
- Department of Neuroscience and Ophthalmology, LSU Neuroscience Center, Louisiana State University Health Sciences Center, New Orleans, LA, USA
As research into micro RNA (miRNA) speciation, complexity, and biological activity progresses, it is becoming clear that a selective subset of all so far characterized miRNAs utilized by human cells and tissues is under the transcriptional control of the pro-inflammatory and immune system-linked transcription factor NF-κB (Sen and Baltimore, 1986; Lukiw and Bazan, 1998; Taganov et al., 2006; Lukiw, 2007, 2012a,b,c; Bazzoni et al., 2009; Ma et al., 2011; Boldin and Baltimore, 2012; Cremer et al., 2012; Li et al., 2012; Lukiw and Alexandrov, 2012; Zhao et al., 2013). The inducible up-regulation of NF-κ B-sensitive miRNAs is by virtue of single, and often multiple, NF-κB-DNA binding recognition sites in the regulatory regions of miRNA-containing genes that drive RNA polymerase II-mediated transcription of pre-miRNA species (Ambros, 2004; Taganov et al., 2006; Baltimore et al., 2008; Cui et al., 2010; Guo et al., 2010; Bredy et al., 2011; Lukiw, 2012a). In neurodegenerative disease research, stress-triggered up-regulation of these NF-κ B-induced miRNAs appear to be playing pathogenic roles in the down-regulation of brain-essential messenger RNAs (mRNA), and the initiation and propagation of pathological gene expression programs that are, for example, characteristic of the Alzheimer's disease (AD) process (Figure 1). NF-κB-mediated up-regulated miRNAs and down-regulated mRNA targets thereby form a highly integrated, pathogenic NF-κ B-miRNA-mRNA signaling network that can explain much of the observed neuropathology in AD, including deficits in phagocytosis (Niemitz, 2012; Zhao et al., 2013), NF-κ B-mediated innate-immune signaling and chronic inflammation (Cui et al., 2010; Heneka et al., 2010; Lukiw and Bazan, 2010; Lukiw et al., 2012), impairments in neurotransmitter packaging and release, neurotrophism and amyloidogenesis (Xu et al., 2009; Lukiw, 2012a,b,c). Under homeostatic conditions, NF-κ B activation involves a coordinated, sequential, and self-limiting sequence of events controlled by positive and negative regulatory mechanisms, however, this does not appear to be the case in early, moderate or especially advanced stages of sporadic AD. In sporadic AD, once initiated, NF-κ B-mediated disruption of homeostatic gene expression can be self-perpetuating due, in part, to the chronic re-activation of NF-κ B activities via up-regulation of interleukin-1β receptor associated kinase-2 (IRAK-2) signaling pathways (Cui et al., 2010). Selective inhibition of the actions of NF-κ B and specific NF-κ B-sensitive miRNAs therefore seems a plausible therapeutic strategy towards neutralizing their combined effects in sporadic AD, and related progressive, age-related neurological diseases with an innate-immune and inflammatory component.
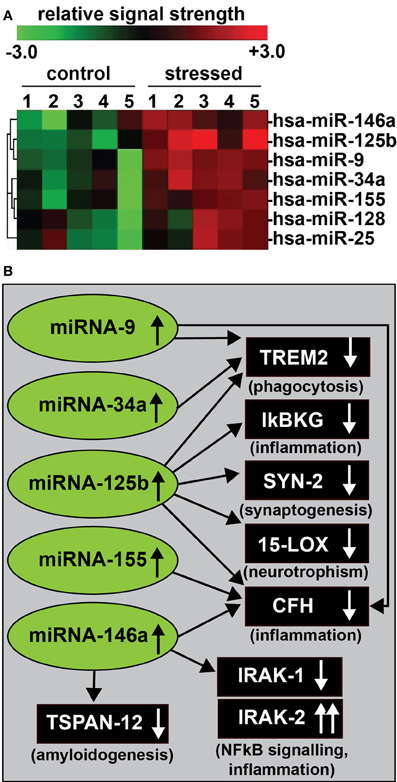
Figure 1. (A) In recent experiments human primary neuronal-glial (HNG) cells were treated with an AD-relevant, NF-κ B-inducing cocktail of amyloid beta 42 (Aβ 42) and interleukin-1beta (IL-1β), and inducible miRNAs were analyzed using miRNA arrays (Lukiw, 2012a). Confirmation of miRNA induction and NF-κ B sensitivity was obtained (1) using LED-Northern dot blot and/or RT-PCR analysis; (2) by inhibition of this induction using specific NF-κ B inhibitors CAPE, CAY10512, and PDTC and (3) by bioinformatics analysis of functional NF-κ B binding sites in the promoters of the genes that encode these inducible miRNAs (Lukiw et al., 2008; Cui et al., 2010; Lukiw, 2012a). A small family of 5 miRNAs—miRNA-9, miRNA-34a, miRNA-125b, miRNA-146a, and miRNA-155—appear to be up-regulated in high quality total RNA isolated from short post-mortem AD brains; note that hsa-miR-128 and miR-25 are variably up-regulated; N = 6; (B) These findings in part define a highly interactive network of NF-κ B-sensitive, up-regulated miRNAs in stressed human brain cells and AD hippocampus that can explain much of the observed neuropathology in AD including deficits in phagocytosis (TREM2), innate-immune signaling and chronic inflammation (IkBKG, CFH, IRAK-1, and IRAK-2), impairments in neurotransmitter packaging and release (SYN-2), neurotrophism (15-LOX), and amyloidogenesis (TSPAN12) (see references in text); these up-regulated miRNAs and down-regulated mRNAs form a highly integrated, pathogenic miRNA-mRNA signaling network resulting in gene expression deficits in sporadic AD that may be self-perpetuating due to chronic re-activation of NF-κB stimulation via IRAK-2 pathways (Cui et al., 2010; Lukiw, 2012a,b,c). Inhibition of the NF-κB initiator or individual blocking of the pathogenic induction of these five miRNAs may provide novel therapeutic approaches for the clinical management of AD, however, what NF-κ B or miRNA inhibition strategies, or whether they can be utilized either alone or in combination, remain open to question. Preliminary data has indicated that these approaches may neutralize this chronic, inducible, progressive pathogenic gene expression program to re-establish brain cell homeostasis, and ultimately be of novel pharmacological use in the clinical management of AD.
The brain-ubiquitous transcription factor NF-κ B comprises a family of heterodimeric DNA-binding proteins, for example the relatively common p50–p65 complex, that normally reside in a “resting state” in the cytoplasm (Sen and Baltimore, 1986; Lukiw and Bazan, 1998; Mattson and Camandola, 2001; Yamamoto and Gaynor, 2001; Lukiw, 2012a,b). In general, cytoplasmic NF-κ B activation is stimulated via a wide array of physiological stressors including ionizing radiation, viral infection, neurotoxic metals, elevations in reactive oxygen species (ROS), inflammatory cytokines and chemokines, Aβ 42 peptides, hypoxia, and other forms of physiological stress (Baltimore et al., 2008; Pogue et al., 2009, 2011; Boldin and Baltimore, 2012). This activation is largely mediated by the serine- phosphorylation of a family of NF-κB inhibitory subunits, non-covalently bound to the NF-κ B heterodimer, and collectively known as the inhibitors of kappaB (Iκ B). Iκ B phosphorylation is followed, in turn, by Iκ B degeneration, in part through cooperation with NF-κ B essential modulator (NEMO) proteins (Baltimore et al., 2008; Shifera, 2010). NF-κ B heterodimers next translocate to the nucleus to target binding sites homologous to the canonical DNA sequence 5′-GGGACTTTCC-3′ in the regulatory regions of NF-κ B-sensitive genes (Meffert and Baltimore, 2005; Wong et al., 2011). Ever since NF-κ B's original description in 1986 several hundred NF-κ B inhibitors, both natural and synthetic, have been developed to inhibit NF-κ B-activation in this complex signaling network (Sen and Baltimore, 1986; Mattson and Camandola, 2001; Aggarwal and Shishodia, 2004; Meffert and Baltimore, 2005; Gilmore and Herscovitch, 2006; Nam, 2006; Gilmore and Garbati, 2011). Indeed, inhibition of NF-κ B can involve multifaceted aspects of NF-κ B activation via blocking of NF-κB signaling at various control points, from inhibition of the kinases that phosphorylate Iκ B (thus preventing activation of NF-κ B), to the deacetylation of the NF-κ B p65 subunit, to the proteasome-mediated degradation of the Iκ B, to the translocation of NF-κ B to the nucleus, to the prevention of NF-κ B binding to DNA recognition sites using decoy and/or antisense strategies (Meffert and Baltimore, 2005; Gilmore and Herscovitch, 2006; Nam, 2006; Yang et al., 2007; Gilmore and Garbati, 2011; Lukiw et al., 2012). Importantly, constitutive NF-κ B signaling is critically homeostatic to many aspects of normal brain function, and essential to the control of cell proliferation, apoptosis, innate, and adaptive immunity, the inflammatory response and related stress responses. NF-κ B activation is also an important part of a cellular recovery process that may protect brain cells against oxidative-stress or brain trauma-induced apoptosis and induced neurodegeneration, hence antagonism of NF-κ B may reduce its intrinsic potential for neuroprotective activity (Lukiw and Bazan, 1998; Yang et al., 2007; Lukiw, 2012a,b,c). Since NF-κ B may have conflicting effects in different brain cell types it is important to assess anti-NF-κ B (or anti-miRNA) inhibition strategies in individual cell types that constitute susceptible tissues. For example, pro-inflammatory NF-κ B-sensitive miRNAs such as miRNA-146a have been shown to be of varying basal abundance, and be variably induced, in different human primary brain and retinal cell types (Alexandrov et al., 2011; Li et al., 2011). Another interesting example of interactive NF-κ B and miRNA effects are the differential regulation of the interleukin-1 receptor associated kinases IRAK-1 and IRAK-2 through NF-κ B actions (Cui et al., 2010; Flannery and Bowie, 2010). In the regulation of IRAK-1 expression, NF-κ B has been shown to induce miRNA-146a which, by virtue of a miRNA-146a recognition feature in the IRAK-1 3′-UTR, down-regulates IRAK-1 expression while at the same time activating transcription of IRAK-2, by virtue of NF-κ B binding in the IRAK-2 upstream 5′ regulatory region (Cui et al., 2010). Importantly the IRAK-1 gene is devoid of NF-κ B recognition features in its 5′ regulatory region, while the IRAK-2 3′-UTR has no recognition feature for miRNA-146a. Hence anti-NF-κ B strategies may affect the pleiotrophic regulation of expression of even highly homologous genes, such as IRAK-1 and IRAK-2 that function in the innate immune response, the sensing of pathogens and the initiation of immunity.
An analogous example of the perceived perils of wide-spectrum NF-κ B inhibition can be taken from neurooncology, and the broad therapeutic strategies of using the common alkylating anti-neoplastic drug temozolamide (TMZ). TMZ attaches an alkyl group to the guanine base of DNA, resulting in base pair stabilization or cross-linking, the inhibition of cell growth and stimulation of apoptosis, and has been widely used to treat glioblastoma and other cancers (Thomas et al., 2013). As the guanines of all genomic DNA are susceptible to alkylation, the use of TMZ is tolerated because high rates of cell proliferation renders cancer cells more sensitive to alkyl modification. However, the guanines of other cell DNAs that are naturally rapidly dividing may also be alkylated leading to off-target neurotoxic and neurological effects (Lai et al., 2008; Nagasawa et al., 2012; Thomas et al., 2013). Similarly, global anti-NF-κ B therapeutic strategies may be more effective when NF-κ B is widely over stimulated in highly progressive neuropathological situations such as AD (Lukiw and Bazan, 1998; Yang et al., 2007; Lukiw, 2013). Put another way, NF-κB-inhibition strategies may be maximized only when NF-κ B stimulated effects have surpassed a critical threshold that are en masse seriously detrimental to the normal homeostatic function of brain cells and tissues. However, it is still problematic and pharmacologically difficult to minimize the complicating effects of NF-κ B inhibition on essential, low-level homeostatic NF-κ B function (Nam, 2006; Lukiw, 2012b).
On the other hand, anti-miRNA (antagomir, AM) approaches are much more target-efficient and a stabilized anti-ribonucleotide, anti-miRNA sequence 100% homologous, and complementary to the target miRNA will inhibit that target miRNA 95% or more with extremely high efficacy (Lukiw et al., 2008; Cui et al., 2010). Interestingly, tailoring imperfections into base-pair complementarity between the anti-miRNA ribonucleotides and the target miRNA could result in partial inhibition of the miRNA with a tailored “throttle-down” of miRNA-mediated effects, and could be used as a therapeutic strategy of “variable efficacy.” It is clear that the appropriate clinical application of these anti-NF-κ B or anti-miRNA strategies requires a more intimate understanding of the specific NF-κ B and miRNA mechanisms responsible for their activation pathway in different brain cell types, definition of the optimal point of intervention in the NF-κ B or miRNA activation pathway, and the characterization of potent and specific inhibitors of the chosen NF-κ B or miRNA targets. Another formidable challenge lies in the implementation of innovative anti-NF-κ B and anti-miRNA strategies without incurring prohibitive off-target toxicity from combinatorial approaches. Indeed, as has been found from cancer research, rigorous anti-NF-κ B strategies may have long term health consequences and appropriate cautions should be exercised (Gilmore and Herscovitch, 2006; Gilmore and Garbati, 2011; Nagasawa et al., 2012; Thomas et al., 2013).
Lastly, virtually each day is now providing increased insight into the induction of NF-κ B and miRNA in response to physiological stressors, including the mechanisms that regulate the bioavailability of pro-inflammatory transcription factors and miRNAs in immune health and disease. The inducers of NF-κ B also appear to be the inducers of a highly selective sub-family of NF-κ B-sensitive miRNAs (Lukiw, 2012a,b,c). These in turn drive the expression of a characteristic AD phenotype that includes an up-regulation in inflammatory signaling, amyloidogenesis, impairment in microglial cell-mediated phagocytosis, and deficits in synaptic and neurotrophic signaling (Figure 1). Because NF-κ B can variably regulate gene expression directly by transcriptional activation and indirectly by miRNA-mediated post-transcriptional down-regulation it will be important to further map out specific NF-κ B-miRNA-mRNA signaling pathways to ascertain at what point anti-NF-κ B or anti-miRNA-based strategies may be best prescribed to fit the individual pathological situation. While NF-κ B up-regulation helps define a subset of inducible NF-κ B-sensitive pre-miRNA genes, there may be other transcription factors, or combinations of transcription factors that define the induction of other small families of pre-miRNAs. Hence a general rule to follow may be that a highly interactive network of NF-κ B or other transcription factor-sensitive up-regulated miRNAs in stressed human brain cells define an epigenetic expression program that is amenable to strategic manipulation at some point, to eventually normalize the dynamic balance between inducible neurodegeneration or cell survival signals.
Acknowledgments
Thanks are extended to Drs. C. Eicken, P. Dua, and C. Hebel for miRNA array work and initial data interpretation, and to D. Guillot and A. I. Pogue for expert technical assistance. Research on miRNA in the Lukiw laboratory involving metal neurotoxicity, the innate-immune response in AD, amyloidogenesis and neuroinflammation was supported through an Alzheimer Association Investigator-Initiated Research Grant IIRG-09-131729 and NIH NIA Grants AG18031 and AG038834.
References
Aggarwal, B. B., and Shishodia, S. (2004). Suppression of the nuclear factor-kB activation pathway by spice-derived phytochemicals. Ann. N.Y. Acad. Sci. 1030, 434–441.
Alexandrov, P. N., Pogue, A., Bhattacharjee, S., and Lukiw, W. J. (2011). Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer's disease. Neuroreport 22, 623–627.
Baltimore, D., Boldin, M. P., O'Connell, R. M., Rao, D. S., and Taganov, K. D. (2008). MicroRNAs: new regulators of immune cell development and function. Nat. Immunol. 9, 839–845.
Bazzoni, F., Rossato, M., Fabbri, M., Gaudiosi, D., Mirolo, M., Mori, L., et al. (2009). Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. U.S.A. 106, 5282–5287.
Boldin, M. P., and Baltimore, D. (2012). MicroRNAs, new effectors and regulators of NF-κ B. Immunol. Rev. 246, 205–220.
Bredy, T. W., Lin, Q., Wei, W., Baker-Andresen, D., and Mattick, J. S. (2011). MicroRNA regulation of neural plasticity and memory. Neurobiol. Learn Mem. 96, 89–94.
Cui, J. G., Li, Y. Y., Zhao, Y., Bhattacharjee, S., and Lukiw, W. J. (2010). Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by miRNA-146a and NF-κB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 285, 38951–38960.
Cremer, T. J., Fatehchand, K., Shah, P., Gillette, D., Patel, H., Marsh, R. L., et al. (2012). miR-155 induction by microbes/microbial ligands requires NF-κ B-dependent de novo protein synthesis. Front. Cell Infect. Microbiol. 2:73. doi: 10.3389/fcimb.2012.00073
Flannery, S., and Bowie, A. G. (2010). The interleukin-1 receptor-associated kinases: critical regulators of innate immune signaling. Biochem. Pharmacol. 80, 1981–1991.
Gilmore, T. D., and Garbati, M. R. (2011). Inhibition of NF-κ B signaling as a strategy in disease therapy. Curr. Top. Microbiol. Immunol. 349, 245–263.
Gilmore, T. D., and Herscovitch, M. (2006). Inhibitors of NF-κB signaling: 785 and counting. Oncogene 25, 6887–6899.
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840.
Heneka, M. T., O'Banion, M. K., Terwel, D., and Kummer, M. P. (2010). Neuroinflammatory processes in Alzheimer's disease. J. Neural. Transm. 117, 919–947.
Lai, A., Filka, E., McGibbon, B., Nghiemphu, P. L., Graham, C., Yong, W. H., et al. (2008). Phase II pilot study of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme: interim analysis of safety and tolerability. Int. J. Radiat. Oncol. Biol. Phys. 71, 1372–1380.
Li, J., Wang, K., Chen, X., Meng, H., Song, M., Wang, Y., et al. (2012). Transcriptional activation of microRNA-34a by NF-κB in human esophageal cancer cells. BMC. Mol. Biol. 13:4. doi: 10.1186/1471-2199-13-4
Li, Y. Y., Cui, J. G., Dua, P., Pogue, A. I., Bhattacharjee, S., and Lukiw, W. J. (2011). Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 499, 109–113.
Lukiw, W. J. (2007). Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport 18, 297–300.
Lukiw, W. J. (2008). Emerging amyloid beta (Aβ) peptide modulators for the treatment of Alzheimer's disease (AD). Expert Opin. Emerg. Drugs 13, 255–271.
Lukiw, W. J. (2012a). NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp. Neurol. 235, 484–490.
Lukiw, W. J. (2012b). NF-κ B-regulated, proinflammatory miRNAs in Alzheimer's disease. Alzheimers Res. Ther. 4, 47–55.
Lukiw, W. J. (2012c). Evolution and complexity of micro RNA in the human brain. Front Genet. 3:166. doi: 10.3389/fgene.2012.00166
Lukiw, W. J. (2013). Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer's disease (AD). Expert Opin. Emerg. Drugs. doi: 10.1517/14728214.2012.672559. [Epub ahead of print].
Lukiw, W. J., and Alexandrov, P. N. (2012). Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer's disease (AD) brain. Mol. Neurobiol. 46, 11–19.
Lukiw, W. J., Alexandrov, P. N., Zhao, Y., Hill, J. M., and Bhattacharjee, S. (2012). Spreading of Alzheimer's disease inflammatory signaling through soluble micro-RNA. Neuroreport 23, 621–626.
Lukiw, W. J., and Bazan, N. G. (1998). Strong NF-κB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer's disease superior temporal lobe neocortex. J. Neurosci. Res. 53, 583–592.
Lukiw, W. J., and Bazan, N. G. (2010). Inflammatory, apoptotic, and survival gene signaling in Alzheimer's disease. A review on the bioactivity of neuroprotectin D1 and apoptosis. Mol. Neurobiol. 42, 10–16.
Lukiw, W. J., Zhao, Y., and Cui, J. G. (2008). An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human neural cells. J. Biol. Chem. 283, 31315–31322.
Ma, X., Becker Buscaglia, L. E., Barker, J. R., and Li, Y. (2011). MicroRNAs in NF-κB signaling. J. Mol. Cell Biol. 3, 159–166.
Mattson, M. P., and Camandola, S. (2001). NF-κB in neuronal plasticity and neurodegenerative disorders. J. Clin. Invest. 107, 247–254.
Meffert, M. K., and Baltimore, D. (2005). Physiological functions for brain NF-κB. Trends Neurosci. 28, 37–43.
Nagasawa, D. T., Chow, F., Yew, A., Kim, W., Cremer, N., and Yang, I. (2012). Temozolomide and other potential agents for the treatment of glioblastoma multiforme. Neurosurg. Clin. N. Am. 23, 307–322.
Pogue, A. I., Li, Y. Y., Cui, J. G., Zhao, Y., Kruck, T. P., Percy, M. E., et al. (2009). Characterization of an NF-κB-regulated, miRNA-146a-mediated down-regulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J. Inorg. Biochem. 103, 1591–1595.
Pogue, A. I., Percy, M. E., Cui, J. G., Li, Y. Y., Bhattacharjee, S., Hill, J. M., et al. (2011). Up-regulation of NF-κB-sensitive miRNA-125b and miRNA-146a in metal sulfate-stressed human astroglial (HAG) primary cell cultures. J. Inorg. Biochem. 105, 1434–1437.
Shifera, A. S. (2010). Protein-protein interactions involving NEMO that promote the activation of NF-κB. J. Cell Physiol. 223, 558–561.
Sen, R., and Baltimore, D. (1986). Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 47, 921–928.
Taganov, K. D., Boldin, M. P., Chang, K. J., and Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486.
Thomas, R. P., Recht, L., and Nagpal, S. (2013). Advances in the management of glioblastoma: the role of temozolomide and MGMT testing. Clin. Pharmacol. 5, 1–9.
Wong, D., Teixeira, A., Oikonomopoulos, S., Humburg, P., Lone, I. N., Saliba, D., et al. (2011). Extensive characterization of NF-κ B binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 12, R70.
Xu, D., Sharma, C., and Hemler, M. E. (2009). Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 23, 3674–3681.
Yamamoto, Y., and Gaynor, R. B. (2001). Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 1, 287–296.
Yang, L., Tao, L. Y., and Chen, X. P. (2007). Roles of NF-κB in CNS damage and repair. Neurosci. Bull. 23, 307–313.
Citation: Lukiw WJ (2013) Antagonism of NF-κB-up-regulated micro RNAs (miRNAs) in sporadic Alzheimer's disease (AD)—anti-NF-κB vs. anti-miRNA strategies. Front. Genet. 4:77. doi: 10.3389/fgene.2013.00077
Received: 05 April 2013; Accepted: 16 April 2013;
Published online: 01 May 2013.
Edited by:
Subbaya Subramanian, University of Minnesota, USAReviewed by:
Subbaya Subramanian, University of Minnesota, USACopyright © 2013 Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence:d2x1a2l3QGxzdWhzYy5lZHU=