Abstract
The hepatitis B virus (HBV) genome forms a covalently closed circular DNA (cccDNA) minichromosome that persists in the nucleus of virus-infected hepatocytes. HBV cccDNA serves as the template for viral mRNA synthesis and is subject to epigenetic regulation by several mechanisms, including DNA methylation and histone acetylation. Recently, microRNAs (miRNAs), a class of small non-coding RNAs, were also directly connected to the epigenetic machinery through a regulatory loop. Epigenetic modifications have been shown to affect miRNA expression, and a sub-group of miRNAs (defined as epi-miRNAs) can directly target effectors of the epigenetic machinery. In this review, we will summarize recent findings on the epigenetic mechanisms controlling HBV cccDNA function, primarily focusing on the epi-miRNA functions operating in HBV replication. Investigation of the epigenetic regulation of HBV replication may help to discover novel potential therapeutic targets for drug development with the goal to eradicate the HBV cccDNA pool in hepatocytes.
INTRODUCTION
Hepatitis B virus (HBV) infection is a global health problem that causes a wide spectrum of liver diseases, including acute or chronic HBV infection. Acute HBV infections either resolve or progress to chronicity. Chronic hepatitis B (CHB) is associated with chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC; McMahon, 2009). It is estimated that more than 350 million patients worldwide are chronically infected with HBV, with the majority of these patients living in the Asia-Pacific region. More than one million deaths occur each year as a direct consequence of CHB (Dienstag, 2008). Medical intervention using antiviral nucleoside/nucleotide analogs and interferon (IFN) was established to treat chronically infected patients (Pardo et al., 2007). However, currently available therapies do not lead to the termination of HBV infection in the majority of patients (Mailliard and Gollan, 2006). There is a consensus that the improved understanding of the HBV–host interaction is a prerequisite for new antiviral therapeutic strategies. Recently, many aspects pertaining to the epigenetic mechanisms responsible for viral persistence and clearance during HBV replication have been addressed, including methylation of viral DNA, acetylation of histone complexes, and microRNA (miRNA) regulation. These topics are described in this review.
HBV cccDNA STRUCTURE AND ITS ROLE IN HBV INFECTION
Hepatitis B virus is the prototype member of the family Hepadnaviridae and has a partially double-stranded DNA genome of approximately 3.2 kb in length. The viral genome harbors seven open reading frames, coding for the viral polymerase, HBV core, and e antigens (HBcAg and HBeAg); the regulatory HBx protein; and the preS/S gene encoding the three surface antigens (LHBsAg, MHBsAg, and SHBsAg). The genome also contains a number of regulatory elements (Seeger and Mason, 2000). The entry of HBV virions is likely initiated through a non-specific interaction with negatively charged glycans at the surface of hepatocytes (Schulze et al., 2007; Bremer et al., 2009) followed by specific binding to the sodium-taurocholate cotransporting polypeptide (NTCP) receptor by a specific sequence (2-48aa) located in the preS1 domain of the LHBsAg protein (Yan et al., 2012). After uncoating, the HBV capsid is transported by the cellular machinery to the nuclear pore. The open circular form of HBV genomic DNA is then converted to a covalently closed circular DNA (cccDNA) molecule in the nucleus. This process requires that the covalently attached viral polymerase is removed from the negative DNA strand by a proteinase and that the positive strand DNA is completed by the cellular replicative machinery so that it matches the negative strand to covalently join the two ends to form a circular, supercoiled molecule (Gao and Hu, 2007).
In the nucleus, HBV cccDNA is incorporated into the host chromatin and exists as an individual minichromosome with a “beads-on-a-string” structure, which is revealed by electron microscopy (Bock et al., 1994; Newbold et al., 1995). This minichromosome has been shown to consist of both histone and non-histone proteins. By immunoblotting with HBcAg, the histone proteins H3 and H2B were the most prominent species, while lower levels of H4, H2A, and H1 were also detectable (Bock et al., 2001). Using the cccDNA-ChIP assay, the group of Massimo Levrero has confirmed the recruitment of the H3 and H4 histones along with the HBcAg and HBx proteins to the cccDNA minichromosome. Using the same approach, several cellular transcription factors (CREB, ATF, YY1, STAT1, and STAT2) and chromatin modifying enzymes (PCAF, p300/CBP, HDAC1, SIRT1, and EZH2) have been shown to bind to the cccDNA in human hepatoma cells containing replicating HBV (Pollicino et al., 2006; Belloni et al., 2009, 2012). The histone acetyltransferases (HATs) p300/CBP and PCAF and the histone deacetylases (HDACs) HDAC1 and SIRTl were shown to be recruited with different kinetics onto HBV cccDNA, implying that HBV cccDNA-bound histones may be subjected to regulatory post-translational modifications (Levrero et al., 2009).
Because cccDNA is the transcriptional template of the virus (Quasdorff and Protzer, 2010), it is required for the maintenance of HBV infection. Unlike HBV transcripts and replicative intermediates, cccDNA is very stable in quiescent hepatocytes and is responsible for the persistence of infection during the natural course of chronic HBV infection and during prolonged antiviral therapy (Werle-Lapostolle et al., 2004). The cccDNA may persist for many years in the liver of patients, even after successful antiviral treatment and reinforcement of immunologic control (Zoulim, 2005). Currently, little is known about the mechanism of HBV cccDNA maintenance in the nuclei of hepatocytes. However, it has been shown that the cccDNA can be eliminated when infected hepatocytes are removed by immune cell-mediated killing or other non-cytopathic mechanisms (Murray et al., 2005) and replaced by cell turnover (Lutgehetmann et al., 2010).
REGULATION OF HBV cccDNA TRANSCRIPTION BY EPIGENETIC MODIFICATION
HISTONE ACETYLATION AND METHYLATION
Recently, it was proposed that the functionality of HBV cccDNA might be controlled by epigenetic mechanisms, regulating its transcriptional activity and HBV replication. Histones and non-histone proteins either bind directly to the cccDNA or are indirectly recruited to viral minichromosomes through protein–protein interactions. Thereby, the acetylation and deacetylation of cccDNA-bound histones may regulate HBV transcription. Exploring a ChIP assay using anti-acetylated-H3 or -H4 antibodies, Pollicino et al. (2006) found that HBV replication is indeed regulated by the acetylation status of H3/H4 histones bound to the viral cccDNA, both in cell-based replication systems and in the liver of chronically HBV infected patients. The co-recruitment of PCAF and p300/CBP parallels viral replication in vitro, whereas HDAC1 recruitment onto the HBV cccDNA correlates with low HBV replication in vitro and with low viremia in vivo. The importance of epigenetic modifications of cccDNA-bound histones in the regulation of HBV replication is further confirmed by experiments exploring the class I and class III HDAC inhibitors trichostatin A (TSA), valproate, and nicotinamide (NAM). These HDAC inhibitors induce an evident increase of both cccDNA-bound acetylated H4 and HBV replication. Another study demonstrated a similar role for the acetylation of cccDNA-bound histones, as well as a role for methylation and phosphorylation of these proteins (Gong et al., 2011).
A recent study demonstrated that in cultured hepatoma cells with HBV replication and in mouse models with repopulated human hepatocytes, administration of IFN-α resulted in the active recruitment of the transcriptional corepressors HDAC1, SIRT1, and polycomb repressor complexes 2 (EZH2 and YY1) to HBV cccDNA as well as the hypoacetylation/hypermethylation of cccDNA-bound histones. IFN-α treatment also reduced the binding of the transcription factors STAT1 and STAT2 to the IFN-sensitive response element on active cccDNA (Belloni et al., 2012). These observations suggested that IFN-α could epigenetically regulate HBV replication, and the hypoacetylation/hypermethylation of histones was associated with decreased replication of HBV. Furthermore, it was shown that small molecules that inhibit p300 and PCAF or activation of SIRT1/2 and EZH2 could induce an “active epigenetic suppression” of the HBV cccDNA minichromosome to suppress HBV replication (Palumbo et al., 2013).
HBV DNA METHYLATION
In addition to post-translational modification of histones, methylation of the CpG islands on HBV genomic DNA also contributes to the regulation of HBV gene expression (Mogul et al., 2011; Rivenbark et al., 2012). It has been shown that early integrated HBV DNA is methylated in HCC cells (Miller and Robinson, 1983; Chen et al., 1988). The non-integrated HBV DNA (Vivekanandan et al., 2008b) and cccDNA (Guo et al., 2009) could also be methylated in liver tissues from patients. Currently, at least six CpG islands have been identified in the HBV genome, including three conventional regions overlapping the start site of the HBV S gene (island 1), the region encompassing enhancer I and the X gene promoter (island 2), and the Sp1 promoter and start codon of the P gene (island 3; Zhang et al., 2013b). Methylation of CpG islands 1 and 2 was found in HBV DNA extracted from liver biopsies from CHB patients, suggesting that increased methylation of HBV DNA may decrease the production of viral proteins (Vivekanandan et al., 2008b). The hypermethylation of island 2 was correlated with low levels or absence of HBsAg production (Vivekanandan et al., 2008a), as well as reduced HBeAg expression (Guo et al., 2009). It was shown that individuals with occult HBV infection, which is characterized by the persistence of HBV DNA in the liver of individuals who test negative for the HBsAg, had a higher degree of methylation in island 2 compared to non-occult CHB patients (Vivekanandan et al., 2008a). Another study with a cohort of cirrhosis patients did not find an association between the methylation status of HBV cccDNA and HBsAg expression in liver tissues, but confirmed that a higher methylation density was associated with lower viral load, lower RNA copies per cccDNA, and lower virion productivity (Kim et al., 2011).
Consistent with these findings, transfection of methylated HBV DNA in HepG2 cells resulted in reduced HBV mRNA levels, decreased intracellular HBsAg and core HBcAg expression, and decreased secretion of HBV viral proteins into cell supernatants. Furthermore, an in vitro equivalent of cccDNA showed decreased viral protein production in HepG2 cells after DNA methylation (Vivekanandan et al., 2009). After transfection of HBV DNA into HepG2 cells, an inverse relationship between methylated HBV DNA and viral mRNA levels was observed in dependence on the upregulation of host DNA methyltransferase (DNMT). Cotransfection with DNMT3a and HBV DNA was associated with decreased production of HBsAg and HBeAg, as well as host proteins implicated in carcinogenesis (Vivekanandan et al., 2010). These data from cell culture experiments suggest that HBV DNA methylation is associated with down regulation of viral protein production.
INTERPLAY BETWEEN HBV, miRNAs, AND THE EPIGENETIC MACHINERY
miRNAs PLAY A PIVOTAL ROLE IN THE EPIGENETIC REGULATION NETWORK
MicroRNAs are approximately 22 nucleotide-long non-coding RNAs that are emerging as key players in regulating gene expression in eukaryotes, influencing various biological processes such as development, infection, immunity, and carcinogenesis (Ambros, 2004). The biogenesis and mechanisms of action of these tiny but potent molecules have been described in detail (Bartel, 2004). Briefly, miRNAs are transcribed from the host genome and generated by Drosha- and Dicer-mediated enzymatic cleavage. Mature miRNAs are engaged in either translational arrest or degradation of targeted transcripts through imperfect base pairing with the 3′-untranslated region (UTR) or the coding region of the target transcripts. Currently, more than 2000 miRNAs have been identified in human organs (Griffiths-Jones et al., 2008). The expression profiles of these miRNAs in different cells or tissues may exhibit temporal or tissue-specific patterns (Skalsky and Cullen, 2010).
Many studies have shown that a set of miRNAs play a pivotal role in the epigenetic regulation network (Chuang and Jones, 2007; Iorio et al., 2010). Epigenetic modifications, such as promoter methylation or histone acetylation, have been demonstrated to affect miRNA expression and are potentially responsible for the aberrant miRNA regulation observed in cancer (Baer et al., 2013). Along with the epigenetic regulation of miRNA expression, many miRNAs themselves can regulate the expression of components of the epigenetic machinery, creating a highly controlled feedback mechanism. A number of the miRNAs related to epigenetic regulation were defined as so-called “epi-miRNAs.” For example, DNMT1 overexpression was responsible for the hypermethylation of the miR-148a and miR-152 promoters. As a direct target of miR-148a and miR-152, DNMT1 was inversely related to the expression levels of miR-148a and miR-152 (Chen et al., 2013). Similarly, miR-1 and miR-449a, which could be induced by 5-AzaC/TSA treatment (Datta et al., 2008) or by HDAC1-3 knock down (Buurman et al., 2012) in HCC cells, directly targeted HDAC4 (Chen et al., 2006) and HDAC1 (Noonan et al., 2009), respectively.
HBV INFECTION AFFECTS miRNA EXPRESSION
Although the viral miRNAs encoded by HBV have not been verified, the products of HBV were shown to alter miRNA expression profiles. In chronic HBV infection or HBV-related HCC, the miRNA profiles in liver tissue or serum levels from numerous studies are controversial and complicated (Ura et al., 2009; Hou et al., 2011; Liu et al., 2011; Wang et al., 2012b). For instance, it was reported that subviral HBsAg circulating in the blood of HBV carriers could carry liver-specific miRNAs (miR-27a, miR-30b, miR-122, miR-126, and miR-145) as well as immune regulatory miRNAs (miR-106b and miR-223) that were involved in hepatocarcinogenesis and HBV persistence (Novellino et al., 2012). In another study, three miRNAs (miR-122, miR-22, and miR-99a) were upregulated at least 1.5-fold in the sera of HBV-infected patients (Hayes et al., 2012). The highly liver-enriched, abundantly expressed miR-122 was consistently upregulated in HBV infected patients, and miR-145 could be used as a candidate tumor suppressive miRNA in the early steps of HBV-related hepatocarcinogenesis (Gao et al., 2011).
Recently, molecular studies have revealed that the HBx protein, which is essential for virus replication in vivo, induced epigenetic changes, including aberrations in DNA methylation, histone modifications, and miRNA expression. HBx expression has been found to be associated with alterations in the host miRNA profile through different epigenetic mechanisms (Tian et al., 2013). MiRNAs upregulated by HBx include miRNA-29a and miR-143 (Zhang et al., 2009; Kong et al., 2011). HBx-downregulated miRNAs include miR-101, miR-122, miR-132, miR-148a, miR-152, let-7, and the miR-16 family (Huang et al., 2010; Wang et al., 2010; Wu et al., 2011; Song et al., 2013; Wei et al., 2013a, b; Xu et al., 2013). In addition, HBx was shown to activate HBV transcription through opposition to the protein phosphatase 1 and HDAC1 complex on the HBV cccDNA (Cougot et al., 2012), or down-regulate DNMT3A expression through miR-101 induction (Wei et al., 2013b). Loss of HBx reduced recruitment of p300, caused rapid hypoacetylation of the cccDNA-bound histones and increased early recruitment of SIRT1 and HDAC1, accompanied by lower HBV replication (Belloni et al., 2009).
CELLULAR miRNAs INHIBIT HBV REPLICATION BY DIRECT BINDING
As HBV produces different transcripts during its life cycle, the transcripts are proposed to be targeted by cellular miRNAs. In a screen for cellular miRNAs affecting HBV replication, Zhang et al. (2010) employed a loss-of-function approach by transfecting antagomirs targeting 328 human miRNAs into HepG2 cells. Two miRNAs, miR-199a-3p and miR-210, were shown to suppress HBsAg expression. The direct effect of these two miRNAs on HBV RNA transcripts was validated by GFP reporter assay (Zhang et al., 2010). In addition, Russo’s group found that miR-125a-5p is able to interfere with HBsAg expression, thus reducing the amount of secreted HBsAg (Potenza et al., 2011). Recently, many cancer-related miRNAs, including miR-15a/miR-16-1 (Wang et al., 2013a), the miR-17-92 cluster (Jung et al., 2013), and miR-224 (Scisciani et al., 2011), were shown to target HBV mRNAs directly by luciferase reporter assay and inhibit HBV replication (summarized in Figure 1). Notably, the expression of these miRNAs was also linked to epigenetic regulation, as well as to promoter methylation (Dakhlallah et al., 2013) and histone acetylation (Zhang et al., 2013a; Wang et al., 2013b).
FIGURE 1
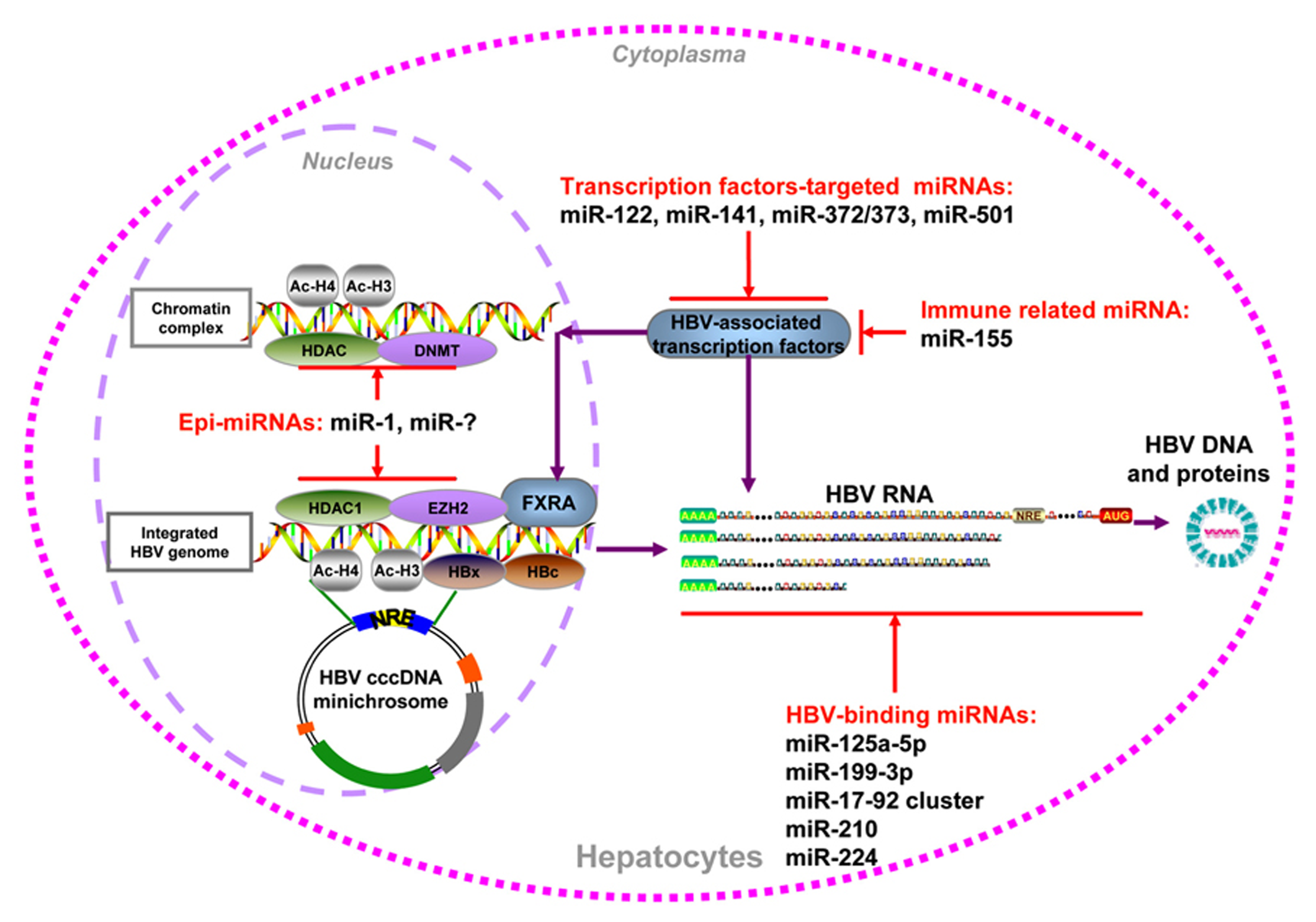
Summary of cellular miRNAs effect on HBV replication. HBV DNA could be integrated into host genome or form cccDNA minichromosome in the hepatocytes nucleus. It serves as the temple for viral transcription, viral DNA, and antigens production. The miRNAs which regulate HBV replication through epigenetic mechanism, transcription factors regulation, immune regulation, and direct targeting viral transcripts were indicated.
CELLULAR miRNAs REGULATE HBV REPLICATION INDIRECTLY
In addition to direct targeting, some cellular miRNAs, including epi-miRNAs, were found to be capable of inhibiting or stimulating HBV replication by indirectly regulating cellular transcription factors. It was shown that the transcription of HBV cccDNA was tightly regulated by a number of liver-enriched transcription factors and nuclear receptors through the recognition of HBV promoter/enhancer elements (Quasdorff and Protzer, 2010). miR-122 may exert its effect on HBV indirectly via downregulation of its target cyclin G1, thus interrupting the interaction between cyclin G1 and p53 and abrogating p53-mediated inhibition of HBV replication (Wang et al., 2012a). miR-372 and -373 are upregulated in HBV-infected liver tissues and promote HBV gene expression through a pathway involving the transcription factor nuclear factor I/B (Guo et al., 2011). The higher expression of miR-501 in HCC tissues could enhance HBV replication partially by targeting HBXIP (Jin et al., 2013). In contrast, miR-141 significantly suppresses HBV expression and replication in HepG2 cells. Bioinformatic analysis and experimental assays indicate that peroxisome proliferator-activated receptor alpha is a relevant target of miR-141 during this process (Hu et al., 2013). For immune-related miRNAs, miR-155 enhances innate antiviral immunity by promoting the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway through the targeting of SOCS1, mildly inhibiting HBV infection in human hepatoma cells (Su et al., 2011).
By screening a set of cellular miRNAs, our group found that epigenetically regulated miR-1 over-expression resulted in a marked increase in HBV replication, accompanied with upregulated HBV transcription, antigen expression, and progeny secretion. HDAC4, the cellular target of miR-1, was able to suppress HBV replication. The expression of nuclear receptor farnesoid X receptor alpha (FXRA) was increased by miR-1, leading to the enhanced transcriptional activity of the HBV core promoter (Zhang et al., 2011). Furthermore, another epi-miRNA that targets HDAC1, miR-449a, had an even higher capacity for enhancing HBV replication but a lower level of induction of FXRA (Zhang et al., unpublished data). Additionally, both of these two defined epi-miRNAs could inhibit the G1/S cell cycle transition and promote cell differentiation by increasing the expression of hepatocyte-specific factors, which may be beneficial for HBV replication (Zhang et al., 2011). Collectively, host epi-miRNAs can modulate HBV replication by regulating cellular epigenetic factors or specific transcription factors that directly bind to the HBV cccDNA minichromosome (summarized in Figure 1).
CONCLUSION AND PERSPECTIVE
In this review, we summarize the available information about the epigenetic mechanisms involved in the regulation of HBV cccDNA function. Notably, miRNAs could be considered part of a multilevel regulatory mechanism aimed to precisely modulate HBV replication and gene expression, likely in the response to the changing hepatic microenvironment. Considerably, many cellular miRNAs indirectly influence the HBV life cycle by regulating the expression of relevant cellular proteins and may play important roles in hepatitis B pathogenesis. Future studies need to be performed to elucidate the regulatory loop involving miRNAs and the cccDNA epigenetic machinery and certainly to investigate how to translate these findings into clinical applications.
Statements
Acknowledgments
This work was supported in part by grants from the Deutsche Forschungs-gemeinschaft (Transregio TRR60 and GRK1045/2).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
- 5-AzaC
5-azacytidine
- cccDNA
covalently closed circular DNA
- CHB
chronic hepatitis B
- DNMT
DNA methyltransferase
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HDAC
histone deacetylase
- IFN
interferon
- miRNA
microRNA
- SIRT1
sirtuin 1
- TSA
trichostatin A.
References
1
Ambros V. (2004). The functions of animal microRNAs.Nature431350–355. 10.1038/nature02871
2
Baer C. Claus R. Plass C. (2013). Genome-wide epigenetic regulation of miRNAs in cancer.Cancer Res.73473–477. 10.1158/0008-5472.CAN-12-3731
3
Bartel D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function.Cell116281–297. 10.1016/S0092-8674(04)00045-5
4
Belloni L. Allweiss L. Guerrieri F. Pediconi N. Volz T. Pollicino T. et al (2012). IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome.J. Clin. Invest.122529–537. 10.1172/JCI58847
5
Belloni L. Pollicino T. De Nicola F. Guerrieri F. Raffa G. Fanciulli M. et al (2009). Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function.Proc. Natl. Acad. Sci. U.S.A.10619975–19979. 10.1073/pnas.0908365106
6
Bock C. T. Schranz P. Schroder C. H. Zentgraf H. (1994). Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell.Virus Genes8215–229. 10.1007/BF01703079
7
Bock C. T. Schwinn S. Locarnini S. Fyfe J. Manns M. P. Trautwein C. et al (2001). Structural organization of the hepatitis B virus minichromosome.J. Mol. Biol.307183–196. 10.1006/jmbi.2000.4481
8
Bremer C. M. Bung C. Kott N. Hardt M. Glebe D. (2009). Hepatitis B virus infection is dependent on cholesterol in the viral envelope.Cell. Microbiol.11249–260. 10.1111/j.1462-5822.2008.01250.x
9
Buurman R. Gurlevik E. Schaffer V. Eilers M. Sandbothe M. Kreipe H. et al (2012). Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells.Gastroenterology143811–820.e15. 10.1053/j.gastro.2012.05.033
10
Chen J. F. Mandel E. M. Thomson J. M. Wu Q. Callis T. E. Hammond S. M. et al (2006). The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation.Nat. Genet.38228–233. 10.1038/ng1725
11
Chen J. Y. Hsu H. C. Lee C. S. Chen D. S. Zuckerman A. J. Harrison T. J. (1988). Detection of hepatitis B virus DNA in hepatocellular carcinoma: methylation of integrated viral DNA.J. Virol. Methods19257–263. 10.1016/0166-0934(88)90020-1
12
Chen Y. Song Y. X. Wang Z. N. (2013). The microRNA-148/152 family: multi-faceted players.Mol. Cancer1243 10.1186/1476-4598-12-43
13
Chuang J. C. Jones P. A. (2007). Epigenetics and microRNAs.Pediatr. Res.6124R–29R. 10.1203/pdr.0b013e3180457684
14
Cougot D. Allemand E. Riviere L. Benhenda S. Duroure K. Levillayer F. et al (2012). Inhibition of PP1 phosphatase activity by HBx: a mechanism for the activation of hepatitis B virus transcription.Sci. Signal.5ra1 10.1126/scisignal.2001906
15
Dakhlallah D. Batte K. Wang Y. Cantemir-Stone C. Z. Yan P. Nuovo G. et al (2013). Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis.Am. J. Respir. Crit. Care Med.187397–405. 10.1164/rccm.201205-0888OC
16
Datta J. Kutay H. Nasser M. W. Nuovo G. J. Wang B. Majumder S. et al (2008). Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis.Cancer Res.685049–5058. 10.1158/0008-5472.CAN-07-6655
17
Dienstag J. L. (2008). Hepatitis B virus infection.N. Engl. J. Med.3591486–1500. 10.1056/NEJMra0801644
18
Gao P. Wong C. C. Tung E. K. Lee J. M. Wong C. M. Ng I. O. (2011). Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis.J. Hepatol.541177–1184. 10.1016/j.jhep.2010.09.023
19
Gao W. Hu J. (2007). Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein.J. Virol.816164–6174. 10.1128/JVI.02721-06
20
Gong Q. Chen S. Guo J. Sun H. Zheng G. Liu Q. et al (2011). Chromosome remodeling related to hepatitis B virus replication in HepG2 cells.DNA Cell Biol.30347–354. 10.1089/dna.2010.1172
21
Griffiths-Jones S. Saini H. K. Van Dongen S. Enright A. J. (2008). miRBase: tools for microRNA genomics.Nucleic Acids Res.36D154–D158. 10.1093/nar/gkm952
22
Guo H. Liu H. Mitchelson K. Rao H. Luo M. Xie L. et al (2011). MicroRNAs-372/373 promote the expression of hepatitis B virus through the targeting of nuclear factor I/B.Hepatology54808–819. 10.1002/hep.24441
23
Guo Y. Li Y. Mu S. Zhang J. Yan Z. (2009). Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication.J. Med. Virol.811177–1183. 10.1002/jmv.21525
24
Hayes C. N. Akamatsu S. Tsuge M. Miki D. Akiyama R. Abe H. et al (2012). Hepatitis B virus-specific miRNAs and Argonaute2 play a role in the viral life cycle.PLoS ONE7:e47490. 10.1371/journal.pone.0047490
25
Hou J. Lin L. Zhou W. Wang Z. Ding G. Dong Q. et al (2011). Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma.Cancer Cell19232–243. 10.1016/j.ccr.2011.01.001
26
Hu W. Wang X. Ding X. Li Y. Zhang X. Xie P. et al (2013). MicroRNA-141 represses HBV replication by targeting PPARA.PLoS ONE7:e34165. 10.1371/journal.pone.0034165
27
Huang J. Wang Y. Guo Y. Sun S. (2010). Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1.Hepatology5260–70. 10.1002/hep.23660
28
Iorio M. V. Piovan C. Croce C. M. (2010). Interplay between microRNAs and the epigenetic machinery: an intricate network.Biochim. Biophys. Acta1799694–701. 10.1016/j.bbagrm.2010.05.005
29
Jin J. Tang S. Xia L. Du R. Xie H. Song J. et al (2013). MicroRNA-501 promotes HBV replication by targeting HBXIP.Biochem. Biophys. Res. Commun.4301228–1233. 10.1016/j.bbrc.2012.12.071
30
Jung Y. J. Kim J. W. Park S. J. Min B. Y. Jang E. S. Kim N. Y. et al (2013). c-Myc-mediated overexpression of miR-17-92 suppresses replication of hepatitis B virus in human hepatoma cells.J. Med. Virol.85969–978. 10.1002/jmv.23534
31
Kim J. W. Lee S. H. Park Y. S. Hwang J. H. Jeong S. H. Kim N. et al (2011). Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection.Intervirology54316–325. 10.1159/000321450
32
Kong G. Zhang J. Zhang S. Shan C. Ye L. Zhang X. (2011). Upregulated microRNA-29a by hepatitis B virus X protein enhances hepatoma cell migration by targeting PTEN in cell culture model.PLoS ONE6:e19518. 10.1371/journal.pone.0019518
33
Levrero M. Pollicino T. Petersen J. Belloni L. Raimondo G. Dandri M. (2009). Control of cccDNA function in hepatitis B virus infection.J. Hepatol.51581–592. 10.1016/j.jhep.2009.05.022
34
Liu A. M. Zhang C. Burchard J. Fan S. T. Wong K. F. Dai H. et al (2011). Global regulation on microRNA in hepatitis B virus-associated hepatocellular carcinoma.OMICS15187–191. 10.1089/omi.2010.0098
35
Lutgehetmann M. Volz T. Kopke A. Broja T. Tigges E. Lohse A.W. et al (2010). In vivo proliferation of hepadnavirus-infected hepatocytes induces loss of covalently closed circular DNA in mice.Hepatology5216–24. 10.1002/hep.23611
36
Mailliard M. E. Gollan J. L. (2006). Emerging therapeutics for chronic hepatitis B.Annu. Rev. Med.57155–166. 10.1146/annurev.med.57.121304.131422
37
McMahon B. J. (2009). The natural history of chronic hepatitis B virus infection.Hepatology49S45–S55. 10.1007/s12072-008-9112-z
38
Miller R. H. Robinson W. S. (1983). Integrated hepatitis B virus DNA sequences specifying the major viral core polypeptide are methylated in PLC/PRF/5 cells.Proc. Natl. Acad. Sci. U.S.A.802534–2538. 10.1073/pnas.80.9.2534
39
Mogul D. Torbenson M. Schwarz K. B. (2011). Epigenetic regulation of hepatitis B virus infection.Curr. Hepat. Rep.10277–284. 10.1007/s11901-011-0113-3
40
Murray J. M. Wieland S. F. Purcell R. H. Chisari F. V. (2005). Dynamics of hepatitis B virus clearance in chimpanzees.Proc. Natl. Acad. Sci. U.S.A.10217780–17785. 10.1073/pnas.0508913102
41
Newbold J. E. Xin H. Tencza M. Sherman G. Dean J. Bowden S. et al (1995). The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes.J. Virol.693350–3357.
42
Noonan E. J. Place R. F. Pookot D. Basak S. Whitson J. M. Hirata H. et al (2009). miR-449a targets HDAC-1 and induces growth arrest in prostate cancer.Oncogene281714–1724. 10.1038/onc.2009.19
43
Novellino L. Rossi R. L. Bonino F. Cavallone D. Abrignani S. Pagani M. et al (2012). Circulating hepatitis B surface antigen particles carry hepatocellular microRNAs.PLoS ONE7:e31952. 10.1371/journal.pone.0031952
44
Palumbo G. Belloni L. Valente S. Rotili D. Pediconi N. Mai A. et al (2013). Suppression of hepatitis B virus (HBV) transcription and replication by small molecules that target the epigenetic control of nuclear cccDNA minichromosome.J. Hepatol.58S25 10.1016/S0168-8278(13)60058-6
45
Pardo M. Bartolome J. Carreno V. (2007). Current therapy of chronic hepatitis B.Arch. Med. Res.38661–677. 10.1016/j.arcmed.2006.12.013
46
Pollicino T. Belloni L. Raffa G. Pediconi N. Squadrito G. Raimondo G. et al (2006). Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones.Gastroenterology130823–837. 10.1053/j.gastro.2006.01.001
47
Potenza N. Papa U. Mosca N. Zerbini F. Nobile V. Russo A. (2011). Human microRNA hsa-miR-125a-5p interferes with expression of hepatitis B virus surface antigen.Nucleic Acids Res.395157–5163. 10.1093/nar/gkr067
48
Quasdorff M. Protzer U. (2010). Control of hepatitis B virus at the level of transcription.J. Viral Hepat.17527–536. 10.1111/j.1365-2893.2010.01315.x
49
Rivenbark A. G. Stolzenburg S. Beltran A. S. Yuan X. Rots M. G. Strahl B. D. et al (2012). Epigenetic reprogramming of cancer cells via targeted DNA methylation.Epigenetics7350–360. 10.4161/epi.19507
50
Schulze A. Gripon P. Urban S. (2007). Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans.Hepatology461759–1768. 10.1002/hep.21896
51
Scisciani C. Belloni L. Guerrieri F. Levrero M. Pediconi N. (2011). mir-224 is a direct target of hbx and modulates hbv replication.J. Hepatol.54S44410.1016/S0168-8278(11)61123-9
52
Seeger C. Mason W. S. (2000). Hepatitis B virus biology.Microbiol. Mol. Biol. Rev.6451–68. 10.1128/MMBR.64.1.51-68.2000
53
Skalsky R. L. Cullen B. R. (2010). Viruses, microRNAs, and host interactions.Annu. Rev. Microbiol.64123–141. 10.1146/annurev.micro.112408.134243
54
Song K. Han C. Zhang J. Lu D. Dash S. Feitelson M. et al (2013). Epigenetic regulation of miR-122 by PPARgamma and hepatitis B virus X protein in hepatocellular carcinoma cells.Hepatology.10.1002/hep.26514[Epub ahead of print].
55
Su C. Hou Z. Zhang C. Tian Z. Zhang J. (2011). Ectopic expression of microRNA-155 enhances innate antiviral immunity against HBV infection in human hepatoma cells.Virol. J.8354 10.1186/1743-422X-8-354
56
Tian Y. Yang W. Song J. Wu Y. Ni B. (2013). Hepatitis B virus x protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis.Mol. Cell. Biol.332810–2816. 10.1128/MCB.00205-13
57
Ura S. Honda M. Yamashita T. Ueda T. Takatori H. Nishino R. et al (2009). Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma.Hepatology491098–1112. 10.1002/hep.22749
58
Vivekanandan P. Daniel H. D. Kannangai R. Martinez-Murillo F. Torbenson M. (2010). Hepatitis B virus replication induces methylation of both host and viral DNA.J. Virol.844321–4329. 10.1128/JVI.02280-09
59
Vivekanandan P. Kannangai R. Ray S. C. Thomas D. L. Torbenson M. (2008a). Comprehensive genetic and epigenetic analysis of occult hepatitis B from liver tissue samples.Clin. Infect. Dis.461227–1236. 10.1086/529437
60
Vivekanandan P. Thomas D. Torbenson M. (2008b). Hepatitis B viral DNA is methylated in liver tissues.J. Viral Hepat.15103–107. 10.1111/j.1365-2893.2007.00905.x
61
Vivekanandan P. Thomas D. Torbenson M. (2009). Methylation regulates hepatitis B viral protein expression.J. Infect. Dis.1991286–1291. 10.1086/597614
62
Wang S. Qiu L. Yan X. Jin W. Wang Y. Chen L. et al (2012a). Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1)-modulated P53 activity.Hepatology55730–741. 10.1002/hep.24809
63
Wang W. Zhao L. J. Tan Y. X. Ren H. Qi Z. T. (2012b). Identification of deregulated miRNAs and their targets in hepatitis B virus-associated hepatocellular carcinoma.World J. Gastroenterol.185442–5453. 10.3748/wjg.v18.i38.5442
64
Wang Y. Jiang L. Ji X. Yang B. Zhang Y. Fu X. D. (2013a). Hepatitis B viral RNA directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16-1 in hepatocytes.J. Biol. Chem.28818484–18493. 10.1074/jbc.M113.458158
65
Wang Y. Toh H. C. Chow P. Chung A. Y. Meyers D. J. Cole P. A. et al (2013b). MicroRNA-224 is up-regulated in hepatocellular carcinoma through epigenetic mechanisms.FASEB J.263032–3041. 10.1096/fj.11-201855
66
Wang Y. Lu Y. Toh S. T. Sung W. K. Tan P. Chow P. et al (2010). Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3.J. Hepatol.5357–66. 10.1016/j.jhep.2009.12.043
67
Wei X. Tan C. Tang C. Ren G. Xiang T. Qiu Z. et al (2013a). Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma.Cell. Signal.251037–1043. 10.1016/j.cellsig.2013.01.019
68
Wei X. Xiang T. Ren G. Tan C. Liu R. Xu X. et al (2013b). miR-101 is down-regulated by the hepatitis B virus x protein and induces aberrant DNA methylation by targeting DNA methyltransferase 3A.Cell. Signal.25439–446. 10.1016/j.cellsig.2012.10.013
69
Werle-Lapostolle B. Bowden S. Locarnini S. Wursthorn K. Petersen J. Lau G. et al (2004). Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy.Gastroenterology1261750–1758. 10.1053/j.gastro.2004.03.018
70
Wu G. Yu F. Xiao Z. Xu K. Xu J. Tang W. et al (2011). Hepatitis B virus X protein downregulates expression of the miR-16 family in malignant hepatocytes in vitro.Br. J. Cancer105146–153. 10.1038/bjc.2011.190
71
Xu X. Fan Z. Kang L. Han J. Jiang C. Zheng X. et al (2013). Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis.J. Clin. Invest.123630–645. 10.1172/JCI64265
72
Yan H. Zhong G. Xu G. He W. Jing Z. Gao Z. et al (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus.eLife1e00049 10.7554/eLife.00049
73
Zhang G. L. Li Y. X. Zheng S. Q. Liu M. Li X. Tang H. (2010). Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210.Antiviral Res.88169–175. 10.1016/j.antiviral.2010.08.008
74
Zhang X. Chen X. Lin J. Lwin T. Wright G. Moscinski L. C. et al (2013a). Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas.Oncogene313002–3008. 10.1038/onc.2011.470
75
Zhang Y. Li C. Zhu H. Kang Y. Liu H. Wang J. et al (2013b). Comparative analysis of CpG islands among HBV genotypes.PLoS ONE8:e56711. 10.1371/journal.pone.0056711
76
Zhang X. Liu S. Hu T. He Y. Sun S. (2009). Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression.Hepatology50490–499. 10.1002/hep.23008
77
Zhang X. Zhang E. Ma Z. Pei R. Jiang M. Schlaak J. F. et al (2011). Modulation of hepatitis B virus replication and hepatocyte differentiation by microRNA-1.Hepatology531476–1485. 10.1002/hep.24195
78
Zoulim F. (2005). New insight on hepatitis B virus persistence from the study of intrahepatic viral cccDNA.J. Hepatol.42302–308. 10.1016/j.jhep.2004.12.015
Summary
Keywords
hepatitis B virus, microRNA, epigenetic regulation, histone deacetylases, DNA methyltransferase
Citation
Zhang X, Hou J and Lu M (2013) Regulation of hepatitis B virus replication by epigenetic mechanisms and microRNAs. Front. Genet. 4:202. doi: 10.3389/fgene.2013.00202
Received
30 May 2013
Accepted
22 September 2013
Published
14 October 2013
Volume
4 - 2013
Edited by
Silvia Carolina Galvan, Universidad Nacional Autónoma de México, Mexico
Reviewed by
Nejat Dalay, Istanbul University Oncology Institute, Turkey; Sushma S. Iyengar, University of Southern California, USA
Copyright
© Zhang, Hou and Lu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mengji Lu, Institute of Virology, University Hospital of Essen, University of Duisburg Essen, Hufelandstrasse 55, 45122 Essen, Germany e-mail: mengji.lu@uni-due.de
This article was submitted to Epigenomics and Epigenetics, a section of the journal Frontiers in Genetics.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.