



- 1Departamento de Microbiologia, Imunologia e Parasitologia, Universidade Federal de São Paulo, São Paulo, Brazil
- 2Laboratório de Genômica Evolutiva e Biocomplexidade, Universidade Federal de São Paulo, São Paulo, Brazil
- 3Departamento de Medicina, Disciplina de Infectologia, Universidade Federal de São Paulo, São Paulo, Brazil
The cell invasion mechanism of Trypanosoma cruzi has similarities with some intracellular bacterial taxa especially regarding calcium mobilization. This mechanism is not observed in other trypanosomatids, suggesting that the molecules involved in this type of cell invasion were a product of (1) acquisition by horizontal gene transfer (HGT); (2) secondary loss in the other trypanosomatid lineages of the mechanism inherited since the bifurcation Bacteria-Neomura (1.9 billion to 900 million years ago); or (3) de novo evolution from non-homologous proteins via convergent evolution. Similar to T. cruzi, several bacterial genera require increased host cell cytosolic calcium for intracellular invasion. Among intracellular bacteria, the mechanism of host cell invasion of genus Salmonella is the most similar to T. cruzi. The invasion of Salmonella occurs by contact with the host's cell surface and is mediated by the type III secretion system (T3SS) that promotes the contact-dependent translocation of effector proteins directly into host's cell cytoplasm. Here we provide evidence of distant sequence similarities and structurally conserved domains between T. cruzi and Salmonella spp T3SS proteins. Exhaustive database searches were directed to a wide range of intracellular bacteria and trypanosomatids, exploring sequence patterns for comparison of structural similarities and Bayesian phylogenies. Based on our data we hypothesize that T. cruzi acquired genes for calcium mobilization mediated invasion by ancient HGT from ancestral Salmonella lineages.
Introduction
The protist Trypanosoma cruzi is a heteroxenic parasite and the causative agent of Chagas disease which represents an important public health problem in Latin America (WHO, 2010). Differently from other mammal infecting trypanosomatids, only T. cruzi can actively invade non-phagocytic host cells (Shi et al., 2004; El-Sayed et al., 2005b; Sibley, 2011). The cellular invasion mechanism of T. cruzi is remarkably similar to invasion mechanisms found in intracellular bacterial genera such as Shigella and Salmonella, especially regarding cellular calcium mobilization. Because these mechanisms are not observed in other trypanosomatids (Docampo and Moreno, 1996; Burleigh and Woolsey, 2002; Shi et al., 2004; El-Sayed et al., 2005b; Sibley, 2011) three possible explanations for the origin of T. cruzi calcium-dependent invasion mechanism can be conjectured: (1) the acquisition by horizontal gene transfer (HGT), (2) secondary loss in non-T. cruzi trypanosomatids, or (3) parallel or convergent evolution from non-homologous T. cruzi surface proteins.
The “TriTryps” sequencing genome project revealed bacterial kinase genes such as ribulokinase and galactokinases in T. cruzi and Leishmania major genome (El-Sayed et al., 2005b), consistent with the idea that these kinases were probably acquired by HGT from bacteria to trypanosomatids. Also, the hypothesis of HGT was tested to explain the similarity between T. cruzi trans-sialidases and bacterial sialidases (Briones et al., 1995). As a matter of fact, Opperdoes and Mitchels propose that the acquisition of a large number of foreign genes from viruses and bacteria was necessary for the evolution of trypanosomatids (Opperdoes and Michels, 2007).
Similarly to T. cruzi, increased host cell cytosolic calcium is required for intracellular invasion of several bacterial genera. Among intracellular bacteria, the mechanism of host cell invasion of genus Salmonella shares the highest similarities with T. cruzi (Clerc et al., 1989; Burleigh and Andrews, 1995; Collazo and Galán, 1997; Dramsi and Cossart, 1998; Suárez and Rüssmann, 1998; Burleigh and Woolsey, 2002; Andrade and Andrews, 2004; TranVan Nhieu et al., 2004). The invasion of Salmonella occurs by contact with the host's cell surface and is mediated by the type III secretion system (T3SS) that promotes the contact-dependent translocation of effector proteins directly into host's cell cytoplasm (Dramsi and Cossart, 1998; Mirold et al., 2001; Cossart and Sansonetti, 2004; TranVan Nhieu et al., 2004).
Here we performed exhaustive database searches directed to a wide range of intracellular bacteria and trypanosomatids, exploring sequence patterns and predicted secondary structures for comparison to detect even distant or marginal similarities between sequences and structures of T. cruzi that could be even remotely conserved with bacterial T3SSs. These conserved structures could be indicative of HGT or an extreme case of convergent evolution very specific in the T. cruzi lineage and completely absent in other trypanosomatids.
Methods
Database Mining
Searches for genes similar to T. cruzi involved in intracellular bacterial invasion
Nucleotide sequences of genes encoding proteins SipD, SopB, SopD, and SopE2, present in all strains of genus Salmonella (Mirold et al., 2001) obtained in GeneDB (http://www.genedb.org/Homepage in September/2009), were used as BLASTN queries (Cummings et al., 2002) in completed intracellular bacterial (facultative or obligate) genome (http://www.genedb.org/Homepage in September/2009). New searches were performed in T. cruzi CL-Brener genome database (http://www.genedb.org/Homepage/Tcruzi in October/2009) using the nucleotide sequences from 57 strains of 11 genera and 28 intracellular bacterial species (including Salmonella typhi) obtained in the former search (Data Sheet 1 in Supplemental Data).
Searches for T. cruzi proteins similar to T3SS effector proteins from different bacteria
Amino acid sequences of proteins SipD, SopB, SopD, and SopE2 were submitted to BLASTP (Cummings et al., 2002) in the T. cruzi CL-Brener protein database (http://www.genedb.org/Homepage/Tcruzi in September/2009). Only the sequences of proteins whose role in calcium mobilization during T. cruzi invasion is currently known were selected (Moreno et al., 1994; Acosta-Serrano et al., 2001; Villalta et al., 2008) (Figure 1A). The amino acid sequences from T3SS proteins of Escherichia coli (EHEC O157:H7) str. EDL933, Salmonella enterica (serovar Typhi) str. CT18, Shigella flexneri (serotype 2a) str. 301, Pseudomonas aeruginosa PAO1, and Yersinia pestis CO92, downloaded from the Virulence Factors Database (http://www.mgc.ac.cn/VFs/ in March/2010) were also submitted to BLASTP (http://www.genedb.org/Homepage/Tcruzi in March/2010), being selected only the first 15 sequences according to their lower E-values. The amino acid consensus sequences of T. cruzi proteins retrieved from BLASTP, TcCLB.508221.420, TcCLB.510693.150, TcCLB.511089.90, and TcCLB.506611.20 (from this point forward designated as 420, 150, 90, and 20, respectively) were manually mapped and submitted again to BLASTP in the T. cruzi genome database GeneDB (http://www.genedb.org/Homepage/ in March/2010) and TriTrypDB—Esmeraldo-like and Non-Esmeraldo-like (http://tritrypdb.org/tritrypdb in April/2010), being selected only the first 15 non-redundant sequences according to their lower E-values (Figure 1B).
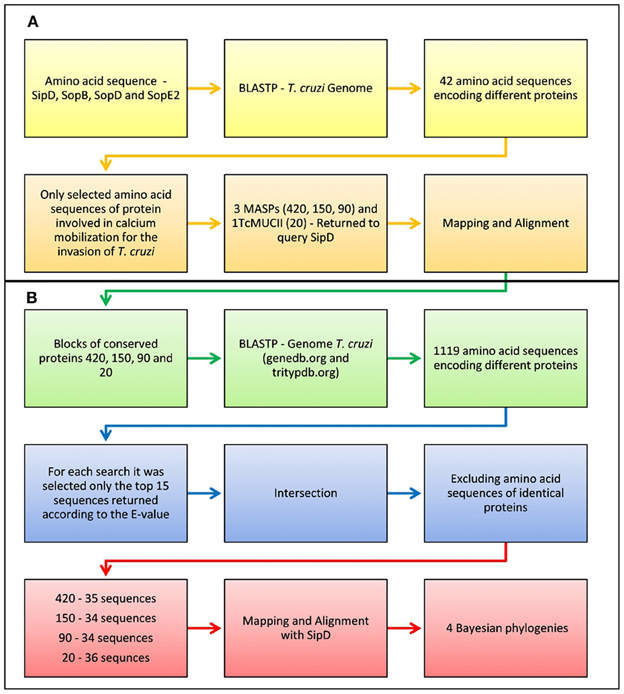
Figure 1. Flowchart of the pipeline used in the analysis of sequence similarities between bacteria and trypanosomatids. (A) Only the sequences of proteins whose role in calcium mobilization during T. cruzi invasion is currently known were selected. The amino acid sequences from T3SS proteins of Escherichia coli (EHEC O157:H7) str. EDL933, Salmonella enterica (serovar Typhi) str. CT18, Shigella flexneri (serotype 2a) str. 301, Pseudomonas aeruginosa PAO1, and Yersinia pestis CO92, downloaded from the Virulence Factors Database (http://www.mgc.ac.cn/VFs/ in March/2010) were also submitted to BLASTP (http://www.genedb.org/Homepage/Tcruzi in March/2010), being selected only the first 15 sequences according to their lower E-values. (B) The amino acid consensus sequences of T. cruzi proteins retrieved from BLASTP, TcCLB.508221.420, TcCLB.510693.150, TcCLB.511089.90, and TcCLB.506611.20 (designated as 420, 150, 90, and 20, respectively) were manually mapped and submitted again to BLASTP in the T. cruzi genome database GeneDB (http://www.genedb.org/Homepage/ in March/2010) and TriTrypDB—Esmeraldo-like and Non-Esmeraldo-like (http://tritrypdb.org/tritrypdb in April/2010), being selected only the first 15 non-redundant sequences according to their lower E-values.
Similarity searches in different protists
Amino acid sequence of S. typhi SipD was used as query in numerous searches with BLASTP in the genome database of Bodo saltans, Trypanosoma brucei gambiense, T. brucei 427, T. brucei 927, Trypanosoma congolense, T. cruzi, Trypanosoma vivax, Leishmania mexicana, L. major strain Friedlin, Leishmania braziliensis and Leishmania infantum in GeneDB and TritrypDB (http://www.genedb.org/Homepage/ and http://tritrypdb.org/tritrypdb in March/2011), Euglena gracilis (txid3039) and Paramecium tetraurelia strain d4-2 (txid412030) (http://blast.ncbi.nlm.nih.gov/Blast.cgi in June/2011). Only the first 15 non-redundant sequences were selected.
Similarities searches of trypanosomatids and S. typhi
Genome sequence of S. typhi CT18 (chromosome, plasmid 1 and 2) was downloaded from NCBI (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi in October/2011) and submitted to BLASTN algorithm in the L. major strain Friedlin, T. brucei strain 927 and T. cruzi strain CL Brener genome databases at GeneDB (http://www.genedb.org/Homepage in November/2011). Sequences encoding ubiquitous proteins such as heat shock and mitochondrial were discarded. Amino acid sequences of proteins SipD, SopB, SopD, and SopE2 of S. typhi were used as query in BLASTP searches in the genome database from L. major strain Friedlin and T. brucei strain 927 at GeneDB (http://www.genedb.org/Homepage in May/2012).
Protein Sequence Alignments
The amino acid sequences were aligned using ClustalX (Thompson et al., 1997). For exclusive initial pairwise alignments were performed using default settings (matrix: Gonnet 250, gap opening = 10.00, and gap extension = 0.10). Multiple alignments were carried out with the following parameters: pairwise and multiple alignments using gap opening and gap extension = 1.00, being the alignment matrix modified to PAM 350 on the protists and trypanosomatids amino acid alignments. Multiple alignments of trypanosomatids and other protists were made using PAM350 matrix, which is most adequate for highly divergent sequences. This matrix is based on an explicit evolution model which takes into account the observed substitutions in a gobal alignment. Also, three different parameters were tested in multiple alignments: (1) pairwise gap opening (go) = 10.00 and gap extension (ge) = 0.10 and multiple go = 10.00 and ge = 0.20, (2)go = 1.00 and ge = 1.00, and (3) pairwise go = 35.00 and ge = 0.75 and multiple go = 15.00 and ge = 0.30. After evaluation of alignments with different parameters we chose go = 1.00 and ge = 1.00 because it maximized the number of conserved blocks. With other parameters the only blocks formed were between proteins in the same gene family where aminoacids are conserved. Also, parameters of type (3) above, yielded poor alignments with several blocks of unaligned sequences. This was used as preliminary approach and that is why it was not included in the manuscript. Therefore, Bayesian trees were not inferred using parameters as described in (1) and (3). For the loopback multiple alignments (420, 150, 90, and 20) the go and ge were both set to 1.00. The matrix was the Gonnet 250 because these were related sequences from the same organism in its majority from the same gene family (MASPs). Alignments were manually checked and adjusted using the Seaview4 sequence editor (Gouy et al., 2010).
In silico Analysis of Deduced Amino Acid Sequences
Secondary structure of proteins 420, 150, 90, 20, and SipD were analyzed using Geneious v5.5 (Drummond et al., 2011) with GOR1 method and idc = 3 (Garnier et al., 1978). Protein domain searches were performed in Pfam database (Finn et al., 2010). Sequences were also submitted to prediction servers at CBS (http://www.cbs.dtu.dk/services) for signal peptide (SP), transmembrane domains, function, and subcellular localization and Post-translational modifications such as N and O-glycosylation. Prediction of GPI-anchor sites (glycosylphosphatidylinositol) was performed by servers GPI-SOM (Fankhauser and Mäser, 2005) and PredGPI (Pierleoni et al., 2008). The membrane proteins were predicted using Mem Type-2L server (Chou and Shen, 2007). The presence of signal sequence of T3SS effector proteins was predicted at Modlab server (Löwer and Schneider, 2009).
Codon Usage and GC Content Analysis
Codon usage analysis was carried out with nucleotide sequences encoding for S. typhi SipD and T. cruzi proteins 420, 150, 90, 20, and actin (TcCLB.510573.10) using The Sequence Manipulation Suite (Stothard, 2000). The GC content was analyzed using the same sequences and also with their respective upstream and downstream intergenic regions using Geneious v5.5 (Drummond et al., 2011).
Sequence Variability
Sequence variability was measured using Shannon entropy (Shannon, 1948) with BioEdit v.7 program (Hall, 1999) for each position of the amino acid alignment from full sequences obtained in loopback searches and alignment with the conserved amino acid blocks used in Bayesian phylogenetic trees. Values obtained in nits were converted to bits by calculating the base 2 log of nit values.
Phylogenetic Inference
Phylogenetic trees were inferred from amino acid sequence alignments retrieved from BLASTP (Data Sheet 1 in Supplemental Data) and from alignments generated from database searches of different protists (B. saltans, E. gracilis, L. mexicana, L. major, L. braziliensis e L. infantum, P. tetraurelia T. brucei gambiense, T. brucei 427, T. brucei 927, T. cruzi, T. congolense, and T. vivax), using MrBayes v3.1.2 (Huelsenbeck et al., 2001). MCMC algorithm started from a random tree, estimating the amino acids substitution model. Trees were inferred from 3 × 107 generations sampling a tree in every 100 generation until the standard deviation from split frequencies were under 0.01. The parameters and the trees were summarized by wasting at least 25% of the samples obtained (burnin). The consensus trees were then used to determine the posterior probabilities values. All phylogenetic trees were then formatted with the FigTree v1.3.1 program (http://tree.bio.ed.ac.uk/software/figtree/).
Results and Discussion
Proteins Involved in Intracellular Invasion Similar to T. cruzi Proteins
Among all bacterial genera analyzed (Data Sheet 1 in Supplemental Data), positive BLASTN results were obtained only for genera Bordetella, Chlamydophila, and Shigella. These sequences, along with sequences encoding proteins SipD, SopB, SopD e SopE2 of S. typhi were used as queries for searches in the T. cruzi genome database. A total of 689 open reading frames (ORFs) were retrieved. Sequences whose in silico translation included frameshifts and/or unrelated amino acids, were excluded. Only amino acid sequences obtained by BLASTP were used for further analysis.
BLASTP searches were then performed using as queries the amino acid sequences of the S. typhi effector proteins SipD, SopB, SopD, and SopE2 against the L. major, T. brucei, and T. cruzi genome database, yielding 21, 24, and 42 sequences, respectively. From these sequences, we performed predictions to determine their possible locations and functions (Data Sheet 3 in Supplemental Data). We show that the number of T. cruzi amino acid sequences potentially involved in the invasion mechanism was superior to other trypanosomatids. Two sequences with the potential to be on the parasite surface were found both in L. major and in T. brucei (Data Sheet 3 in Supplemental Data). However, they were not analyzed further because they are classified as hypothetical or pseudogenes and because it is already known that both parasites do not mobilize intracellular calcium during invasion and thus cannot actively invade host cells (Shi et al., 2004; El-Sayed et al., 2005b; Sibley, 2011). Prediction analysis of T. cruzi BLASTP results output showed that 9 sequences had the potential to be involved in host cell invasion (Data Sheet 3 in Supplemental Data). Among those, only the putative sequences of mucins and/or mucin associated surface proteins (MASP) (420, 150, 90, and 20) were selected because of their already known involvement with calcium mobilization during T. cruzi cell invasion (Moreno et al., 1994; Acosta-Serrano et al., 2001; Villalta et al., 2008). We discarded search hits of proteins whose involvement in T. cruzi cell invasion has not yet been demonstrated to increase the chance to detect marginal similarities among proteins associated with this mechanism (Figure 1B). Positive database search results were only obtained with protein SipD. This protein is known to increase the level of proteins secreted by the T3SS and plays a crucial role in Salmonella host cell invasion. Its absence causes the complete impairment of effector proteins translocation and hinders the invasion process (Kubori and Galán, 2002). T. cruzi MASPs and mucins and bacterial SipD are expressed on cell surface even before invasion, although these can also be found in the cytosol and are intimately involved with mechanisms of pathogenicity (Acosta-Serrano et al., 2001; Kubori and Galán, 2002; Eswarappa et al., 2008; Villalta et al., 2008; De Pablos et al., 2011). These data suggest the homology among SipD, MASPs, and mucins, and also suggest that their functions in calcium mobilization might be conserved (Henikoff and Henikoff, 1992).
In an attempt to find proteins similar to MASPs and mucins in other T3SS bacteria and not restrict the analysis to proteins associated with calcium mobilization of genus Salmonella, we performed new searches against the T. cruzi genome database with amino acid sequences from different bacterial T3SS (Data Sheet 4 in Supplemental Data). These searches revealed a considerable number of MASPs and mucins (Table 1). Our results are consistent with the hypothesis of HGT of T3SS genes to T. cruzi because BLAST results of MASPs and mucins are not unique to Salmonella queries. However, because the percentage of MASPs returned by searches with Salmonella was significantly higher, sequences from other genera were not further analyzed (Table 1). Also, when comparing the invasion mechanisms associated with different T3SS, Salmonella shows the highest similarity with T. cruzi. Both organisms can invade non-phagocytic cells, use inositol 1,4,5-trisphosphate (IP3) to elevate intracellular calcium and consequently induce cytoskeleton rearrangement and remain inside vacuoles during the first stages of cell invasion (Clerc et al., 1989; Burleigh and Andrews, 1995; Collazo and Galán, 1997; Dramsi and Cossart, 1998; Suárez and Rüssmann, 1998; Burleigh and Woolsey, 2002; Andrade and Andrews, 2004; TranVan Nhieu et al., 2004). Although other bacteria share some of these mechanisms, genus Salmonella shares most of the observed features. The host cell invasion mechanism of Shigella is relatively similar to Salmonella (Dramsi and Cossart, 1998) and involves T3SS proteins (Espina et al., 2006; Parsot, 2009) but differs from T. cruzi because it does not exclusively depend on intracellular calcium mobilization and does not remain in vacuoles during the first stages of invasion (Clerc et al., 1989; Collazo and Galán, 1997).
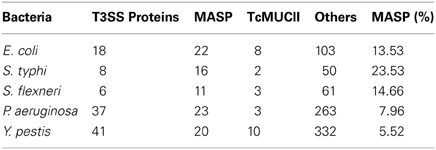
Table 1. Database searches using amino acid sequences of the T3SS proteins of different bacteria.
To verify if the marginal sequence similarities between bacteria and T. cruzi are specific to genes encoding T3SS proteins, searches using the whole S. typhi genome as query were performed against the genome databases from different members of Trypanosomatidae (Table 2). These searches returned a large number of sequences coding for common proteins shared by all classes of eukaryotic organisms such as mitochondrial and heat shock proteins. These searches also returned several genes encoding hypothetical proteins and stage-specific proteins of each parasite (data not shown). However, these genes were not considered as positive hits for possible “trace-homologies” that could be involved with infectivity, because negative results were obtained when predictions for subcellular localization, SP, and GPI anchoring were performed with their deduced amino acid sequence (data not shown), suggesting that these putative proteins are possibly not secreted or present on the cell surface. These results are supported by the fact that T. cruzi adhesion and invasion does not seem to be simple i.e., involving a single ligand-receptor interaction. Trypomastigotes exploit a huge palette of surface glycoproteins, secreted proteases, and agonist signaling to actively manipulate the host cell invasion (Burleigh and Andrews, 1995; Di Noia et al., 1998; Acosta-Serrano et al., 2001; Burleigh and Woolsey, 2002; Buscaglia et al., 2006; Yoshida, 2006; Villalta et al., 2008). As expected, searches in the T. cruzi genome database using the whole S. typhi genome returned several sequences that encode proteins involved in host cell adhesion/invasion such as DGF-1 (Dispersed Gene Family 1) and MASPs (Moreno et al., 1994; Acosta-Serrano et al., 2001; Villalta et al., 2008; Kawashita et al., 2009) (Data Sheet 2 in Supplemental Data).
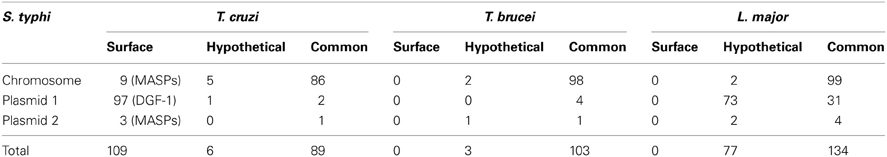
Table 2. Comparative genome analysis of S. typhi and trypanosomatids.
Amino Acid Sequences Similarities
The complete amino acid sequences of S. typhi SipD and of T. cruzi MASPs and mucins (420, 150, 90, and 20) were aligned. As expected, due to the high rate of divergence among sequences, it resulted in few conserved blocks and positions embedded in highly divergent domains (data not shown). However, the mapping of local amino acid residues (local alignment) resulted in an alignment with good quality (pairwise identity, identical sites and similarities above 13, 16, and 29%, respectively) (Table 3) showing potential homologous positions (Figure 2). Alignments often provide important insights into protein functional mechanisms being the pairwise alignment of blocks a better option to perform homology searches (Henikoff and Henikoff, 1992; Batzoglou, 2005). SipD has residues important for Salmonella invasion. Although most of functional residues are located at the C-terminal, the portion of N-terminal which aligns with the T. cruzi proteins also has important sites, both by decreasing the invasion itself and by involvement with bile salts that suppress the Salmonella invasion (Wang et al., 2010; Chatterjee et al., 2011). Although most of the transferred genes are non-functional in the recipient genome, Woolfit et al. (2009) suggest that independently of the direction of the HGT, transferred genes may remain functional. These propositions are supported by different authors that argue that these genes are really important in the adaptation to new niches, to originate novel functions and for virulence (Opperdoes and Michels, 2007; Keeling and Palmer, 2008; Andersson, 2009; Cohen et al., 2011).
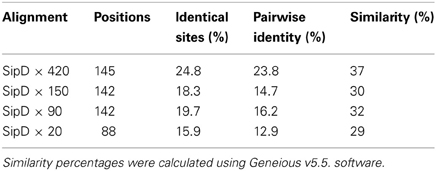
Table 3. Sequence similarities between Salmonella SipD and T. cruzi MASPs and mucin.

Figure 2. Similarity between Salmonella typhi SipD and T. cruzi proteins. The identity and similarity between the aligned sequences. Red represents identical residues and green indicates conservative changes. Local amino acid sequences were initially aligned using ClustalX (Thompson et al., 1997). Pairwise alignments were performed with default settings (see Methods) and adjusted manually in Seaview sequence editor (Gouy et al., 2010). (A), (B), (C), and (D) refers to the local alignment of the amino acid sequence of the protein SipD with MASPs 420, 150, 90, and mucin 20, respectively.
In silico Analysis of Protein Structure and Motifs
To verify possible homologies (“trace-homologies”) between T. cruzi and Salmonella proteins and also address the possible structural and functional properties shared by them, amino acid sequences were analyzed by different prediction methods. Searches for known sequence motifs and domains from manually curated databases using the amino acid sequences of proteins 420, 150, 90, and 20 from T. cruzi and the sequence of S. typhi SipD, showed that no characterized domains or motifs are present (data not shown). However, our predictions showed that SipD is part of the IpaD family, effector proteins from Shigella that share similar functional roles with SipD (Espina et al., 2006; Parsot, 2009).
As expected, SipD does not present a canonical SP because proteins from the T3SS are secreted through a sec-independent mechanism (Büttner and Bonas, 2002). The proteins 420, 150, 90, and 20 from T. cruzi present potential cleavage sites in positions 21 and 22, 25 and 26, 26 and 27, and 24 and 25, respectively. More importantly, the fact that the possible signal sequences in these proteins remain outside amino acid blocks that aligns with SipD (Figure 3) suggests that these residues are not cleaved during secretion. Predictions also suggest that proteins 420 and 90 possess possible transmembrane helices between positions 7 and 29, overlapping with their signal sequences. According to Bendtsen et al. (2004), transmembrane helices must be disregarded in these cases because signal sequences interfere with these predictions, leading to false positives. In addition, it is known that MASPs are GPI-anchored (Acosta-Serrano et al., 2001; Buscaglia et al., 2006) and that GPI-anchored proteins lack the transmembrane domains (Elortza et al., 2003).
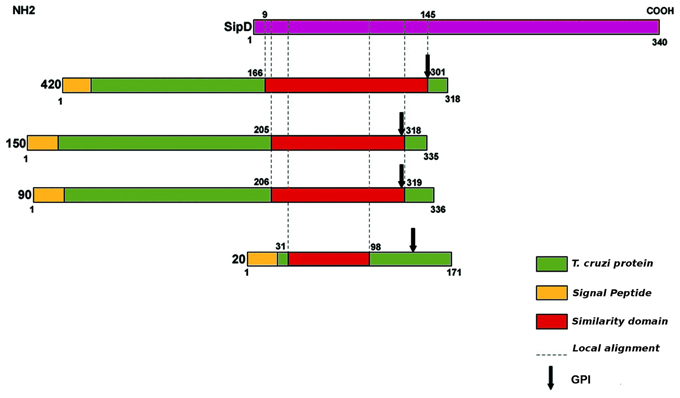
Figure 3. Schematic illustration of amino acid sequence similarity between SipD (purple) and T. cruzi proteins (green). Protein domain searches were performed in Pfam database (Finn et al., 2010). Sequences were also analysed at CBS (http://www.cbs.dtu.dk/services) for signal peptide (SP), transmembrane domains, function, and subcellular localization, and Post-translational modifications such as N and O-glycosylation. GPI-anchor sites (glycosylphosphatidylinositol) was predicted by GPI-SOM (Fankhauser and Mäser, 2005) and PredGPI (Pierleoni et al., 2008). The membrane proteins were predicted using Mem Type-2L server (Chou and Shen, 2007). The presence of signal sequence of T3SS effector proteins was predicted by Modlab (Löwer and Schneider, 2009).
We also found potential GPI anchoring sites in T. cruzi proteins 420, 150, 90, and 20 in positions 291, 305, 306, and 145, respectively. As a negative control, the amino acid sequence of SipD was used in this prediction. These data confirm our results because it is already known that MASPs and mucins are GPI-anchored proteins (Acosta-Serrano et al., 2001; Buscaglia et al., 2006). The potential GPI anchor sites of putative MASPs 420, 150, and 90 are localized at the end of the amino acid sequences that align with SipD. On the other hand, the predicted GPI-anchor site of putative mucin 20 differs from other proteins (Figure 3), suggesting a potential specialized and/or functional role of this specific site in these MASPs, and supporting their involvement with host-parasite interactions (Elortza et al., 2003; Epting et al., 2010).
In addition to the comparative results obtained with SipD, putative post translational modifications were analyzed (Table 4). Not surprisingly, the predictions are consistent with already known characteristics of this protein class (Acosta-Serrano et al., 2001; Buscaglia et al., 2006; Bartholomeu et al., 2009).
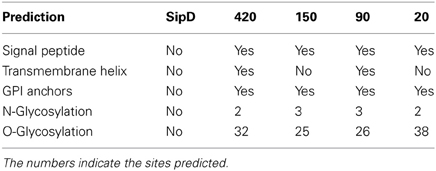
Table 4. Predictions of protein sequence features.
The comparison of protein structures is important to reveal evolutionary relationships among proteins. Protein families tend to be structurally conserved and these structures may be maintained even when sequences have diverged beyond any recognizable similarity (Orengo et al., 1997; Wieser and Niranjan, 2009; Joseph et al., 2011). To verify if the putative T. cruzi proteins and S. typhi SipD possess conserved secondary structural domains, their local amino acid sequences were analyzed. These local conserved residues are, in general, rare in regions containing sequences of amino acids forming beta-sheets and rich in alpha-helices and coil structures (Figure 4). The secondary structure of SipD maintains a similarity of approximately 30–45% with T. cruzi proteins (Table 5). Considering the phylogenetic distance between these organisms, it is reasonable to propose that these levels of secondary structure similarities might indicate homology. However, the quantification of secondary structure predictions should be taken carefully because the current software works with a confidence level of approximately 70% (Garnier et al., 1978; Creighton, 1990; Joseph et al., 2011). Nevertheless, our data indicate that the secondary structures of the conserved amino acid regions of T. cruzi and S. typhi are more conserved than the primary structure (Table 1), mostly because the secondary structure can be maintained even in regions where amino acids are not identical, via conservative amino acid substitutions.
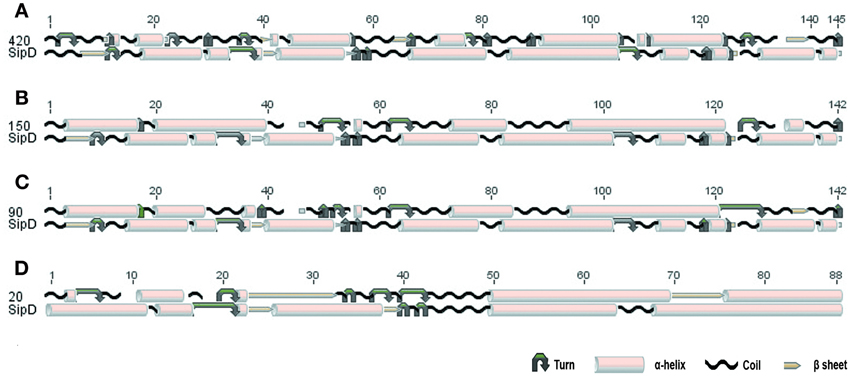
Figure 4. Conserved secondary structure of the aligned blocks of proteins (A) 420, (B) 150, (C) 90, and (D) 20 of T. cruzi with the SipD. Secondary structure of proteins 420, 150, 90, 20, and SipD were analyzed using the GOR1 method and idc = 3 (Garnier et al., 1978).
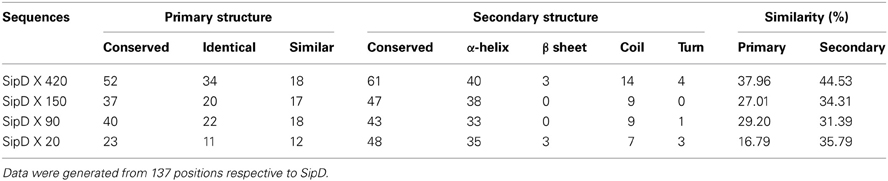
Table 5. Comparison of primary and secondary structure similarities.
Horizontal Gene Transfer and Invasion Mechanisms
Although HGT is recognized as an important evolutionary mechanism, its impact has been neglected and confused with mere phylogenetic noise in favor of a vertical signal resulting from the transmission of information from ancestors to descendants (Comas et al., 2006).
In view of the amino acid similarities and function, shared by S. typhi and T. cruzi proteins here presented and because this parasite is the only trypanosomatid that can actively invade host cells (Docampo and Moreno, 1996; Burleigh and Woolsey, 2002; Shi et al., 2004; El-Sayed et al., 2005b; Sibley, 2011), we propose the hypothesis of ancient HGT for the origin of calcium-dependent invasion mechanism of T. cruzi. It can be speculated that these ancient HGT events might have occurred by: (1) the ingestion of blood contaminated with Salmonella spp. or some other T3SS intracellular bacteria by species of Triatominae and the insertion of bacterial genes into the T. cruzi genome or (2) insertions and/or gene exchange by endosymbiotic bacteria. We also do not exclude that other trypanosomatids lost their ability to invade since the Bacteria-Neomura bifurcation (secondary loss). Nevertheless, the occurrence of multiple HGT events from bacterial endosymbionts in plants to trypanosomatids described by Hannaert et al. (2003) and by the possible occurrence of HGT in trypanosomatids originated from bacteria present in the intestine of Triatominae (Opperdoes and Michels, 2007).
Here we examine three possibilities of HGT, summarized two different scenarios, monophyletic (Figure 5A) and paraphyletic (Figure 5B). Although most studies agree with the monophyly of the trypanosomatids, this issue remains controversial (Simpson et al., 2006; Leonard et al., 2011). Firstly, we supposed that this event might have occurred at point 1, being the genes transferred from one ancestor to all trypanosomatids. Therefore, all trypanosomatids would carry genes involved in calcium-dependent host cell invasion, but during evolution these genes could have been lost or silenced. Secondly, if HGT occurred at the point 2, genes would be present only in T. cruzi and T. brucei spp. (Figure 5A) or if we consider the trypanosomatids family tree presented in Figure 5B, genes would be present only in T. cruzi and Leishmania spp. Finally, if the transfer occurred at the point 3, only T. cruzi would have acquired the genes to actively invade host cell. Among these three hypotheses, we believe that the third has the highest likelihood due to the relative similarity of the host cell invasion mechanisms of bacteria, such as Salmonella, and T. cruzi (Clerc et al., 1989; Burleigh and Andrews, 1995; Collazo and Galán, 1997; Dramsi and Cossart, 1998; Suárez and Rüssmann, 1998; Burleigh and Woolsey, 2002; Andrade and Andrews, 2004; TranVan Nhieu et al., 2004) and absence of even remotely similar sequences in T. brucei and Leishmania. In addition, this is the most parsimonious hypothesis because it involves only one acquisition whereas the other hypotheses involve one acquisition and at least one secondary loss (Figure 4). This hypothesis is also supported by computational predictions (Data Sheet 3 in Supplemental Data), by the highly superior number of sequences obtained in database searches within T. cruzi genome database and by the potential of these sequences to be involved in invasion mechanisms. Although in small numbers, searches against the genome of L. major and T. brucei also returned 2 amino acid sequences. This may suggest that HGT occurred in a trypanosomatid common ancestor and that other trypanosomatids have lost this mechanism. The vertical inheritance would imply a loss dating to the bifurcation Bacteria-Neomura between 1.9 billion and 900 million years ago (Proterozoic Eon) (Cavalier-Smith, 1998).
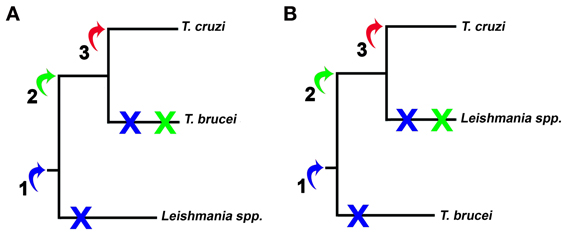
Figure 5. Representation of HGT hypothesized in this work. The arrows and numbers represent the possible insertion of bacterial genes. (A) and (B) represent the branching order of the trees of trypanosomatids considered in this work. Early HGT implies one acquisition (blue arrow) and two losses (blue X), while HGT in the Trypanosoma genus would imply one acquisition and one loss (green arrow and X). The most parsimonious hypothesis, or late HGT in T. cruzi, implies only one acquisition and no character loss (red arrow).
There are different ways to detect patterns and signs of HGT events. In general they are based on bio-computational analysis, including homology searches, codon usage, and GC content analysis and phylogenetic inference (Cohen and Pupko, 2010; Li et al., 2011). Most commonly these approaches search for the distribution of atypical genes in different organisms and may include the identification of: (a) genes with highly restricted distributions, present in isolated taxa but absent from closely related species, (b) highly similar genes, and (c) genes whose phylogenies are incongruent with the relationships inferred from other genes in their respective genomes (Gogarten et al., 2002). Nonetheless, most methods used to evidence HGT are based on recent events, since ancient HGT events are harder to detect and genes may lose ancestor signatures through evolution. Phylogenetic inference of a broad range of sequences, though, may reveal ancient HGTs (McDonald et al., 2012), being considered as gold-standards.
Parametric analysis such as codon usage and GC content profiles are preferentially used to detect recent HGT events (Becq et al., 2010). We analyzed the codon usage profiles of nucleotide sequences encoding the putative T. cruzi proteins and Salmonella SipD. These analyses were performed with the four-fold degenerated amino acids only. These results did not strongly indicate the occurrence of HGT, but it is noticeable that the codon usage pattern of actin differ from other T. cruzi genes (Figure 6), suggesting a possible HGT event. Although SipD has a different codon usage profile in comparison to T. cruzi genes, this cannot be considered a negative result, since highly divergent genes tend to lose features from their ancestors (Philippe and Douady, 2003; McDonald et al., 2012). Additionally, transferred genes tend to behave homogeneously, similar to genes from the receptor organism. Thus, codon usage analyses are not sensitive enough to distinguish ancient HGT (Koski et al., 2001; Philippe and Douady, 2003). Therefore, if we look carefully it is possible to note that the frequencies of G and C levels in third codon positions are relatively close among genes encoding the T. cruzi proteins 420, 150, 90, and 20 and S. typhi SipD, in comparison to values of T. cruzi actin gene, mainly for the amino acids alanine (ALA), proline (PRO), and threonine (THR) (Figure 6). Usually vertically inherited genes are adapted to the codon usage characteristic of their original genome and expression level. On the other hand, horizontally acquired genes frequently have atypical G and C base compositions (Karberg et al., 2011). Together these results support the hypothesis that these T. cruzi genes were acquired by HGT, because they have different sequence features when compared to the actin gene.
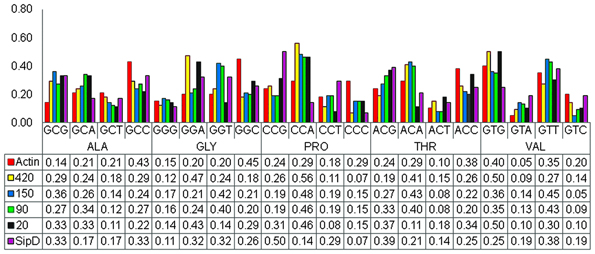
Figure 6. Codon usage profiles. The pattern of codon usage was obtained from the nucleotide sequences coding for proteins SipD, 420, 150, 90, 20, and the actin gene within The Sequence Manipulation Suite (Stothard, 2000). The charts were plotted with the Excel program. The abscissa indicates the four-fold degenerated amino acids and the ordinate represents the codon frequencies. Bars represent each codon used by the respective gene, and the values below the chart indicate the frequency of each codon in the respective genes.
Gene fixation in the HGT receptor organism requires a progressive compatibility of GC content and codon usage (Medrano-Soto et al., 2004). This criterion is used in the analysis of T. cruzi and S. typhi genes in this study, both with approximately 51% GC content (Parkhill et al., 2001; El-Sayed et al., 2005a). However, most methods identify horizontally transferred genes based on the identification of atypical GC content in DNA sequences (Becq et al., 2010; Karberg et al., 2011). The presence of atypical GC content in intergenic regions may reveal horizontally transferred genome islands (Kurup et al., 2010). Our results demonstrated that some values were in proximity to the GC content of intergenic and coding regions of each gene, except for the intergenic regions of actin (Table 6). It is known that MASPs and mucins, as well as some other surface proteins, unique to T. cruzi, are encoded by non-sintenic islands (El-Sayed et al., 2005b). Although we have not observed atypical GC content in intergenic regions between the possible genes acquired by horizontal transfer, we do not consider this as a negative result for a possible HGT event, particularly because methods to identify atypical sequences are limited to detection of recent transfers (Gogarten et al., 2002) and also because intergenic regions showed lower GC content than the other regions (Table 6). Gene content varies along a genome, and the number of members in each gene family. The difference in gene repertoire between the genomes of the same family and/or species is generally attributed to gene loss or HGT (Daubin and Ochman, 2004). Thus, we can assume that T. cruzi may have acquired a large number of foreign genes, since the size of its genome is approximately 20 Mb greater than the genomes of T. brucei and L. major, and MASPs and mucins are encoded within large genomic islands (El-Sayed et al., 2005b).
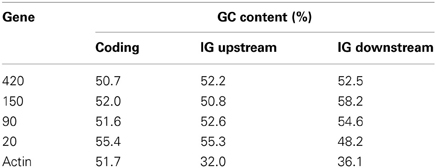
Table 6. GC content of T. cruzi genes and intergenic regions (IG).
Entropy analysis was used here as means to study HGT because HGT per se is a source of disorder in the receptor genome. Gene exchange among organisms, populations and species causes extensive genome instability, increase mutation frequency, and affects gene expression (Chia and Goldenfeld, 2011). Functional proteins (less entropic) are usually more conserved than non-functional proteins (more entropic) (Albà and Castresana, 2007) and therefore it is expected that lower entropy in conserved functional blocks as opposed to non-functional blocks. In the 4 alignments obtained with the sequences from loopback searches there are 21 different characters (20 different amino acids and gaps). The maximum entropy in this case is 4.3 bits. Thus, positions with entropies higher than 2.0 bits were considered variable, while entropies lower than 2.0 bits were considered conserved (Kawashita et al., 2009). In general, our data shows that these aligned amino acid blocks are well conserved, as indicated by the low entropy values (Figure 7).
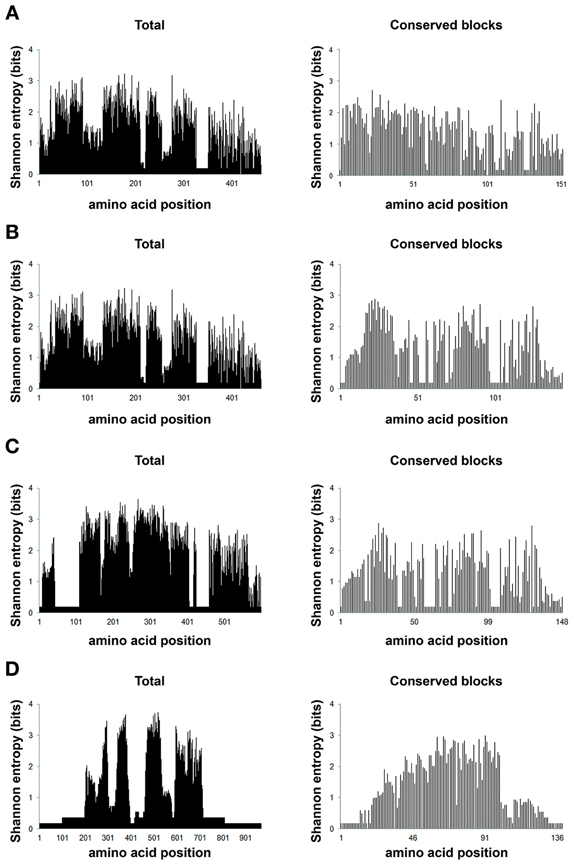
Figure 7. Positional entropy. Shannon information entropy values for the eight different amino acid alignments (full sequences and conserved amino acid blocks) were plotted according to the values generated from BioEdit (Hall, 1999). The chart (A) (420), is represented by alignments with 35 sequences, 460 positions (total) and 34 positions (blocks); (B) (150), by 34 sequences, 460 (total) and 144 (blocks) positions; (C) (90), 34 sequences, 598 (total) and 148 (blocks) positions; (D) (20) represented by alignments with 36 sequences and 967 (total) and 139 (blocks) positions. The abscissa represents the positions in each alignment and the ordinate represents the entropy values in bits for each alignment position.
To obtain a congruent analysis that could establish evolutionary relationships between S. typhi SipD and putative T. cruzi MASPs and one mucin, a larger number of amino acid sequences were obtained (Brown, 2003) by performing new searches within the T. cruzi genome database, using the conserved amino acid blocks from proteins 420, 150, 90, and 20 as queries. This type of approach reduces the false positives and increases the chance to find new sequences that could not be discovered by searches with the primary query. The amino acid sequences (Data Sheet 2 in Supplemental Data) and sequences obtained from database searches of different protists were aligned and submitted to Bayesian phylogenetic inferences. A total of six multiple alignments were generated (one for each T. cruzi proteins), comprising up to 36 sequences which included the S. typhi SipD, with up to 152 positions, and other 2 alignments, one comprising 179 sequences with 368 positions (different protists) and the other with 139 sequences and 444 positions (only trypanosomatids), obtained by searches in different protein databases. Apart from the phylogenetic inference obtained with the putative mucin 20, which showed a large polytomy (Figure 8D), all phylogenetic trees inferred with the MASPs (420, 150, and 90) showed the formation of a cluster comprising S. typhi SipD and several T. cruzi proteins, with posterior probabilities above 0.79 (Figure 8), suggesting a common evolutionary origin. Interestingly, a common feature of trees obtained from the alignments 420, 150, and 90 is that some putative family members of MASPs were closer to SipD than other members within the same family, indicating the presence of different groups of MASPs with distinct phylogenetic distances in relation to SipD. The sequences of putative MASPs of the inference 420 (TcCLB.510693.91 and TcCLB.510693.280) for example, were more divergent in comparison to the rest of MASPs family and forms an outgroup (Figure 8A). SipD, although more divergent than all the others proteins in the alignments, did not cluster as outgroup. The MASP (TcCLB.510693.190) that clustered with SipD (Figure 8A) was recently described by dos Santos et al. (2012) as MASP16 being highly expressed in bloodstream trypomastigote and myoblast cells. Therefore, MASP16 well as other MASPs may be involved in the invasion mechanism and calcium mobilization of T. cruzi, suggesting a possible homology and analogy of these MASPs with SipD.
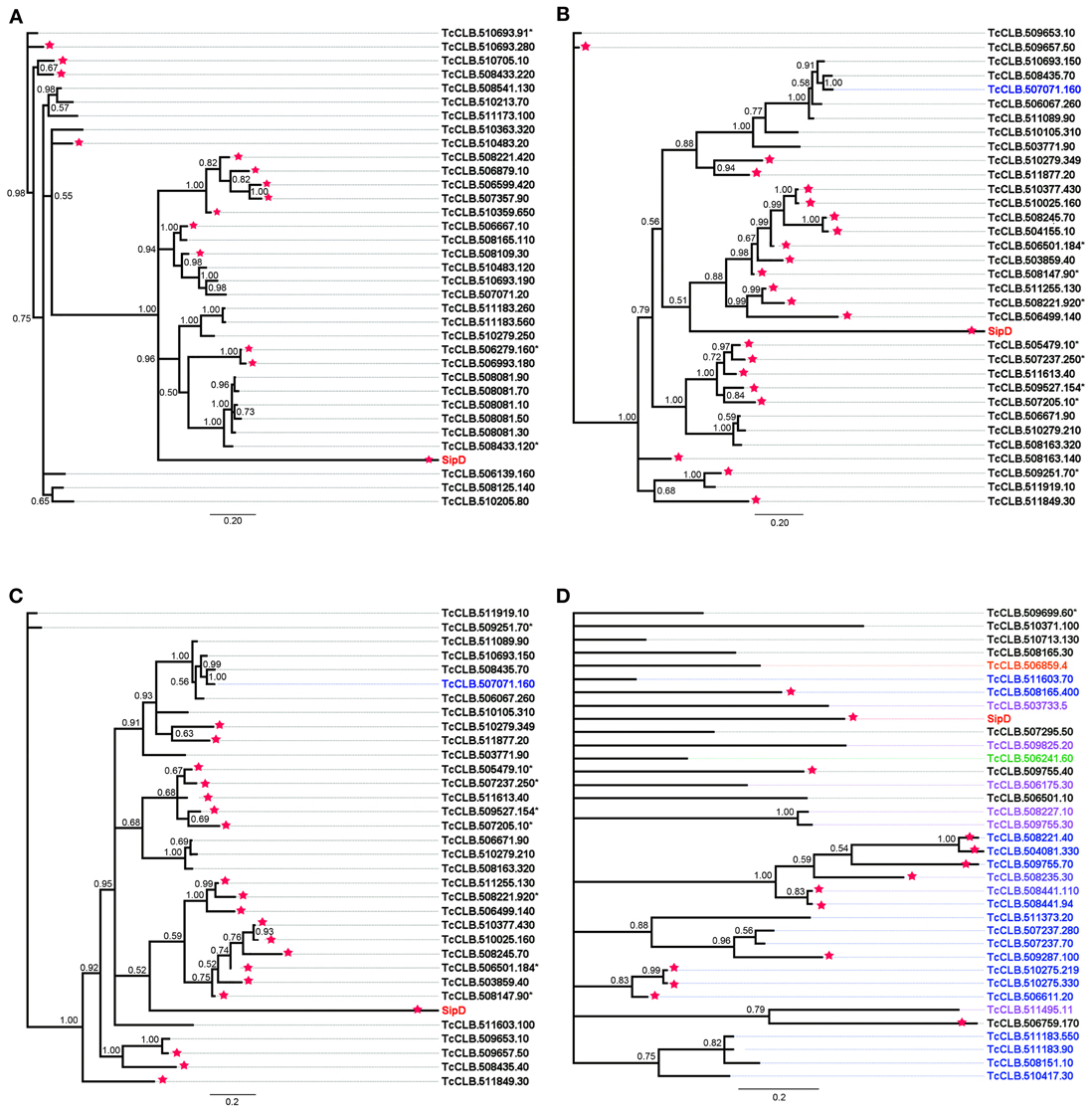
Figure 8. Bayesian phylogeny of MASPs, mucins, and Salmonella SipD. Trees were inferred with the conserved amino acid blocks obtained by loopback searches. The tree's named 420 (A), 150 (B), and 20 (D) were calculated from 1 × 107 generations and the tree 90 (C) were calculated from 1.5 × 107 generations. Numbers in branches represents the posterior probabilities. Letters and numbers on the right side represent GeneDB and TriTrypDB proteins access codes. Different colors indicate the types of proteins, black: MASP, blue: mucins and red: SipD (other colors, check Data Sheet 3 in Supplemental Data). Asterisks and stars within the codes represent pseudogenes and positive predictions for T3SS proteins, respectively.
The phylogeny inferred using amino acid sequences of different protists was used to test if earlier branching organisms such as Euglena gracilis, Paramecium tetraurelia, and Bodo saltans would cluster together with SipD (Figure 9). A SipD clade with posterior probability 0.90 comprises one Paramecium sequence, one Euglena sequence and a polytomus subclade including several trypanosomatids. For this analysis the Bayesian inference was used to obtain several phylogenies in two runs with convergent LnL scores after the burn-in, around 3 × 107 generations (Figure 10). The resulting phylogeny is the MrBayes “sumt” consensus of trees with converging maximum LnL scores.
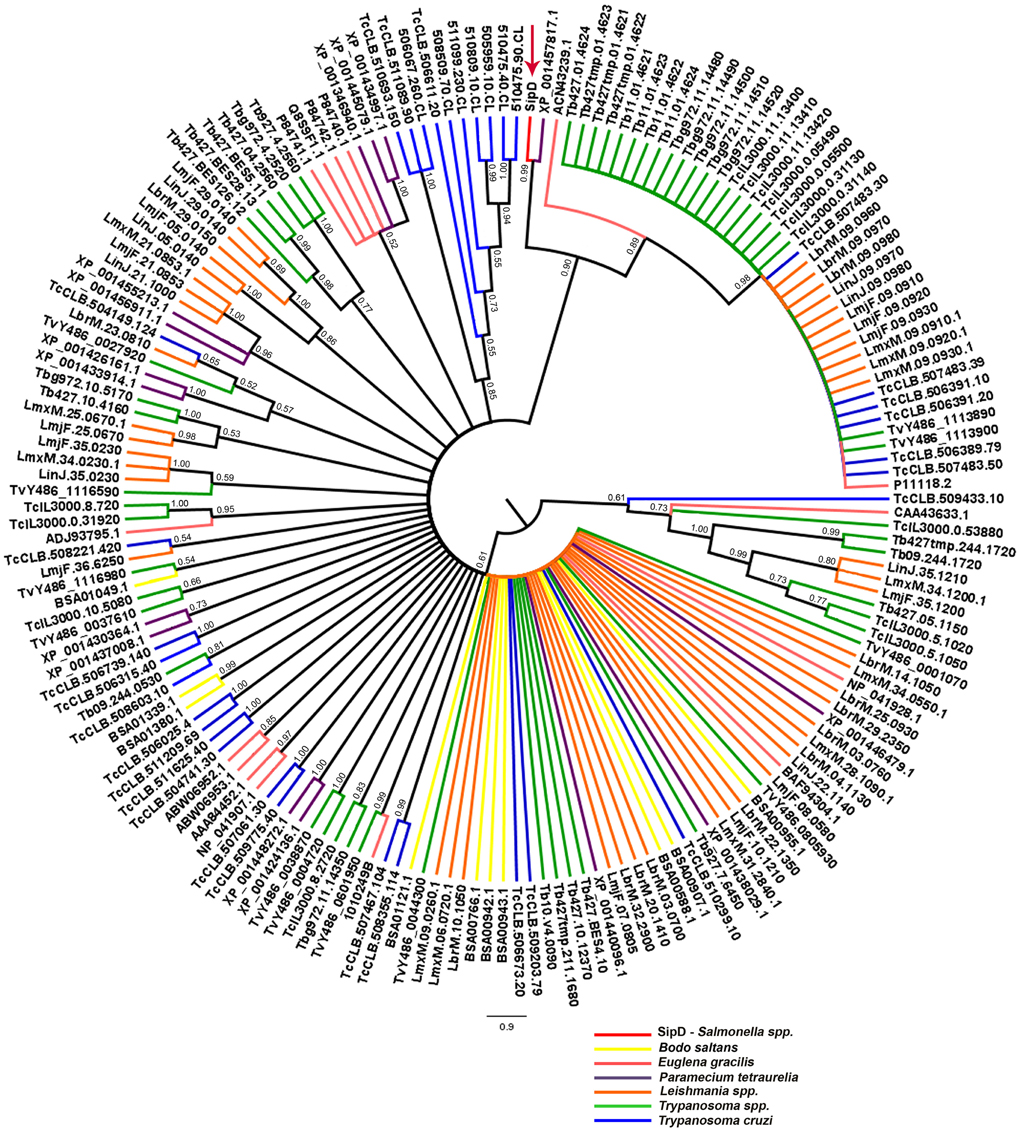
Figure 9. Bayesian phylogeny with different protists and SipD. Trees were inferred with the conserved amino acid blocks obtained by BLASTP of different protists and were calculated from 3 × 107 generations. Trees are depicted as midpoint rooted. Branches colored according to genus of protists and numbers in branches represent the posterior probabilities of nodes. Letters and numbers along the branches represent GeneDB, TriTrypDB, and NCBI access codes. Arrow indicates the position of the Salmonella SipD.

Figure 10. Burn-in plot of the Bayesian inference of different protists and SipD. The abscissa represents the generations in the search and the ordinate the LnL scores of trees. Two runs are depicted.
To resolve the polytomy observed in the Bayesian tree in Figure 9 a phylogeny including only amino acid sequences of trypanosomatids was inferred (Figure 11). It was observed that SipD is closer to T. cruzi with posterior probability 1.00 (Figure 11). This result supports our hypothesis of HGT from intracellular bacteria, more specifically from Salmonella spp to T. cruzi, because even with a large number of sequences from different trypanosomatids, SipD still clustered with T. cruzi sequences.
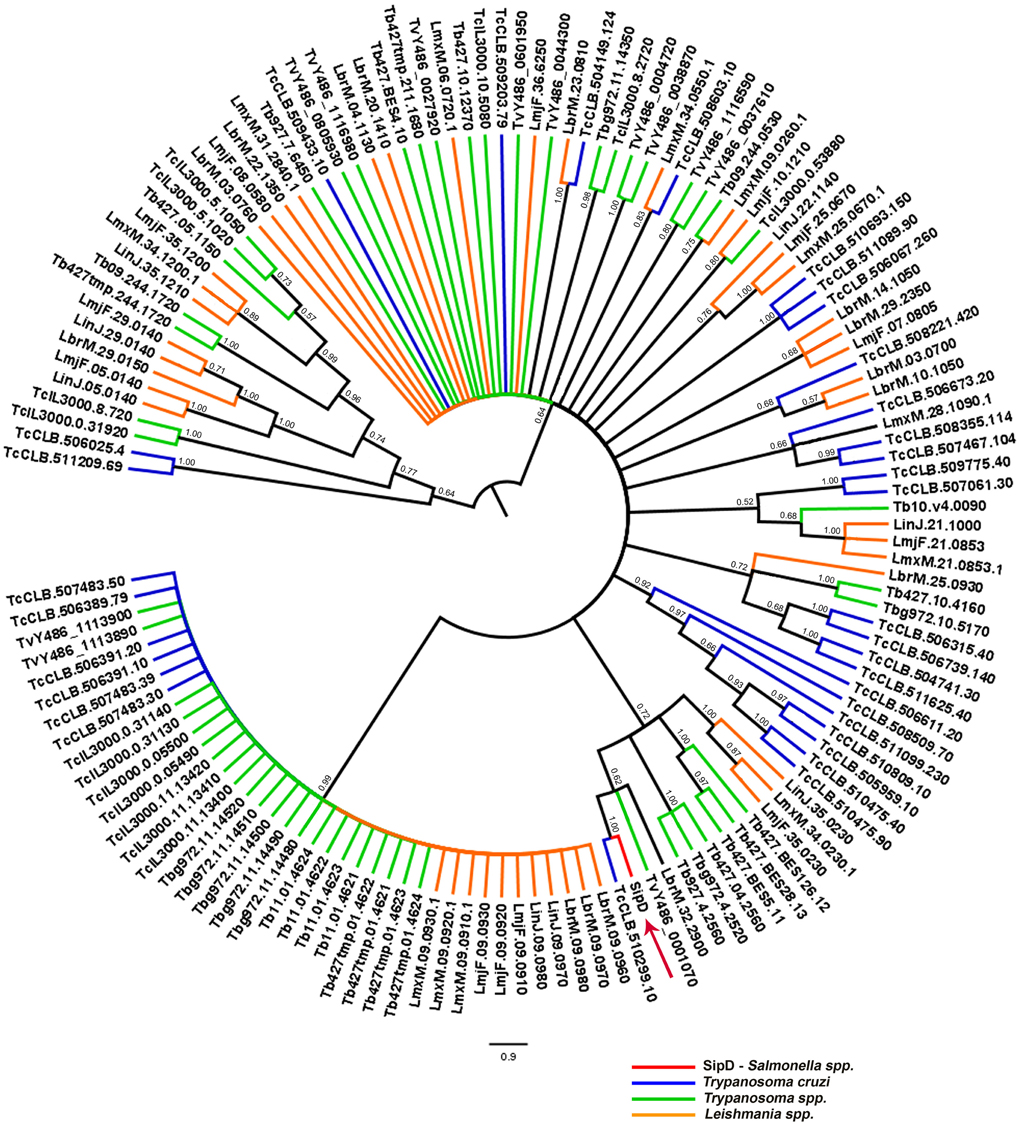
Figure 11. Bayesian phylogeny of trypanosomatids and SipD. Trees were inferred with the conserved amino acid blocks obtained by BLASTP of trypanosmatids and were calculated from 2 × 107 generations. Trees are depicted as midpoint rooted. Branches colored according to genera and numbers in branches represent the posterior probabilities of nodes. Letters and numbers along the branches represent GeneDB and TriTrypDB proteins access codes. Arrow indicates the position of the Salmonella SipD.
The accuracy with which phylogenies can be reconstructed, and by which HGTs can be detected, depends on the degree of divergence (Gogarten et al., 2002; Brown, 2003) and for highly divergent sequences, the number of amino acid substitutions may be saturated, resulting in loss of phylogenetic signal (Gogarten et al., 2002; Philippe and Douady, 2003; Mayrose et al., 2004). Of note, recently it has been shown that L. tarentolae expressing two different proteins of the MASP family trigger intracellular calcium transients in HeLa cells, presumably by injury to the cell membrane (Choi et al., 2012). This observation is consistent with our prediction of functional analogy with Salmonella SipD and the HGT here proposed.
Conclusions
Our results are consistent with the hypothesis that genes involved in host cell invasion were horizontally transferred from S. typhi to T. cruzi in early evolutionary history of T. cruzi. Because of the marginal sequence similarities involved and long divergence dates, our data cannot rule out extreme convergent evolution. Nevertheless, the acquisition of ancestral T3SS from Salmonella might have contributed to the pathogenicity and singular invasion mechanisms among trypanosomatids that allowed it to actively invade host cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Thais F. Bartelli for careful review of the manuscript. Danielle C. F. Silva and Richard C. Silva received fellowships from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil and Renata C. Ferreira received a postdoctoral fellowship from FAPESP, Brazil. This work was supported by grants to Marcelo R. S. Briones from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Brazil; from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil and the International Program of the Howard Hughes Medical Institute (HHMI).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Statistical_Genetics_and_Methodology/10.3389/fgene.2013.00143/abstract
References
Acosta-Serrano, A., Almeida, I. C., Freitas-Junior, L. H., Yoshida, N., and Schenkman, S. (2001). The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol. Biochem. Parasitol. 114, 143–150. doi: 10.1016/S0166-6851(01)00245-6
Albà, M. M., and Castresana, J. (2007). On homology searches by protein Blast and the characterization of the age of genes. BMC Evol. Biol. 7:53. doi: 10.1186/1471-2148-7-53
Andersson, J. O. (2009). Gene transfer and diversification of microbial eukaryotes. Annu. Rev. Microbiol. 63, 177–193. doi: 10.1146/annurev.micro.091208.073203
Andrade, L. O., and Andrews, N. W. (2004). Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J. Exp. Med. 200, 1135–1143. doi: 10.1084/jem.20041408
Bartholomeu, D. C., Cerqueira, G. C., Leão, A. C. A., daRocha, W. D., Pais, F. S., Macedo, C., et al. (2009). Genomic organization and expression profile of the mucin-associated surface protein (masp) family of the human pathogen Trypanosoma cruzi. Nucleic Acids Res. 37, 3407–3417. doi: 10.1093/nar/gkp172
Batzoglou, S. (2005). The many faces of sequence alignment. Brief. Bioinform. 6, 6–22. doi: 10.1093/bib/6.1.6
Becq, J., Churlaud, C., and Deschavanne, P. (2010). A benchmark of parametric methods for horizontal transfers detection. PLoS ONE 5:e9989. doi: 10.1371/journal.pone.0009989
Bendtsen, J. D., Nielsen, H., von Heijne, G., and Brunak, S. (2004). Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. doi: 10.1016/j.jmb.2004.05.028
Briones, M. R., Egima, C. M., Eichinger, D., and Schenkman, S. (1995). Trans-sialidase genes expressed in mammalian forms of Trypanosoma cruzi evolved from ancestor genes expressed in insect forms of the parasite. J. Mol. Evol. 41, 120–131. doi: 10.1007/BF00170663
Brown, J. R. (2003). Ancient horizontal gene transfer. Nat. Rev. Genet. 4, 121–132. doi: 10.1038/nrg1000
Burleigh, B. A., and Andrews, N. W. (1995). The mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu. Rev. Microbiol. 49, 175–200. doi: 10.1146/annurev.mi.49.100195.001135
Burleigh, B. A., and Woolsey, A. M. (2002). Cell signalling and Trypanosoma cruzi invasion. Cell. Microbiol. 4, 701–711. doi: 10.1046/j.1462-5822.2002.00226.x
Buscaglia, C. A., Campo, V. A., Frasch, A. C. C., and Di Noia, J. M. (2006). Trypanosoma cruzi surface mucins: host-dependent coat diversity. Nat. Rev. Microbiol. 4, 229–236. doi: 10.1038/nrmicro1351
Büttner, D., and Bonas, U. (2002). Port of entry–the type III secretion translocon. Trends Microbiol. 10, 186–192. doi: 10.1016/S0966-842X(02)02331-4
Cavalier-Smith, T. (1998). A revised six-kingdom system of life. Biol. Rev. Camb. Philos. Soc. 73, 203–266. doi: 10.1017/S0006323198005167
Chatterjee, S., Zhong, D., Nordhues, B. A., Battaile, K. P., Lovell, S., and De Guzman, R. N. (2011). The crystal structures of the Salmonella type III secretion system tip protein SipD in complex with deoxycholate and chenodeoxycholate. Protein Sci. 20, 75–86. doi: 10.1002/pro.537
Chia, N., and Goldenfeld, N. (2011). Statistical mechanics of horizontal gene transfer in evolutionary ecology. J. Stat. Phys. 142, 1287–1301. doi: 10.1007/s10955-010-0112-8
Choi, J., Fernandes, M. C., Cai, Q., Cerqueira, G., Sheng, Z., and El-Sayed, N. M. (2012). “From genomes to host-pathogen infectomes: models in profiling host-pathogen interactions,” in Proceedings of the XXVIII Annual Meeting of the Brazilian Society of Protozoology, (pCaxambu).
Chou, K.-C., and Shen, H.-B. (2007). MemType-2L: a web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem. Biophys. Res. Commun. 360, 339–345. doi: 10.1016/j.bbrc.2007.06.027
Clerc, P. L., Berthon, B., Claret, M., and Sansonetti, P. J. (1989). Internalization of Shigella flexneri into HeLa cells occurs without an increase in cytosolic Ca2+ concentration. Infect. Immun. 57, 2919–2922.
Cohen, O., Gophna, U., and Pupko, T. (2011). The complexity hypothesis revisited: connectivity rather than function constitutes a barrier to horizontal gene transfer. Mol. Biol. Evol. 28, 1481–1489. doi: 10.1093/molbev/msq333
Cohen, O., and Pupko, T. (2010). Inference and characterization of horizontally transferred gene families using stochastic mapping. Mol. Biol. Evol. 27, 703–713. doi: 10.1093/molbev/msp240
Collazo, C. M., and Galán, J. E. (1997). The invasion-associated type-III protein secretion system in Salmonella–a review. Gene 192, 51–59. doi: 10.1016/S0378-1119(96)00825-6
Comas, I., Moya, A., Azad, R. K., Lawrence, J. G., and Gonzalez-Candelas, F. (2006). The evolutionary origin of Xanthomonadales genomes and the nature of the horizontal gene transfer process. Mol. Biol. Evol. 23, 2049–2057. doi: 10.1093/molbev/msl075
Cossart, P., and Sansonetti, P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248. doi: 10.1126/science.1090124
Cummings, L., Riley, L., Black, L., Souvorov, A., Resenchuk, S., Dondoshansky, I., et al. (2002). Genomic BLAST: custom-defined virtual databases for complete and unfinished genomes. FEMS Microbiol. Lett. 216, 133–138. doi: 10.1111/j.1574-6968.2002.tb11426.x
Daubin, V., and Ochman, H. (2004). Bacterial genomes as new gene homes: the genealogy of ORFans in E. coli. Genome Res. 14, 1036–1042. doi: 10.1101/gr.2231904
De Pablos, L. M., González, G. G., Solano Parada, J., Seco Hidalgo, V., Díaz Lozano, I. M., Gómez Samblás, M. M., et al. (2011). Differential expression and characterization of a member of the mucin-associated surface protein family secreted by Trypanosoma cruzi. Infect. Immun. 79, 3993–4001. doi: 10.1128/IAI.05329-11
Di Noia, J. M., D'Orso, I., Aslund, L., Sánchez, D. O., and Frasch, A. C. (1998). The Trypanosoma cruzi mucin family is transcribed from hundreds of genes having hypervariable regions. J. Biol. Chem. 273, 10843–10850.
Docampo, R., and Moreno, S. N. (1996). The role of Ca2+ in the process of cell invasion by intracellular parasites. Parasitol. Today 12, 61–65. doi: 10.1016/0169-4758(96)80656-9
dos Santos, S. L., Freitas, L. M., Lobo, F. P., Rodrigues-Luiz, G. F., Mendes, T. A., Oliveira, A. C. S., et al. (2012). The MASP Family of Trypanosoma cruzi: changes in gene expression and antigenic profile during the acute phase of experimental infection. PLoS Negl. Trop. Dis. 6:e1779. doi: 10.1371/journal.pntd.0001779
Dramsi, S., and Cossart, P. (1998). Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 14, 137–166. doi: 10.1146/annurev.cellbio.14.1.137
Drummond, A. J., Bruxton, S., Cheung, M., Cooper, A., Duran, C., Field, M., et al. (2011). Geneious v5.4. Available online at: http://www.geneious.com.
Elortza, F., Nühse, T. S., Foster, L. J., Stensballe, A., Peck, S. C., and Jensen, O. N. (2003). Proteomic analysis of glycosylphosphatidylinositol-anchored membrane proteins. Mol. Cell. Proteomics 2, 1261–1270. doi: 10.1074/mcp.M300079-MCP200
El-Sayed, N. M., Myler, P. J., Bartholomeu, D. C., Nilsson, D., Aggarwal, G., Tran, A.-N., et al. (2005a). The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309, 409–415. doi: 10.1126/science.1112631
El-Sayed, N. M., Myler, P. J., Blandin, G., Berriman, M., Crabtree, J., Aggarwal, G., et al. (2005b). Comparative genomics of trypanosomatid parasitic protozoa. Science 309, 404–409. doi: 10.1126/science.1112181
Epting, C. L., Coates, B. M., and Engman, D. M. (2010). Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp. Parasitol. 126, 283–291. doi: 10.1016/j.exppara.2010.06.023
Espina, M., Olive, A. J., Kenjale, R., Moore, D. S., Ausar, S. F., Kaminski, R. W., et al. (2006). IpaD localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect. Immun. 74, 4391–4400. doi: 10.1128/IAI.00440-06
Eswarappa, S. M., Janice, J., Nagarajan, A. G., Balasundaram, S. V., Karnam, G., Dixit, N. M., et al. (2008). Differentially evolved genes of Salmonella pathogenicity islands: insights into the mechanism of host specificity in Salmonella. PLoS ONE 3:e3829. doi: 10.1371/journal.pone.0003829
Fankhauser, N., and Mäser, P. (2005). Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics 21, 1846–1852. doi: 10.1093/bioinformatics/bti299
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., et al. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222. doi: 10.1093/nar/gkp985
Garnier, J., Osguthorpe, D. J., and Robson, B. (1978). Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 120, 97–120. doi: 10.1016/0022-2836(78)90297-8
Gogarten, J. P., Doolittle, W. F., and Lawrence, J. G. (2002). Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19, 2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. 41, 95–98.
Hannaert, V., Saavedra, E., Duffieux, F., Szikora, J.-P., Rigden, D. J., Michels, P. A. M., et al. (2003). Plant-like traits associated with metabolism of Trypanosoma parasites. Proc. Natl. Acad. Sci. U.S.A. 100, 1067–1071. doi: 10.1073/pnas.0335769100
Henikoff, S., and Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 89, 10915–10919. doi: 10.1073/pnas.89.22.10915
Huelsenbeck, J. P., Ronquist, F., Nielsen, R., and Bollback, J. P. (2001). Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294, 2310–2314. doi: 10.1126/science.1065889
Joseph, A. P., Srinivasan, N., and de Brevern, A. G. (2011). Improvement of protein structure comparison using a structural alphabet. Biochimie 93, 1434–1445. doi: 10.1016/j.biochi.2011.04.010
Karberg, K. A., Olsen, G. J., and Davis, J. J. (2011). Similarity of genes horizontally acquired by Escherichia coli and Salmonella enterica is evidence of a supraspecies pangenome. Proc. Natl. Acad. Sci. U.S.A. 108, 20154–20159. doi: 10.1073/pnas.1109451108
Kawashita, S. Y., Da Silva, C. V., Mortara, R. A., Burleigh, B. A., and Briones, M. R. S. (2009). Homology, paralogy and function of DGF-1, a highly dispersed Trypanosoma cruzi specific gene family and its implications for information entropy of its encoded proteins. Mol. Biochem. Parasitol. 165, 19–31. doi: 10.1016/j.molbiopara.2008.12.010
Keeling, P. J., and Palmer, J. D. (2008). Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618. doi: 10.1038/nrg2386
Koski, L. B., Morton, R. A., and Golding, G. B. (2001). Codon bias and base composition are poor indicators of horizontally transferred genes. Mol. Biol. Evol. 18, 404–412. doi: 10.1093/oxfordjournals.molbev.a003816
Kubori, T., and Galán, J. E. (2002). Salmonella type III secretion-associated protein InvE controls translocation of effector proteins into host cells. J. Bacteriol. 184, 4699–4708. doi: 10.1128/JB.184.17.4699-4708.2002
Kurup, K., Mary, S., and Rafi, Z. A. (2010). Intergenics: a tool for extraction of intergenicregions. Bioinformation 5, 83–84. doi: 10.6026/97320630005083
Leonard, G., Soanes, D. M., and Stevens, J. R. (2011). Resolving the question of trypanosome monophyly: a comparative genomics approach using whole genome data sets with low taxon sampling. Infect. Genet. Evol. 11, 955–959. doi: 10.1016/j.meegid.2011.03.005
Li, Z.-W., Shen, Y.-H., Xiang, Z.-H., and Zhang, Z. (2011). Pathogen-origin horizontally transferred genes contribute to the evolution of Lepidopteran insects. BMC Evol. Biol. 11:356. doi: 10.1186/1471-2148-11-356
Löwer, M., and Schneider, G. (2009). Prediction of type III secretion signals in genomes of gram-negative bacteria. PLoS ONE 4:e5917. doi: 10.1371/journal.pone.0005917
Mayrose, I., Graur, D., Ben-Tal, N., and Pupko, T. (2004). Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol. Biol. Evol. 21, 1781–1791. doi: 10.1093/molbev/msh194
McDonald, T. R., Dietrich, F. S., and Lutzoni, F. (2012). Multiple horizontal gene transfers of ammonium transporters/ammonia permeases from prokaryotes to eukaryotes: toward a new functional and evolutionary classification. Mol. Biol. Evol. 29, 51–60. doi: 10.1093/molbev/msr123
Medrano-Soto, A., Moreno-Hagelsieb, G., Vinuesa, P., Christen, J. A., and Collado-Vides, J. (2004). Successful lateral transfer requires codon usage compatibility between foreign genes and recipient genomes. Mol. Biol. Evol. 21, 1884–1894. doi: 10.1093/molbev/msh202
Mirold, S., Ehrbar, K., Weissmüller, A., Prager, R., Tschäpe, H., Rüssmann, H., et al. (2001). Salmonella host cell invasion emerged by acquisition of a mosaic of separate genetic elements, including Salmonella pathogenicity island 1 (SPI1), SPI5, and sopE2. J. Bacteriol. 183, 2348–2358. doi: 10.1128/JB.183.7.2348-2358.2001
Moreno, S. N., Silva, J., Vercesi, A. E., and Docampo, R. (1994). Cytosolic-free calcium elevation in Trypanosoma cruzi is required for cell invasion. J. Exp. Med. 180, 1535–1540. doi: 10.1084/jem.180.4.1535
Opperdoes, F. R., and Michels, P. A. M. (2007). Horizontal gene transfer in trypanosomatids. Trends Parasitol. 23, 470–476. doi: 10.1016/j.pt.2007.08.002
Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T., Swindells, M. B., and Thornton, J. M. (1997). CATH–a hierarchic classification of protein domain structures. Structure 5, 1093–1108. doi: 10.1016/S0969-2126(97)00260-8
Parkhill, J., Dougan, G., James, K. D., Thomson, N. R., Pickard, D., Wain, J., et al. (2001). Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413, 848–852. doi: 10.1038/35101607
Parsot, C. (2009). Shigella type III secretion effectors: how, where, when, for what purposes? Curr. Opin. Microbiol. 12, 110–116. doi: 10.1016/j.mib.2008.12.002
Philippe, H., and Douady, C. J. (2003). Horizontal gene transfer and phylogenetics. Curr. Opin. Microbiol. 6, 498–505. doi: 10.1016/j.mib.2003.09.008
Pierleoni, A., Martelli, P. L., and Casadio, R. (2008). PredGPI: a GPI-anchor predictor. BMC Bioinformatics 9:392. doi: 10.1186/1471-2105-9-392
Shannon, C. E. (1948). A mathematical theory of communication. Bell System Technical Journal 27, 379–423.
Shi, M., Wei, G., Pan, W., and Tabel, H. (2004). Trypanosoma congolense infections: antibody-mediated phagocytosis by Kupffer cells. J. Leukoc. Biol. 76, 399–405. doi: 10.1189/jlb.1003500
Sibley, L. D. (2011). Invasion and intracellular survival by protozoan parasites. Immunol. Rev. 240, 72–91. doi: 10.1111/j.1600-065X.2010.00990.x
Simpson, A. G. B., Stevens, J. R., and Lukes, J. (2006). The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 22, 168–174. doi: 10.1016/j.pt.2006.02.006
Stothard, P. (2000). The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102; 1104.
Suárez, M., and Rüssmann, H. (1998). Molecular mechanisms of Salmonella invasion: the type III secretion system of the pathogenicity island 1. Int. Microbiol. 1, 197–204.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
TranVan Nhieu, G., Clair, C., Grompone, G., and Sansonetti, P. (2004). Calcium signalling during cell interactions with bacterial pathogens. Biol. Cell 96, 93–101. doi: 10.1016/j.biolcel.2003.10.006
Villalta, F., Madison, M. N., Kleshchenko, Y. Y., Nde, P. N., and Lima, M. F. (2008). Molecular analysis of early host cell infection by Trypanosoma cruzi. Front. Biosci. 13, 3714–3734. doi: 10.2741/2961
Wang, Y., Nordhues, B. A., Zhong, D., and De Guzman, R. N. (2010). NMR characterization of the interaction of the Salmonella type III secretion system protein SipD and bile salts. Biochemistry 49, 4220–4226. doi: 10.1021/bi100335u
WHO. (2010). WHO Chagas Disease (American trypanosomiasis). Available online at: http://www.who.int/mediacentre/factsheets/fs340/en/index.html (Accessed May 31, 2012).
Wieser, D., and Niranjan, M. (2009). Remote homology detection using a kernel method that combines sequence and secondary-structure similarity scores. In Silico Biol. 9, 89–103.
Woolfit, M., Iturbe-Ormaetxe, I., McGraw, E. A., and O'Neill, S. L. (2009). An ancient horizontal gene transfer between mosquito and the endosymbiotic bacterium Wolbachia pipientis. Mol. Biol. Evol. 26, 367–374.
Keywords: horizontal gene transfer (HGT), evolution, Trypanosoma cruzi, Salmonella spp., Type III secretion system (T3SS)
Citation: Silva DCF, Silva RC, Ferreira RC and Briones MRS (2013) Examining marginal sequence similarities between bacterial type III secretion system components and Trypanosoma cruzi surface proteins: horizontal gene transfer or convergent evolution? Front. Genet. 4:143. doi: 10.3389/fgene.2013.00143
Received: 19 March 2013; Paper pending published: 22 May 2013;
Accepted: 13 July 2013; Published online: 16 August 2013.
Edited by:
Vladimir Bajic, King Abdullah University of Science and Technology, Saudi ArabiaReviewed by:
Alexander V. Favorov, Johns Hopkins University, USAJianhua Guo, Northeast Normal University, China
Copyright © 2013 Silva, Silva, Ferreira and Briones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo R. S. Briones, Departamento de Microbiologia, Imunologia e Parasitologia, Universidade Federal de São Paulo, Rua Pedro de Toledo 669 4° andar, São Paulo, SP, CEP 04023-062, Brazil e-mail:bWFyY2Vsby5icmlvbmVzQHVuaWZlc3AuYnI=