
 Aleš Belič2
Aleš Belič2
- 1Faculty of Computer Sciences and Informatics, University of Ljubljana, Ljubljana, Slovenia
- 2Faculty of Electrical Engineering, University of Ljubljana, Ljubljana, Slovenia
- 3Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, Germany
- 4University of Tuebingen, Tuebingen, Germany
- 5Centre for Functional Genomics and Bio-Chips, Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
Non-alcoholic fatty liver disease (NAFLD), the hepatic manifestation of the metabolic syndrome, is a complex multifactorial disease characterized by metabolic deregulations that include accumulation of lipids in the liver, lipotoxicity, and insulin resistance. The progression of NAFLD to non-alcoholic steatohepatitis and cirrhosis, and ultimately to carcinomas, is governed by interplay of pro-inflammatory pathways, oxidative stress, as well as fibrogenic and apoptotic cues. As the liver is the major organ of biotransformation, deregulations in hepatic signaling pathways have effects on both, xenobiotic and endobiotic metabolism. Several major nuclear receptors involved in the transcription and regulation of phase I and II drug metabolizing enzymes and transporters also have endobiotic ligands including several lipids. Hence, hepatic lipid accumulation in steatosis and NAFLD, which leads to deregulated activation patterns of nuclear receptors, may result in altered drug metabolism capacity in NAFLD patients. On the other hand, genetic and association studies have indicated that a malfunction in drug metabolism can affect the prevalence and severity of NAFLD. This review focuses on the complex interplay between NAFLD pathogenesis and drug metabolism. A better understanding of these relationships is a prerequisite for developing improved drug dosing algorithms for the pharmacotherapy of patients with different stages of NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in Western countries with a wide disease spectrum. It ranges from the hepatic accumulation of lipids known as steatosis, non-alcoholic steatohepatitis (NASH) wherein steatosis is accompanied by inflammation and can further progress to cirrhosis and hepatocellular carcinoma. NAFLD is the hepatic manifestation of the metabolic syndrome as it is frequently associated with obesity, insulin resistance, hyperglycemia, hypertension, and dyslipidemia (Anderson and Borlak, 2008; Lewis and Mohanty, 2010; Cohen et al., 2011).
The accumulation of hepatic triglycerides due to an imbalance between free fatty acids uptake, de novo lipogenesis, oxidation, esterification, and secretion, is a hallmark of hepatic steatosis (Donnelly et al., 2005). Initially the progression of NAFLD to NASH was described as a two-hit event, with the accumulation of hepatic triglycerides being the first hit, which in turn triggers the second hit – inflammation and oxidative stress (Crespo et al., 2001; Browning and Horton, 2004; Sunny et al., 2011; Wree et al., 2011). An extended hypothesis proposed a reduced capacity of hepatic regeneration and the detrimental effects of free fatty acid lipotoxicity as the third hit (Feldstein et al., 2004; Day, 2006).
Several other factors contribute to the induction of NAFLD and/or to its progression. These include compromised adipose tissue storage and function (Calvert et al., 2007), upregulation of inflammatory factors, and reactive oxygen species (ROS; Crespo et al., 2001; Browning and Horton, 2004; Wree et al., 2011), the interaction of the intestinal microbial populations with the host through the inflammasome (Henao-Mejia et al., 2012), downregulation of the endoplasmic reticulum stress and protein ubiquitination pathways (HSPA5, USP25), and gene expression changes in cell development, morphology, movement, death, and antigen presentation pathways (Gawrieh et al., 2010). Insulin resistance also plays a detrimental role in the pathogenesis of NAFLD. Polymorphisms that may potentially explain this effect were identified in the insulin receptor-substrate 1 (IRS-1; Gly172Arg; Dongiovanni et al., 2010).
As a multifactorial disorder, NAFLD is characterized by interactions between genetic and environmental factors, thus proving it difficult to understand its manifestations (Fon Tacer and Rozman, 2011; Lorbek and Rozman, 2012). Moreover, the scarcity of robust non-invasive diagnostic methods represents an obstacle in accurately determining the prevalence of NAFLD (Ratziu et al., 2011). By current estimations, NAFLD has a prevalence rate of 6–35% with a median of 20%, depending on the population studied and the method of assessment (Vernon et al., 2011; Chalasani et al., 2012). Ethnic differences in the prevalence of NAFLD also exist, with a lower frequency in African–Americans compared to Hispanic-Americans (Wagenknecht et al., 2009; Stepanova et al., 2010). It occurs in children (Roberts, 2007) and adults of all age groups, however conflicting observations have been made regarding the gender-specific risk of NAFLD (Bedogni et al., 2005; Chen et al., 2008b). The prevalence of NASH is much lower, affecting 2–5% of the population (Neuschwander-Tetri and Caldwell, 2003; Vernon et al., 2011), however, its frequency escalates with increasing age, body mass index (BMI), more severe forms of insulin resistance, hypertriglyceridemia, and poor liver function. Increased progression to NASH is observed in Hispanics, possibly due to the large-scale adaptation of western lifestyles (Browning et al., 2004). Obese individuals (BMI > 30 kg/m2) are at a higher risk of developing NAFLD, with a prevalence of 85–98% for NAFLD and >25% for NASH (Machado et al., 2006). Furthermore, diabetes mellitus (Type 2 Diabetes, T2D) is a major determinant of NAFLD with a 70% prevalence of NAFLD in some diabetic populations (Targher et al., 2006; Leite et al., 2009). However, NAFLD also occurs in approximately 18% of normal weight non-diabetic populations (de Alwis and Day, 2008).
Due to the lack of targeted drugs, NAFLD patients are usually treated by cholesterol-lowering statins, fibrates, or anti-diabetics such as thiozolidinediones, sulfonylureas, etc. (Rozman and Monostory, 2010). The controversy regarding the NAFLD patients’ benefits versus the potential harm due to liver toxicity is however, a matter of investigations and vivid debates.
NAFLD and Drug Disposition
Liver is the major organ of endogenous and xenobiotic metabolism. In healthy livers, the metabolic processes are in homeostasis. A long-term disturbance of one or more metabolic pathways can provoke liver diseases. The intracellular accumulation of xeno- and endo-biotics is potentially toxic and is regulated at several levels including uptake, biotransformation, and elimination by drug metabolizing enzymes (DMEs). DMEs are classified as phase I, mainly cytochrome P450s (CYPs) that catalyze hydroxylation reactions, or phase II enzymes that are involved in conjugation reactions. Human phase I DME subfamilies CYP3A and CYP2C account for 50% of all hepatic CYPs and metabolize a large number of diverse drugs, e.g., lovastatin, tamoxifen, and R-warfarin. Phase II DMEs include UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), N-acetyltransferases, and several other transferases that transform compounds into more excretable forms. Transporters such as organic anion transporting polypeptide (OATP) and ATP-binding cassette (ABC)-transporters are responsible for the hepatocellular uptake and excretion of xenobiotics. The expression of phase I and II DMEs and transporters is regulated by a battery of nuclear receptors in a coordinated manner (Aleksunes and Klaassen, 2012).
Non-alcoholic fatty liver disease patients show differences in drug metabolism and its regulatory components, as summarized in Tables 1– 3. Compared to normal subjects, a pediatric NAFLD population exhibited altered glucuronidation of acetaminophen, a commonly used analgesic and antipyretic agent known to cause acute hepatic failure (Barshop et al., 2011). Although the pharmacokinetic profile of acetaminophen in both the normal and NAFLD subjects was unaltered, this study highlighted changes in the biotransformation of the drug and the possibility of compensation by other excretory pathways in the presence of NAFLD. Similarly, the metabolism of ezetimibe, an intestinal cholesterol-uptake blocker, is altered in NASH patients due to differential localization of ABCC2 and ABCB1 efflux transporters, hence, resulting in plasma retention of the active glucuronide metabolite of ezetimibe (Hardwick et al., 2012b). Studies have also indicated a reduced efficacy of certain treatments in NAFLD condition, such as the novel oral hypoglycemic sitagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor. T2D patients with NAFLD have increased serum DPP-4 activity, an enzyme that inhibits incretins such as glucagon-like peptide 1 (GLP-1), and thus, reduced efficacy of sitagliptin (Firneisz et al., 2010; Iwasaki et al., 2012).
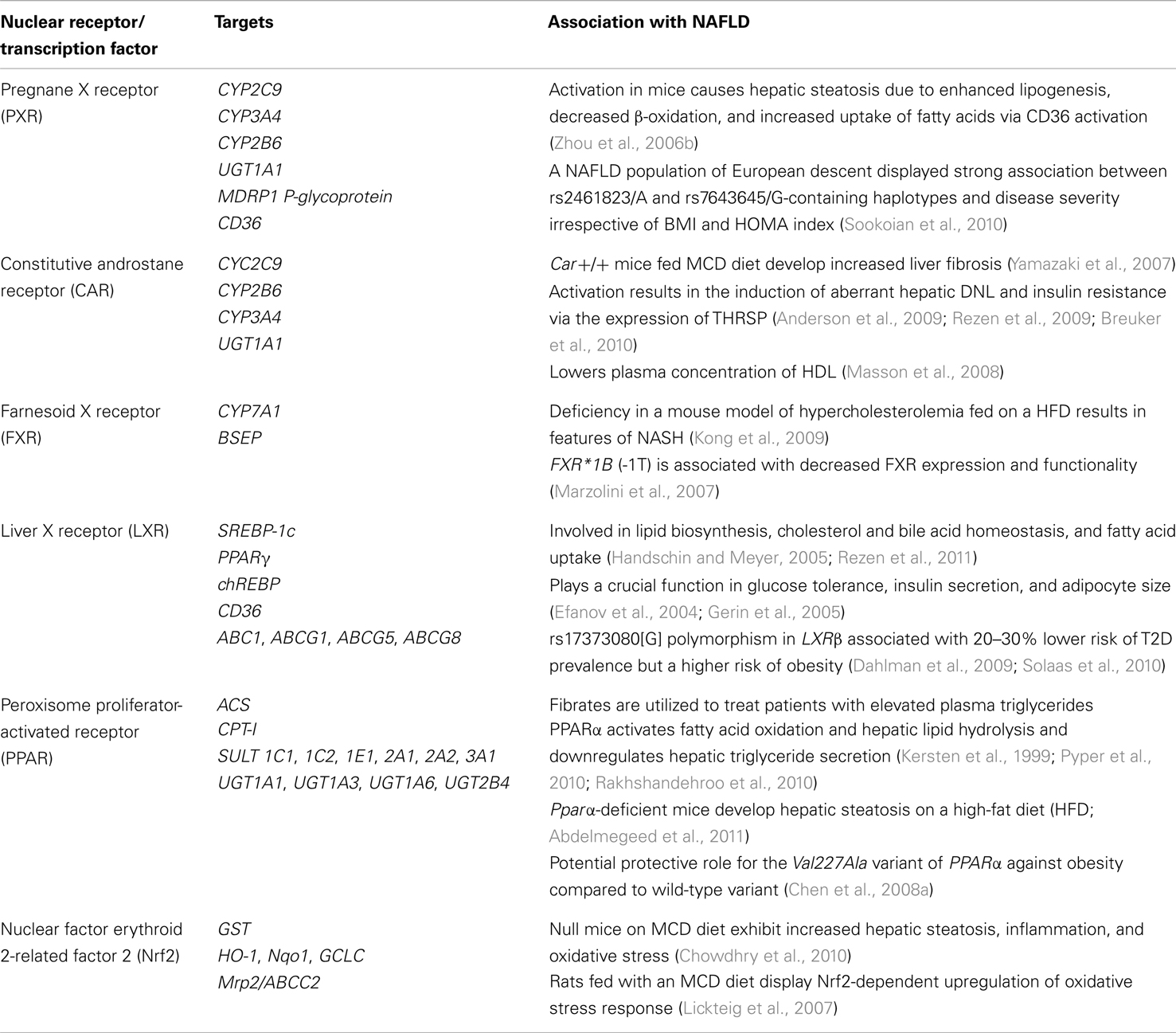
Table 1. Nuclear receptors and transcription factors in interaction between NAFLD and drug metabolism.
Several drug metabolizing CYPs are downregulated in genetically modified (e.g., leptin deficient ob/ob, dysfunctional leptin receptor db/db mice) and diet-induced [e.g., high-fat diet, methionine- and choline-deficient (MCD) diet] NAFLD animal models and in patients with characteristics of the metabolic syndrome (Buechler and Weiss, 2011; Ghose et al., 2011). Genome wide association and candidate gene studies have identified single nucleotide polymorphisms (SNPs) in DMEs that associate with NAFLD prevalence, progression, or severity, highlighting the role of altered drug metabolism in NAFLD pathogenesis (Anstee et al., 2011; Lake et al., 2011). Thus, there is ample evidence for altered xenobiotic metabolism and efficacy in NAFLD patients.
A function of CYPs in hepatic lipid homeostasis is indicated by their broad role in microsomal oxidation, cholesterol biosynthesis, and their activation by lipids. Further proof of this role was observed from studies in the liver conditional CYPs reductase microsomal flavoprotein NADPH: CYP oxidoreductase (Por) null mice. POR is an essential redox partner of the microsomal CYPs. The liver conditional Por knockout mice display hepatomegaly, hepatic steatosis, and a reduced capacity of drug metabolism (Gu et al., 2003). One of the CYPs, CYP51, is an essential enzyme of cholesterol synthesis (Keber et al., 2011). Cholesterol and its metabolites are also known to contribute to lipotoxicity and inflammation-mediated progression of NAFLD to NASH (Caballero et al., 2009). The cholesterol synthesis pathway responds to feedback regulation by cholesterol, TNF-α stimulation (Tacer et al., 2007), and xenobiotics, such as statins (Rezen et al., 2008, 2009; Rozman and Monostory, 2010), thus emphasizing the hepatic crosstalk between endobiotic and xenobiotic metabolism and inflammation. Endobiotics and xenobiotics activate various nuclear receptors and thus influence the expression of genes involved in the various hepatic metabolic pathways (Gao and Xie, 2010). The interplay between metabolism of endobiotics and xenobiotics is a frequent cause of drug side effects that can now be explained at the molecular level (Hafner et al., 2011; Rezen, 2011).
Nuclear Receptors Regulating DMEs
Pregnane X receptor
Pregnane X receptor (PXR, NR1I2) is a ligand-activated nuclear receptor that upon activation forms a heterodimer with retinoid X receptor (RXR) and regulates the expression of a wide range of DMEs (Tolson and Wang, 2010). Apart from being activated by xenobiotics, it also responds to endobiotics including bile acids (Xie et al., 2001) and steroid hormones (di Masi et al., 2009). In mice, PXR activation results in hepatic steatosis due to enhanced sterol regulatory element-binding protein-1c (SREBP-1c)-independent lipogenesis, decreased β-oxidation, and increased uptake of fatty acids via fatty acid translocase (FAT/CD36) activation (Zhou et al., 2006b). The activation of CD36 by PXR in mice occurs directly or via the activation of peroxisome proliferator-activated receptor gamma (PPARγ; Tontonoz et al., 1998; Zhou et al., 2006b). PXR also plays a role in glucose metabolism (Gao and Xie, 2012). It inhibits gluconeogenesis by inactivating hepatocyte nuclear factor-4 (HNF-4) and forkhead box protein O1 (FOXO1), both of which are positive regulators of gluconeogenic genes (Bhalla et al., 2004; Kodama et al., 2004). Genetic association studies in a European NAFLD population indicated an association of the PXR rs2461823[A] and rs7643645[G]-containing haplotypes and disease severity, irrespective of BMI and homeostatic model assessment (HOMA) index (Sookoian et al., 2010). Although PXR activation increases steatosis, PXR-dependent counteraction of inflammation by inhibition of nuclear factor kappa-B (NFκB) has also been documented in human and mouse samples (Zhou et al., 2006a). In support of the previous statement, rats treated with a PXR activator pregnenolone-16α-carbonitrile (PCN) displayed reduced hepatic fibrosis and necrosis in response to a fibrogenesis-inducing agent carbon tetrachloride (CCl4; Marek et al., 2005). It is thus plausible that PXR polymorphisms associated with NAFLD may affect disease severity by lowering PXR activity, thus resulting in increased susceptibility to NASH. Another link between PXR and NAFLD is through the modulation of DMEs. PXR induces the expression of CYP2C9 (Gerbal-Chaloin et al., 2002), which metabolizes the anti-diabetic drug rosiglitazone known to reduce liver fat. Since rosiglitazone is used in NAFLD patients with hyperglycemia and IR (Ratziu et al., 2008), patients with PXR gene variants may theoretically suffer from aberrant rosiglitazone metabolism. PXR is thus a potential pharmacogenetic marker for thiazolidinedione treatments. Furthermore, PXR is a strong modulator of CYP3A4, the major phase I DME in humans. Several SNPs that affect the expression of CYP3A4 reside within the PXR coding, non-coding, and promoter regions (Zhang et al., 2008). Since many NAFLD patients are treated with drugs metabolized by CYP3A4, further pharmacogenetic evaluation of patients with these PXR variants is required (Table 1).
Constitutive androstane receptor
Constitutive androstane receptor (CAR, NR1I3) is also a key regulator of xenobiotic and endobiotic metabolism (Moore et al., 2000; Handschin and Meyer, 2005). Upon ligand activation, CAR is translocated to the nucleus where it binds to DNA elements of DME genes such as CYP2B6, CYP3A4 CYP2Cs, and others (Sueyoshi and Negishi, 2001; Gerbal-Chaloin et al., 2002; Faucette et al., 2006; Chen and Goldstein, 2009), as well as phase II enzymes involved in glucuronidation (Sugatani et al., 2005), sulfation, and drug transport (Tolson and Wang, 2010). Several studies also indicate a role of CAR in energy homeostasis (Wada et al., 2009). Hence, the activation of CAR for regulation of energy homeostasis may affect drug metabolism (Hafner et al., 2011). SREBP-1c, which is upregulated in hepatic steatosis, inhibits CAR and thus, may further contribute to aberrant xenobiotic and endobiotic metabolism (Roth et al., 2008). Car+/+ mice fed with MCD diet, known to induce NASH (Rinella et al., 2008), developed enhanced liver fibrosis due to lipid peroxidation, inducible nitric oxide synthase (iNOS), and increased CYP induction compared to Car−/− mice (Yamazaki et al., 2007). However, no difference in hepatic lipid accumulation was observed between Car+/+ and Car−/− mice, indicating that CAR may be involved in later stages of NAFLD progression and hepatocarcinogenesis (Takizawa et al., 2011). Furthermore, observations in Car−/− mice and human hepatocytes highlight the role of CAR activation in the induction of aberrant hepatic de novo lipogenesis and insulin resistance by enhancing the expression of thyroid hormone-responsive spot 14 protein (THRSP; Anderson et al., 2009; Breuker et al., 2010). Activation of CAR in mice with 1,4-Bis [2-(3,5-dichloropyridyloxy)] benzene (TCPOBOP) increased serum triglycerides and hepatic fatty acid synthesis and repressed adaptation to hyperlipidemia, which is expected to favor the development of NAFLD (Rezen et al., 2009). Contradictory observations arise from other mouse models, where CAR has been linked to improved fatty liver due to decreased lipogenesis, increased β-oxidation, improved glucose tolerance, and insulin sensitivity (Dong et al., 2009; Gao et al., 2009). In mice, CAR also regulates cholesterol and bile acid metabolism by lowering plasma high-density lipoprotein (HDL) and reverse cholesterol transport, possibly via downregulation of apolipoprotein A1 (ApoA1; Masson et al., 2008). CAR polymorphisms have not yet been linked to metabolic diseases; however, its role in glucose and lipid metabolism and its functional redundancy with PXR highlights that the CAR gene is an important candidate for NAFLD association studies (Rezen et al., 2009).
FXR and LXR
Farnesoid X receptor (FXR) and Liver X receptor (LXR) are not major regulators of xenobiotic metabolism, but they play an important role in the metabolism of cholesterol and bile acids (Rezen et al., 2011). FXR/NR1H4 is the predominant regulator of bile acid synthesis and secretion, thereby lowering hepatic cholesterol levels. The first and rate-limiting step of bile acid synthesis from cholesterol is catalyzed by cholesterol 7α-hydroxylase (CYP7A1). The activation of FXR in primary human and mouse hepatocytes results in decreased transcription of CYP7A1 due to an indirect negative feedback mechanism (Goodwin et al., 2000; Holt et al., 2003). Furthermore, activated FXR upregulates the expression of CYP3A4, which hydroxylates some common bile acids into more soluble forms (Gnerre et al., 2004), as well as the bile salt export pump (BSEP; Ananthanarayanan et al., 2001; Plass et al., 2002; Song et al., 2008). Deficiency of FXR in an Ldlr−/− mouse model of hypercholesterolemia on high-fat diet results in features of NASH, such as macrosteatosis, hepatocyte ballooning, and inflammation (Kong et al., 2009). A common SNP, FXR*1B (-1T), has been identified in the sequence flanking the start codon of FXR in European, African, Chinese, and Hispanic-American populations. It associates with decreased FXR expression and functionality, which may contribute to the pathogenesis of metabolic disorders (Marzolini et al., 2007). However, association of this FXR polymorphism with NAFLD in humans has not yet been identified.
Liver X receptor plays important roles in lipid biosynthesis as well as cholesterol and bile acid homeostasis (Handschin and Meyer, 2005; Rezen et al., 2011). Typical activators of LXR are oxysterols such as 22(R)-hydroxycholesterol (22(R)-HC), 24(S), 25-epoxycholesterol, and 25-hydroxycholesterol (Jakobsson et al., 2012). LXR activation not only increases cholesterol catabolism but also hepatic lipogenesis through activation of SREBP-1c, PPARγ, or carbohydrate response element-binding protein (chREBP; Lee et al., 2008). LXR and PXR share many target genes (Boergesen et al., 2012) and both regulate the uptake of fatty acids into hepatocytes via expression of FAT CD36 (Zhou et al., 2006b). Although LXR activates Cyp7a1 expression during bile acid synthesis in rodents, it does not have an effect on human CYP7A1 expression (Goodwin et al., 2003). In humans, the LXRα isoform is mainly expressed in tissues involved in lipid metabolism, where it regulates the expression of cholesterol transporter genes, such as the ABC transporters ABC1 (Schwartz et al., 2000), ABCG1 (Sabol et al., 2005), ABCG5 (Repa et al., 2002), and ABCG8 (Repa et al., 2002). LXRβ, the ubiquitously expressed isoform and the only isoform present in pancreatic β-cells, does not play a role in cholesterol homeostasis (Alberti et al., 2001) but has a crucial role in glucose tolerance, insulin secretion, and adipocyte size (Efanov et al., 2004; Gerin et al., 2005). The rs17373080[G] polymorphism in LXRβ associates with a 20–30% lower risk of T2D but with a higher risk of obesity, as observed in two independent studies (Dahlman et al., 2009; Solaas et al., 2010). This is in accordance with data on Lxrβ-null mice that display a lean phenotype with glucose intolerance (Gerin et al., 2005). According to the best of our knowledge, LXR polymorphisms have not yet been linked to NAFLD.
Peroxisome proliferator-activated receptors
Peroxisome proliferator-activated receptors (PPARs) are transcription factors that are activated by endogenous ligands, such as fatty acids, and synthetic ligands, such as the hypolipidaemic fibrates and the insulin-sensitizing thiozolidinediones. Like PXR and CAR, they form heterodimers with RXR and transactivate numerous target genes with vital roles in metabolism by binding to PPAR response elements (PPRE; Nielsen et al., 2008; van der Meer et al., 2010). The identified subtypes PPARα, PPARγ, and PPARβ/δ have different tissue-specificities and functions (Kallwitz et al., 2008). Whilst PPARγ is highly expressed in adipose tissue and functions in adipocyte differentiation, PPARα functions as a major regulator of lipid and glucose metabolism in the liver. PPARβ/δ is ubiquitously expressed with a wide array of functions. PPARα-agonists, the fibrates, are utilized to treat patients with elevated plasma triglycerides (Sirtori and Franceschini, 1988) due to the ability of PPARα to activate fatty acid oxidation and hepatic lipid hydrolysis by regulating acyl CoA synthetase (Acs), carnitine palmitoyl transferase I (Cpt-I), mitochondrial β-oxidation enzymes, and hepatic lipases in addition to downregulating apoCIII and decreasing hepatic triglyceride secretion (Kersten et al., 1999; Pyper et al., 2010; Rakhshandehroo et al., 2010). Accordingly, Pparα-deficient mice develop hepatic steatosis on a high-fat diet (Abdelmegeed et al., 2011).
A case-control study of NAFLD patients highlighted a potentially protective role for the Val227Ala variant of PPARα against obesity compared to subjects with the wild-type receptor (Chen et al., 2008a). The adipocyte differentiation regulator, PPARγ, plays an important role in lipid homeostasis and insulin sensitivity by enhancing fatty acid and insulin-dependent glucose uptake in adipose tissue (Kallwitz et al., 2008). Moreover, novel PPARγ agonists acting mainly on adipose PPARγ prevent formation of steatotic livers in mice by improving insulin resistance, upregulating adiponectin, and downregulating leptin expression and secretion (Zheng et al., 2011). PPARγ is expressed at low levels in the liver but is upregulated in rodent fatty livers, contributing to hepatic triglyceride accumulation with a protective effect to dyslipidemia and insulin resistance in other tissues (Gavrilova et al., 2003). Upregulation of hepatic PPARγ is also observed in obese NAFLD patients (Pettinelli and Videla, 2011).
PPARα affects the expression of several phase II enzymes such as SULTs and UGT (Runge-Morris and Kocarek, 2009) as well as of CYP3A4 and several other CYPs in humans (Rakhshandehroo et al., 2009; Klein et al., 2012). Contrasting observations have been made regarding the regulation of DMEs by PPARα in human versus mouse. While treatment of human primary hepatocytes with the selective PPARα agonist WY14 643 resulted in the activation of several drug metabolizing CYPs including CYP3A4, CYP2B6, CYP2C8, and CYP1A2, none of the mouse gene orthologs were regulated (Rakhshandehroo et al., 2009). Downregulation of CYP3A4 in the presence of PPARα variants that result in decreased hepatic PPARα protein levels was also found by genetic association analysis and confirmed in a human atorvastatin volunteer study (Klein et al., 2012). Moreover, activation of PPARα by agonists downregulates the expression of representatives genes of the Sult 1, 2, 3, and 5 families, specifically in female rats (Alnouti and Klaassen, 2008). However, in human hepatocytes activation of PPARα resulted in the upregulation of SULT2A1 via a functional PPRE, further emphasizing species and gender differences in the functionality of PPARα (Fang et al., 2005). Protein and mRNA levels of UGT1A1 increased upon PPARα activation in rat and human hepatocytes (Jemnitz et al., 2000; Richert et al., 2003). Additionally, UGT1A3, UGT1A4, and UGT1A6 are upregulated in human hepatocytes and transgenic mice carrying the human UGT1 locus (Senekeo-Effenberger et al., 2007). UGT2B4 is also enhanced after treatment of human hepatocytes with PPARα agonists (Barbier et al., 2003). Functional PPREs have been identified in the 5′-flanking regions of UGT1A1, UGT1A3, UGT1A6, and UGT2B4 genes, thus providing evidence that these genes are direct targets of PPAR (Barbier et al., 2003; Senekeo-Effenberger et al., 2007). The widespread use of drugs metabolized by SULTs and UGTs such as hormonal contraceptives, acetaminophen, β2-adrenergic agonists, anti-depressants, and non-steroidal anti-inflammatory drugs highlights the implications of altered PPARα activation on xenobiotic metabolism in NAFLD patients.
Nuclear factor erythroid 2-related factor 2
Nuclear Factor Erythroid 2-related factor 2 (NRF2) is a transcription factor that responds to oxidative/electrophilic stimuli by releasing from its repressor Kelch-like ECH associating protein 1 (Keap1) in the cytosol, translocating to the nucleus, binding to antioxidant response elements (AREs) upstream of numerous phase II DME genes, and genes involved in redox balance and oxidative stress response [e.g., heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase-1 (NQO1)] and activating their transcription (Wu et al., 2012). NRF2 also regulates the glutathione synthesis enzyme, glutamate cysteine ligase catalytic (GCLC). The expression of a canalicular biliary efflux transporter, multidrug resistance protein 2 (MRP2/ABCC2) and sinusoidal transporters, MRP3 and MRP4 is also regulated by NRF2 in mouse liver and HepG2 cells in response to oxidative stress and xenobiotics, thus providing further evidence that phase II enzymes and efflux transporters are regulated simultaneously (Vollrath et al., 2006; Aleksunes et al., 2008). MRP2 is involved in the excretion of reduced and oxidized glutathione and hence plays an important role in detoxification and against oxidative stress. Nrf2-null mice on MCD diet exhibit increased hepatic steatosis accompanied by inflammation and oxidative stress (Chowdhry et al., 2010). Similarly, in rats on MCD diet, the NRF2-dependent genes involved in the oxidative stress response were upregulated (Lickteig et al., 2007). Thus, NRF2 appears to have a crucial role in the pathogenesis of NAFLD.
Thus, it is evident that as many of the lipids that accumulate in obesity and steatosis, such as fatty acids, cholesterol, or bile acids, are endogenous ligands of nuclear receptors, their deregulation may not only exacerbate the deregulated metabolic processes in NAFLD patients but also result in deregulated xenobiotic metabolism.
Phase I DMEs
CYP3A
The CYP3A sub-family of DMEs plays a predominant role in the metabolism of statins. Statins, in monotherapy and in combination with other lipid-lowering drugs or antioxidants, are beneficial in NAFLD patients by improving dyslipidemia (Athyros et al., 2011; Fon Tacer and Rozman, 2011). The inter-individual variability in the response to statins varies in NAFLD patients based on their risk for cardiovascular diseases (Maroni et al., 2011). Moreover, the CYP3A4 drug metabolizing activity is also a factor influencing inter-individual variability and hence, is relevant to NAFLD patients undergoing statin therapy. The level of CYP3A protein correlates negatively with the severity of steatosis in humans (Kolwankar et al., 2007). No changes were found in the CYP3A4 mRNA level in human fatty liver samples at various stages of NAFLD progression, however a trend of decreasing activity and protein levels was observed (Fisher et al., 2009b). In another study, CYP3A4 activity significantly decreased in macrosteatotic fatty livers and cultured human hepatocytes treated with fatty acids (Donato et al., 2006, 2007). CYP3A4 also displays sexual dimorphism with approximately twofold elevated expression in premenopausal women (Wolbold et al., 2003), who display a more favorable lipid profile compared to men (Williams, 2004). An intron 6 polymorphism in CYP3A4 (rs35599367[T]) results in decreased expression and activity of CYP3A4, with carriers of the T allele requiring significantly lower doses of statins (Elens et al., 2011; Wang et al., 2011). In accordance with the reduced CYP3A4 expression in NAFLD, studies to determine the association of the rs35599367 CYP3A4 polymorphism in NAFLD cohorts will enable the elucidation of statin dose selection in these patients. Moreover, genetic variants in other factors implicated in NAFLD, endobiotic, and xenobiotic metabolism such as PXR, PPARα, and POR have also been associated with altered CYP3A4 expression and activity (Zhang et al., 2008; Gomes et al., 2009; Klein et al., 2012). These studies emphasize the high level of variability in responses to statin treatments and may provide a basis for dose selection in NAFLD patients based on CYP3A4 status (Table 2).

Table 2. Phase I drug metabolizing enzymes implicated in the pathogenesis of NAFLD.
CYP2C9
CYP2C9 is the most abundant CYP of the CYP2C sub-family in human liver microsomes, accounting for the metabolism of a large number of clinically important drugs, especially some with a narrow therapeutic index, such as warfarin. The expression of CYP2C9 is coordinated by nuclear receptors such as CAR and PXR in association with nuclear factors and coactivators such as hepatocyte nuclear factor-4 alpha (HNF-4α) and PPARγ coactivator-1 alpha (PGC-1α), which is also involved in energy homeostasis (Chen and Goldstein, 2009). CYP2C9 has been closely associated with adverse drug reactions. Its mRNA and enzyme activity increase with NAFLD progression, hypoxia, and at later stages of NASH in humans (Fisher et al., 2009b). Previous observations linking CYP2C9 with arachidonic acid metabolism and vasocontraction in hypoxic conditions (Pokreisz et al., 2006) may possibly provide an explanation for elevated CYP2C9 in progressive NAFLD. Approximately 50 variants have been identified in the CYP2C9 gene to date, with the CYP2C9*2 and CYP2C9*3 loss-of-function alleles as the most important. Heterozygotes and homozygotes for these polymorphisms are common in Caucasians, with frequencies of approximately 10–17 (CYP2C9*2) and 7% (CYP2C9*3). Both polymorphic alleles were associated with increased response to anti-diabetic sulfonylurea drugs and an increased glycemic response in T2D patients (Zhou et al., 2010). As NAFLD patients are treated with sulfonylureas, genotyping is clinically relevant. Further studies are needed to identify the association of the CYP2C9*2 and CYP2C9*3 variants with adverse drug reactions such as hypoglycemia and weight gain resulting from sulfonylurea treatment.
CYP2E1
CYP2E1, a fatty acid (Ω-1)-hydroxylase, catalyzes the oxidation of many low molecular weight molecules, including ethanol and acetone, a product of fatty acid oxidation. An important catalytic feature of CYP2E1 is the generation of ROS such as superoxide anion radical and hydrogen peroxide as a result of uncoupling of oxygen consumption with NADPH oxidation and as a by-product of lipid peroxidation (Robertson et al., 2001; Caro and Cederbaum, 2004). It is also involved in the biotransformation of xenobiotics such as acetaminophen, resulting in the generation of toxic reactive metabolites (Aubert et al., 2011). NAFLD and NASH patients and the MCD diet-fed rat model of NASH display enhanced expression of CYP2E1, which is in contrast to all other drug metabolizing CYPs, and elevated lipid peroxidation (Weltman et al., 1996, 1998; Videla et al., 2004) with increased localization to hepatic areas with oxidative stress injuries. Obese females with steatosis and NASH display elevated CYP2E1 protein levels and a positive correlation between the c2 allele of Rsa1/Pst1 polymorphisms in CYP2E1 and liver injury (Varela et al., 2008). Mice with silenced diacylglycerol acyltransferase 2 (Dgat2) on MCD diet display elevated Cyp2e1 expression that correlates with increased lipid peroxidation and oxidative damage, thus highlighting the role of CYP2E1 in the progression to NASH in response to increased hepatic free fatty acids (Yamaguchi et al., 2007). An upregulation in CYP2E1 has also been associated with leptinemia, reduced adiponectin levels, and insulin resistance in NAFLD (Aubert et al., 2011). This phenomenon is reversed in patients who have undergone bariatric surgery with resulting decreases in weight and hepatic steatosis (Bell et al., 2010). With the robust cellular protection mechanisms intact, increases in pro-oxidant molecules and CYP2E1 are counteracted by increased levels of glutathione (GSH). However, most NASH rodent models display lower GSH, indicating defects in the oxidative stress response pathways in progressive NAFLD. Nitrosylation of antioxidant enzymes superoxide dismutase (SOD) and catalase (CAT) is crucial in a Cyp2e1-overexpressing mouse model of NAFLD (Kathirvel et al., 2010) because increased levels of iNOS generates reactive nitrogen species (RNS), which nitrosylate antioxidant enzymes and decrease their activity. Thus, CYP2E1 polymorphisms that associate with the progression of NAFLD to NASH may possibly trigger the combined detrimental effects of both ROS and RNS, which in combination with toxic metabolites from xenobiotic biotransformation may result in further aggravated liver injury in NAFLD patients.
Observations in a NAFLD pediatric population have indicated a direct correlation between lipid peroxidation and disease severity irrespective of CYP2E1 levels; however, the small sample size of this study and the possibility of alternative mechanisms of lipid peroxidation in early onset hepatic steatosis cannot be excluded (Bell et al., 2011). Other groups have also previously indicated decreased CYP2E1 mRNA and protein levels and no changes in CYP2E1 activity at progressive stages of NAFLD (Fisher et al., 2009b; Mitsuyoshi et al., 2009). Thus, no conclusive role of CYP2E1 in NAFLD can be described. Interestingly, CYP4A genes seem to compensate for microsomal lipid oxidation in the absence of CYP2E1 as observed in Cyp2e1-null mice that display lipid peroxidation and increased expression of Cyp4a10 and Cyp4a14 (Leclercq et al., 2000; Hardwick et al., 2009). This observation may possibly explain the absence of changes in CYP2E1 activity in some NAFLD populations.
CYP4A
CYP4A enzymes ω-hydroxylate fatty acids into dicarboxylic acids that are preferentially oxidized by peroxisomes. Genes of the CYP4A sub-family are induced by PPARα-agonists and in conditions of fasting. The enhanced activity of CYP4A results in increased production of ROS, thus contributing to steatohepatitis. In contrast, PPARα agonists prevent NASH by increasing β-oxidation. Moreover, Pparα-null mice on MCD diet are more prone to developing NASH in the absence of Cyp4a induction (Ip et al., 2003). These observations suggest that the anti-steatotic effects of PPARα may be more potent than its activation of CYP4A genes, hence overriding the ROS-generating effects of CYP4A. In the absence of PPARα alternative oxidative stress mechanisms may act as causal factors.
Phase II DMEs
Phase II DMEs are conjugative, detoxification enzymes that transform substrates into more excretable inactive forms or on the other hand may also be involved in bioactivation. Glutathione-S-transferases (GSTs) are present as different isoforms Alpha, Mu, and Pi and conjugate electrophilic compounds with reduced GSH (Hayes et al., 2005). While a GST A and P are upregulated with disease progression in the livers of NAFLD patients, GST M is significantly downregulated, thus highlighting the differential regulation of GST isoforms in NAFLD progression; however, the overall GST activity was decreased in these samples (Hardwick et al., 2010). GSTM2, GSTM4, and GSTM5 mRNA levels are expressed at lower levels in patients with steatosis and NASH (Younossi et al., 2005). GSTs play a significant role in controlling oxidative stress by conjugating harmful by-products of oxidative stress with GSH (Hayes et al., 2005). Decreased GST activity in progressive NAFLD samples was accompanied by a reduced pool of GSH, highlighting the depleted ability to combat oxidative stress, a causal factor for NASH (Hardwick et al., 2010). The antioxidant, S-adenosyl-L-methionine (SAM) provides the cysteine moiety for the generation of GSH. Several rodent studies have indicated a decrease in SAM on a high-fat diet (Kwon do et al., 2009; Buechler and Weiss, 2011). Furthermore, GSTs have a lower expression in Caucasians compared to African–Americans, who have a lower prevalence of NAFLD (Stepanova et al., 2010). The GSTM1-null genotype, shown to confer a higher risk of T2D, is also present at a higher frequency in NAFLD subjects compared to control (Hori et al., 2007, 2009). Thus, decreased activity of GSTs play a plausible role in NAFLD progression as a result of increased damage by oxidative stress (Table 3).
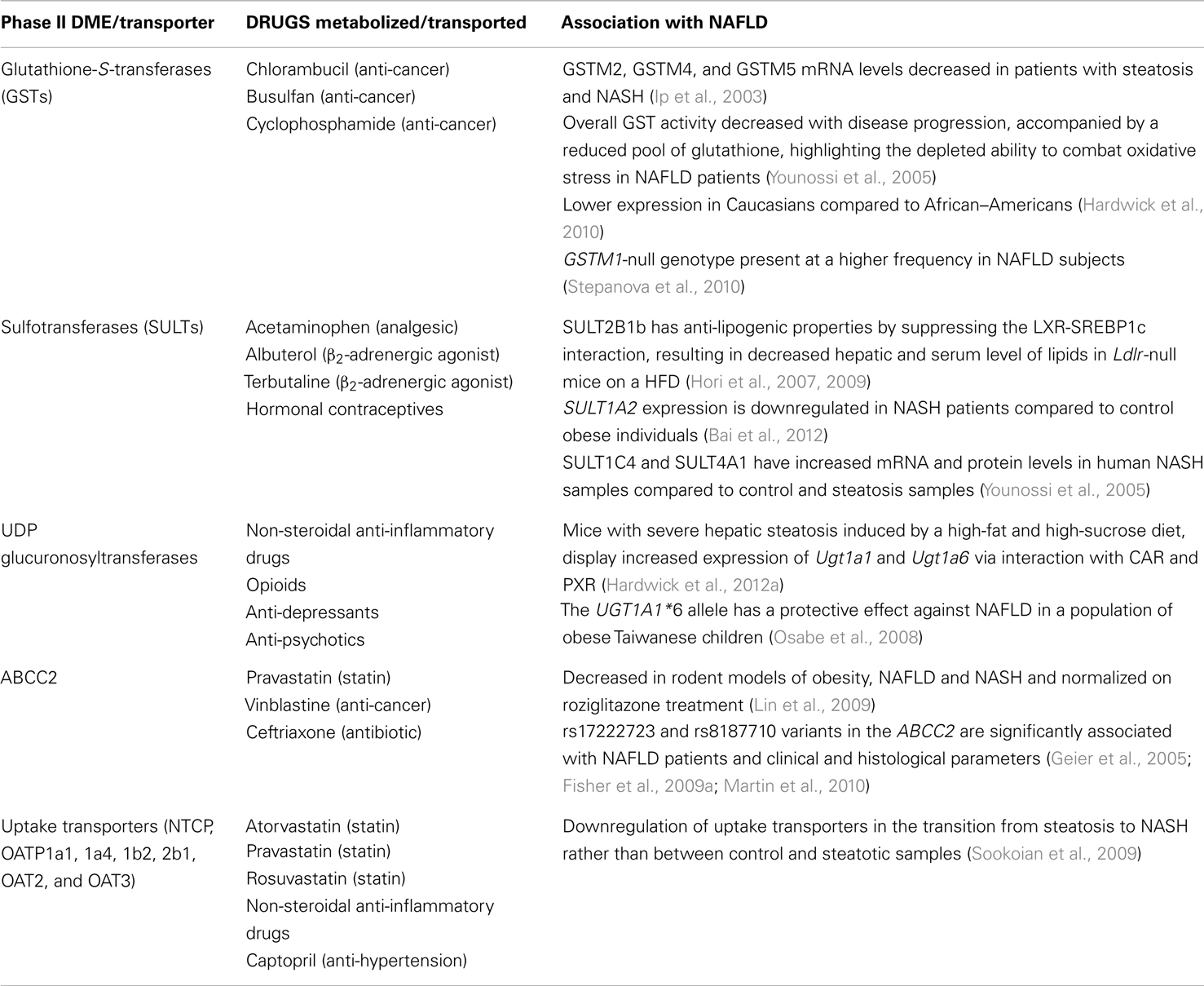
Table 3. Phase II drug metabolizing enzymes and transporters implicated in the pathogenesis of NAFLD.
Sulfotransferases are involved in sulfation of several endogenous steroids and xenobiotics. The sulfation of oxysterols by SULT2B1b has anti-lipogenic properties by suppressing the LXR-SREBP-1c interaction, resulting in significantly lower hepatic and serum lipids as observed in low-density lipoprotein receptor (Ldlr)-null mice on a high-fat diet (Bai et al., 2012). Moreover, SULT1A2 gene expression is downregulated in NASH patients compared to control obese individuals (Younossi et al., 2005). SULT2A1 is upregulated by PPARα agonists in primary human hepatocytes, but not in rat hepatocytes, due to the presence of a PPRE in the 5′ region of the gene (Fang et al., 2005; Runge-Morris and Kocarek, 2009). Thus, downregulation of PPARα observed in NAFLD may have implications in the altered expression of SULT2A1. However, only two SULT isoforms, SULT1C4 and SULT4A1, whose regulation and function are largely unknown, have increased mRNA and protein levels in human NASH samples compared to control and steatosis samples (Hardwick et al., 2012a). A previous association of SULT4A1 in deregulated metabolic homeostasis makes it a good candidate for further studies in the context of NAFLD (Kiba et al., 2009).
UDP glucuronosyltransferases are involved in the glucuronidation of 40–70% of all clinical drugs in humans (Wells et al., 2004). UGT1A1, 1A3, 1A4, 1A6, and 2B4 are induced by PPARα-agonists in primary human hepatocytes and PPREs have been identified in these genes (Runge-Morris and Kocarek, 2009). Mice on high-fat and high-sucrose diet, which develop severe hepatic steatosis, display elevated expression of Ugt1a1 and Ugt1a6 mediated by CAR and PXR (Osabe et al., 2008). A study in a pediatric NAFLD population identified UGT1A1 as a risk factor for NAFLD. The UGT1A1*6 allele in the coding region has a protective effect against NAFLD in obese Taiwanese children (Lin et al., 2009). UGT1A1 is involved in the glucuronidation of heme after breakdown to bilirubin. The ability of bilirubin to oxidize ROS may provide protection against the progression of NAFLD. Additionally, the high prevalence of unconjugated hyperbilirubinemia was detected in NAFLD patients (25.4%) that were diagnosed with less severe forms of NAFLD (Kumar et al., 2012). However, the absence of changes in glucuronidation activity in human steatosis and NASH liver samples warrants the need for further studies to investigate the role of UGTs in NAFLD (Hardwick et al., 2012a).
Transporters
Solute carrier transporters are uptake transporters that transport molecules from the blood into the hepatocyte. Studies in rat and human samples have indicated a coordinated downregulation of uptake transporter genes in NASH, such as the sodium/bile acid transporter (NTCP), organic anion transporting polypeptide 1a1 (OATP1a1), 1a4, 1b2, 2b1, OAT2, and OAT3. The expression of these transporters is significantly altered in the transition from steatosis to NASH rather than between control and steatotic samples (Fisher et al., 2009a; Lake et al., 2011). These changes appear to be hepatoprotective to prevent the accumulation of toxic intermediates and xenobiotics in the diseased liver. However, they have major implications in therapeutic regimens in NAFLD patients in terms of dose selection and side effects of drugs due to excessive accumulation (Table 3).
Transporters on the hepatocyte canalicular membranes are involved in the secretion of several endobiotics and xenobiotics via the bile. ABC-transporters are the most extensively studied and are altered in steatotic and NASH livers (Buechler and Weiss, 2011). Of particular interest is Mrp2/Abcc2, which is decreased in several rodent models of obesity, NAFLD and NASH and is normalized upon rosiglitazone treatment (Geier et al., 2005; Fisher et al., 2009a; Martin et al., 2010). Furthermore, the rs17222723 and rs8187710 variants in ABCC2 significantly associate with NAFLD and clinical and histological parameters (Sookoian et al., 2009). Decreased levels of ABCC2 protein may result in hampered secretion of bile, leading to the accumulation of cholesterol and drug-related toxicities. As mentioned previously, this may result from impaired NRF2 function.
Conclusion and Future Directions
The high prevalence of NAFLD is concerning in terms of general population health and also drug treatment regimens. A recent study in mice has identified that the feed-forward cycle of continuous exposure to high-fat diet over two generations leads to a significantly higher degree of obesity, NAFLD, insulin and leptin resistance, and epigenetic modifications resulting in increased lipogenesis and ER stress in future generations (Li et al., 2011). If these observations are also true for humans, the rising epidemics of obesity and NAFLD will expand exponentially in the absence of serious efforts to tackle these conditions.
With the widespread prevalence of NAFLD, the proportion of patients with steatotic livers undergoing drug therapies for various disorders has also increased. The variability of drug treatment responses in these patients highlights the need for personalized therapeutic regimens. As detailed in this review, several components of the drug metabolism pathway are significantly affected in the presence of NAFLD. Similarly, genetic variations in DMEs and nuclear receptors associate with NAFLD with either positive or negative prognosis. Hence, inter-dependent interactions and common confounding factors exist between the pathogenesis of NAFLD and altered drug metabolism. As a majority of the DMEs are also involved in the metabolism of steroids and other lipids, polymorphisms in DMEs resulting in non-functional proteins may further aggravate the prognosis of NAFLD. The utility of identified genetic associations to determine NAFLD disease susceptibility, improve drug sensitivity or prevent adverse drug reactions holds great potential. Further efforts to characterize DMEs and identify risk factors for adverse drug reactions or treatment efficacies in NAFLD populations may lead to the utilization of innovative interdisciplinary strategies to provide a better insight into the pharmacokinetic profile of drugs and their efficacy. Although the implementation of these findings in the clinic is still a long-term goal with hurdles to pass, novel technologies and increasing interest in this field continues to increase our understanding of NAFLD and its interactions with drug metabolism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the FP7 FightingDrugFailure ITN Marie Curie grant #238132 and by the German BMBF (Virtual Liver grant 0315755 to Ulrich M. Zanger).
References
Abdelmegeed, M. A., Yoo, S. H., Henderson, L. E., Gonzalez, F. J., Woodcroft, K. J., and Song, B. J. (2011). PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 141, 603–610.
Alberti, S., Schuster, G., Parini, P., Feltkamp, D., Diczfalusy, U., Rudling, M., et al. (2001). Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXR beta-deficient mice. J. Clin. Invest. 107, 565–573.
Aleksunes, L. M., and Klaassen, C. D. (2012). Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPAR-, and Nrf2-null mice. Drug Metab. Dispos. 40, 1366–1379.
Aleksunes, L. M., Slitt, A. L., Maher, J. M., Augustine, L. M., Goedken, M. J., Chan, J. Y., et al. (2008). Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol. Appl. Pharmacol. 226, 74–83.
Alnouti, Y., and Klaassen, C. D. (2008). Regulation of sulfotransferase enzymes by prototypical microsomal enzyme inducers in mice. J. Pharmacol. Exp. Ther. 324, 612–621.
Ananthanarayanan, M., Balasubramanian, N., Makishima, M., Mangelsdorf, D. J., and Suchy, F. J. (2001). Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 276, 28857–28865.
Anderson, G. W., Zhu, Q. H., Metkowski, J., Stack, M. J., Gopinath, S., and Mariash, C. N. (2009). The Thrsp null mouse (Thrsp(tm1cnm)) and diet-induced obesity. Mol. Cell. Endocrinol. 302, 99–107.
Anderson, N., and Borlak, J. (2008). Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev. 60, 311–357.
Anstee, Q. M., Daly, A. K., and Day, C. P. (2011). Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim. Biophys. Acta 1812, 1557–1566.
Athyros, V. G., Tziomalos, K., Daskalopoulos, G. N., Karagiannis, A., and Mikhailidis, D. P. (2011). Statin-based treatment for cardiovascular risk and non-alcoholic fatty liver disease. Killing two birds with one stone? Ann. Med. 43, 167–171.
Aubert, J., Begriche, K., Knockaert, L., Robin, M. A., and Fromenty, B. (2011). Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 35, 630–637.
Bai, Q., Zhang, X., Xu, L., Kakiyama, G., Heuman, D., Sanyal, A., et al. (2012). Oxysterol sulfation by cytosolic sulfotransferase suppresses liver X receptor/sterol regulatory element binding protein-1c signaling pathway and reduces serum and hepatic lipids in mouse models of nonalcoholic fatty liver disease. Metabolism 61, 836–845.
Barbier, O., Duran-Sandoval, D., Pineda-Torra, I., Kosykh, V., Fruchart, J. C., and Staels, B. (2003). Peroxisome proliferator-activated receptor alpha induces hepatic expression of the human bile acid glucuronidating UDP-glucuronosyltransferase 2B4 enzyme. J. Biol. Chem. 278, 32852–32860.
Barshop, N. J., Capparelli, E. V., Sirlin, C. B., Schwimmer, J. B., and Lavine, J. E. (2011). Acetaminophen pharmacokinetics in children with nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 52, 198–202.
Bedogni, G., Miglioli, L., Masutti, F., Tiribelli, C., Marchesini, G., and Bellentani, S. (2005). Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 42, 44–52.
Bell, L. N., Molleston, J. P., Morton, M. J., Klipsch, A., Saxena, R., Vuppalanchi, R., et al. (2011). Hepatic lipid peroxidation and cytochrome P-450 2E1 in pediatric nonalcoholic fatty liver disease and its subtypes. J. Clin. Gastroenterol. 45, 800–807.
Bell, L. N., Temm, C. J., Saxena, R., Vuppalanchi, R., Schauer, P., Rabinovitz, M., et al. (2010). Bariatric surgery-induced weight loss reduces hepatic lipid peroxidation levels and affects hepatic cytochrome P-450 protein content. Ann. Surg. 251, 1041–1048.
Bhalla, S., Ozalp, C., Fang, S., Xiang, L., and Kemper, J. K. (2004). Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J. Biol. Chem. 279, 45139–45147.
Boergesen, M., Pedersen, T., Gross, B., Van Heeringen, S. J., Hagenbeek, D., Bindesbøll, C., et al. (2012). Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor (in mouse liver reveals extensive sharing of binding sites. Mol. Cell. Biol. 32, 852–867.
Breuker, C., Moreau, A., Lakhal, L., Tamasi, V., Parmentier, Y., Meyer, U., et al. (2010). Hepatic expression of thyroid hormone-responsive spot 14 protein is regulated by constitutive androstane receptor (NR1I3). Endocrinology 151, 1653–1661.
Browning, J. D., and Horton, J. D. (2004). Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152.
Browning, J. D., Szczepaniak, L. S., Dobbins, R., Nuremberg, P., Horton, J. D., Cohen, J. C., et al. (2004). Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40, 1387–1395.
Buechler, C., and Weiss, T. S. (2011). Does hepatic steatosis affect drug metabolizing enzymes in the liver? Curr. Drug Metab. 12, 24–34.
Caballero, F., Fernandez, A., De Lacy, A. M., Fernandez-Checa, J. C., Caballeria, J., and Garcia-Ruiz, C. (2009). Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 50, 789–796.
Calvert, V. S., Collantes, R., Elariny, H., Afendy, A., Baranova, A., Mendoza, M., et al. (2007). A systems biology approach to the pathogenesis of obesity-related nonalcoholic fatty liver disease using reverse phase protein microarrays for multiplexed cell signaling analysis. Hepatology 46, 166–172.
Caro, A. A., and Cederbaum, A. I. (2004). Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 44, 27–42.
Chalasani, N., Younossi, Z., Lavine, J. E., Diehl, A. M., Brunt, E. M., Cusi, K., et al. (2012). The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55, 2005–2023.
Chen, S., Li, Y., Li, S., and Yu, C. (2008a). A Val227Ala substitution in the peroxisome proliferator activated receptor alpha (PPAR alpha) gene associated with non-alcoholic fatty liver disease and decreased waist circumference and waist-to-hip ratio. J. Gastroenterol. Hepatol. 23, 1415–1418.
Chen, Z. W., Chen, L. Y., Dai, H. L., Chen, J. H., and Fang, L. Z. (2008b). Relationship between alanine aminotransferase levels and metabolic syndrome in nonalcoholic fatty liver disease. J. Zhejiang Univ. Sci. B. 9, 616–622.
Chen, Y., and Goldstein, J. A. (2009). The transcriptional regulation of the human CYP2C genes. Curr. Drug Metab. 10, 567–578.
Chowdhry, S., Nazmy, M. H., Meakin, P. J., Dinkova-Kostova, A. T., Walsh, S. V., Tsujita, T., et al. (2010). Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 48, 357–371.
Cohen, J. C., Horton, J. D., and Hobbs, H. H. (2011). Human fatty liver disease: old questions and new insights. Science 332, 1519–1523.
Crespo, J., Cayon, A., Fernandez-Gil, P., Hernandez-Guerra, M., Mayorga, M., Dominguez-Diez, A., et al. (2001). Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 34, 1158–1163.
Dahlman, I., Nilsson, M., Gu, H. F., Lecoeur, C., Efendic, S., Ostenson, C. G., et al. (2009). Functional and genetic analysis in type 2 diabetes of Liver X receptor alleles – a cohort study. BMC Med. Genet. 10:27. doi:10.1186/1471-2350-10-27
de Alwis, N. M., and Day, C. P. (2008). Non-alcoholic fatty liver disease: the mist gradually clears. J. Hepatol. 48(Suppl. 1), S104–S112.
di Masi, A., De Marinis, E., Ascenzi, P., and Marino, M. (2009). Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol. Aspects Med. 30, 297–343.
Donato, M. T., Jimenez, N., Serralta, A., Mir, J., Castell, J. V., and Gomez-Lechon, M. J. (2007). Effects of steatosis on drug-metabolizing capability of primary human hepatocytes. Toxicol. In vitro 21, 271–276.
Donato, M. T., Lahoz, A., Jimenez, N., Perez, G., Serralta, A., Mir, J., et al. (2006). Potential impact of steatosis on cytochrome P450 enzymes of human hepatocytes isolated from fatty liver grafts. Drug Metab. Dispos. 34, 1556–1562.
Dong, B., Saha, P. K., Huang, W. D., Chen, W. L., Abu-Elheiga, L. A., Wakil, S. J., et al. (2009). Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. U.S.A. 106, 18831–18836.
Dongiovanni, P., Valenti, L., Rametta, R., Daly, A. K., Nobili, V., Mozzi, E., et al. (2010). Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 59, 267–273.
Donnelly, K. L., Smith, C. I., Schwarzenberg, S. J., Jessurun, J., Boldt, M. D., and Parks, E. J. (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351.
Efanov, A. M., Sewing, S., Bokvist, K., and Gromada, J. (2004). Liver X receptor activation stimulates insulin secretion via modulation of glucose and lipid metabolism in pancreatic beta-cells. Diabetes 53, S75–S78.
Elens, L., Becker, M. L., Haufroid, V., Hofman, A., Visser, L. E., Uitterlinden, A. G., et al. (2011). Novel CYP3A4 intron 6 single nucleotide polymorphism is associated with simvastatin-mediated cholesterol reduction in the Rotterdam Study. Pharmacogenet Genomics 21, 861–866.
Fang, H. L., Strom, S. C., Cai, H., Falany, C. N., Kocarek, T. A., and Runge-Morris, M. (2005). Regulation of human hepatic hydroxysteroid sulfotransferase gene expression by the peroxisome proliferator-activated receptor alpha transcription factor. Mol. Pharmacol. 67, 1257–1267.
Faucette, S. R., Sueyoshi, T., Smith, C. M., Negishi, M., Lecluyse, E. L., and Wang, H. (2006). Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J. Pharmacol. Exp. Ther. 317, 1200–1209.
Feldstein, A. E., Werneburg, N. W., Canbay, A., Guicciardi, M. E., Bronk, S. F., Rydzewski, R., et al. (2004). Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40, 185–194.
Firneisz, G., Varga, T., Lengyel, G., Feher, J., Ghyczy, D., Wichmann, B., et al. (2010). Serum dipeptidyl peptidase-4 activity in insulin resistant patients with non-alcoholic fatty liver disease: a novel liver disease biomarker. PLoS ONE 5:e12226. doi:10.1371/journal.pone.0012226
Fisher, C. D., Lickteig, A. J., Augustine, L. M., Oude Elferink, R. P., Besselsen, D. G., Erickson, R. P., et al. (2009a). Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur. J. Pharmacol. 613, 119–127.
Fisher, C. D., Lickteig, A. J., Augustine, L. M., Ranger-Moore, J., Jackson, J. P., Ferguson, S. S., et al. (2009b). Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 37, 2087–2094.
Fon Tacer, K., and Rozman, D. (2011). Nonalcoholic fatty liver disease: focus on lipoprotein and lipid deregulation. J. Lipids 2011. doi: 10.1155/2011/783976
Gao, J., He, J. H., Zhai, Y. G., Wada, T. R., and Xie, W. (2009). The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 284, 25984–25992.
Gao, J., and Xie, W. (2010). Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab. Dispos. 38, 2091–2095.
Gao, J., and Xie, W. (2012). Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol. Sci. 33, 552–558.
Gavrilova, O., Haluzik, M., Matsusue, K., Cutson, J. J., Johnson, L., Dietz, K. R., et al. (2003). Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 278, 34268–34276.
Gawrieh, S., Baye, T. M., Carless, M., Wallace, J., Komorowski, R., Kleiner, D. E., et al. (2010). Hepatic gene networks in morbidly obese patients with nonalcoholic fatty liver disease. Obes. Surg. 20, 1698–1709.
Geier, A., Dietrich, C. G., Grote, T., Beuers, U., Prufer, T., Fraunberger, P., et al. (2005). Characterization of organic anion transporter regulation, glutathione metabolism and bile formation in the obese Zucker rat. J. Hepatol. 43, 1021–1030.
Gerbal-Chaloin, S., Daujat, M., Pascussi, J. M., Pichard-Garcia, L., Vilarem, M. J., and Maurel, P. (2002). Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J. Biol. Chem. 277, 209–217.
Gerin, I., Dolinsky, V. W., Shackman, J. G., Kennedy, R. T., Chiang, S. H., Burant, C. F., et al. (2005). LXR beta is required for adipocyte growth, glucose homeostasis, and beta cell function. J. Biol. Chem. 280, 23024–23031.
Ghose, R., Omoluabi, O., Gandhi, A., Shah, P., Strohacker, K., Carpenter, K. C., et al. (2011). Role of high-fat diet in regulation of gene expression of drug metabolizing enzymes and transporters. Life Sci. 89, 57–64.
Gnerre, C., Blättler, S., Kaufmann, M. R., Looser, R., and Meyer, U. A. (2004). Regulation of CYP3A4 by the bile acid receptor FXR: evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 14, 635–645.
Gomes, A. M., Winter, S., Klein, K., Turpeinen, M., Schaeffeler, E., Schwab, M., et al. (2009). Pharmacogenomics of human liver cytochrome P450 oxidoreductase: multifactorial analysis and impact on microsomal drug oxidation. Pharmacogenomics 10, 579–599.
Goodwin, B., Jones, S. A., Price, R. R., Watson, M. A., McKee, D. D., Moore, L. B., et al. (2000). A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6, 517–526.
Goodwin, B., Watson, M. A., Kim, H., Miao, J., Kemper, J. K., and Kliewer, S. A. (2003). Differential regulation of rat and human CYP7A1 by the nuclear oxysterol receptor liver X receptor-alpha. Mol. Endocrinol. 17, 386–394.
Gu, J., Weng, Y., Zhang, Q. Y., Cui, H., Behr, M., Wu, L., et al. (2003). Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 278, 25895–25901.
Hafner, M., Rezen, T., and Rozman, D. (2011). Regulation of hepatic cytochromes p450 by lipids and cholesterol. Curr. Drug Metab. 12, 173–185.
Handschin, C., and Meyer, U. A. (2005). Regulatory network of lipid-sensing nuclear receptors: roles for CAR, PXR, LXR, and FXR. Arch. Biochem. Biophys. 433, 387–396.
Hardwick, J. P., Osei-Hyiaman, D., Wiland, H., Abdelmegeed, M. A., and Song, B. J. (2009). PPAR/RXR regulation of fatty acid metabolism and fatty acid omega-hydroxylase (CYP4) isozymes: implications for prevention of lipotoxicity in fatty liver disease. PPAR Res. 2009, 952734.
Hardwick, R. N., Ferreira, D. W., More, V. R., Lake, A. D., Lu, Z., Manautou, J. E., et al. (2012a). Altered UDP-glucuronosyltransferase (UGT) and sulfotransferase (SULT) expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. doi:10.1124/dmd.112.048439
Hardwick, R. N., Fisher, C. D., Street, S. M., Canet, M. J., and Cherrington, N. J. (2012b). Molecular mechanism of altered ezetimibe disposition in nonalcoholic steatohepatitis. Drug Metab. Dispos. 40, 450–460.
Hardwick, R. N., Fisher, C. D., Canet, M. J., Lake, A. D., and Cherrington, N. J. (2010). Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 38, 2293–2301.
Hayes, J. D., Flanagan, J. U., and Jowsey, I. R. (2005). Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 45, 51–88.
Henao-Mejia, J., Elinav, E., Jin, C., Hao, L., Mehal, W. Z., Strowig, T., et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185.
Holt, J. A., Luo, G. Z., Billin, A. N., Bisi, J., McNeill, Y. Y., Kozarsky, K. F., et al. (2003). Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 17, 1581–1591.
Hori, M., Oniki, K., Nakagawa, T., Takata, K., Mihara, S., Marubayashi, T., et al. (2009). Association between combinations of glutathione-S-transferase M1, T1 and P1 genotypes and non-alcoholic fatty liver disease. Liver Int. 29, 164–168.
Hori, M., Oniki, K., Ueda, K., Goto, S., Mihara, S., Marubayashi, T., et al. (2007). Combined glutathione S-transferase T1 and M1 positive genotypes afford protection against type 2 diabetes in Japanese. Pharmacogenomics 8, 1307–1314.
Ip, E., Farrell, G. C., Robertson, G., Hall, P., Kirsch, R., and Leclercq, I. (2003). Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 38, 123–132.
Iwasaki, T., Tomeno, W., Yoneda, M., Inamori, M., Shirakawa, J., Imajo, K., et al. (2012). Non-alcoholic fatty liver disease adversely affects the glycemic control afforded by sitagliptin. Hepatogastroenterology 59, 1522–1525.
Jakobsson, T., Treuter, E., Gustafsson, J., and Steffensen, K. R. (2012). Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol. Sci. 33, 394–404.
Jemnitz, K., Veres, Z., Monostory, K., and Vereczkey, L. (2000). Glucuronidation of thyroxine in primary monolayer cultures of rat hepatocytes: in vitro induction of UDP-glucuronosyltranferases by methylcholanthrene, clofibrate, and dexamethasone alone and in combination. Drug Metab. Dispos. 28, 34–37. doi:10.1155/2011/783976
Kallwitz, E. R., McLachlan, A., and Cotler, S. J. (2008). Role of peroxisome proliferators-activated receptors in the pathogenesis and treatment of nonalcoholic fatty liver disease. World J. Gastroenterol. 14, 22–28.
Kathirvel, E., Chen, P., Morgan, K., French, S. W., and Morgan, T. R. (2010). Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. J. Gastroenterol. Hepatol. 25, 1136–1143.
Keber, R., Motaln, H., Wagner, K. D., Debeljak, N., Rassoulzadegan, M., Acimovic, J., et al. (2011). Mouse knockout of the cholesterogenic cytochrome P450 lanosterol 14alpha-demethylase (Cyp51) resembles Antley-Bixler syndrome. J. Biol. Chem. 286, 29086–29097.
Kersten, S., Seydoux, J., Peters, J. M., Gonzalez, F. J., Desvergne, B., and Wahli, W. (1999). Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498.
Kiba, T., Kintaka, Y., Suzuki, Y., Nakata, E., Ishigaki, Y., and Inoue, S. (2009). Ventromedial hypothalamic lesions change the expression of neuron-related genes and immune-related genes in rat liver. Neurosci. Lett. 455, 14–16.
Klein, K., Thomas, M., Winter, S., Nussler, A. K., Niemi, M., Schwab, M., et al. (2012). PPARA: a novel genetic determinant of CYP3A4 in vitro and in vivo. Clin. Pharmacol. Ther. 91, 1044–1052.
Kodama, S., Koike, C., Negishi, M., and Yamamoto, Y. (2004). Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol. Cell. Biol. 24, 7931–7940.
Kolwankar, D., Vuppalanchi, R., Ethell, B., Jones, D. R., Wrightow, S. A., Hall, S. D., et al. (2007). Association between nonalcoholic hepatic steatosis and hepatic cytochrome P-450 3A activity. Clin. Gastroenterol. Hepatol. 5, 388–393.
Kong, B., Luyendyk, J. P., Tawfik, O., and Guo, G. L. (2009). Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 328, 116–122.
Kumar, R., Rastogi, A., Maras, J. S., and Sarin, S. K. (2012). Unconjugated hyperbilirubinemia in patients with non-alcoholic fatty liver disease: a favorable endogenous response. Clin. Biochem. 45, 272–274.
Kwon do, Y., Jung, Y. S., Kim, S. J., Park, H. K., Park, J. H., and Kim, Y. C. (2009). Impaired sulfur-amino acid metabolism and oxidative stress in nonalcoholic fatty liver are alleviated by betaine supplementation in rats. J. Nutr. 139, 63–68.
Lake, A. D., Novak, P., Fisher, C. D., Jackson, J. P., Hardwick, R. N., Billheimer, D. D., et al. (2011). Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 39, 1954–1960.
Leclercq, I. A., Farrell, G. C., Field, J., Bell, D. R., Gonzalez, F. J., and Robertson, G. R. (2000). CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Invest. 105, 1067–1075.
Lee, J. H., Zhou, J., and Xie, W. (2008). PXR and LXR in hepatic steatosis: a new dog and an old dog with new tricks. Mol. Pharm. 5, 60–66.
Leite, N. C., Salles, G. F., Araujo, A. L., Villela-Nogueira, C. A., and Cardoso, C. R. (2009). Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 29, 113–119.
Lewis, J. R., and Mohanty, S. R. (2010). Nonalcoholic fatty liver disease: a review and update. Dig. Dis. Sci. 55, 560–578.
Li, J., Huang, J., Li, J. S., Chen, H., Huang, K., and Zheng, L. (2011). Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J. Hepatol. 56, 900–907.
Lickteig, A. J., Fisher, C. D., Augustine, L. M., and Cherrington, N. J. (2007). Genes of the antioxidant response undergo upregulation in a rodent model of nonalcoholic steatohepatitis. J. Biochem. Mol. Toxicol. 21, 216–220.
Lin, Y. C., Chang, P. F., Hu, F. C., Chang, M. H., and Ni, Y. H. (2009). Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics 124, e1221–e1227.
Lorbek, G., and Rozman, D. (2012). “Cholesterol and inflammation at the crossroads of non-alcoholic fatty liver disease (NAFLD) and atherogenesis,” in Atherogenesis, ed. S. Parthasarathy (Rijeka: InTech), 281–304.
Machado, M., Marques-Vidal, P., and Cortez-Pinto, H. (2006). Hepatic histology in obese patients undergoing bariatric surgery. J. Hepatol. 45, 600–606.
Marek, C. J., Tucker, S. J., Konstantinou, D. K., Elrick, L. J., Haefner, D., Sigalas, C., Murray, G.I., et al. (2005). Pregnenolone-16alpha-carbonitrile inhibits rodent liver fibrogenesis via PXR (pregnane X receptor)-dependent and PXR-independent mechanisms. Biochem. J. 387, 601–608.
Maroni, L., Guasti, L., Castiglioni, L., Marino, F., Contini, S., Macchi, V., et al. (2011). Lipid targets during statin treatment in dyslipidemic patients affected by nonalcoholic fatty liver disease. Am. J. Med. Sci. 342, 383–387.
Martin, I. V., Schmitt, J., Minkenberg, A., Mertens, J. C., Stieger, B., Mullhaupt, B., et al. (2010). Bile acid retention and activation of endogenous hepatic farnesoid-X-receptor in the pathogenesis of fatty liver disease in ob/ob-mice. Biol. Chem. 391, 1441–1449.
Marzolini, C., Tirona, R. G., Gervasini, G., Poonkuzhali, B., Assem, M., Lee, W., et al. (2007). A common polymorphism in the bile acid receptor farnesoid X receptor is associated with decreased hepatic target gene expression. Mol. Endocrinol. 21, 1769–1780.
Masson, D., Qatanani, M., Sberna, A. L., Xiao, R., De Barros, J. P. P., Grober, J., et al. (2008). Activation of the constitutive androstane receptor decreases HDL in wild-type and human apoA-I transgenic mice. J. Lipid Res. 49, 1682–1691.
Mitsuyoshi, H., Yasui, K., Harano, Y., Endo, M., Tsuji, K., Minami, M., et al. (2009). Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol. Res. 39, 366–373.
Moore, L. B., Parks, D. J., Jones, S. A., Bledsoe, R. K., Consler, T. G., Stimmel, J. B., et al. (2000). Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 275, 15122–15127.
Neuschwander-Tetri, B.A., and Caldwell, S. H. (2003). Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology 37, 1202–1219.
Nielsen, R., Pedersen, T. A., Hagenbeek, D., Moulos, P., Siersbaek, R., Megens, E., et al. (2008). Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 22, 2953–2967.
Osabe, M., Sugatani, J., Fukuyama, T., Ikushiro, S., Ikari, A., and Miwa, M. (2008). Expression of hepatic UDP-glucuronosyltransferase 1A1 and 1A6 correlated with increased expression of the nuclear constitutive androstane receptor and peroxisome proliferator-activated receptor alpha in male rats fed a high-fat and high-sucrose diet. Drug Metab. Dispos. 36, 294–302.
Pettinelli, P., and Videla, L. A. (2011). Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 96, 1424–1430.
Plass, J. R. M., Mol, O., Heegsma, J., Geuken, M., Faber, K. N., Jansen, P. L. M., et al. (2002). Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 35, 589–596.
Pokreisz, P., Fleming, I., Kiss, L., Barbosa-Sicard, E., Fisslthaler, B., Falck, J. R., et al. (2006). Cytochrome P450 epoxygenase gene function in hypoxic pulmonary vasoconstriction and pulmonary vascular remodeling. Hypertension 47, 762–770.
Pyper, S. R., Viswakarma, N., Yu, S., and Reddy, J. K. (2010). PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 8, e002.
Rakhshandehroo, M., Hooiveld, G., Müller, M., and Kersten, S. (2009). Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 4:e6796. doi:10.1371/journal.pone.0006796
Rakhshandehroo, M., Knoch, B., Muller, M., and Kersten, S. (2010). Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010. doi: 10.1155/2010/612089
Ratziu, V., Giral, P., Jacqueminet, S., Charlotte, F., Hartemann-Heurtier, A., Serfaty, L., et al. (2008). Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled fatty liver improvement with rosiglitazone therapy (FLIRT) trial. Gastroenterology 135, 100–110.
Ratziu, V., Voiculescu, M., and Poynard, T. (2011). Touching some firm ground in the epidemiology of NASH. J. Hepatol. 56, 23–25.
Repa, J. J., Berge, K. E., Pomajzl, C., Richardson, J. A., Hobbs, H., and Mangelsdorf, D. J. (2002). Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 277, 18793–18800.
Rezen, T. (2011). The impact of cholesterol and its metabolites on drug metabolism. Expert Opin. Drug Metab. Toxicol. 7, 387–398.
Rezen, T., Juvan, P., Fon Tacer, K., Kuzman, D., Roth, A., Pompon, D., et al. (2008). The sterolgene v0 cDNA microarray: a systemic approach to studies of cholesterol homeostasis and drug metabolism. BMC Genomics 9:76. doi:10.1186/1471-2164-9-76
Rezen, T., Rozman, D., Pascussi, J. M., and Monostory, K. (2011). Interplay between cholesterol and drug metabolism. Biochim. Biophys. Acta 1814, 146–160.
Rezen, T., Viola, T., Lovgren-Sandblom, A., Bjorkhem, I., Meyer, U. A., and Rozman, D. (2009). Effect of CAR activation on selected metabolic pathways in normal and hyperlipidemic mouse livers. BMC Genomics 10:384. doi:10.1186/1471-2164-10-384
Richert, L., Lamboley, C., Viollon-Abadie, C., Grass, P., Hartmann, N., Laurent, S., et al. (2003). Effects of clofibric acid on mRNA expression profiles in primary cultures of rat, mouse and human hepatocytes. Toxicol. Appl. Pharmacol. 191, 130–146.
Rinella, M. E., Elias, M. S., Smolak, R. R., Fu, T., Borensztajn, J., and Green, R. M. (2008). Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 49, 1068–1076.
Roberts, E. A. (2007). Pediatric nonalcoholic fatty liver disease (NAFLD): a “growing” problem? J. Hepatol. 46, 1133–1142.
Robertson, G., Leclercq, I., and Farrell, G. C. (2001). Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G1135–G1139.
Roth, A., Looser, R., Kaufmann, M., and Meyer, U. A. (2008). Sterol regulatory element binding protein 1 interacts with pregnane X receptor and constitutive androstane receptor and represses their target genes. Pharmacogenet. Genomics 18, 325–337.
Rozman, D., and Monostory, K. (2010). Perspectives of the non-statin hypolipidemic agents. Pharmacol. Ther. 127, 19–40.
Runge-Morris, M., and Kocarek, T. A. (2009). Regulation of sulfotransferase and UDP-glucuronosyltransferase gene expression by the PPARs. PPAR Res. 2009, 728941.
Sabol, S. L., Brewer, H. B., and Santamarina-Fojo, S. (2005). The human ABCG1 gene: identification of LXR response elements that modulate expression in macrophages and liver. J. Lipid Res. 46, 2151–2167.
Schwartz, K., Lawn, R. M., and Wade, D. P. (2000). ABC1 gene expression and ApoA-I-mediated cholesterol efflux are regulated by LXR. Biochem. Biophys. Res. Commun. 274, 794–802.
Senekeo-Effenberger, K., Chen, S., Brace-Sinnokrak, E., Bonzo, J. A., Yueh, M. F., Argikar, U., et al. (2007). Expression of the human UGT1 locus in transgenic mice by 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (WY-14643) and implications on drug metabolism through peroxisome proliferator-activated receptor alpha activation. Drug Metab. Dispos. 35, 419–427.
Sirtori, C. R., and Franceschini, G. (1988). Effects of fibrates on serum lipids and atherosclerosis. Pharmacol. Ther. 37, 167–191.
Solaas, K., Legry, V., Retterstol, K., Berg, P. R., Holven, K. B., Ferrieres, J., et al. (2010). Suggestive evidence of associations between liver X receptor beta polymorphisms with type 2 diabetes mellitus and obesity in three cohort studies: HUNT2 (Norway), MONICA (France) and HELENA (Europe). BMC Med. Genet. 11:144. doi:10.1186/1471-2350-11-144
Song, X. L., Kaimal, R., Yan, B. F., and Deng, R. T. (2008). Liver receptor homolog 1 transcriptionally regulates human bile salt export pump expression. J. Lipid Res. 49, 973–984.
Sookoian, S., Castano, G., Gianotti, T. F., Gemma, C., and Pirola, C. J. (2009). Polymorphisms of MRP2 (ABCC2) are associated with susceptibility to nonalcoholic fatty liver disease. J. Nutr. Biochem. 20, 765–770.
Sookoian, S., Castano, G. O., Burgueno, A. L., Gianotti, T. F., Rosselli, M. S., and Pirola, C. J. (2010). The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease. Pharmacogenet. Genomics 20, 1–8.
Stepanova, M., Hossain, N., Afendy, A., Perry, K., Goodman, Z. D., Baranova, A., et al. (2010). Hepatic gene expression of Caucasian and African-American patients with obesity-related non-alcoholic fatty liver disease. Obes. Surg. 20, 640–650.
Sueyoshi, T., and Negishi, M. (2001). Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu. Rev. Pharmacol. Toxicol. 41, 123–143.
Sugatani, J., Nishitani, S., Yamakawa, K., Yoshinari, K., Sueyoshi, T., Negishi, M., et al. (2005). Transcriptional regulation of human UGT1A1 gene expression: activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol. Pharmacol. 67, 845–855.
Sunny, N. E., Parks, E. J., Browning, J. D., and Burgess, S. C. (2011). Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 14, 804–810.
Tacer, K. F., Kuzman, D., Seliskar, M., Pompon, D., and Rozman, D. (2007). TNF-alpha interferes with lipid homeostasis and activates acute and proatherogenic processes. Physiol. Genomics 31, 216–227.
Takizawa, D., Kakizaki, S., Horiguchi, N., Yamazaki, Y., Tojima, H., and Mori, M. (2011). Constitutive active/androstane receptor promotes hepatocarcinogenesis in a mouse model of non-alcoholic steatohepatitis. Carcinogenesis 32, 576–583.
Targher, G., Bertolini, L., Padovani, R., Rodella, S., Zoppini, G., Zenari, L., et al. (2006). Relations Between carotid artery wall thickness and liver histology in subjects with nonalcoholic fatty liver disease. Diabetes Care 29, 1325–1330.
Tolson, A. H., and Wang, H. B. (2010). Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 62, 1238–1249.
Tontonoz, P., Nagy, L., Alvarez, J. G., Thomazy, V. A., and Evans, R. M. (1998). PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93, 241–252.
van der Meer, D. L., Degenhardt, T., Väisänen, S., De Groot, P. J., Heinäniemi, M., De Vries, S. C., et al. (2010). Profiling of promoter occupancy by PPARalpha in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res. 38, 2839–2850.
Varela, N. M., Quinones, L. A., Orellana, M., Poniachik, J., Csendes, A., Smok, G., et al. (2008). Study of cytochrome P450 2E1 and its allele variants in liver injury of nondiabetic, nonalcoholic steatohepatitis obese women. Biol. Res. 41, 81–92.
Vernon, G., Baranova, A., and Younossi, Z. M. (2011). Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 34, 274–285.
Videla, L. A., Rodrigo, R., Orellana, M., Fernandez, V., Tapia, G., Quinones, L., et al. (2004). Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 106, 261–268.
Vollrath, V., Wielandt, A. M., Iruretagoyena, M., and Chianale, J. (2006). Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 395, 599–609.
Wada, T., Gao, J., and Xie, W. (2009). PXR and CAR in energy metabolism. Trends Endocrinol. Metabol. 20, 273–279.
Wagenknecht, L. E., Scherzinger, A. L., Stamm, E. R., Hanley, A. J., Norris, J. M., Chen, Y. D., et al. (2009). Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity (Silver Spring) 17, 1240–1246.
Wang, D., Guo, Y., Wrighton, S. A., Cooke, G. E., and Sadee, W. (2011). Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 11, 274–286.
Wells, P. G., Mackenzie, P. I., Chowdhury, J. R., Guillemette, C., Gregory, P. A., Ishii, Y., et al. (2004). Glucuronidation and the UDP-glucuronosyltransferases in health and disease. Drug Metab. Dispos. 32, 281–290.
Weltman, M. D., Farrell, G. C., Hall, P., Ingelman-Sundberg, M., and Liddle, C. (1998). Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 27, 128–133.
Weltman, M. D., Farrell, G. C., and Liddle, C. (1996). Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 111, 1645–1653.
Wolbold, R., Klein, K., Burk, O., Nussler, A. K., Neuhaus, P., Eichelbaum, M., et al. (2003). Sex is a major determinant of CYP3A4 expression in human liver. Hepatology 38, 978–988.
Wree, A., Kahraman, A., Gerken, G., and Canbay, A. (2011). Obesity affects the liver – the link between adipocytes and hepatocytes. Digestion 83, 124–133.
Wu, K. C., Cui, J. Y., and Klaassen, C. D. (2012). Effect of graded Nrf2 activation on phase-I and -II drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 7:e39006. doi:10.1371/journal.pone.0039006
Xie, W., Radominska-Pandya, A., Shi, Y., Simon, C. M., Nelson, M. C., Ong, E. S., et al. (2001). An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. U.S.A. 98, 3375–3380.
Yamaguchi, K., Yang, L., McCall, S., Huang, J., Yu, X. X., Pandey, S. K., et al. (2007). Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45, 1366–1374.
Yamazaki, Y., Kakizaki, S., Horiguchi, N., Sohara, N., Sato, K., Takagi, H., et al. (2007). The role of the nuclear receptor constitutive androstane receptor in the pathogenesis of non-alcoholic steatohepatitis. Gut 56, 565–574.
Younossi, Z. M., Baranova, A., Ziegler, K., Del Giacco, L., Schlauch, K., Born, T. L., et al. (2005). A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology 42, 665–674.
Zhang, B., Xie, W., and Krasowski, M. D. (2008). PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 9, 1695–1709.
Zheng, H., Li, S., Ma, L., Cheng, L., Deng, C., Chen, Z., et al. (2011). A novel agonist of PPAR-gamma based on barbituric acid alleviates the development of non-alcoholic fatty liver disease by regulating adipocytokine expression and preventing insulin resistance. Eur. J. Pharmacol. 659, 244–251.
Zhou, C., Tabb, M. M., Nelson, E. L., Grun, F., Verma, S., Sadatrafiei, A., et al. (2006a). Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Invest. 116, 2280–2289.
Zhou, J., Zhai, Y., Mu, Y., Gong, H., Uppal, H., Toma, D., et al. (2006b). A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 281, 15013–15020.
Keywords: NAFLD, xenobiotic metabolism, nuclear receptors, phase I and II enzymes, transporters
Citation: Naik A, Belič A, Zanger UM and Rozman D (2013) Molecular interactions between NAFLD and xenobiotic metabolism. Front. Gene. 4:2. doi: 10.3389/fgene.2013.00002
Received: 15 October 2012; Accepted: 03 January 2013;
Published online: 22 January 2013.
Edited by:
José A. Agúndez, University of Extremadura, SpainReviewed by:
Erik Eliasson, Karolinska Institute, SwedenUrs A. Meyer, University of Basel, Switzerland
Copyright: © 2013 Naik, Belič, Zanger and Rozman. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Damjana Rozman, Centre for Functional Genomics and Bio-Chips, Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Zaloska 4, SI-1000 Ljubljana, Slovenia. e-mail:ZGFtamFuYS5yb3ptYW5AbWYudW5pLWxqLnNp