” mice and PMCA mutants (Prasad et al., 2007), show that at least some PMCAs play a more specific role in local Ca2+ handling. In addition, a growing number of specific PMCA-interacting proteins have been identified with regulatory, targeting, and signaling functions. These findings support a new paradigm, whereby PMCAs are not only responsible for global Ca2+ homeostasis but are dynamic participants in spatially defined Ca2+ signaling. The main regulator of PMCA function is Ca2+ calmodulin (Ca2+-CaM; Werth et al., 1996). In the absence of CaM, the pumps are autoinhibited by a mechanism that involves the binding of their C-terminal tail to the two major intracellular loops. Activation requires binding of Ca2+-CaM to the C-terminal tail and a conformational change that displaces the autoinhibitory tail from the major catalytic domain. Release of autoinhibition may be facilitated by means other than CaM binding, including by acidic phospholipids, protein kinase A- or C-mediated phosphorylation of specific (Ser/Thr) residues in the C-terminal tail (Werth et al., 1996), partial proteolytic cleavage of the tail (e.g., by calpain or caspases), or dimerization via the C-terminal tail (for a detailed review see Di Leva et al., 2008). Different PMCA isoforms show significant differences in their regulation by kinases and CaM. Interestingly, loss of PMCA function was reported to lead to an increase in the levels of intracellular Ca2+, causing apoptotic death of cerebellar and spinal cord neurons (Kurnellas et al., 2007).
INTRACELLULAR CALCIUM HOMEOSTASIS IN NEURONS
Ca2+homeostasis in the ER
The ER, a complex system of endomembranes, is present in all neurons and extends from the nucleus to the soma, dendrites, and dendritic spines, and down the axon to the presynaptic terminals. Particularly relevant for neuronal function is the ability of the ER to act as a dynamic Ca2+ store, able to actively accumulate Ca2+ and to release it in response to physiological stimulation. As such, the ER contains a variety of channels, buffers, and sensors dedicated to Ca2+ homeostasis (Figure 1; Table 1). In general, Ca2+ exits the ER through several types of Ca2+ release channels, such as inositol 3-phosphate (InsP3) receptors, ryanodine receptors (RyR), nicotinic acid adenine dinucleotide phosphate (NAADP) receptors, and polycystin-2 channels [the relative of transient receptor potential (TRP) proteins]. In neurons, the NAADP receptors were reported to exist in brain microsome preparations (Bak et al., 1999) and Ca2+ release from these channels was described in neurons from the buccal ganglion of aplysia (Chameau et al., 2001), yet their relevance in vertebrate neurons remains unclear. Regarding the TRPs, although they are expressed by neurons, there is so far no evidence for their involvement in Ca2+ homeostasis in these cells. Therefore, in neurons, Ca2+ exit from the ER occurs mainly through the inositol 3-phosphate receptors (IP3-Rs) and the Ca2+ activated RyR, both forming large tetrameric channel proteins. Both receptor families are comprised of multiple members that display distribution patterns that are both temporally and spatially regulated in neurons. For example, there are three RyRs, all of which can be activated by Ca2+ on the cytosolic side with differential sensitivities (RyR1 > RyR2 > RyR3). All three members have been detected in neurons, with distinct patterns that change during development and postnatal growth. For example, postnatally, RyR1 is highly expressed in cerebellar Purkinje cells, RyR3 in the hippocampus, striatum, and diencephalon, while many neurons co-express more than one RyR isoform (Hakamata et al., 1992; Lai et al., 1992; Furuichi et al., 1994; for review also see Berridge, 1998; Hertle and Yeckel, 2007). Regarding their sub-cellular localization, RyRs have been seen in all parts of neurons, including the soma, axons, dendrites, and even the spine apparatus of excitatory neurons. Similarly, there are three InsP3R isoforms with different sensitivities to Ca2+, and further diversity may arise from alternative splicing of InsP3R1. InsP3R1 is the main isoform in neurons in the brain, while InsP3R3 is mainly found in the spinal cord and in glial cells (Berridge, 1998).
Propagating Ca2+ waves is the most dramatic expression of Ca2+ release from the ER, reflecting the Ca2+-induced Ca2+ release (CICR) mode, where elevated cytoplasmic Ca2+ induces further Ca2+ release. Ca2+ waves in neurons were described more recently, after the notion had first been established using Xenopus oocytes (Lechleiter et al., 1991; Parker and Ivorra, 1991). Given the functional compartmentalization of neurons, Ca2+ waves take up different properties depending on their spatial localization and neuronal type diversity. For example, synaptically activated Ca2+ waves preferentially initiate at branch points of dendrites (Nakamura et al., 2002; Larkum et al., 2003; Fitzpatrick et al., 2009) and are mediated by the IP3-Rs (Nakamura et al., 1999). Such waves have been observed in pyramidal neurons of the rodent CA1 and CA3 regions of the hippocampus (Miller et al., 1996; Kapur et al., 2001), in layers 2 and 3 of the cortex (Larkum et al., 2003; Hagenston et al., 2008) and principal neurons of the amygdala (Power and Sah, 2008), all regions heavily involved in memory and learning. Relevant to the cognitive decline and memory loss associated with aging, synaptically induced Ca2+ waves are functionally linked to synaptic plasticity, a process known to require a rise in the postsynaptic concentration of Ca2+. More specifically, there are several cases where synaptically activated Ca2+ release from stores was shown to induce LTP (Yeckel et al., 1999), though it remains controversial as one study challenged this conclusion (Mellor and Nicoll, 2001).
In addition to the channels discussed above, some studies have suggested that presenilin 1 and 2, beyond constituting the proteases in the γ-secretase complex, also function as Ca2+ leak channels in the ER, either by themselves, or indirectly by increasing the activity of IP3-Rs and RyRs (Pack-Chung et al., 2000). In particular, presenilin 2 was shown to interact with sorcin, a cytoplasmic calcium-binding protein that modulates the activity of RyRs (Pack-Chung et al., 2000). Interestingly, in some mutations of presenilin 1 and 2 that are responsible for familial Alzheimer’s disease, disruption of intracellular Ca2+ homeostasis by the ER is the major measurable cellular consequence (Nelson et al., 2010), as discussed later on.
Calcium uptake into the ER lumen results from the function of Ca2+ pumps of the P-type sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) family. This family includes three members (SERCA1–3), as well as two splice isoforms of SERCA2. While SERCA2b is ubiquitously expressed, SERCA2a and SERCA3 are found almost exclusively in cerebellar Purkinje neurons. Inhibition of the SERCA pumps results in a relatively slow emptying of ER Ca2+ stores, with Ca2+ exiting the ER through poorly described pathways (Camello et al., 2002). Ca2+ buffering in the ER lumen is achieved by specific Ca2+-binding proteins. In neurons, the most abundant of these is calreticulin and calsequestrin, while some others such as endoplasmin, BiP/grp78, and proteins of the CREC family also participate in Ca2+ buffering. A main difference of ER Ca2+ buffers is that, unlike their cytosolic counterparts, they have a low affinity for Ca2+ to allow the maintenance of high intra-ER Ca2+ levels.
Ca2+ homeostasis in the Golgi
Ca2+ uptake in the Golgi apparatus involves two groups of Ca2+ pumps: the well characterized SERCAs, discussed above, and a less characterized group of ATPases that were described as secretory-pathway Ca2+-ATPases (SPCAs; Shull, 2000; Figure 1; Table 1). The SPCAs in addition supply the Golgi lumen with Mn2+, which is needed for many enzymatic reactions in this compartment. Mammalian SPCA was originally cloned from rat using a probe derived from sequences of the ATP-binding site of SERCA1 and SERCA2 (Gunteski-Hamblin et al., 1992). The corresponding human gene (ATP2C1) was described by two independent groups (Hu et al., 2000; Sudbrak et al., 2000). Alternative processing of ATP2C1 results in four SPCA1 proteins with C-termini differing in length and specific amino acid sequence (Hu et al., 2000; Sudbrak et al., 2000; Fairclough et al., 2003), SPCA1a, SPCA1b, SPCA1c, and SPCA1d. Ishikawa et al. (1998) later described a second human SPCA isoform, named SPCA2. Its human gene (ATP2C2) was independently described in 2005 by two groups (Vanoevelen et al., 2005; Xiang et al., 2005). The widespread expression pattern of SPCA1 and the observation that homozygous loss of a functional ATP2C1 gene do not seem to be viable suggest that SPCA1 is a housekeeping enzyme. The tissue and cellular expression of SPCA2 appears to be more restricted than that of SPCA1, and based on mRNA data it is expressed in the brain, among other tissues (Vanoevelen et al., 2005; Xiang et al., 2005). It is now well established using a range of different cell types that the endogenous SPCA1 is specifically located in the Golgi compartment (Behne et al., 2003; Van Baelen et al., 2003; Reinhardt et al., 2004; Ramos-Castaneda et al., 2005). The relative contribution of SERCAs and SPCAs to the total uptake of Ca2+ into the Golgi apparatus seems to be cell-type-dependent. The highest dependence on SPCAs occurs in human keratinocytes (Callewaert et al., 2003). This finding is important for explaining the physiopathology of the skin-related Hailey–Hailey disease.
While the potentially specific roles of SPCAs in neurons are poorly understood, our own recent findings (Kourtis et al., 2012) suggest that SPCA1 is both necessary and sufficient in mediating the neuroprotective function of heat preconditioning in a model of heat stroke-induced neurodegeneration. Notably, this mechanism is evolutionarily conserved as it is preserved from C. elegans to mammals. This finding invites the speculation that SPCAs may have a more general neuroprotective role, whose relevance to other forms of neurodegeneration and aging remains to be examined.
Ca2+ homeostasis by mitochondria
Beyond their main role in the cell to produce NADH and ATP, it is now well accepted that mitochondria also function as Ca2+ buffers (Figure 1; Table 1). As proton pumping creates an inside-negative membrane potential in mitochondria, Ca2+ tends to be drawn into the mitochondrial matrix following its electrochemical gradient. This influx is mainly achieved by the mitochondrial Ca2+ uniporter whose conductance is dependent on both intracellular Ca2+ concentration and energy demand. At high cytosolic Ca2+ concentrations and low ATP/ADP ratio more Ca2+ is conducted, whereas at low cytosolic Ca2+ concentration and high ATP/ADP ratio less Ca2+ is conducted. Intricately enough, increasing mitochondrial Ca2+ concentration activates the enzymes of the Krebs cycle, thus causing increased ATP production. As mitochondrial Ca2+ buffering is more energy efficient compared to expelling Ca2+ through the plasma membrane or into the ER, this mechanism is considered of high relevance for neurons in situations when ATP and oxygen demands reach high levels, such as in the case of repeated axon potentials (Contreras et al., 2010).
Calcium is expelled from the mitochondrial matrix into the cytosol mainly by the mitochondrial sodium calcium exchanger (NCX; three Na+ for one Ca2+), in conditions of low ATP demand and oxygen consumption, or through a mitochondrial proton/Ca2+ exchanger (two or more H+ per Ca2+). Indirect experiments with isolated mitochondria under pathological conditions or Ca2+ overload suggest an additional, higher conductance route, through the transient opening of the mitochondrial permeability transition pore (mPTP). However, the physiological relevance of mPTP in Ca2+ homeostasis remains controversial and is not supported by genetic ablation studies (Ichas et al., 1997; Baines et al., 2005). In addition to its contribution in disease, which is discussed later, new roles for mitochondrial Ca2+ homeostasis are also emerging for normal neuron physiology. For example, it was recently described that olfactory sensory neurons require mitochondrial Ca2+ mobilization in order to encode intensity (Fluegge et al., 2012). Therefore, aberrant mitochondrial Ca2+ homeostasis in these neurons converts them into simple signal detectors and impairs their function in olfaction.
Calcium buffers and sensors
A large set of proteins with ability to bind Ca2+ specifically and reversibly provide yet another level of control in Ca2+ homeostasis by acting as sensors or buffers (Figure 1; Table 1). A large family of these Ca2+-binding proteins is the one containing EF-hand Ca2+ binding domains. These motifs consist of two 10–12 residue long alpha helices, oriented perpendicularly against each other, separated by a 12-residue long loop region. EF-hand domains often exist as multiple pairs generating a wide structural and functional variability within this large family of proteins (Kretsinger, 1980). A prominent member of this family, calmodulin, serves as a Ca2+ sensor that translates graded changes of intracellular Ca2+ concentration into a graded signaling response by interacting with various Ca2+-sensitive enzymes.
Another set of EF-hand-containing proteins, represented by calretinin, calbindin, and parvalbumin, function as Ca2+ buffers. These proteins are predominantly expressed by the inhibitory GABAergic interneurons of the central nervous system in specific patterns, therefore contributing to the diversification of these interneurons into distinct subtypes (Van Brederode et al., 1990). A multitude of studies has demonstrated that these proteins modulate the Ca2+ levels locally in the presynaptic active zone or at postsynaptic densities. Moreover, they are thought to actively and differentially participate in modulating neuronal vulnerability to different types of stress. In hippocampal primary cultures, neurons expressing calbindin are less vulnerable to oxidative stress-induced apoptosis because they recover Ca2+ concentration more effectively after stimulation, whereas in cortical neurons this is true for calretinin-containing neurons (Mattson et al., 1991). Similarly, genetic over-expression of parvalbumin in mice rescues motorneurons from injury-induced cell death (Dekkers et al., 2004).
It is generally thought that the transduction of the Ca2+ signal by EF-hand proteins consists a series of conformational changes that occur after Ca2+ has become bound. However, it is important to also mention that there are some exceptions, as no significant conformational changes after Ca2+ binding have been described for at least two of the EF-hand proteins, such as parvalbumin itself and calbindin, which are thus likely to act instead only as temporal Ca2+ buffers. Although most EF-hand proteins reside in the cytosol (and in the nucleoplasm), reticulocalbin is localized in the lumen of the ER (Tachikui et al., 1997). On the other hand, Cab45 (Scherer et al., 1996) and nucleobindin are localized in the Golgi apparatus (Lin et al., 1998) and glycerophosphate dehydrogenase (Pilstrom and Kiessling, 1972) and Aralar are located on the outer face of the inner mitochondrial membrane (del Arco and Satrustegui, 1998; Del Arco et al., 2000).
Another group of Ca2+-binding proteins, collectively known as intracellular neuronal calcium sensors (NCS; Braunewell and Gundelfinger, 1999; Burgoyne and Weiss, 2001), includes five subfamilies: the recoverins and guanylyl cyclase activating proteins (GCAPs), which are primarily expressed in retinal photoreceptor cells and have established roles in the regulation of photo-transduction; the frequenins, visinin-like and Kv-channel-interacting proteins (KChIPs), which are widely expressed in central neurons. One key feature of most NCS is N-terminal acylation: several members of the family are N-terminally myristoylated. Binding of Ca2+ to recoverin, and presumably to other NCS proteins, changes their conformation, exposing the myristoyl residue and hydrophobic portions of the molecule, making them available for membrane (or target protein) interaction. The Ca2+-myristoyl switch could be a mechanism that affects the compartmentation of signaling cascades in neurons and/or the transmission of Ca2+ signals to their membranes (Braunewell and Gundelfinger, 1999; Burgoyne and Weiss, 2001). Although the functions of the last three families are not clearly defined, it has been shown that they interact with multiple target proteins and with nucleic acids as well (Carrion et al., 1999). KChIP3 encodes the protein calsenilin, shown recently to interact with presenilin 1 and 2, two proteins whose mutations result in familial Alzheimer’s disease (AD; Buxbaum et al., 1998; Buxbaum, 2004). Relevant to the neurodegenerative phenotype of AD pathology, this interaction was shown to modulate the proteolytic processing of presenilins. In addition, two other NCS proteins, recoverin and GCAP1 have been involved in degenerative diseases of the retina. Mutations in the GCAP gene have been associated with autosomal dominant cone dystrophy. One of the defects has been related to constitutive activation of guanylyl cyclase that is not properly inactivated by high levels of Ca2+, characteristic of physiological dark conditions, eventually leading to degeneration of cone cells (Dizhoor et al., 1998; Sokal et al., 1998). The other condition [GCAP1(P50L); Sokal et al., 2000] is a milder form of autosomal dominant cone dystrophy in which the mutation reduces the Ca2+-binding ability of GCAP1. Recoverin has been identified as the autoantigen in a degenerative disease of the retina called cancer-associated retinopathy (CAR), in which patients lose vision due to degeneration of photoreceptors (Polans et al., 1991; Polans et al., 1995).
BRAIN AGING AND THE “CALCIUM HYPOTHESIS”
The potential contribution of altered Ca2+ homeostasis at least to some aspects of brain aging and neurodegeneration was first put forward by Khachaturian in the 1980s, with the formulation of the “Ca2+ hypothesis of aging” (Gibson and Peterson, 1987; Disterhoft et al., 1994; Khachaturian, 1994). Early findings in the field that corroborated this hypothesis examined the major transport pathways of Ca2+ during aging and found that at least in some types of neurons, such as the principal cells in the hippocampal CA1 region, there is an increased Ca2+ influx mediated by increased VOCC activity in aged neurons (Landfield and Pitler, 1984; Thibault and Landfield, 1996). Similarly, Ca2+ extrusion through the PMCA was found to be decreased in aged neurons (Michaelis et al., 1996). Subsequently, the focus shifted toward the intracellular mechanisms of Ca2+ homeostasis and their deregulation during aging. Several studies demonstrated that there is an increased release of Ca2+ from the ER stores through both the InsP3 and RyR receptors (Thibault et al., 2007), leading to the proposal that release from the RyR receptor may be a useful biomarker of neuronal aging. Below, we will consider in more detail findings that relate to two key elements of aging: aberrant synaptic plasticity and neurodegeneration.
ROLE OF CALCIUM IN SYNAPTIC PLASTICITY AND NEURONAL EXCITABILITY DURING AGING
Aging of the brain is manifested in humans by a progressive cognitive decline associated with weakening of the ability to process new information and of the executive function. The most dramatic effect is notably observed on the function of episodic memory, including spatial memory. The cognitive decline associated with normal aging is not attributed to significant neuronal loss (Gallagher et al., 1996), but is rather thought to result from changes in synaptic connectivity and plasticity. There is a general consensus that memory and learning are molecularly encoded by mechanisms controlling synaptic plasticity in several brain areas. Among these, the afferent pathways of the hippocampus are the most relevant, but other areas such as the amygdale, the visual, somatosensory and prefrontal cortices, and the subiculum also play important roles in processing, integration, and consolidation of new information. Using mainly the hippocampus, numerous studies have deciphered a major role for Ca2+ in the two major forms of synaptic plasticity, LTP (Bliss and Collingridge, 1993) and long-term depression (LTD). LTP represents an increase in synaptic transmission, induced by pattern stimulation of afferent fibers and it is the main process proposed to underlie memory formation. On the other hand, LTD is a means of decreasing synaptic strength, contributing to the loss of synaptic contacts and associated with increased forgetfulness during aging (Foster, 1999,2007; Zhou et al., 2004; Shinoda et al., 2005). Age-related changes in LTP and LTD underline the functional significance of altered synaptic plasticity for cognitive function (Foster and Norris, 1997; Foster, 1999; Foster and Kumar, 2002).
Relevant to the role of Ca2+ deregulation in memory loss, the critical event leading to induction of LTP appears to be the large influx of calcium ions into the postsynaptic spine. Importantly, LTP is blocked by injection of intracellular Ca2+ chelators such as EGTA (Lynch et al., 1983) or BAPTA (Mulkey and Malenka, 1992) and conversely, LTP is induced when the postsynaptic cell is loaded with calcium (Malenka et al., 1988). Therefore, it is well established that a significant elevation of postsynaptic Ca2+ concentration is both necessary and sufficient for the induction of hippocampal LTP (Bliss and Collingridge, 1993). In contrast, a modest rise in Ca2+ concentration results in induction of LTD through activation of protein phosphatases that dephosphorylate AMPA receptors (Artola and Singer, 1993; Lisman, 1989,1994). Due to the differential level of Ca2+ fluctuation involved in the generation of the various forms of synaptic plasticity, the stimulation patterns for the induction of LTP and LTD constitute high- and low-frequency stimulation, respectively.
In general, the effect of aging on synaptic plasticity can be summarized by several key observations: First, the threshold for induction of LTP increases such that higher stimulation frequencies or more induction sessions are required in older animals in order to achieve the same level of potentiation. Second, the threshold for induction of LTD is lowered in aged animals, facilitating its prevalence. Furthermore, the maintenance of LTP is disrupted such that the enhanced transmission decays more rapidly in aged animals. In contrast, LTD and depotentiation, or erasure of LTP, are increased in aged animals due to a lowering of the threshold stimulation needed for induction of synaptic depression (Norris et al., 1996; Foster and Norris, 1997; Kamal et al., 2000; Vouimba et al., 2000). Thus, the age-related decline in synaptic transmission (Barnes, 1994) may reflect a shift in the LTP/LTD balance, with insufficient LTP induction and maintenance and excessive synaptic depression (Foster et al., 2001).
In most of the synapses that support LTP (in the hippocampus and elsewhere), the postsynaptic increase in calcium is mediated through the activation of the NMDA receptor. As already mentioned earlier, NMDA receptor activation allows the influx of calcium only when the receptor is occupied by L-glutamate and concomitantly the postsynaptic membrane is depolarized. Emerging evidence indicates that the synaptic plasticity shift during aging results from changes in the source of Ca2+ such that Ca2+ influx through NMDARs is reduced (Lehohla et al., 2008; Bodhinathan et al., 2010) and Ca2+ influx through L-type VDCCs is increased (Barnes, 1994; Norris et al., 1996; Thibault and Landfield, 1996; Shankar et al., 1998; Potier et al., 2000). The increase could arise from altered gene or protein expression (Herman et al., 1998), or phosphorylation changes of the L-type Ca2+ channels (Norris et al., 2002; Davare and Hell, 2003). Interestingly, the L-type Ca2+ channel blocker nimodipine counteracts age-related learning impairment in rabbits (Deyo et al., 1989; Kowalska and Disterhoft, 1994), rodents (Levere and Walker, 1992), non-human primates (Sandin et al., 1990), and elderly patients with dementia (Ban et al., 1990; Tollefson, 1990).
Additionally, aged neurons show a multitude of defects in Ca2+ homeostasis, including enhanced release of Ca2+ from the ER (Kumar and Foster, 2004; Gant et al., 2006), diminished Ca2+ extrusion through the plasma membrane ATPase (Michaelis et al., 1996; Gao et al., 1998), reduced cellular Ca2+ buffering capacity due to impairment of the SERCA pumps (Murchison and Griffith, 1999), and diminished mitochondrial Ca2+ sink capability (Murchison and Griffith, 1999; Xiong et al., 2002). The overall result is an increase of Ca2+ loads which negatively impact neuronal excitability (Landfield and Pitler, 1984; Khachaturian, 1989; Matthews et al., 2009). Moreover, such an increase in intracellular Ca2+ concentration increases the threshold frequency for induction of LTP (Shankar et al., 1998; Ris and Godaux, 2007), and enhances the susceptibility to induction of LTD (Norris et al., 1996; Kumar and Foster, 2005), ultimately explaining the age-associated deficits in learning and memory. In line with this notion, administration of the cell permeable Ca2+ chelator BAPTA, ameliorates impaired presynaptic cytosolic and mitochondrial Ca2+ dynamics in hippocampal CA1 synapses of old rats (Tonkikh and Carlen, 2009), and enhances spatial learning (Tonkikh et al., 2006).
In the context of LTP induction, a key early finding was the observation that postsynaptic entry of calcium leads to activation of Ca2+/calmodulin complex-dependent kinase II (CaMKII), one of the most abundant proteins in neurons comprising 1–2% of the total protein. Although it is expressed both pre- and postsynaptically, its expression is particularly high in the postsynaptic density, where it is ideally located to respond to changes in calcium concentration. There are more than 30 isoforms of CaMKII and numerous substrates, many of which are located in the postsynaptic density (Fink and Meyer, 2002). CaMKII is generally considered a mediator of primary importance in linking transient calcium signals to neuronal plasticity. Importantly, observations by Silva et al. (1992a),b,(c) indicated that deletion of the CaMKII gene in mice results in impaired LTP and aberrant spatial memory. Moreover, activation of CaMKII is significantly reduced in aged hippocampal neurons (Mullany et al., 1996). The data obtained from studies on rodents have to a large extent, been paralleled by similar findings in other organisms, indicating that several models expressing various forms of synaptic plasticity exhibit a requirement for CaMKII activation. For instance, CaMKII knockout in Drosophila exhibits impaired associative learning, while motor and sensory systems remain unaffected (Joiner and Griffith, 1999). Similarly, knockout of unc-43 (a gene encoding the CaMKII analog in C. elegans) affects the stability of synapses and general neuronal physiology, ultimately culminating in altered function of olfactory neurons (Sagasti et al., 2001).
Beyond activating the CaMKII signaling cascade, Ca2+ also acts as a second messenger that is responsible for the activity-dependent transcription of several key genes (West et al., 2001). The products of these genes are necessary in order to convert the effects of transient stimuli into long-term changes in brain function, a process that is required for the formation of memories. Of the neural-selective activity-dependent genes, brain-derived neurotrophic factor (BDNF) is activated by calcium influx through L-type VOCCs (L-VOCCs) acting on the transcription of BDNF from promoter III (West et al., 2001). BDNF is among the most relevant calcium targets for the modulation of memory. BDNF transcription is up-regulated dramatically by membrane depolarization in vitro (Ghosh et al., 1994; Tao et al., 1998) and by induction of LTP, and associative learning (Ernfors et al., 1991; Patterson et al., 1992; Tokuyama et al., 2000). Moreover, loss of BDNF is associated with impaired LTP among other synaptic defects. It is also well established that BDNF transcription is largely decreased during aging (Tapia-Arancibia et al., 2008), and that epigenetic induction of BDNF transcription in aged subjects significantly ameliorates the cognitive and memory defects associated with aging (Zeng et al., 2011). A summary of the perturbations of Ca2+ homeostasis associated with nervous system aging is shown in Table 2.
Table 2
| ca2+ deregulation associated with aging of the nervous system | Reference |
|---|---|
| Increased ca2+ influx mediated by voltage-dependent calcium channels | Landfield and Pitler (1984), Thibault and Landfield (1996) |
| Decreased ca2+ extrusion through the plasma membrane pump (PMCA) | Michaelis et al. (1996), Gao et al. (1998) |
| Increased release of ca2+ from the ER stores through both the InsP3 and RyR receptors | Thibault et al. (2007) |
| Reduced ca2+ influx through NMDARs | Lehohla et al. (2008), Bodhinathan et al. (2010) |
| Increased ca2+ influx through L-type VDCCs | Barnes (1994), Norris et al. (1996), Thibault and Landfield (1996), Shankar et al. (1998), Potier et al. (2000) |
| Phosphorylation changes of the L-type ca2+ channels | Norris et al. (2002), Davare and Hell (2003) |
| Increased release of ca2+ from the ER | Gant et al. (2006), Kumar and Foster (2004) |
| Impairment of the SERCA pumps | Murchison and Griffith (1999) |
| Diminished mitochondrial ca2+ sink capability | Murchison and Griffith (1999), Xiong et al. (2002) |
| Reduced activation of CaMKII in hippocampal neurons | Mullany et al. (1996) |
| Reduced ca2+-dependent transcription of genes such as BDNF | Tapia-Arancibia et al. (2008) |
Perturbations of Ca2+ homeostasis in the aging nervous system.
ROLE OF CALCIUM IN AGING-RELATED NEURODEGENERATION
Aging is the greatest risk factor for the development of neurodegenerative disorders. These include a diverse collection of pathologies characterized by the late onset and gradual loss of specific neuronal subpopulations in motor, sensory, or cognitive systems. Despite major intrinsic differences in the etiology of each disorder, deregulated Ca2+ homeostasis has emerged as a common underlying mechanism of neuronal loss in AD, Parkinson’s (PD) diseases, amyotrophic lateral sclerosis (ALS), and other neurodegenerative disorders (Mattson, 2007; Bezprozvanny, 2009).
Alterations of Ca2+ homeostasis may be in some cases directly responsible for neuronal death. Persistently increased levels of intracellular Ca2+ can result in severe phenotypes in neurons, culminating to neuronal death and degeneration (Siman et al., 1989; Celsi et al., 2009). This process is often specifically mediated or even initiated by the diminished capacity of mitochondria to buffer Ca2+. An example where there is ample evidence that altered mitochondrial Ca2+ homeostasis mediates neuronal loss is ALS, an adult onset disease, with incidence increasing with age. ALS is characterized by selective and progressive degeneration of motorneurons in the spinal cord and brain, leading to weakness, atrophy, and paralysis of voluntary muscles. Mutations in superoxide dismutase (SOD1) are the most common genetic factors responsible for about 20% of familial ALS cases (Rosen et al., 1993). SOD1 is a ubiquitously expressed enzyme that converts superoxide to hydrogen peroxide in order to protect cells against oxidative stress. While there is still no consensus as to how mutant SOD1 causes selective toxicity to motorneurons, increasing evidence suggests that the mechanisms largely concentrate on the dysfunction of ER and mitochondrial Ca2+ homeostasis (Bacman et al., 2006; Hervias et al., 2006; Magrane et al., 2009; Shi et al., 2010).
At the level of the ER, a recent paper implicates the Ca2+ buffering protein calreticulin in the death of motorneurons in a model of ALS (Bernard-Marissal et al., 2012). More specifically, fast fatigable motorneurons selectively activate an ER stress response that drives their early degeneration, while a subset of mSOD1 motorneurons shows exacerbated sensitivity to activation of the motorneuron-specific Fas (transmembrane TNF receptor superfamily member 6) and nitric oxide (NO) pathway. However, the links between the two mechanisms and the molecular basis of their cellular specificity remained unclear. This paper demonstrates that Fas activation causes reduced levels of calreticulin specifically in mSOD1 motorneurons. Decreased expression of calreticulin is both necessary and sufficient to trigger SOD1(G93A) motorneuron death through the Fas/NO signaling pathway, and represents an early event that precedes muscle denervation and is restricted to vulnerable motor pools.
At the mitochondrial level, altered Ca2+ handling also appears early on, before motorneuron degeneration is manifested, suggesting that it is actively involved in disease pathogenesis. SOD1, which is a predominantly cytosolic protein, also localizes to the ER and mitochondria (Jaarsma et al., 2001; Okado-Matsumoto and Fridovich, 2001; Higgins et al., 2002; Mattiazzi et al., 2002), predominantly in the intermembrane space and less so on the outer membrane (Pasinelli et al., 2004; Vande Velde et al., 2008) and matrix (Vijayvergiya et al., 2005). By mechanisms that are still poorly understood, mutant SOD1 induces increased Ca2+ uptake by mitochondria, as convincingly demonstrated in mitochondria isolated from the brain and spinal cord of SOD1 mutant mice (Damiano et al., 2006). This defect appears to be neuron-specific, as liver cells from the same mutants retain unaffected mitochondrial Ca2+ homeostasis. Impaired Ca2+ handling by mitochondria is thought to be the primary cause of the abnormally high concentration of intracellular Ca2+ observed in ALS motorneurons (Carri et al., 1997; Kruman et al., 1999), making them vulnerable to degeneration (Kim et al., 2002,2007).
Mitochondrial Ca2+ overload is associated with activation of cell death pathways (Bernardi et al., 1999) and is observed in many pathological conditions in addition to ALS (Honda and Ping, 2006; Norenberg and Rao, 2007). The mechanisms responsible for Ca2+ overload are not entirely clear; however, their elucidation could provide a base for significant pharmacological interventions in the future. Theoretically, defects of the mitochondrial NCX could be involved in causing Ca2+ overload in ALS, although this putative mechanism remains to be directly explored. Another potential factor contributing to Ca2+ overload could be the functional and physical link between mitochondria and ER. Transfer of Ca2+ from the large stores in the ER to mitochondria depends on the relative positioning of these two organelles, and it is thought to occur at Ca2+ “hotspots”, sites where ER and mitochondrial membranes are in close physical contact (Rizzuto et al., 1999). Shortening the distance between the two organelles was shown to result in increased accumulation of Ca2+ in mitochondria, causing cell death (Csordas et al., 2006). Since mutant SOD1 accumulates both in ER (Kikuchi et al., 2006; Urushitani et al., 2006) and mitochondrial (Liu et al., 2004) membranes, it is plausible that the structure of these calcium hotspots is altered in mutant neurons, leading to abnormal handling of Ca2+ between the two organelles.
Whatever the mechanism of the increased Ca2+ accumulation in mitochondria, activation of cell death by mitochondrial Ca2+ overload involves the opening of the mPTP, followed by release of cytochrome c, and downstream activation of apoptosis. Cytochrome c released into the cytosol can further propagate apoptotic signaling by binding to the IP3-R on the ER, desensitizing its autoinhibition by calcium and thus causing further calcium release from ER stores (Boehning et al., 2003). Ablation of cyclophilin D (CypD), a modulatory component of the mPTP, delays the opening of mPTP (Basso et al., 2005) and has a protective effect against neuronal death in models of ischemia (Baines et al., 2005; Schinzel et al., 2005). In ALS, it was also reported that loss of CypD in SOD1 mutant mice delays the onset of the disease and significantly extends lifespan (Martin et al., 2009). Moreover, two studies using the immunosuppressant cyclosporin A, which binds to CypD to inhibit mPTP, in mutant SOD1 mice, suggest that inhibition of mPTP may be of benefit to ALS (Keep et al., 2001; Kirkinezos et al., 2004).
Another mechanism whereby Ca2+ contributes to the activation of cell death is by stimulating the production of mitochondrial reactive oxygen species (ROS). Oxidative stress caused by the damaging effect of ROS to proteins, lipids, and DNA, is a common feature of aging-related diseases, including ALS (Floyd and Hensley, 2002; Lin and Beal, 2006). Mitochondrial dysfunction (Wei, 1998), and particularly mitochondrial Ca2+ overload (Petrosillo et al., 2004), increases ROS production. In particular, increased levels of mitochondrial Ca2+ enhance cytochrome c release through a mechanism involving ROS-mediated oxidation of cardiolipin (Vercesi et al., 1997; Iverson and Orrenius, 2004). Notably, lipid peroxidation (Mattiazzi et al., 2002) and dissociation of cytochrome c from the mitochondrial inner membrane (Kirkinezos et al., 2005) have been reported in mutant SOD1 mice, but also in PD (Beal, 2003), and AD (Green and Kroemer, 2004;Lin and Beal, 2006; Kawamoto et al., 2012; Lee et al., 2012a).
Alzheimer’s disease is perhaps the most widespread neurodegenerative disorder of the elderly, with most familiar cases attributed to several mutations in presenilin 1 and 2, genes whose protein products are responsible for the proteolytic cleavage of the amyloid precursor peptide (APP). The mechanism by which presenilin mutations cause AD involves increased production of Aβ1–42 which aggregates and damages neurons. This view has been recently expanded by emerging findings suggesting that perturbed ER Ca2+ homeostasis significantly contributes to the dysfunction and degeneration of neurons in AD (Kipanyula et al., 2012). For example, recent work indicates that there is impaired Ca2+ uptake by mitochondria in the dentate gyrus of a mouse model of AD (Lee et al., 2012b). This can be explained to some extent by the novel role proposed by at least two groups for presenilins as regulators of Ca2+ homeostasis in the ER (Pack-Chung et al., 2000; Yoo et al., 2000). Interestingly, mutations in presenilin 1 that cause early onset familial AD, increase the pool of ER Ca2+ available for release, and enhance Ca2+ release from the ER through IP3- and RyR receptors (Chan et al., 2000; Guo et al., 1996,1999; Cheung et al., 2010; Leissring et al., 2000). Future research should clarify the specific contributions of perturbed ER Ca2+ handling to the cellular events that underlie synaptic dysfunction and neuronal degeneration in AD. While elevated pools of ER Ca2+ resulting from mutations in presenilins have been widely documented in a range of cell culture and animal models, the molecular basis of this alteration remains unknown and is potentially a key field for the development of novel pharmacological targets.
In addition to direct effects on neuronal survival, altered Ca2+ homeostasis is also likely to contribute to the initiation or progression of the neurodegenerative process by enhancing neuronal vulnerability to metabolic and other stressors (Toescu and Verkhratsky, 2004; Toescu and Vreugdenhil, 2010). One such example is the population of basal forebrain cholinergic neurons, a group of neurons that are selectively vulnerable to pathology and loss early in AD, as well as in a number of other neurodegenerative disorders of the elderly. In the primate, including man, these neurons are rich in the Ca2+ buffer protein calbindin. Notably, there is a substantial loss of calbindin in the course of normal aging and a further loss in AD(Iacopino and Christakos, 1990). Significantly, cholinergic neurons that had lost their calbindin in the course of normal aging were those that selectively degenerated in AD, while calbindin-containing neighboring neurons were virtually resistant to the process of tangle formation, a hallmark of the disease (Riascos et al., 2011). Another study reported that over-expression of calbindin in presenilin 1 mutant neurons was sufficient to prevent apoptosis (Guo et al., 1998). Similarly, a dramatic reduction in the Ca2+ buffering protein calbindin levels has been described in brains of PD patients (Iacopino and Christakos, 1990) and dopaminergic (DA) neurons expressing higher levels of calbindin, or other Ca2+ buffers such as calretinin and parvalbumin, were shown to be resistant to degeneration in PD (Yamada et al., 1990; Tsuboi et al., 2000). These findings are consistent with earlier findings suggesting that calbindin-positive hippocampal neurons are more resistant against oxidative stress (Mattson et al., 1991), although other Ca2+ buffer proteins seem to confer resistance to stress in different neuronal subpopulations. Understanding the mechanisms underlying such an instructive function of Ca2+ buffer proteins is of great importance as there may be a yet unidentified crosstalk with major signaling cascades. More work in this direction would greatly enhance our ability to selectively intervene in order to modulate the vulnerability of distinct neuronal populations.
Similar to ALS and AD, PD is another case where Ca2+ deregulation has recently attracted a lot of attention. PD is characterized by motor defects resulting from the selective loss of DA neurons in the substantia nigra and intracellular accumulation of cell aggregates known as Lewy bodies, mostly composed of α-synuclein. The idea that mitochondria could be directly involved in the pathogenesis of PD comes from the early accidental observation that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), an inhibitor of the mitochondrial respiratory chain complex I, causes Parkinson-like symptoms (Langston and Ballard, 1983). Later on, it was also demonstrated that DA neurons from PD patients show massive accumulation of mitochondrial DNA (mtDNA) deletions that impair the function of the respiratory chain complexes (Exner et al., 2012), thus increasing the probability of dysfunctions in these organelles.
Some clues as to the selective vulnerability of this population arise from the fact that DA neurons of the substantia nigra display unusual physiological properties. First, unlike most other neurons in the brain, they are autonomously active, generating regular action potentials in the absence of synaptic input (Grace and Bunney, 1983). This pacemaking activity is thought to maintain physiological levels of dopamine in regions they innervate, particularly the striatum (Romo and Schultz, 1990). To drive this pacemaking activity, these neurons rely, at least in part, on a rare form of L-type Ca2+ channels (Bonci et al., 1998; Ping and Shepard, 1996; Puopolo et al., 2007) comprised of the Cav1.3 pore-forming subunit (Striessnig et al., 2006; Chan et al., 2007). This leads to typically elevated intracellular Ca2+ concentrations under physiological conditions (Wilson and Callaway, 2000; Chan et al., 2007). Second, DA neurons of the substantia nigra display an elaborate axonal network (Matsuda et al., 2009), supporting orders of magnitude more synapses compared to a cortical pyramidal neuron (Arbuthnott and Wickens, 2007). As a result, the mitochondrial density in their somatic and dendritic regions is very low compared to other neuronal types (Liang et al., 2007). Taken together, these characteristics are thought to contribute to an intrinsic state of increased metabolic stress, where increased load of intracellular Ca2+ is met by a depleted mitochondrial network.
Additional genetic factors could increase the rate at which mitochondrial Ca2+ homeostasis is compromised in these already vulnerable neurons. At least 13 gene loci and 9 genes have been linked to both autosomal dominant and recessive forms of PD (Lesage and Brice, 2009). Mutations in three proteins encoded by these genes, namely, parkin (PARK2), DJ-1 (PARK7), and PINK1 (PARK6), are associated with recessive early onset forms of PD, whereas mutations in α-synuclein (PARK1–4) and LRRK2 (PARK8) are responsible for dominant forms of familial PD. Mitochondrial dysfunction has been described for mutants of all these genes (Lesage and Brice, 2009).
Recent papers have started to explore in more detail the possibility of Ca2+ handling by the PD-related proteins. DJ-1 is a multitask protein that, in addition to its main role as an antioxidant (Taira et al., 2004), is also involved in maintaining cytosolic basal Ca2+ concentration values to permit depolarization-induced Ca2+ release from the sarcoplasmic reticulum in muscle cells (Shtifman et al., 2011). Moreover, DJ-1 was shown to protect DA neurons from Ca2+-induced mitochondrial uncoupling and ROS production during physiological pacemaking (Guzman et al., 2010).
Regarding α-synuclein, it has been described that it can modulate Ca2+ influx from the extracellular milieu by enhancing the plasma membrane ion permeability (Danzer et al., 2007) either through their direct insertion into the plasma membrane and the formation of a pore (Lashuel et al., 2002) or through the modulation of plasma membrane Ca2+ permeability (Furukawa et al., 2006). The actual mechanisms through which α-synuclein aggregation and Ca2+ dysfunction influence each other are not clear, however, a functional interplay is unambiguous: Increased intracellular Ca2+ promotes α-synuclein aggregation, which in turn could promote intracellular Ca2+ increase (Nath et al., 2011). A recent study suggests that using its C-terminal domain, α-synuclein controls mitochondrial calcium homeostasis by enhancing ER–mitochondria interactions (Cali et al., 2012). As these results were obtained in vitro using non-neuronal cell lines, their relevance to DA neuron physiology and pathology remains to be examined.
As to PINK1, its direct role in regulating cellular, and most specifically mitochondrial Ca2+ fluxes, has been recently proposed starting with the observation that the co-expression of mutant PINK1 in a cellular model of PD-expressing mutated α-synuclein exacerbated the observed mitochondrial defects, that is, increased mitochondrial size with loss of cristae and reduced ATP levels (Marongiu et al., 2009). The proposed mechanisms of PINK1 action was based on a deregulation of mitochondrial Ca2+ influx. As by blocking mitochondrial Ca2+ uptake, it was possible to restore the original phenotype (Marongiu et al., 2009), thus suggesting that mutant PINK1 could reinforce α-synuclein pathology by acting on converging pathways affecting mitochondrial function. Other studies have further investigated the role of PINK1 in mitochondrial Ca2+ metabolism, but the results are controversial. In one case, it was proposed that PINK1 absence caused an impairment of mitochondrial Ca2+ efflux, probably affecting the mitochondrial Na+/Ca2+ exchanger activity and thus resulting in mitochondrial Ca2+ overload, ROS production, and impaired respiration (Gandhi et al., 2009). In another very recent study, PINK1 depletion has instead been shown to impair mitochondrial Ca2+ uptake and consequently to affect energy metabolism (Heeman et al., 2011). However, consistently, numerous reports showed that PINK1-deficient cells have impaired mitochondrial membrane potential and enhanced sensitivity to the toxic effects of mitochondrial complex I inhibitors (Wood-Kaczmar et al., 2008), as well as enhanced Ca2+ vulnerability (Akundi et al., 2011).
OUTLOOK
Given the fundamental importance of Ca2+ homeostasis in the biology of all cells, it is not completely surprising that more and more studies suggest that deregulated Ca2+ is actively involved in the course of normal aging and in diverse pathological conditions. A general message arising from these studies is that in the nervous system Ca2+ signaling and homeostasis should be examined in view of the amazing cellular diversity exhibited by the nervous system. The machinery controlling Ca2+ homeostasis is similarly diverse among neurons, uniquely suited to the needs of each neuronal subtype. Taken together, the intrinsic differences of neurons in morphology, connectivity, proteome and Ca2+ homeostatic machinery are very likely to collectively and synergistically contribute to the selective vulnerability of distinct neuronal populations to different causes of senescence. The more we understand the interplay of Ca2+ homeostatic mechanisms with the intrinsic qualities of different neurons, the closer we will get to developing cell-specific therapies.
Statements
Acknowledgments
Work in the authors’ laboratory is funded by grants from the European Research Council (ERC), the European Commission Framework Programmes, and the Greek Ministry of Education. Vassiliki Nikoletopoulou is supported by an EMBO long-term postdoctoral fellowship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potentialconflict of interest.
References
1
Akundi R. S. Huang Z. Eason J. Pandya J. D. Zhi L. Cass W. A. et al (2011). Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice.PLoS ONE6e16038 10.1371/journal.pone.0016038
2
Arbuthnott G. W. Wickens J. (2007). Space, time and dopamine.Trends Neurosci.3062–69.
3
Artola A. Singer W. (1993). Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation.Trends Neurosci.16480–487.
4
Atluri P. P. Regehr W. G. (1998). Delayed release of neurotransmitter from cerebellar granule cells.J. Neurosci.188214–8227.
5
Bacman S. R. Bradley W. G. Moraes C. T. (2006). Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target? Mol. Neurobiol.33113–131.
6
Baines C. P. Kaiser R. A. Purcell N. H. Blair N. S. Osinska H. Hambleton M. A. et al (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death.Nature434658–662.
7
Bajjalieh S. M. Scheller R. H. (1995). The biochemistry of neurotransmitter secretion.J. Biol. Chem.2701971–1974.
8
Bak J. White P. Timar G. Missiaen L. Genazzani A. A. Galione A. (1999). Nicotinic acid adenine dinucleotide phosphate triggers Ca2+ release from brain microsomes.Curr. Biol.9751–754.
9
Baker P. F. Allen T. J. (1984). The voltage-sensitivity of Na–Ca exchange in the squid axon.Prog. Clin. Biol. Res.16889–94.
10
Ban T. A. Morey L. Aguglia E. Azzarelli O. Balsano F. Marigliano V. et al (1990). Nimodipine in the treatment of old age dementias.Prog. Neuropsychopharmacol. Biol. Psychiatry14525–551.
11
Barnes C. A. (1994). Normal aging: regionally specific changes in hippocampal synaptic transmission.Trends Neurosci.1713–18.
12
Barrett E. F. Stevens C. F. (1972). The kinetics of transmitter release at the frog neuromuscular junction.J. Physiol.227691–708.
13
Basso E. Fante L. Fowlkes J. Petronilli V. Forte M. A. Bernardi P. (2005). Properties of the permeability transition pore in mitochondria devoid of cyclophilin D.J. Biol. Chem.28018558–18561.
14
Beal M. F. (2003). Mitochondria, oxidative damage, and inflammation in Parkinson’s disease.Ann. N. Y. Acad. Sci.991120–131.
15
Behne M. J. Tu C. L. Aronchik I. Epstein E. Bench G. Bikle D. D. et al (2003). Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2Ca2+ stores.J. Invest. Dermatol.121688–694.
16
Bernard-Marissal N. Moumen A. Sunyach C. Pellegrino C. Dudley K. Henderson C. E. et al (2012). Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS.J. Neurosci.324901–4912.
17
Bernardi P. Scorrano L. Colonna R. Petronilli V Di Lisa F. (1999). Mitochondria and cell death.Mechanistic aspects and methodological issues. Eur. J. Biochem.264687–701.
18
Berridge M. J. (1998). Neuronal calcium signaling.Neuron2113–26.
19
Bezprozvanny I. (2009). Calcium signaling and neurodegenerative diseases.Trends Mol. Med.1589–100.
20
Blaustein M. P. (1988). Sodium/calcium exchange and the control of contractility in cardiac muscle and vascular smooth muscle.J. Cardiovasc. Pharmacol. 12(Suppl.5)S56–S68.
21
Blaustein M. P. Juhaszova M. Golovina V. A. Church P. J. Stanley E. F. (2002). Na/Ca exchanger and PMCA localization in neurons and astrocytes: functional implications.Ann. N. Y. Acad. Sci.976356–366.
22
Bliss T. V. Collingridge G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus.Nature36131–39.
23
Bodhinathan K. Kumar A. Foster T. C. (2010). Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling.J. Neurophysiol.1042586–2593.
24
Boehning D. Patterson R. L. Sedaghat L. Glebova N. O. Kurosaki T. Snyder S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis.Nat. Cell Biol.51051–1061.
25
Bonci A. Grillner P. Mercuri N. B. Bernardi G. (1998). L-type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons.J. Neurosci.186693–6703.
26
Braunewell K. H. Gundelfinger E. D. (1999). Intracellular neuronal calcium sensor proteins: a family of EF-hand calcium-binding proteins in search of a function.Cell Tissue Res.2951–12.
27
Burgoyne R. D. Weiss J. L. (2001). The neuronal calcium sensor family of Ca2+-binding proteins.Biochem. J.3531–12.
28
Buxbaum J. D. (2004). A role for calsenilin and related proteins in multiple aspects of neuronal function.Biochem. Biophys. Res. Commun.3221140–1144.
29
Buxbaum J. D. Choi E. K. Luo Y. Lilliehook C. Crowley A. C. Merriam D. E. et al (1998). Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment.Nat. Med.41177–1181.
30
Cali T. Ottolini D. Negro A. Brini M. (2012). Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum–mitochondria interactions.J. Biol. Chem.28717914–17929.
31
Callewaert G. Parys J. B. De Smedt H. Raeymaekers L. Wuytack F. Vanoevelen J. et al (2003). Similar Ca(2+)-signaling properties in keratinocytes and in COS-1 cells overexpressing the secretory-pathway Ca(2+)-ATPase SPCA1.Cell Calcium34157–162.
32
Camello C. Lomax R. Petersen O. H. Tepikin A. V. (2002). Calcium leak from intracellular stores – the enigma of calcium signalling.Cell Calcium32355–361.
33
Carafoli E. Longoni S. (1987). The plasma membrane in the control of the signaling function of calcium.Soc. Gen. Physiol. Ser.4221–29.
34
Carri M. T. Ferri A. Battistoni A. Famhy L. Gabbianelli R. Poccia F. et al (1997). Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells.FEBS Lett.414365–368.
35
Carrion A. M. Link W. A. Ledo F. Mellstrom B. Naranjo J. R. (1999). DREAM is a Ca2+-regulated transcriptional repressor.Nature39880–84.
36
Catterall W. A. Nunoki K. Lai Y. De Jongh K. Thomsen W. Rossie S. (1990). Structure and modulation of voltage-sensitive sodium and calcium channels.Adv. Second Messenger Phosphoprotein Res.2430–35.
37
Celsi F. Pizzo P. Brini M. Leo S. Fotino C. Pinton P. et al (2009). Mitochondria, calcium and cell death: a deadly triad in neurodegeneration.Biochim. Biophys. Acta1787335–344.
38
Chalfie M. Wolinsky E. (1990). The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans.Nature345410–416.
39
Chameau P. Van de Vrede Y. Fossier P. Baux G. (2001). Ryanodine-, IP3- and NAADP-dependent calcium stores control acetylcholine release.Pflugers Arch.443289–296.
40
Chan C. S. Guzman J. N. Ilijic E. Mercer J. N. Rick C. Tkatch T. et al (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease.Nature4471081–1086.
41
Chan S. L. Mayne M. Holden C. P. Geiger J. D. Mattson M. P. (2000). Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons.J. Biol. Chem.27518195–18200.
42
Cheung K. H. Mei L. Mak D. O. Hayashi I. Iwatsubo T. Kang D. E. et al (2010). Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons.Sci. Signal.3ra22.
43
Contreras L. Drago I. Zampese E. Pozzan T. (2010). Mitochondria: the calcium connection.Biochim. Biophys. Acta1797607–618.
44
Csordas G. Renken C. Varnai P. Walter L. Weaver D. Buttle K. F. et al (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria.J. Cell Biol.174915–921.
45
Damiano M. Starkov A. A. Petri S. Kipiani K. Kiaei M. Mattiazzi M. et al (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice.J. Neurochem.961349–1361.
46
Danzer K. M. Haasen D. Karow A. R. Moussaud S. Habeck M. Giese A. et al (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding.J. Neurosci.279220–9232.
47
Davare M. A. Hell J. W. (2003). Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging.Proc. Natl. Acad. Sci. U.S.A.10016018–16023.
48
Dekkers J. Bayley P. Dick J. R. Schwaller B. Berchtold M. W. Greensmith L. (2004). Over-expression of parvalbumin in transgenic mice rescues motoneurons from injury-induced cell death.Neuroscience123459–466.
49
Del Arco A. Agudo M. Satrustegui J. (2000). Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues.Biochem. J. 345(Pt3)725–732.
50
del Arco A. Satrustegui J. (1998). Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain.J. Biol. Chem.27323327–23334.
51
Deyo R. A. Straube K. T. Moyer J. R. Jr. Disterhoft J. F. (1989). Nimodipine ameliorates aging-related changes in open-field behaviors of the rabbit.Exp. Aging Res.15169–175.
52
Di Leva F. Domi T. Fedrizzi L. Lim D. Carafoli E. (2008). The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation.Arch. Biochem. Biophys.47665–74.
53
DiPolo R. Beauge L. (1983). The calcium pump and sodium-calcium exchange in squid axons.Annu. Rev. Physiol.45313–324.
54
Disterhoft J. F. Moyer J. R. Jr. Thompson L. T. (1994). The calcium rationale in aging and Alzheimer’s disease.Evidence from an animal model of normal aging. Ann. N. Y. Acad. Sci.747382–406.
55
Dizhoor A. M. Boikov S. G. Olshevskaya E. V. (1998). Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1.Possible role in causing human autosomal dominant cone degeneration. J. Biol. Chem.27317311–17314.
56
Dunlap K. Luebke J. I. Turner T. J. (1995). Exocytotic Ca2+ channels in mammalian central neurons.Trends Neurosci.1889–98.
57
Ernfors P. Bengzon J. Kokaia Z. Persson H. Lindvall O. (1991). Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis.Neuron7165–176.
58
Ertel E. A. Campbell K. P. Harpold M. M. Hofmann F. Mori Y. Perez-Reyes E. et al (2000). Nomenclature of voltage-gated calcium channels.Neuron25533–535.
59
Exner N. Lutz A. K. Haass C. Winklhofer K. F. (2012). Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences.EMBO J.313038–3062.
60
Fairclough R. J. Dode L. Vanoevelen J. Andersen J. P. Missiaen L. Raeymaekers L. et al (2003). Effect of Hailey–Hailey disease mutations on the function of a new variant of human secretory pathway Ca2+/Mn2+-ATPase (hSPCA1).J. Biol. Chem.27824721–24730.
61
Fink C. C. Meyer T. (2002). Molecular mechanisms of CaMKII activation in neuronal plasticity.Curr. Opin. Neurobiol.12293–299.
62
Fitzpatrick J. S. Hagenston A. M. Hertle D. N. Gipson K. E. Bertetto-D’Angelo L. Yeckel M. F. (2009). Inositol-1,4,5-trisphosphate receptor-mediated Ca2+ waves in pyramidal neuron dendrites propagate through hot spots and cold spots.J. Physiol.5871439–1459.
63
Floyd R. A. Hensley K. (2002). Oxidative stress in brain aging.Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging23795–807.
64
Fluegge D. Moeller L. M. Cichy A. Gorin M. Weth A. Veitinger S. et al (2012). Mitochondrial Ca(2+) mobilization is a key element in olfactory signaling.Nat. Neurosci.15754–762.
65
Foster T. C. (1999). Involvement of hippocampal synaptic plasticity in age-related memory decline.Brain Res. Brain Res. Rev.30236–249.
66
Foster T. C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain.Aging Cell6319–325.
67
Foster T. C. Kumar A. (2002). Calcium dysregulation in the aging brain.Neuroscientist8297–301.
68
Foster T. C. Norris C. M. (1997). Age-associated changes in Ca(2+)-dependent processes: relation to hippocampal synaptic plasticity.Hippocampus7602–612.
69
Foster T. C. Sharrow K. M. Masse J. R. Norris C. M. Kumar A. (2001). Calcineurin links Ca2+ dysregulation with brain aging.J. Neurosci.214066–4073.
70
Furuichi T. Furutama D. Hakamata Y. Nakai J. Takeshima H. Mikoshiba K. (1994). Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain.J. Neurosci.144794–4805.
71
Furukawa K. Matsuzaki-Kobayashi M. Hasegawa T. Kikuchi A. Sugeno N. Itoyama Y. et al (2006). Plasma membrane ion permeability induced by mutant alpha-synuclein contributes to the degeneration of neural cells.J. Neurochem.971071–1077.
72
Gallagher M. Landfield P. W. McEwen B. Meaney M. J. Rapp P. R. Sapolsky R. et al (1996). Hippocampal neurodegeneration in aging.Science274484–485.
73
Gandhi S. Wood-Kaczmar A. Yao Z. Plun-Favreau H. Deas E. Klupsch K. et al (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death.Mol. Cell.33627–638.
74
Gant J. C. Sama M. M. Landfield P. W. Thibault O. (2006). Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release.J. Neurosci.263482–3490.
75
Gao J. Cohen I. S. Mathias R. T. Baldo G. J. (1998). The inhibitory effect of beta-stimulation on the Na/K pump current in guinea pig ventricular myocytes is mediated by a cAMP-dependent PKA pathway.Pflugers Arch.435479–484.
76
Ghosh A. Carnahan J. Greenberg M. E. (1994). Requirement for BDNF in activity-dependent survival of cortical neurons.Science2631618–1623.
77
Gibson G. E. Peterson C. (1987). Calcium and the aging nervous system.Neurobiol. Aging8329–343.
78
Goda Y. Stevens C. F. (1994). Two components of transmitter release at a central synapse.Proc. Natl. Acad. Sci. U.S.A.9112942–12946.
79
Grace A. A. Bunney B. S. (1983). Intracellular and extracellular electrophysiology of nigral dopaminergic neurons-2.Action potential generating mechanisms and morphological correlates. Neuroscience10317–331.
80
Green D. R. Kroemer G. (2004). The pathophysiology of mitochondrial cell death.Science305626–629.
81
Gunteski-Hamblin A. M. Clarke D. M. Shull G. E. (1992). Molecular cloning and tissue distribution of alternatively spliced mRNAs encoding possible mammalian homologues of the yeast secretory pathway calcium pump.Biochemistry317600–7608.
82
Guo Q. Fu W. Sopher B. L. Miller M. W. Ware C. B. Martin G. M. et al (1999). Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice.Nat. Med.5101–106.
83
Guo Q. Christakos S. Robinson N Mattson M.P. (1998). Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: reduced oxidative stress and preserved mitochondrial function.Proc. Natl. Acad. Sci. U.S.A.953227–3232.
84
Guo Q. Furukawa K. Sopher B. L. Pham D. G. Xie J. Robinson N. et al (1996). Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide.Neuroreport8379–383.
85
Guzman J. N. Sanchez-Padilla J. Wokosin D. Kondapalli J. Ilijic E. Schumacker P. T. et al (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1.Nature468696–700.
86
Hagenston A. M. Fitzpatrick J. S. Yeckel M. F. (2008). MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons.Cereb. Cortex18407–423.
87
Hagler D. J. Jr., GodaY. (2001). Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons.J. Neurophysiol.852324–2334.
88
Hakamata Y. Nakai J. Takeshima H. Imoto K. (1992). Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain.FEBS Lett.312229–235.
89
Heeman B. Van den Haute C. Aelvoet S. A. Valsecchi F. Rodenburg R. J. Reumers V. et al (2011). Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance.J. Cell Sci.1241115–1125.
90
Herman J. P. Chen K. C. Booze R. Landfield P. W. (1998). Up-regulation of alpha1D Ca2+ channel subunit mRNA expression in the hippocampus of aged F344 rats.Neurobiol. Aging19581–587.
91
Hertle D. N. Yeckel M. F. (2007). Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus.Neuroscience150625–638.
92
Hervias I. Beal M. F. Manfredi G. (2006). Mitochondrial dysfunction and amyotrophic lateral sclerosis.Muscle Nerve33598–608.
93
Higgins C. M. Jung C. Ding H. Xu Z. (2002). Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS.J. Neurosci.22RC215.
94
Honda H. M. Ping P. (2006). Mitochondrial permeability transition in cardiac cell injury and death.Cardiovasc. Drugs Ther.20425–432.
95
Hu Z. Bonifas J. M. Beech J. Bench G. Shigihara T. Ogawa H. et al (2000). Mutations in ATP2C1, encoding a calcium pump, cause Hailey–Hailey disease.Nat. Genet.2461–65.
96
Hubbard J. I. (1963). Repetitive stimulation at the mammalian neuromuscular junction, and the mobilization of transmitter.J. Physiol.169641–662.
97
Iacopino A. M. Christakos S. (1990). Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases.Proc. Natl. Acad. Sci. U.S.A.874078–4082.
98
Ichas F. Jouaville L. S. Mazat J. P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals.Cell891145–1153.
99
Ishikawa K. Nagase T. Suyama M. Miyajima N. Tanaka A. Kotani H. et al (1998). Prediction of the coding sequences of unidentified human genes.X. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res.5169–176.
100
Iverson S. L. Orrenius S. (2004). The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis.Arch. Biochem. Biophys.42337–46.
101
Jaarsma D. Rognoni F. van Duijn W. Verspaget H. W. Haasdijk E. D. Holstege J. C. (2001). CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations.Acta Neuropathol.102293–305.
102
Joiner M. A. Griffith L. C. (1999). Mapping of the anatomical circuit of CaM kinase-dependent courtship conditioning in Drosophila.Learn. Mem.6177–192.
103
Juhaszova M. Church P. Blaustein M. P. Stanley E. F. (2000). Location of calcium transporters at presynaptic terminals.Eur. J. Neurosci.12839–846.
104
Kamal A. Biessels G. J. Duis S. E. Gispen W. H. (2000). Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing.Diabetologia43500–506.
105
Kapur A. Yeckel M. Johnston D. (2001). Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway.Neuroscience10759–69.
106
Kawamoto Y. Ito H. Ihara M. Takahashi R. (2012). Immunohistochemical localization of X-linked inhibitor of apoptosis protein in brainstem-type and cortical Lewy bodies.Neuroreport23162–167.
107
Keep M. Elmer E. Fong K. S. Csiszar K. (2001). Intrathecal cyclosporin prolongs survival of late-stage ALS mice.Brain Res.894327–331.
108
Khachaturian Z. S. (1989). Calcium, membranes, aging, and Alzheimer’s disease.Introduction and overview. Ann. N. Y. Acad. Sci.5681–4.
109
Khachaturian Z. S. (1994). Calcium hypothesis of Alzheimer’s disease and brain aging.Ann. N. Y. Acad. Sci.7471–11.
110
Kikuchi H. Almer G. Yamashita S. Guegan C. Nagai M. Xu Z. et al (2006). Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model.Proc. Natl. Acad. Sci. U.S.A.1036025–6030.
111
Kim D. Nguyen M. D. Dobbin M. M. Fischer A. Sananbenesi F. Rodgers J. T. et al (2007). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis.EMBO J.263169–3179.
112
Kim H. J. Kim M. Kim S. H. Sung J. J. Lee K. W. (2002). Alteration in intracellular calcium homeostasis reduces motor neuronal viability expressing mutated Cu/Zn superoxide dismutase through a nitric oxide/guanylyl cyclase cGMP cascade.Neuroreport131131–1135.
113
Kipanyula M. J. Contreras L. Zampese E. Lazzari C. Wong A. K. Pizzo P. et al (2012). Ca(2+) dysregulation in neurons from transgenic mice expressing mutant presenilin 2.Aging Cell11885–893.
114
Kirkinezos I. G. Bacman S. R. Hernandez D. Oca-Cossio J. Arias L. J. Perez-Pinzon M. A. et al (2005). Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice.J. Neurosci.25164–172.
115
Kirkinezos I. G. Hernandez D. Bradley W. G. Moraes C. T. (2004). An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment.J. Neurochem.88821–826.
116
Kourtis N. Nikoletopoulou V. Tavernarakis N. (2012). Small heat shock proteins protect from heat stroke-associated neurodegeneration.Nature10.1038/nature11417.[Epub ahead of print].
117
Kowalska M. Disterhoft J. F. (1994). Relation of nimodipine dose and serum concentration to learning enhancement in aging rabbits.Exp. Neurol.127159–166.
118
Kretsinger R. H. (1980). Crystallographic studies of calmodulin and homologs.Ann. N. Y. Acad. Sci.35614–19.
119
Kruman II Pedersen W. A. Springer J. E. Mattson M. P. (1999). ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis.Exp. Neurol.16028–39.
120
Kumar A. Foster T. C. (2004). Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores.J. Neurophysiol.912437–2444.
121
Kumar A. Foster T. C. (2005). Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging.Brain Res.1031125–128.
122
Kurnellas M. P. Lee A. K. Li H. Deng L. Ehrlich D. J. Elkabes S. (2007). Molecular alterations in the cerebellum of the plasma membrane calcium ATPase 2 (PMCA2)-null mouse indicate abnormalities in Purkinje neurons.Mol. Cell. Neurosci.34178–188.
123
Lai F. A. Liu Q. Y. Xu L. el-Hashem A. Kramarcy N. R. Sealock R. et al (1992). Amphibian ryanodine receptor isoforms are related to those of mammalian skeletal or cardiac muscle.Am. J. Physiol.263C365–C372.
124
Landfield P. W. Pitler T. A. (1984). Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats.Science2261089–1092.
125
Langston J. W. Ballard P. A. Jr. (1983). Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine.N. Engl. J. Med.309–310.
126
Larkum M. E. Watanabe S. Nakamura T. Lasser-Ross N. Ross W. N. (2003). Synaptically activated Ca2+ waves in layer 2/3 and layer 5 rat neocortical pyramidal neurons.J. Physiol.549471–488.
127
Lashuel H. A. Petre B. M. Wall J. Simon M. Nowak R. J. Walz T. et al (2002). Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils.J. Mol. Biol.3221089–1102.
128
Lechleiter J. Girard S. Peralta E. Clapham D. (1991). Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes.Science252123–126.
129
Lee J. C. Shin J. H. Park B. W. Kim G. S. Kim J. C. Kang K. S. et al (2012a). Region-specific changes in the immunoreactivity of SIRT1 expression in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis.Brain Res.143320–28.
130
Lee S. H. Kim K. R. Ryu S. Y. Son S. Hong H. S. Mook-Jung I. et al (2012b). Impaired short-term plasticity in mossy fiber synapses caused by mitochondrial dysfunction of dentate granule cells is the earliest synaptic deficit in a mouse model of Alzheimer’s disease.J. Neurosci.325953–5963.
131
Lehohla M. Kellaway L. Russell V. A. (2008). Effect of ageing on Ca2+ uptake via NMDA receptors into barrel cortex slices of spontaneously hypertensive rats.Metab. Brain Dis.231–8.
132
Leissring M. A. Akbari Y. Fanger C. M. Cahalan M. D. Mattson M. P. LaFerla F. M. (2000). Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice.J. Cell Biol.149793–798.
133
Lesage S. Brice A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors.Hum. Mol. Genet.18R48–R59.
134
Levere T. E. Walker A. (1992). Old age and cognition: enhancement of recent memory in aged rats by the calcium channel blocker nimodipine.Neurobiol. Aging1363–66.
135
Liang C. L. Wang T. T. Luby-Phelps K. German D. C. (2007). Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson’s disease.Exp. Neurol.203370–380.
136
Lin M. T. Beal M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases.Nature443787–795.
137
Lin P. Le-Niculescu H. Hofmeister R. McCaffery J. M. Jin M. Hennemann H. et al (1998). The mammalian calcium-binding protein, nucleobindin (CALNUC), is a Golgi resident protein.J. Cell Biol.1411515–1527.
138
Lisman J. (1989). A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory.Proc. Natl. Acad. Sci. U.S.A.869574–9578.
139
Lisman J. (1994). The CaM kinase II hypothesis for the storage of synaptic memory.Trends Neurosci.17406–412.
140
Liu J. Lillo C. Jonsson P. A. Vande Velde C. Ward C. M. Miller T. M. et al (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria.Neuron435–17.
141
Llinas R. Steinberg I. Z. Walton K. (1981). Presynaptic calcium currents in squid giant synapse.Biophys. J.33289–321.
142
Llinas R. R. (1988). The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function.Science2421654–1664.
143
Lu T. Trussell L. O. (2000). Inhibitory transmission mediated by asynchronous transmitter release.Neuron26683–694.
144
Lynch G. Larson J. Kelso S. Barrionuevo G. Schottler F. (1983). Intracellular injections of EGTA block induction of hippocampal long-term potentiation.Nature305719–721.
145
Ma Z. Siebert A. P. Cheung K. H. Lee R. J. Johnson B. Cohen A. S. et al (2012). Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability.Proc. Natl. Acad. Sci. U.S.A.109E1963–E1971.
146
MacDermott A. B. Mayer M. L. Westbrook G. L. Smith S. J. Barker J. L. (1986). NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones.Nature321519–522.
147
Magrane J. Hervias I. Henning M. S. Damiano M. Kawamata H. Manfredi G. (2009). Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities.Hum. Mol. Genet.184552–4564.
148
Malenka R. C. Kauer J. A. Perkel D. J. Nicoll R. A. (1989). The impact of postsynaptic calcium on synaptic transmission – its role in long-term potentiation.Trends Neurosci.12444–450.
149
Malenka R. C. Kauer J. A. Zucker R. S. Nicoll R. A. (1988). Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission.Science24281–84.
150
Marongiu R. Spencer B. Crews L. Adame A. Patrick C. Trejo M. et al (2009). Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux.J. Neurochem.1081561–1574.
151
Martin L. J. Gertz B. Pan Y. Price A. C. Molkentin J. D. Chang Q. (2009). The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice.Exp. Neurol.218333–346.
152
Marty A. Zimmerberg J. (1989). Diffusion into the patch-clamp recording pipette of a factor necessary for muscarinic current response.Cell. Signal.1259–268.
153
Matsuda W. Furuta T. Nakamura K. C. Hioki H. Fujiyama F. Arai R. et al (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum.J. Neurosci.29444–453.
154
Matthews E. A. Linardakis J. M. Disterhoft J. F. (2009). The fast and slow afterhyperpolarizations are differentially modulated in hippocampal neurons by aging and learning.J. Neurosci.294750–4755.
155
Mattiazzi M. D’Aurelio M. Gajewski C. D. Martushova K. Kiaei M. Beal M. F. et al (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice.J. Biol. Chem.27729626–29633.
156
Mattson M. P. (2007). Calcium and neurodegeneration.Aging Cell6337–350.
157
Mattson M. P. Rychlik B. Chu C. Christakos S. (1991). Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons.Neuron641–51.
158
Mayer M. L. Westbrook G. L. (1987). Permeation and block of N-methyl-\refscd-aspartic acid receptor channels by divalent cations in mouse cultured central neurones.J. Physiol.394501–527.
159
McCormack J. G. Denton R. M. (1990). Intracellular calcium ions and intramitochondrial Ca2+ in the regulation of energy metabolism in mammalian tissues.Proc. Nutr. Soc.4957–75.
160
Mellor J. Nicoll R. A. (2001). Hippocampal mossy fiber LTP is independent of postsynaptic calcium.Nat. Neurosci.4125–126.
161
Michaelis M. L. Bigelow D. J. Schoneich C. Williams T. D. Ramonda L. Yin D. et al (1996). Decreased plasma membrane calcium transport activity in aging brain.Life Sci.59405–412.
162
Miljanich G. P. Ramachandran J. (1995). Antagonists of neuronal calcium channels: structure, function, and therapeutic implications.Annu. Rev. Pharmacol. Toxicol.35707–734.
163
Miller L. D. Petrozzino J. J. Golarai G. Connor J. A. (1996). Ca2+ release from intracellular stores induced by afferent stimulation of CA3 pyramidal neurons in hippocampal slices.J. Neurophysiol.76554–562.
164
Mulkey R. M. Malenka R. C. (1992). Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus.Neuron9967–975.
165
Mullany P. Connolly S. Lynch M. A. (1996). Ageing is associated with changes in glutamate release, protein tyrosine kinase and Ca2+/calmodulin-dependent protein kinase II in rat hippocampus.Eur. J. Pharmacol.309311–315.
166
Murchison D. Griffith W. H. (1999). Age-related alterations in caffeine-sensitive calcium stores and mitochondrial buffering in rat basal forebrain.Cell Calcium25439–452.
167
Nakamura T. Hayashi H. Satoh H. Katoh H. Kaneko M. Terada H. (1999). A single cell model of myocardial reperfusion injury: changes in intracellular Na+ and Ca2+ concentrations in guinea pig ventricular myocytes.Mol. Cell. Biochem.194147–157.
168
Nakamura T. Lasser-Ross N. Nakamura K. Ross W. N. (2002). Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons.J. Physiol.543465–480.
169
Nath S. Goodwin J. Engelborghs Y. Pountney D. L. (2011). Raised calcium promotes alpha-synuclein aggregate formation.Mol. Cell. Neurosci.46516–526.
170
Neher E. Sakaba T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release.Neuron59861–872.
171
Nelson O. Supnet C. Liu H. Bezprozvanny I. (2010). Familial Alzheimer’s disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes.J. Alzheimers Dis.21781–793.
172
Norenberg M. D. Rao K. V. (2007). The mitochondrial permeability transition in neurologic disease.Neurochem. Int.50983–997.
173
Norris C. M. Blalock E. M. Chen K. C. Porter N. M. Landfield P. W. (2002). Calcineurin enhances \refscl-type Ca(2+) channel activity in hippocampal neurons: increased effect with age in culture.Neuroscience110213–225.
174
Norris C. M. Korol D. L. Foster T. C. (1996). Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging.J. Neurosci.165382–5392.
175
Nowak L. Bregestovski P. Ascher P. Herbet A. Prochiantz A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones.Nature307462–465.
176
Okado-Matsumoto A. Fridovich I. (2001). Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria.J. Biol. Chem.27638388–38393.
177
Olivera B. M. Miljanich G. P. Ramachandran J. Adams M. E. (1994). Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins.Annu. Rev. Biochem.63823–867.
178
Pack-Chung E. Meyers M. B. Pettingell W. P. Moir R. D. Brownawell A. M. Cheng I. et al (2000). Presenilin 2 interacts with sorcin, a modulator of the ryanodine receptor.J. Biol. Chem.27514440–14445.
179
Parker I. Ivorra I. (1991). Caffeine inhibits inositol trisphosphate-mediated liberation of intracellular calcium in Xenopus oocytes.J. Physiol.433229–240.
180
Pasinelli P. Belford M. E. Lennon N. Bacskai B. J. Hyman B. T. Trotti D. et al (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria.Neuron4319–30.
181
Patterson S. L. Grover L. M. Schwartzkroin P. A. Bothwell M. (1992). Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs.Neuron91081–1088.
182
Petrosillo G. Ruggiero F. M. Pistolese M. Paradies G. (2004). Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: role of cardiolipin.J. Biol. Chem.27953103–53108.
183
Pilstrom L. Kiessling K. H. (1972). A possible localization of -glycerophosphate dehydrogenase to the inner boundary membrane of mitochondria in livers from rats fed with ethanol.Histochemie32329–334.
184
Ping H. X. Shepard P. D. (1996). Apamin-sensitive Ca(2+)-activated K+ channels regulate pacemaker activity in nigral dopamine neurons.Neuroreport7809–814.
185
Polans A. S. Buczylko J. Crabb J. Palczewski K. (1991). A photoreceptor calcium binding protein is recognized by autoantibodies obtained from patients with cancer-associated retinopathy.J. Cell Biol.112981–989.
186
Polans A. S. Witkowska D. Haley T. L. Amundson D. Baizer L. Adamus G. (1995). Recoverin, a photoreceptor-specific calcium-binding protein, is expressed by the tumor of a patient with cancer-associated retinopathy.Proc. Natl. Acad. Sci. U.S.A.929176–9180.
187
Poncer J. C. McKinney R. A. Gahwiler B. H. Thompson S. M. (1997). Either N- or P-type calcium channels mediate GABA release at distinct hippocampal inhibitory synapses.Neuron18463–472.
188
Potier B. Poindessous-Jazat F. Dutar P. Billard J. M. (2000). NMDA receptor activation in the aged rat hippocampus.Exp. Gerontol.351185–1199.
189
Power J. M. Sah P. (2008). Competition between calcium-activated K+ channels determines cholinergic action on firing properties of basolateral amygdala projection neurons.J. Neurosci.283209–3220.
190
Prasad V. Okunade G. Liu L. Paul R. J. Shull G. E. (2007). Distinct phenotypes among plasma membrane Ca2+-ATPase knockout mice.Ann. N. Y. Acad. Sci.1099276–286.
191
Puopolo M. Raviola E. Bean B. P. (2007). Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons.J. Neurosci.27645–656.
192
Rahamimoff R. Yaari Y. (1973). Delayed release of transmitter at the frog neuromuscular junction.J. Physiol.228241–257.
193
Ramos-Castaneda J. Park Y. N. Liu M. Hauser K. Rudolph H. Shull G. E. et al (2005). Deficiency of ATP2C1, a Golgi ion pump, induces secretory pathway defects in endoplasmic reticulum (ER)-associated degradation and sensitivity to ER stress.J. Biol. Chem.2809467–9473.
194
Reinhardt T. A. Horst R. L. Waters W. R. (2004). Characterization of Cos-7 cells overexpressing the rat secretory pathway Ca2+-ATPase.Am. J. Physiol. Cell Physiol.286C164–C169.
195
Riascos D. de Leon D. Baker-Nigh A. Nicholas A. Yukhananov R. Bu J. et al (2011). Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer’s disease.Acta Neuropathol.122565–576.
196
Ris L. Godaux E. (2007). Synapse specificity of long-term potentiation breaks down with aging.Learn. Mem.14185–189.
197
Rizzuto R. Pinton P. Brini M. Chiesa A. Filippin L. Pozzan T. (1999). Mitochondria as biosensors of calcium microdomains.Cell Calcium26193–199.
198
Romo R. Schultz W. (1990). Dopamine neurons of the monkey midbrain: contingencies of responses to active touch during self-initiated arm movements.J. Neurophysiol.63592–606.
199
Rosen D. R. Siddique T. Patterson D. Figlewicz D. A. Sapp P. Hentati A. et al (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis.Nature36259–62.
200
Sabatini B. L. Regehr W. G. (1996). Timing of neurotransmission at fast synapses in the mammalian brain.Nature384170–172.
201
Sagasti A. Hisamoto N. Hyodo J. Tanaka-Hino M. Matsumoto K. Bargmann C. I. (2001). The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates.Cell105221–232.
202
Sandin M. Jasmin S. Levere T. E. (1990). Aging and cognition: facilitation of recent memory in aged nonhuman primates by nimodipine.Neurobiol. Aging11573–575.
203
Scherer P. E. Lederkremer G. Z. Williams S. Fogliano M. Baldini G. Lodish H. F. (1996). Cab45, a novel (Ca2+)-binding protein localized to the Golgi lumen.J. Cell Biol.133257–268.
204
Schinzel A. C. Takeuchi O. Huang Z. Fisher J. K. Zhou Z. Rubens J. et al (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia.Proc. Natl. Acad. Sci. U.S.A.10212005–12010.
205
Shankar S. Teyler T. J. Robbins N. (1998). Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1.J. Neurophysiol.79334–341.
206
Shi P. Gal J. Kwinter D. M. Liu X. Zhu H. (2010). Mitochondrial dysfunction in amyotrophic lateral sclerosis.Biochim. Biophys. Acta180245–51.
207
Shinoda Y. Kamikubo Y. Egashira Y. Tominaga-Yoshino K. Ogura A. (2005). Repetition of mGluR-dependent LTD causes slowly developing persistent reduction in synaptic strength accompanied by synapse elimination.Brain Res.104299–107.
208
Shtifman A. Zhong N. Lopez J. R. Shen J. Xu J. (2011). Altered Ca2+ homeostasis in the skeletal muscle of DJ-1 null mice.Neurobiol. Aging32125–132.
209
Shull G. E. (2000). Gene knockout studies of Ca2+-transporting ATPases.Eur. J. Biochem.2675284–5290.
210
Silva A. J. Paylor R. Wehner J. M. Tonegawa S. (1992a). Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice.Science257206–211.
211
Silva A. J. Stevens C. F. Tonegawa S. Wang Y. (1992b). Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice.Science257201–206.
212
Silva A. J. Wang Y. Paylor R. Wehner J. M. Stevens C. F. Tonegawa S. (1992c). Alpha calcium/calmodulin kinase II mutant mice: deficient long-term potentiation and impaired spatial learning.Cold Spring Harb. Symp. Quant. Biol.57527–539.
213
Siman R. Noszek J. C. Kegerise C. (1989). Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage.J. Neurosci.91579–1590.
214
Snutch T. P. Reiner P. B. (1992). Ca2+ channels: diversity of form and function.Curr. Opin. Neurobiol.2247–253.
215
Sokal I. Li N. Surgucheva I. Warren M. J. Payne A. M. Bhattacharya S. S. et al (1998). GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy.Mol. Cell.2129–133.
216
Sokal I. Li N. Verlinde C. L. Haeseleer F. Baehr W. Palczewski K. (2000). Ca(2+)-binding proteins in the retina: from discovery to etiology of human disease(1).Biochim. Biophys. Acta1498233–251.
217
Sollner T. Whiteheart S. W. Brunner M. Erdjument-Bromage H. Geromanos S. Tempst P. et al (1993). SNAP receptors implicated in vesicle targeting and fusion.Nature362318–324.
218
Stanley E. F. (1993). Presynaptic calcium channels and the transmitter release mechanism.Ann. N. Y. Acad. Sci.681368–372.
219
Strehler E. E. Filoteo A. G. Penniston J. T. Caride A. J. (2007). Plasma-membrane Ca(2+) pumps: structural diversity as the basis for functional versatility.Biochem. Soc. Trans.35919–922.
220
Strehler E. E. Treiman M. (2004). Calcium pumps of plasma membrane and cell interior.Curr. Mol. Med.4323–335.
221
Striessnig J. Koschak A. Sinnegger-Brauns M. J. Hetzenauer A. Nguyen N. K. Busquet P. et al (2006). Role of voltage-gated \refscl-type Ca2+ channel isoforms for brain function.Biochem. Soc. Trans.34903–909.
222
Sudbrak R. Brown J. Dobson-Stone C. Carter S. Ramser J. White J. et al (2000). Hailey–Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca(2+) pump.Hum. Mol. Genet.91131–1140.
223
Sudhof T. C. (1995). The synaptic vesicle cycle: a cascade of protein–protein interactions.Nature375645–653.
224
Sudhof T. C. (2004). The synaptic vesicle cycle.Annu. Rev. Neurosci.27509–547.
225
Szekely A. M. Costa E. Grayson D. R. (1990). Transcriptional program coordination by N-methyl-\refscd-aspartate-sensitive glutamate receptor stimulation in primary cultures of cerebellar neurons.Mol. Pharmacol.38624–633.
226
Tachikui H. Navet A. F. Ozawa M. (1997). Identification of the Ca(2+)-binding domains in reticulocalbin, an endoplasmic reticulum resident Ca(2+)-binding protein with multiple EF-hand motifs.J. Biochem.121145–149.
227
Taira T. Saito Y. Niki T. Iguchi-Ariga S. M. Takahashi K. Ariga H. (2004). DJ-1 has a role in antioxidative stress to prevent cell death.EMBO Rep.5213–218.
228
Takahashi M. Catterall W. A. (1987). Identification of an alpha subunit of dihydropyridine-sensitive brain calcium channels.Science23688–91.
229
Tao X. Finkbeiner S. Arnold D. B. Shaywitz A. J. Greenberg M. E. (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism.Neuron20709–726.
230
Tapia-Arancibia L. Aliaga E. Silhol M. Arancibia S. (2008). New insights into brain BDNF function in normal aging and Alzheimer disease.Brain Res. Rev.59201–220.
231
Thibault O. Gant J. C. Landfield P. W. (2007). Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store.Aging Cell6307–317.
232
Thibault O. Landfield P. W. (1996). Increase in single \refscl-type calcium channels in hippocampal neurons during aging.Science2721017–1020.
233
Toescu E. C. Verkhratsky A. (2004). Ca2+ and mitochondria as substrates for deficits in synaptic plasticity in normal brain ageing.J. Cell Mol. Med.8181–190.
234
Toescu E. C. Vreugdenhil M. (2010). Calcium and normal brain ageing.Cell Calcium47158–164.
235
Tokuyama W. Okuno H. Hashimoto T. Xin Li Y. Miyashita Y. (2000). BDNF upregulation during declarative memory formation in monkey inferior temporal cortex.Nat. Neurosci.31134–1142.
236
Tollefson G. D. (1990). Short-term effects of the calcium channel blocker nimodipine (Bay-e-9736) in the management of primary degenerative dementia.Biol. Psychiatry271133–1142.
237
Tonkikh A. Janus C. El-Beheiry H. Pennefather P. S. Samoilova M. McDonald P. et al (2006). Calcium chelation improves spatial learning and synaptic plasticity in aged rats.Exp. Neurol.197291–300.
238
Tonkikh A. A. Carlen P. L. (2009). Impaired presynaptic cytosolic and mitochondrial calcium dynamics in aged compared to young adult hippocampal CA1 synapses ameliorated by calcium chelation.Neuroscience1591300–1308.
239
Tsien R. W. Ellinor P. T. Horne W. A. (1991). Molecular diversity of voltage-dependent Ca2+ channels.Trends Pharmacol. Sci.12349–354.
240
Tsien R. W. Lipscombe D. Madison D. V. Bley K. R. Fox A. P. (1988). Multiple types of neuronal calcium channels and their selective modulation.Trends Neurosci.11431–438.
241
Tsuboi K. Kimber T. A. Shults C. W. (2000). Calretinin-containing axons and neurons are resistant to an intrastriatal 6-hydroxydopamine lesion.Brain Res.86655–64.
242
Urushitani M. Sik A. Sakurai T. Nukina N. Takahashi R. Julien J. P. (2006). Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis.Nat. Neurosci.9108–118.
243
Van Baelen K. Vanoevelen J. Callewaert G. Parys J. B. De Smedt H. Raeymaekers L. et al (2003). The contribution of the SPCA1 Ca2+ pump to the Ca2+ accumulation in the Golgi apparatus of HeLa cells assessed via RNA-mediated interference.Biochem. Biophys. Res. Commun.306430–436.
244
Van Brederode J. F. Mulligan K. A. Hendrickson A. E. (1990). Calcium-binding proteins as markers for subpopulations of GABAergic neurons in monkey striate cortex.J. Comp. Neurol.2981–22.
245
Vande Velde C. Miller T. M. Cashman N. R. Cleveland D. W. (2008). Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria.Proc. Natl. Acad. Sci. U.S.A.1054022–4027.
246
Vanoevelen J. Dode L. Van Baelen K. Fairclough R. J. Missiaen L. Raeymaekers L. et al (2005). The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions.J. Biol. Chem.28022800–22808.
247
Vercesi A. E. Kowaltowski A. J. Grijalba M. T. Meinicke A. R. Castilho R. F. (1997). The role of reactive oxygen species in mitochondrial permeability transition.Biosci. Rep.1743–52.
248
Vijayvergiya C. Beal M. F. Buck J. Manfredi G. (2005). Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice.J. Neurosci.252463–2470.
249
Vouimba R. M. Foy M. R. Foy J. G. Thompson R. F. (2000). 17beta-estradiol suppresses expression of long-term depression in aged rats.Brain Res. Bull.53783–787.
250
Wei Y. H. (1998). Oxidative stress and mitochondrial DNA mutations in human aging.Proc. Soc. Exp. Biol. Med.21753–63.
251
Werth J. L. Usachev Y. M. Thayer S. A. (1996). Modulation of calcium efflux from cultured rat dorsal root ganglion neurons.J. Neurosci.161008–1015.
252
West A. E. Chen W. G. Dalva M. B. Dolmetsch R. E. Kornhauser J. M. Shaywitz A. J. et al (2001). Calcium regulation of neuronal gene expression.Proc. Natl. Acad. Sci. U.S.A.9811024–11031.
253
Wilson C. J. Callaway J. C. (2000). Coupled oscillator model of the dopaminergic neuron of the substantia nigra.J. Neurophysiol.833084–3100.
254
Wood-Kaczmar A. Gandhi S. Yao Z. Abramov A. Y. Miljan E. A. Keen G. et al (2008). PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons.PLoS ONE3e2455 10.1371/journal.pone.0002455
255
Xiang M. Mohamalawari D. Rao R. (2005). A novel isoform of the secretory pathway Ca2+,Mn(2+)-ATPase, hSPCA2, has unusual properties and is expressed in the brain.J. Biol. Chem.28011608–11614.
256
Xiong J. Verkhratsky A. Toescu E. C. (2002). Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices.J. Neurosci.2210761–10771.
257
Yamada T. McGeer P. L. Baimbridge K. G. McGeer E. G. (1990). Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K.Brain Res.526303–307.
258
Yeckel M. F. Kapur A. Johnston D. (1999). Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism.Nat. Neurosci.2625–633.
259
Yoo A. S. Cheng I. Chung S. Grenfell T. Z. Lee H. Pack-Chung E. et al (2000). Presenilin-mediated modulation of capacitative calcium entry.Neuron27561–572.
260
Zeng Y. Tan M. Kohyama J. Sneddon M. Watson J. B. Sun Y. E. et al (2011). Epigenetic enhancement of BDNF signaling rescues synaptic plasticity in aging.J. Neurosci.3117800–17810.
261
Zhou Q. Homma K. J. Poo M. M. (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses.Neuron44749–757.
Summary
Keywords
endoplasmic reticulum, Golgi, long-term potentiation, ion channel, mitochondria, neurodegeneration, neurotransmitter, synaptic plasticity
Citation
Nikoletopoulou V and Tavernarakis N (2012) Calcium homeostasis in aging neurons. Front. Gene. 3:200. doi: 10.3389/fgene.2012.00200
Received
22 May 2012
Accepted
19 September 2012
Published
02 October 2012
Volume
3 - 2012
Edited by
Joy Alcedo, Wayne State University, USA
Reviewed by
Joy Alcedo, Wayne State University, USA; QueeLim Ch’Ng, King’s College London, UK
Copyright
© Nikoletopoulou and Tavernarakis.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Nektarios Tavernarakis, Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology – Hellas, Vassilika Vouton, PO Box 1385, Heraklion 71110, Crete, Greece. e-mail: tavernarakis@imbb.forth.gr
This article was submitted to Frontiers in Genetics of Aging, a specialty of Frontiers in Genetics.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.