 Martin J. Allday
Martin J. Allday- Section of Virology, Department of Medicine, Imperial College London, London, UK
Viruses that establish a persistent infection, involving intracellular latency, commonly stimulate cellular DNA synthesis and sometimes cell division early after infection. However, most cells of metazoans have evolved “fail-safe” responses that normally monitor unscheduled DNA synthesis and prevent cell proliferation when, for instance, cell proto-oncogenes are “activated” by mutation, amplification, or chromosomal rearrangements. These cell intrinsic defense mechanisms that reduce the risk of neoplasia and cancer are collectively called oncogenic stress responses (OSRs). Mechanisms include the activation of tumor suppressor genes and the so-called DNA damage response that together trigger pathways leading to cell cycle arrest (e.g., cell senescence) or complete elimination of cells (e.g., apoptosis). It is not surprising that viruses that can induce cellular DNA synthesis and cell division have the capacity to trigger OSR, nor is it surprising that these viruses have evolved countermeasures for inactivating or bypassing OSR. The main focus of this review is how the human tumor-associated Epstein–Barr virus manipulates the host polycomb group protein system to control – by epigenetic repression of transcription – key components of the OSR during the transformation of normal human B cells into permanent cell lines.
Introduction – The Biology of Epstein–Barr Virus (EBV)
Epstein–Barr virus (EBV) is a human gamma-herpesvirus (HHV4) and as such is characterized by a tropism for lymphocytes and an ability to persist life-long in the infected host. Data on persistent EBV infection in humans are consistent with the viral genome residing in a population of long-lived, largely non-dividing memory B cells. To establish persistence, EBV first infects resting (naïve) B cells – probably in tissues of the oropharynx – and transiently drives these to proliferate as activated B-blasts. The expanding B-blast population is thought to then either migrate into, or nucleate the formation of, a germinal center in local lymphoid tissue and therein the cells differentiate to become centroblasts, centrocytes, and finally resting memory B cells that enter the peripheral circulation (reviewed in Thorley-Lawson and Gross, 2004; Roughan and Thorley-Lawson, 2009). While the precise series of events that the EBV-positive B cells undergo to reach the memory compartment is not yet known, it is generally agreed that it involves regulated shut-down of latent EBV gene expression from an initial state called latency III, via latency II, until in quiescent memory B cells no EBV proteins can be detected in a state called latency 0. However, there is still some controversy as to whether or not the differentiation of EBV-infected B-blasts to resting memory B cells can occur anywhere outside the microenvironment of a germinal center (Rowe et al., 2009; Heath et al., 2012; Thorley-Lawson et al., 2013).
In more than 90% of the global population, following primary infection in infancy, EBV establishes an asymptomatic, stable, life-long, persistent infection in this long-lived pool of circulating memory cells. Periodic activation of an infected memory B cell by exposure to cognate antigen or aberrant T cell activity is thought to trigger plasma cell differentiation and concomitant “lytic” viral replication with the production of infectious virus that is released in the oropharynx and shed in saliva (reviewed in Thorley-Lawson and Gross, 2004; Laichalk and Thorley-Lawson, 2005; Roughan and Thorley-Lawson, 2009).
Primary EBV infection can cause the benign self-limiting disease infectious mononucleosis (IM) in some adolescents who were not infected in childhood. Uncontrolled proliferation of infected B cells in the immunocompromised of any age may result in a fatal form of IM, a chronic B lymphoproliferative disease or rarely the development of malignant immunoblastic lymphoma (Williams and Crawford, 2006). In normal individuals EBV-infected B-blasts are targets for EBV-specific cytotoxic T lymphocytes (CTLs) that recognize and destroy the EBV-infected proliferating B-blasts – so an equilibrium is established between B-blast proliferation on the one hand, and their immune-mediated elimination or differentiation to resting memory B cells on the other (Babcock et al., 1999; Thorley-Lawson and Gross, 2004; Hislop et al., 2007). Individuals who are co-infected with malaria or HIV are at increased risk of developing EBV-associated lymphomas, including Burkitt’s lymphoma (BL). EBV is also etiologically linked to subgroups of Hodgkin’s lymphoma (HL) and diffuse large B cell lymphoma (DLBCL), in addition to various non-B cell malignancies (reviewed in Young and Rickinson, 2004).
Infection of resting naïve B cells ex vivo with EBV can also induce the proliferation of the B-blast-like cells that in vivo would differentiate to become memory cells. In vitro these B cells do not differentiate, but are transformed to continuously proliferating permanent lymphoblastoid cell lines (LCLs) that retain the activated B cell phenotype and carry the viral genome as extra-chromosomal episomes. Only the nine latency III-associated proteins six nuclear (EBNAs 1, 2, 3A, 3B, 3C, and LP) and three membrane-associated (LMP1, LMP2A, and 2B) together with several RNA species are expressed from the viral genome (reviewed in Bornkamm and Hammerschmidt, 2001; Young and Rickinson, 2004; Forte and Luftig, 2011). These latency-associated gene products are responsible for activating the quiescent primary cells into the cell cycle, inducing and sustaining their proliferation and maintaining the extrachromosomal episome in these blast-like cells. There is general agreement – that at least in the initial stages after infection – LCL outgrowth recapitulates the early events of establishing latency prior to differentiation and long-term persistence in vivo. EBV may therefore be considered one of the few viruses that initiate and sustain the proliferation of infected cells as a necessary step in their life cycle, in the natural host. Some of the molecular details of how EBV does this in the face of intrinsic barriers to aberrant proliferation are the focus of this review. Specific attention will be paid to the polycomb group (PcG) protein-mediated epigenetic repression of the cyclin-dependent kinase inhibitor p16INK4a and the pro-apoptotic BH3-only inducer of apoptosis BIM.
Oncogenic Stress Responses (OSRs) and Oncogene-Induced Senescence (OIS)
The seminal discovery in 1992 that the Myc proto-oncoprotein can trigger rapid apoptosis as well as cell growth and proliferation, led to the compelling hypothesis that apoptotic pathways must be disabled for oncogenes to promote neoplastic transformation of cells and the development of cancer (Askew et al., 1991; Evan et al., 1992). About 5 years later an equally influential discovery was that oncogenic mutant Ras protein – in addition to activating proliferative signaling pathways – also provokes in normal fibroblasts a cell cycle arrest resembling premature cell senescence. This was associated with the accumulation of tumor suppressors (ts) p53 and p16INK4a (Serrano et al., 1997), and further endorsed the hypothesis that normal mammalian cells possess intrinsic defenses against oncogenic transformation. These observations inspired the concepts of “OSR,” “intrinsic tumor suppression,” and “OIS” and produced many detailed descriptions of mechanisms involving the p53 and retinoblastoma (Rb) tumor suppressor pathways that prevent deregulated oncogenes causing cancer (Figure 1; reviewed in Sherr, 1998, 2012; Lowe et al., 2004; Braig and Schmitt, 2006).
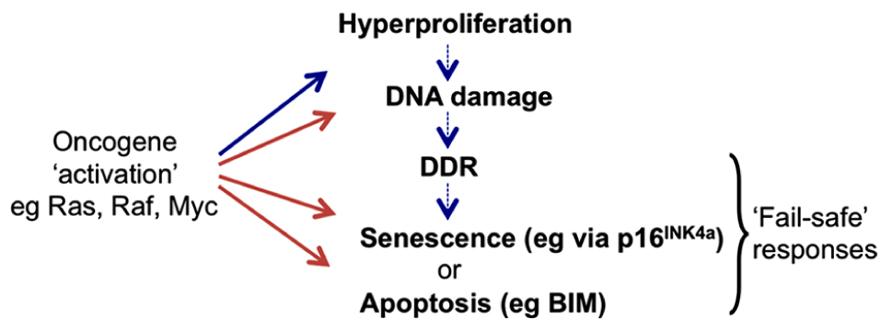
FIGURE 1. Activation of cell proto-oncogenes can lead to oncogenic stress responses (OSR). Oncogene “activation” by mutation or constitutive expression at supra-physiological levels can induce aberrant cell division that may become manifest as rapid cell proliferation (hyperproliferation). However higher vertebrates have evolved cell intrinsic “fail-safe” responses to recognize such cells and so block their proliferation or eliminate them completely. These can include the induction of tumor suppressors (ts) such as p16INK4a that halt the cell cycle and can cause cells to enter a prolonged state of arrest called senescence, or pro-apoptotic proteins such as BIM that can induce programmed cell death (apoptosis). Responses may involve direct activation of the ts genes by oncoproteins or they can result from secondary signaling pathways (DDR) linked to the detection of damaged DNA produced during periods of aberrant DNA replication and cell division.
Relationship between OSR/OIS and DNA Damage Responses (DDRs)
Since cell proto-oncogenes generally control signaling pathways and/or gene networks that link proliferative signals to the cell cycle machinery, when they are deregulated this can result in unscheduled entry into S phase and aberrant DNA synthesis (sometimes referred to as “replicative stress”). As a consequence, oncogene activation can produce the stalling of DNA replication forks that results in damaged DNA – particularly double strand breaks. Such lesions can also be caused by the action of multiple physical and chemical agents and they can trigger, primarily via the ATM/ATR-kinase signaling pathway, the stabilization and activation of p53 and also the induction of 16INK4a. Depending on the physiological and cellular context this leads to DNA repair, cell death, or senescence. This complex response is known as the DDR. It has been proposed that the induction of apoptosis or cell cycle arrest/senescence by oncogenic stress is a general downstream manifestation of the DDR acting as a barrier to cell transformation in vitro and tumor progression in vivo (Di Micco et al., 2006; Bartek et al., 2007; Halazonetis et al., 2008). However, it remains unclear whether all oncogene-mediated stress responses act via the DDR, or whether alternative signaling pathways directly regulate downstream effectors (see for example induction of p16INK4a in response to oncogenic RAS/RAF signaling (Agger et al., 2009; Barradas et al., 2009) and the relationship between MYC and BIM in B cell lymphomas described below). The links between DDR, OSR, and OIS have been extensively reviewed (Braig and Schmitt, 2006; Gil and Peters, 2006; Kim and Sharpless, 2006; Wade and Wahl, 2006; Bartek et al., 2007; Halazonetis et al., 2008).
A common feature of herpesviruses is their capacity to activate DDRs in infected cells (Shirata et al., 2005; Gaspar and Shenk, 2006; Koopal et al., 2007; Tarakanova et al., 2007; Nikitin et al., 2010). Although in some cases this is associated with lytic or productive infection, when the virus has a requirement for rapid replication of its genome prior to virion assembly, at least two gamma-herpesviruses (Kaposi’s Sarcoma associated herpes virus (KSHV, aka HHV8) and EBV) trigger DDRs during the establishment of a latent infection. This is largely because latency-associated viral proteins drive cells into the cell cycle and can induce hyperproliferation, replication errors, and associated DNA damage (Koopal et al., 2007; Nikitin et al., 2010). Moreover, it has been suggested that EBV infection of B cells in vitro may also induce reactive oxygen species (ROS) that can damage DNA (reviewed in Allday, 2009; Gruhne et al., 2009). EBV and KSHV appear to have evolved mechanisms for the attenuation of the DDR to ensure latent infection is maintained. Virus-associated responses involving the DDR have recently been comprehensively reviewed elsewhere (Leidal et al., 2012; Nikitin and Luftig, 2012) and for EBV will be reconsidered below.
The INK4b-ARF-INK4a Locus, p16INK4a, OSR/OIS, Aging, and Cancer
Within the INK4b-ARF-INK4a locus at human chromosome 9p21, CDKN2A encodes two potent tumor suppressors, p16INK4a, and p14ARF (p19ARF in mice); these proteins are critical negative regulators of cell proliferation. Although exons 2 and 3 are shared by INK4a and ARF, the proteins result from differential splicing and are encoded in alternative reading frames (reviewed in Gil and Peters, 2006; Kim and Sharpless, 2006; Sherr, 2012). Adjacent to CDKN2A is a second related gene CDKN2B that encodes a protein closely related to p16INK4a called p15INK4b (Figure 2). The cyclin-dependent kinase (CDK) inhibitor p16INK4a acts on the cyclin D-dependent kinases (CDK4 and CDK6) abrogating their binding to D-type cyclins and so inhibiting CDK4/6-mediated phosphorylation of the Rb protein. By binding CDKs and blocking Rb hyperphosphorylation, increased p16INK4a expression causes a G1 cell cycle arrest and senescence (Gil and Peters, 2006; Kim and Sharpless, 2006; Sherr, 2012). Although the CDK inhibitor p15INK4b has about 85% amino acid similarity to p16INK4a and biochemically behaves in much the same way, in most mammalian cells – for unknown reasons – it has distinct functions. In contrast to the CDK inhibitors, the p14 and p19 ARF proteins regulate the stability of p53 by inactivating MDM2 – a p53-specific ubiquitin ligase that facilitates p53 degradation. The concomitant stabilization and activation of p53 leads to G1 and G2 cell cycle arrest by inducing the CDK regulator p21WAF1 or apoptosis by inducing pro-apoptotic factors such as NOXA and PUMA (Vousden and Prives, 2009; Sherr, 2012).
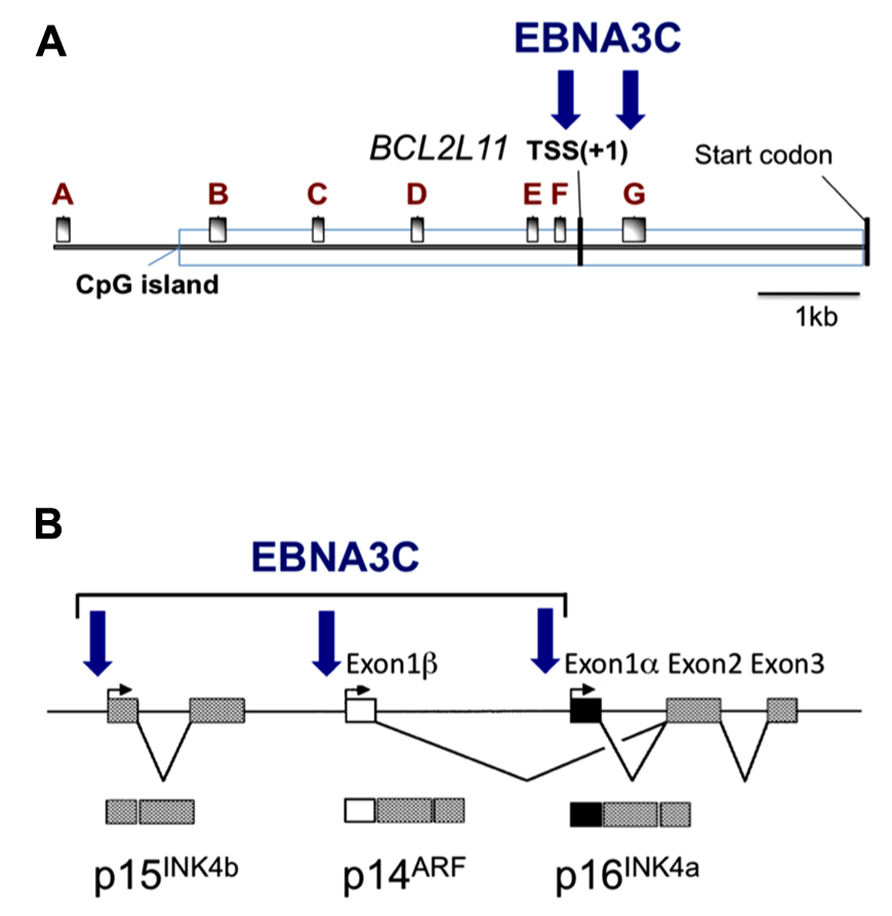
FIGURE 2. Epitope-tagged EBNA3C associates with the promoter for BIM and genes in the INK4b-ARF-INK4a locus. Schematic representations of (A) approximately 9kb of the BCL2L11 (BIM) promoter and (B) approximately 40 kb including the INK4b-ARF-INK4a locus. Vertical arrows indicate the positions where EBNA3C has been detected by chromatin immunoprecipitation (ChIP) analyses using lymphoblastoid cell lines (LCL) established using EBV expressing an epitope-tagged EBNA3C. These same regions of chromatin are marked by the polycomb (PRC2)-mediated modification H3K27me3 when EBNA3A and EBNA3C are present (adapted from Paschos et al., 2012; Skalska et al., 2013). The BCL2L11 gene transcriptional start site (TSS) and the protein products of INK4b-ARF-INK4a are indicated. A–G in (A) mark the positions of RT-PCR primer sets used in (Paschos et al., 2012).
The products of CDKN2A can therefore be key mediators of OSR and potent barriers to the “immortalization” of cells in culture and the development of cancers in vivo. Both p16INK4a and ARF are also progressively up-regulated with tissue aging, when they probably contribute to the aging process by reducing reservoirs of stem cells capable of self-renewal (Kim and Sharpless, 2006; Collado et al., 2007). There is general agreement that p19ARF plays the more important role in all these processes in mice, whereas in human cells p16INK4a is the dominant player. It is therefore not surprising that in a wide variety of human cancers INK4a is inactivated by gene deletion, mutation, or promoter DNA methylation (Gil and Peters, 2006; Kim and Sharpless, 2006; Popov and Gil, 2010). The whole INK4b-ARF-INK4a locus appears to be coordinately regulated epigenetically by polycomb protein complexes generating repressive histone modifications (Gil and Peters, 2006; Popov and Gil, 2010). Although induction of p16INK4A in fibroblasts and epithelial cells is generally associated with cell cycle arrest and senescence, in B cells – which exhibit no obvious characteristics of senescence – there may be some crosstalk between p16INK4a and the apoptotic machinery, since in lymphocytes the default pathway triggered by p16INK4a can be death rather than prolonged cell cycle arrest (Lagresle et al., 2002; Bianchi et al., 2006).
BIM, B Cells, and MYC
BIM (Bcl2-interacting mediator) is a pro-apoptotic member of the BH3-only family of BCL2-like proteins and is encoded by the BCL2L11 gene at human chromosome 2q13. BIM acts as a potent, direct initiator of apoptosis because it binds with high affinity to BCL2 and all the other pro-survival family members to inactivate them. BIM also binds and activates pro-apoptotic BAX to initiate cytochrome-c release from mitochondria (Strasser, 2005; Gavathiotis et al., 2008). BIM is particularly important in the immune system, acting as a major regulator of life-and-death decisions during lymphocyte development including the negative selection of auto-reactive B cells and programmed death of low-affinity antibody-expressing germinal center-derived B cells (Enders et al., 2003; Strasser, 2005; Fischer et al., 2007). Bim-null mice accumulate excess lymphoid and myeloid cells and loss of Bim accelerates B cell lymphomagenesis induced by an Eμ-Myc transgene. Even loss of a single allele accelerates lymphomagenesis significantly, indicating Bim is a haploinsufficient tumor suppressor and that the level of Bim protein is rate-limiting in murine B cell survival (Egle et al., 2004).
Extending this Eμ-Myc-lymphoma model to human B cell lymphomagenesis, the relationship between MYC and BIM in EBV-negative BL was investigated (Dang et al., 2005; Hemann et al., 2005). This study brought into sharp focus the activation of BCL2L11/BIM by MYC, and led to the proposal that MYC-induced apoptosis can be overridden by inactivation of any one of several MYC effectors – including p53, p14ARF, or BIM – causing apoptosis-firing to drop below a critical threshold to allow cell proliferation. It also established that BCL2L11/BIM is a p14ARF/p53-independent target of MYC and that its activation does not require MYC-induced hyperproliferation (Dang et al., 2005; Hemann et al., 2005). Thus BIM is a uniquely important tumor suppressor in cells of the hematopoietic lineage and operationally its activation by MYC is a component of the OSR in B cells. Since MYC is induced and becomes constitutively expressed early after EBV infection of primary human B cells, modulation of BIM expression by EBV is likely to be a contributory factor in B cell transformation and the development of any EBV-associated B cell lymphomas (discussed in more detail below).
Polycomb Group Proteins and Epigenetic Repression
Epigenetic gene regulation is heritable and results from changes in a chromosome without alterations to DNA sequence (Berger et al., 2009). Such changes can be mediated by chemical modifications to chromatin on either DNA or DNA-associated histones and may involve non-coding RNAs. PcG proteins were first identified in Drosophila and are best known as repressors of the homeotic (Hox) transcription factor genes during embryonic development. They are very highly conserved from flies to humans and homologues regulating developmental transitions are found in plants. PcG proteins form multi-protein complexes called polycomb repressive complexes (PRCs) that bind and epigenetically regulate hundreds of genes, predominantly associated with cell-fate decisions and development (reviewed in Bracken and Helin, 2009; Margueron and Reinberg, 2011; Bemer and Grossniklaus, 2012; Simon and Kingston, 2013). They can repress transcription by introducing post-translational covalent modifications on histones in chromatin located in the regulatory regions of target genes. This repression/silencing is stable and heritable so can be described as epigenetic (Berger et al., 2009).
PRC2 is a multi-component complex that mediates tri-methylation at lysine 27 of histone H3 (H3K27me3). In humans the core complex is comprised of three polycomb proteins: suppressor of zeste (SUZ)12, embryonic ectoderm development (EED), and enhancer of zeste (EZH)2. EZH2 contains the catalytic SET domain responsible for lysine methyltransferase activity (Bracken and Helin, 2009; Margueron and Reinberg, 2011; Simon and Kingston, 2013). Other components of PRC2 are histone chaperone RbAp46/48 and recently an ancillary factor, JARID2, has been identified as being essential for recruitment of PRC2 to some polycomb-target genes (Murzina et al., 2008; Landeira et al., 2010; Margueron and Reinberg, 2011; Simon and Kingston, 2013). It remains unclear how in most cases the polycomb proteins are recruited to specific promoters in mammalian cells, although sequence context is probably important and a preference for regions rich in CpG dinucleotides (CpG-islands) has been reported (Ku et al., 2008). However, for most target genes, it remains to be determined whether specificity comes from sequence-specific transcription factors, PRC2-interacting non-coding RNA species, or yet to be identified mechanisms (Bracken and Helin, 2009; Khalil et al., 2009; Gupta et al., 2010; Kanhere et al., 2010; Simon and Kingston, 2013).
H3K27me3 on chromatin attracts the binding of a second complex, PRC1 that mediates the repressive ubiquitinylation at lysine 119 of histone H2A (H2AK119Ub). PRC1 core proteins include chromobox (CBX) proteins, whose chromodomains are thought to recruit the complex to the H3K27me3 mark, and RING finger proteins, such as RING1B, MEL18, and BMI1 that are responsible for the E3 ubiquitin ligase activity that produces H2AK119Ub. PRC1 mediates chromatin compaction and the local formation of heterochromatin (Grau et al., 2011) and together with PRC2, increases the chances of the more stable CpG DNA methylation mark being deposited (reviewed in Cedar and Bergman, 2009). Although recent evidence suggests H3K27me3 is stable and heritable (Simon and Kingston, 2013) this histone modification can be rapidly removed by demethylase enzymes such as JMJD3 (aka KDM6B; Agger et al., 2009; Barradas et al., 2009). Moreover, if a promoter carries H3K27me3 and simultaneously has the activation-associated modification H3K4me3 at the same locus, it is repressed but is described as “bivalent” and thought to be poised for rapid reactivation by removal of H3K27me3; genes with such bivalent domains are common in stem cells (Bernstein et al., 2006; Voigt et al., 2013). Cancer cells and stem cells often share gene expression patterns and multiple reports suggest that polycomb complexes contribute to the aberrant CpG DNA methylation profiles that are critical in the genesis and progression of many diverse cancers (Cedar and Bergman, 2009). The mechanism for this is suggested by the capacity of various polycomb proteins to physically interact with DNA methyl transferases (DNMTs) and recruit them to chromatin. It has been estimated that PcG-target genes are up to 12 times more likely to be aberrantly methylated in cancer than non-targets (Widschwendter et al., 2007).
Epigenetic Regulation of BIM and p16INK4a Expression by EBV
EBNA3A and EBNA3C Cooperate as Oncogenic Repressors of Transcription
The EBV EBNA3 proteins (EBNA3A, EBNA3B, and EBNA3C) are large (>900 aa) latency-associated nuclear proteins that show no significant similarity to known cell or viral factors. Although none of them appears to bind DNA directly, they all bind the cellular DNA-binding factor CBF-1 (aka RBP-JK; reviewed in Bornkamm and Hammerschmidt, 2001; Young and Rickinson, 2004). All three EBNA3s can also interact with cellular factors associated with the covalent modification of histones, the repression of transcription, and gene silencing; for example, EBNA3A and EBNA3C associate with histone deacetylases (HDACs) and the conserved co-repressor CtBP (Radkov et al., 1999; Bornkamm and Hammerschmidt, 2001; Touitou et al., 2001; Hickabottom et al., 2002; Young and Rickinson, 2004). EBNA3A, EBNA3B, and EBNA3C are all robust repressors of transcription when targeted directly to DNA in transient assays (Bain et al., 1996; Cludts and Farrell, 1998 and our unpublished data), and EBNA3A and EBNA3C – but not EBNA3B – are necessary to establish LCLs from purified B cells (Tomkinson and Kieff, 1992; Tomkinson et al., 1993). EBNA3A and EBNA3C also cooperate with oncogenic Ha-Ras in the transformation/immortalization of primary rodent fibroblasts and require the interaction with CtBP to do this (Parker et al., 1996; Touitou et al., 2001; Hickabottom et al., 2002). All the data are therefore consistent with EBNA3A and EBNA3C acting as oncoproteins in the transformation of B cells and in EBV-associated lymphomagenesis. However EBNA3B is unnecessary in these processes, and can even act as a tumor suppressor (White et al., 2012).
Recent microarray gene-expression analyses using LCLs or lymphoma cells infected with recombinant B95.8 strain EBVs that express defined EBNA3 mutants, suggest that together the EBNA3s can regulate >1000 host genes in B cells – often repressing transcription. The regulation of many of these genes seems to require the functional interaction of at least two EBNA3s and in several cases that have been subjected to further analysis, gene repression appears to utilize the host PcG system to inhibit transcription via the H3K27me3 chromatin modification (Hertle et al., 2009; Skalska et al., 2010; White et al., 2010, 2012; Maruo et al., 2011; Zhao et al., 2011; McClellan et al., 2012; Paschos et al., 2012). Two genes repressed by the combined action of EBNA3C, EBNA3A, and PcG proteins – and of particular interest in the context of OSR – encode BIM and p16INK4a.
Repression of BIM Transcription
The first indication that EBNA3A and EBNA3C can cooperate to repress specific host cell genes came using a panel of EBNA3-knockout recombinant B95.8-derived EBVs to infect EBV-negative BL cells. This revealed that expression of both EBNA3A and EBNA3C are necessary to repress transcription of BCL2L11/BIM (Anderton et al., 2008). Subsequently it was found that DNA in a large CpG island located at the 5′ end of BCL2L11/BIM becomes methylated on CpG dinucleotides in EBV-positive BLs (Paschos et al., 2009). However a reduction in BIM expression occurred soon after EBV infection of B cells in culture and did not initially involve detectable CpG methylation, but correlated with the deposition of the polycomb signature H3K27me3 on chromatin proximal to the transcription start site (TSS; Paschos et al., 2009, 2012). Detailed chromatin immunoprecipitation (ChIP) analyses of the chromatin around the BCL2L11/BIM promoter revealed that latent EBV triggers the recruitment of polycomb repressive complex 2 (PRC2) core subunits and the trimethylation of histone H3 lysine 27 (H3K27me3) at this locus. It appears that in uninfected BL cells, RbAp48, and JARID2 already associate with the chromatin proximal to the TSS and that EBV infection is necessary to recruit SUZ12 and EZH2 to establish functional PRC2. Assembly of PRC2 at the locus was absolutely dependent on both EBNA3A and EBNA3C being expressed, and using a recombinant EBV expressing an epitope-tagged EBNA3C, it was shown by ChIP that EBNA3C associates with chromatin near the TSS – it is therefore likely to physically interact with PRC2 (Paschos et al., 2012; Figure 2 and model in Figure 3). Since the activation mark H3K4me3 is largely unaltered at this locus irrespective of H3K27me3- or EBNA3-status the establishment of a “bivalent” chromatin domain is suggested. Consistent with the “poised” nature of these domains, RNA polymerase II (RNA Pol II) occupancy at the BCL2L11/BIM TSS was not altered by EBV. However, further analysis of phospho-serine 5 on RNA Pol II indicated that when EBNA3A and EBNA3C are both expressed they inhibit this phosphorylation step and block the initiation of the BIM transcripts. It was not determined whether this involves the direct action of an EBV protein on the kinase CDK7 or is a consequence of the recruitment of PRC2 and/or PRC1 to this particular locus. B cell lines carrying EBV encoding a conditional EBNA3C-modified estrogen receptor-fusion revealed that this epigenetic repression of BIM was reversible, but took more than 30 days from when EBNA3C was inactivated, emphasizing the stability of these chromatin modifications through rounds of cell division. Lentivirus delivery of shRNAs against PRC2 and PRC1 subunits disrupted EBV repression of BCL2L11/BIM, thus confirming the requirement for PcG complexes (Paschos et al., 2012).
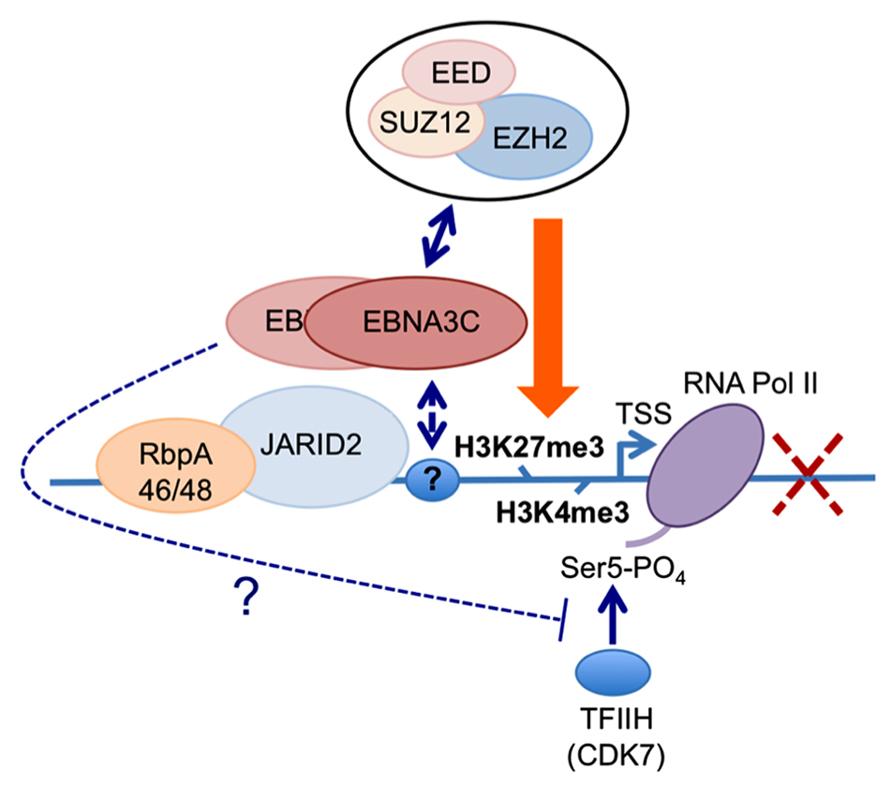
FIGURE 3. Working hypothesis for the role(s) of EBNA3C and EBNA3A in the PRC2-mediated repression of the BIM promoter. The available data indicate that EBNA3C (and EBNA3A) are recruited to regions proximal to the BCL2L11/BIM transcriptional start site (TSS) in EBV-infected B cells (Paschos et al., 2012; our unpublished data and Figure 2). Irrespective of whether EBNA3C or EBNA3A are expressed in these cells, the PRC2-associated factors RbpA46/48 and JARID2 are present at the locus. Similarly the activation mark H3K4me3 and RNA polymerase II (RNA Pol II) occupy the TSS irrespective of which EBNA3s are expressed. Only when both EBNA3C and EBNA3A are present are core components of the PRC2 complex found at this site and the repressive chromatin mark H3K27me3 is detected across the TSS; concomitantly the level of transcription and BIM expression are reduced. The simultaneous presence of both H3K4me3 and H3K27me3 at the locus define it as a “bivalent” or “poised” domain and is consistent with RNA Pol II always being detected. However only in the absence of either EBNA3C or EBNA3A is RNA Pol II phosphorylated on serine residue 5 (RNA Pol II Ser 5), suggesting that in addition to playing a key role in the recruitment of PRC2 core complex, the presence of EBNA3C and EBNA3A might interfere with serine 5 phosphorylation of RNA Pol II and therefore block the initiation of transcription. Since EBNA3A and EBNA3C can be co-immunoprecipitated from infected B cells and both are necessary for repression of BIM (and p16INK4a) expression, in this model we assume they are co-localized at these loci. The identity of the factor(s) responsible for targeting EBNA3C and/or EBNA3A to this particular stretch of chromatin is still unknown, as is the mechanism of interaction with PRC2.
Repression of Transcription from the CDKN2A Locus
Direct evidence that EBNA3C modulates the cell cycle during EBV-mediated transformation of B cells came from Maruo et al. (2006). Using a recombinant Akata strain EBV made conditional for EBNA3C function by fusing EBNA3C with a modified estrogen receptor, they revealed that EBNA3C represses expression of the CDK inhibitor p16INK4A in LCLs. Removing the inducer of EBNA3C activity (4-hydroxytamoxifen, 4HT) from the culture medium resulted in an accumulation of both p16INK4A mRNA and protein, de-phosphorylation of Rb, and concomitant cell cycle arrest (Maruo et al., 2006). Using a similar recombinant virus expressing an EBNA3A-fusion, the same authors showed that inactivation of EBNA3A also resulted in reduced proliferation, although the mechanism was not determined (Maruo et al., 2003). Since EBNA3A and EBNA3C are necessary for the H3K27me3-mediated chromatin manipulation and epigenetic repression of BCL2L11/BIM, and since the CDKN2A locus that encodes p16INK4a had been identified as a target of polycomb-mediated repression in proliferating cells, it was not surprising to discover that the combined action of EBNA3C and EBNA3A repressed CDKN2A in cycling B cells by facilitating the deposition of H3K27me3 across the locus – primarily around the p16INK4a TSS (Skalska et al., 2010). Furthermore, establishing LCLs with recombinant viruses encoding CtBP-binding mutants of EBNA3C and EBNA3A revealed that their interaction with this highly conserved cellular co-repressor was necessary for the efficient deposition of H3K27me3 and repression of p16INK4a expression. ChIP analysis for the epitope-tagged EBNA3C expressed in an LCL revealed EBNA3C at the TSS of p16INK4A and ARF, and also the CDKN2B gene encoding p15INK4b (Figure 2; Skalska et al., 2013). Although it was initially unclear whether the EBNA3C-associated H3K27me3 deposition at CDKN2A was a cause or a consequence of cells exiting from the cell cycle, regulation of the locus by EBNA3C in an Rb-null LCL (Skalska et al., 2010) and in several p16INK4a-null LCLs (Skalska et al., 2013 and see below) unequivocally established that EBNA3C regulation of the locus is independent of the degree of cell proliferation. As with BLC2L11/BIM, B cell lines carrying EBV encoding the conditional EBNA3C-modified estrogen receptor-fusion revealed that this epigenetic repression of CDKN2A was reversible by adding or removing 4HT from the medium. Taken together all these data suggest that EBNA3C (cooperating with EBNA3A) coordinately regulates the whole INK4b-ARF-INK4a locus by directing the recruitment of PRC2 to the three transcriptional start sites. Consistent with this we have recently found that the level of p15INK4b mRNA is coordinately regulated with that of p16INK4a in EBNA3C-conditional LCLs (our unpublished data).
The specific role of p16INK4a as a target for EBNA3C and a major barrier to B cell transformation was further explored making use of an “experiment of nature” in the form of “Leiden” B cells carrying a homozygous genomic deletion that specifically ablates production of functional p16INK4a (Brookes et al., 2002; Hayes et al., 2004). These cells were infected with recombinant B95.8-derived EBVs that express either the conditional EBNA3C or no EBNA3C (Skalska et al., 2013). A comparison of p16-null LCLs with LCLs established from normal B cells showed unequivocally that, if p16INK4a is not functional, then EBNA3C is unnecessary to sustain cell proliferation. Consistent with this – and providing formal proof that p16INK4a is the main target of EBNA3C – it was possible to transform p16-null B cells into stable LCLs with EBV, but without any functional EBNA3C ever having been expressed.
Inhibiting OSR/OIS is Necessary for LCL Outgrowth
A reasonable but speculative explanation for why EBV has evolved a mechanism for suppressing p16INK4a (and BIM) expression became apparent from examining the outcome of attempted transformations of normal B cells with EBNA3C-deficient EBV (Figure 4; Skalska et al., 2013). These experiments revealed that infection with a “wild type” EBV modestly induced p16INK4a transcription in the first few days after infection – when EBNA2 transactivates inducers of cell cycle progression (e.g., MYC and cyclin D2) and a period of hyperproliferation has been described (Sinclair et al., 1994; Spender et al., 1999; Nikitin et al., 2010). It is likely that unscheduled entry into S-phase, is interpreted by the cell as oncogenic stress and activation of p16INK4a transcription is a consequence. When the infecting virus expressed functional EBNA3C (and EBNA3A) there was a halt to the increase of p16INK4a expression from about day 7 onwards. However, if EBNA3C was not expressed or was non-functional (i.e., no 4HT in the medium), transcription from INK4a continued unrestrained and the level of mRNA progressively increased over the next 2–3 weeks, until most of the cells stopped proliferating. Early after infection BIM expression is down-regulated, and very soon (<5 days) reaches a steady state, but if EBNA3C is deleted or functionally inactivated in the infecting EBV – beginning about 4 days post infection – the level of mRNA corresponding to BIM also increases, in parallel with that of p16INK4a. This increase continues for the next week or two until cells arrest or die (Skalska et al., 2013). Largely similar results were obtained with EBNA3A-negative virus (our unpublished data).
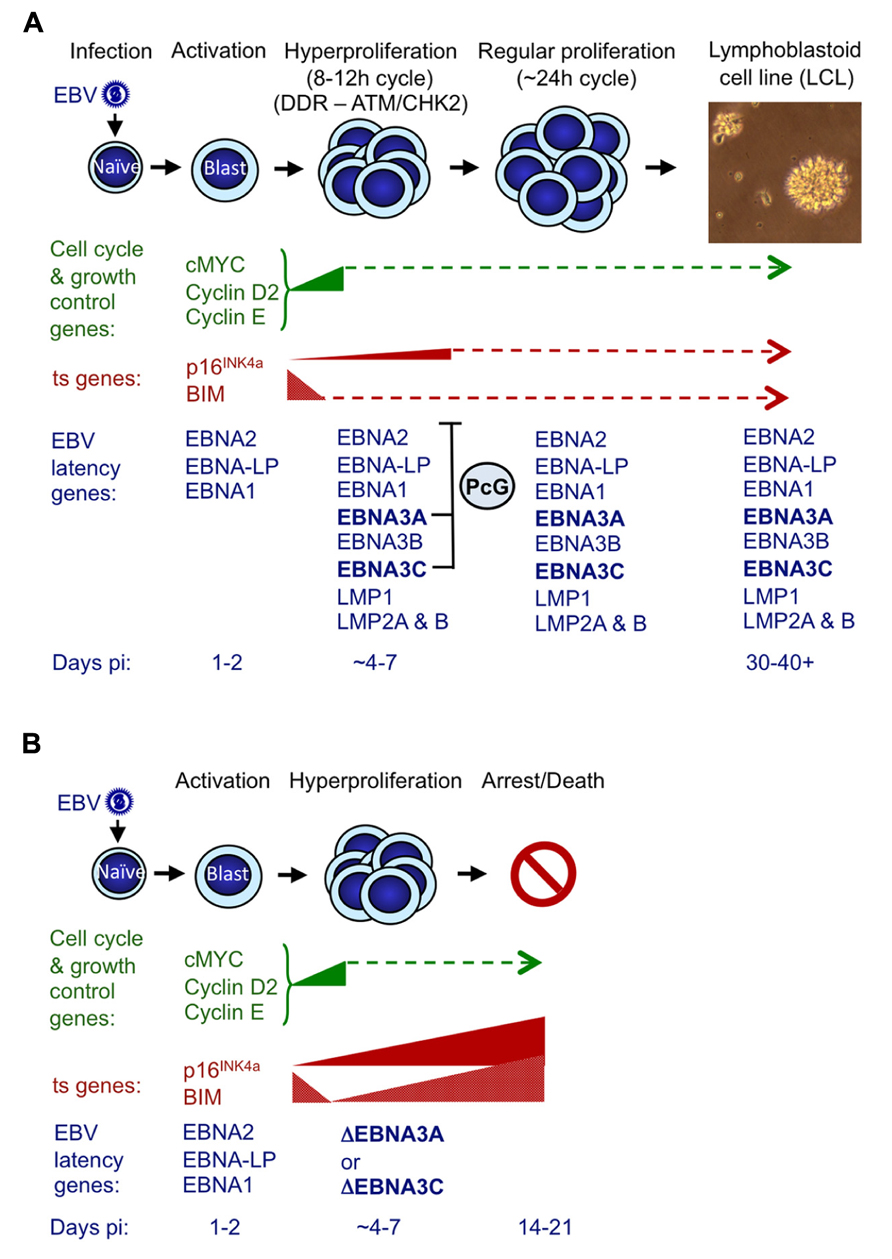
FIGURE 4. Events following infection of primary resting B cells by EBV that initiate transformation into continuously proliferating LCLs. (A) During the first 24–48 h post-infection (pi) with a B95.8-derived EBV, cell genes associated with growth and cell cycle are transactivated and their products (e.g., MYC, cyclin D2, cyclin E) drive cells from G0 to G1, to become enlarged, activated and start proliferating. The whole process is driven by the EBV transactivator protein EBNA2, probably assisted by the co-factor EBNA-LP (Sinclair et al., 1994; Spender et al., 1999; Nikitin et al., 2010). During the next 3–4 days cells undergo rounds of rapid cell division (hyperproliferation) and in some cells this results in damage to DNA that can activate the DNA damage response (DDR) and initiate a signaling cascade involving the kinases ATM and CHK2 (Nikitin et al., 2010). If the full complement of nine EBV latency-associated proteins is expressed, the DDR becomes attenuated (in part by EBNA3C) and cells continue to proliferate to produce polyclonal LCLs that have a population doubling time of about 24 h. Early after infection BIM expression is down-regulated, and although the level of p16INK4a expression increases slightly, this soon reaches a steady state. In both cases we assume that EBNA3A and EBNA3C cooperate by harnessing the polycomb group (PcG) protein system to epigenetically repress (or restrain the transcription of) these ts genes via H3K27me3 (Anderton et al., 2008; Paschos et al., 2012; Skalska et al., 2013). (B) If EBNA3C or EBNA3A are deleted (ΔEBNA3C and ΔEBNA3A) or functionally inactivated in the infecting EBV, beginning about 4–7 days pi, the levels of mRNAs corresponding to p16INK4a and BIM progressively increase and continue to do so for the next week or two until finally most of the cells arrest and/or die (Skalska et al., 2013 and our unpublished data). The PcG-mediated repression of these two ts genes – in particular p16INK4a (see text) – is part of a critical countermeasure evolved by EBV to bypass an intrinsic host defense against oncogenic transformation. If primary B cells are p16INK4a-null, functional EBNA3C is dispensable for the outgrowth of LCLs. This is consistent with p16INK4a being the dominant barrier to outgrowth and subsequent proliferation of LCLs, and the principal requirement of EBNA3C appears to be to restraining transcription of p16INK4a (see text for details and Skalska et al., 2013). The precise relationships between DDR, p16INK4a and EBNA3C/EBNA3A have yet to be defined.
The EBNA3C/3A-mediated epigenetic inhibition of INK4a and BCL2L11/BIM transcription is therefore critical for EBV to bypass an intrinsic host cell defense against oncogenic transformation probably triggered by EBNA2 acting through MYC (summarized in Figure 4; see also Nikitin et al., 2010). Thus expression of both EBNA3C and EBNA3A ensures expansion of the infected B cell population and LCL outgrowth in vitro and in vivo the initiation of latency. Strictly speaking, in this context, EBNA3C and EBNA3A do not actually repress INK4a and BCL2L11/BIM transcription, but rather prevent their activation. This most likely involves the recruitment of PcG protein complexes to the loci, leading to H3K27me3 modifications on chromatin around the TSSs, as is seen in established LCLs; however this has not yet been formally demonstrated in newly infected cells.
Concluding Remarks
Through the combined action of EBNA3C and EBNA3A and their interaction with the cellular PcG protein system, EBV has evolved a very effective countermeasure to OSR/OIS that appears to be critical in its normal life cycle to establish a latent infection and therefore initiate long-term persistence in B cells. In vitro this mechanism neatly overcomes a major early obstacle to cellular “immortalization,” making EBV one of the most potent transforming/immortalizing biological agents to have been identified. By utilizing an epigenetic mode of gene regulation to tackle the problem of OSR/OIS, key target ts genes including INK4a and BCL2L11/BIM are repressed not only in the infected cells, but also in their progeny; furthermore the genes become particularly predisposed to complete silencing by DNA modification. It is self-evident – since EBV stably ablates at least two major barriers to oncogenic transformation – that this will substantially increase the likelihood of EBV-infected B cells undergoing additional genetic and/or epigenetic changes leading to cancer (discussed further in Thorley-Lawson and Allday, 2008; Allday, 2009; Skalska et al., 2010; Paschos et al., 2012). This manipulation of the PcG system to specifically regulate key tumor suppressor genes in B cells makes EBV – to our knowledge – unique among tumor viruses. Now the challenges are to provide complete biochemical descriptions of how the EBNA3 proteins interact with PcG complexes and – employing genome-wide screens such as ChIP-seq – determine the extent of polycomb-mediated epigenetic reprogramming of B cells by EBV.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research performed in my laboratory and described in this review, was generously funded by the Wellcome Trust.
References
Agger, K., Cloos, P. A., Rudkjaer, L., Williams, K., Andersen, G., Christensen, J., et al. (2009). The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176. doi: 10.1101/gad.510809
Allday, M. J. (2009). How does Epstein–Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt’s lymphoma? Semin. Cancer Biol. 19, 366–376. doi: 10.1016/j.semcancer.2009.07.007
Anderton, E., Yee, J., Smith, P., Crook, T., White, R. E., and Allday, M. J. (2008). Two Epstein–Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 27, 421–433. doi: 10.1038/sj.onc.1210668
Askew, D. S., Ashmun, R. A., Simmons, B. C., and Cleveland, J. L. (1991). Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6, 1915–1922.
Babcock, G. J., Decker, L. L., Freeman, R. B., and Thorley-Lawson, D. A. (1999). Epstein–Barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J. Exp. Med. 190, 567–576. doi: 10.1084/jem.190.4.567
Bain, M., Watson, R. J., Farrell, P. J., and Allday, M. J. (1996). Epstein–Barr virus nuclear antigen 3C is a powerful repressor of transcription when tethered to DNA. J. Virol. 70, 2481–2489.
Barradas, M., Anderton, E., Acosta, J. C., Li, S., Banito, A., Rodriguez-Niedenfuhr, M., et al. (2009). Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 23, 1177–1182. doi: 10.1101/gad.511109
Bartek, J., Bartkova, J., and Lukas, J. (2007). DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 26, 7773–7779. doi: 10.1038/sj.onc.1210881
Bemer, M., and Grossniklaus, U. (2012). Dynamic regulation of Polycomb group activity during plant development. Curr. Opin. Plant Biol. 15, 523–529. doi: 10.1016/j.pbi.2012.09.006
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. doi: 10.1101/gad.1787609
Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. doi: 10.1016/j.cell.2006.02.041
Bianchi, T., Rufer, N., MacDonald, H. R., and Migliaccio, M. (2006). The tumor suppressor p16Ink4a regulates T lymphocyte survival. Oncogene 25, 4110–4115. doi: 10.1038/sj.onc.1209437
Bornkamm, G. W., and Hammerschmidt, W. (2001). Molecular virology of Epstein–Barr virus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 437–459. doi: 10.1098/rstb.2000.0781
Bracken, A. P., and Helin, K. (2009). Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 9, 773–784. doi: 10.1038/nrc2736
Braig, M., and Schmitt, C. A. (2006). Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res. 66, 2881–2884. doi: 10.1158/0008-5472.CAN-05-4006
Brookes, S., Rowe, J., Ruas, M., Llanos, S., Clark, P. A., Lomax, M., et al. (2002). INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J. 21, 2936–2945. doi: 10.1093/emboj/cdf289
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. doi: 10.1038/nrg2540
Cludts, I., and Farrell, P. J. (1998). Multiple functions within the Epstein–Barr virus EBNA-3A protein. J. Virol. 72, 1862–1869.
Collado, M., Blasco, M. A., and Serrano, M. (2007). Cellular senescence in cancer and aging. Cell 130, 223–233. doi: 10.1016/j.cell.2007.07.003
Dang, C. V., O’Donnell, K. A., and Juopperi, T. (2005). The great MYC escape in tumorigenesis. Cancer Cell 8, 177–178. doi: 10.1016/j.ccr.2005.08.005
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642. doi: 10.1038/nature05327
Egle, A., Harris, A. W., Bouillet, P., and Cory, S. (2004). Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. U.S.A. 101, 6164–6169. doi: 10.1073/pnas.0401471101
Enders, A., Bouillet, P., Puthalakath, H., Xu, Y., Tarlinton, D. M., and Strasser, A. (2003). Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J. Exp. Med. 198, 1119–1126. doi: 10.1084/jem.20030411
Evan, G. I., Wyllie, A. H., Gilbert, C. S., Littlewood, T. D., Land, H., and Brooks, M. (1992). Induction of apoptosis in fibroblasts by c-myc protein. Cell 69, 119–128. doi: 10.1016/0092-8674(92)90123-T
Fischer, S. F., Bouillet, P., O’Donnell, K., Light, A., Tarlinton, D. M., and Strasser, A. (2007). Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood 110, 3978–3984. doi: 10.1182/blood-2007-05-091306
Forte, E., and Luftig, M. A. (2011). The role of microRNAs in Epstein–Barr virus latency and lytic reactivation. Microbes Infect. 13, 1156–1167. doi: 10.1016/j.micinf.2011.07.007
Gaspar, M., and Shenk, T. (2006). Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 2821–2826. doi: 10.1073/pnas.0511148103
Gavathiotis, E., Suzuki, M., Davis, M. L., Pitter, K., Bird, G. H., and Katz, S. G. (2008). BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081. doi: 10.1038/nature07396
Gil, J., and Peters, G. (2006). Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat. Rev. Mol. Cell Biol. 7, 667–677. doi: 10.1038/nrm1987
Grau, D. J., Chapman, B. A., Garlick, J. D., Borowsky, M., Francis, N. J., and Kingston, R. E. (2011). Compaction of chromatin by diverse polycomb group proteins requires localized regions of high charge. Genes Dev. 25, 2210–2221. doi: 10.1101/gad.17288211
Gruhne, B., Kamranvar, S. A., Masucci, M. G., and Sompallae, R. (2009). EBV and genomic instability: a new look at the role of the virus in the pathogenesis of Burkitt’s lymphoma. Semin. Cancer Biol. 19, 394–400. doi: 10.1016/j.semcancer.2009.07.005
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi: 10.1038/nature08975
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. doi: 10.1126/science.1140735
Hayes, M. J., Koundouris, A., Gruis, N., Bergman, W., Peters, G. G., and Sinclair, A. J. (2004). p16(INK4A)-independence of Epstein–Barr virus-induced cell proliferation and virus latency. J. Gen. Virol. 85, 1381–1386. doi: 10.1099/vir.0.79831-0
Heath, E., Begue-Pastor, N., Chaganti, S., Croom-Carter, D., Shannon-Lowe, C., et al. (2012). Epstein–Barr virus infection of naive B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: implications for virus biology. PLoS Pathog. 8:e1002697. doi: 10.1371/journal.ppat.1002697
Hemann, M. T., Bric, A., Teruya-Feldstein, J., Herbst, A., Nilsson, J. A., Cordon-Cardo, C., et al. (2005). Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436, 807–811. doi: 10.1038/nature03845
Hertle, M. L., Popp, C., Petermann, S., Maier, S., Kremmer, E., Lang, R., et al. (2009). Differential gene expression patterns of EBV infected EBNA-3A positive and negative human B lymphocytes. PLoS Pathog. 5:e1000506. doi: 10.1371/journal.ppat.1000506
Hickabottom, M., Parker, G. A., Freemont, P., Crook, T., and Allday, M. J. (2002). Two nonconsensus sites in the Epstein–Barr virus oncoprotein EBNA3A cooperate to bind the co-repressor carboxyl-terminal-binding protein (CtBP). J. Biol. Chem. 277, 47197–47204. doi: 10.1074/jbc.M208116200
Hislop, A. D., Taylor, G. S., Sauce, D., and Rickinson, A. B. (2007). Cellular responses to viral infection in humans: lessons from Epstein–Barr virus. Annu. Rev. Immunol. 25, 587–617. doi: 10.1146/annurev.immunol.25.022106.141553
Kanhere, A., Viiri, K., Araujo, C. C., Rasaiyaah, J., Bouwman, R. D., Whyte, W. A., et al. (2010). Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 38, 675–688. doi: 10.1016/j.molcel.2010.03.019
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672. doi: 10.1073/pnas.0904715106
Kim, W. Y., and Sharpless, N. E. (2006). The regulation of INK4/ARF in cancer and aging. Cell 127, 265–275. doi: 10.1016/j.cell.2006.10.003
Koopal, S., Furuhjelm, J. H., Jarviluoma, A., Jaamaa, S., Pyakurel, P., Pussinen, C., et al. (2007). Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 3:e140. doi: 10.1371/journal.ppat.0030140
Ku, M., Koche, R. P., Rheinbay, E., Mendenhall, E. M., Endoh, M., Mikkelsen, T. S., et al. (2008). Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 4:e1000242. doi: 10.1371/journal.pgen.1000242
Lagresle, C., Gardie, B., Eyquem, S., Fasseu, M., Vieville, J. C., Pla, M., et al. (2002). Transgenic expression of the p16(INK4a) cyclin-dependent kinase inhibitor leads to enhanced apoptosis and differentiation arrest of CD4–CD8- immature thymocytes. J. Immunol. 168, 2325–2331.
Laichalk, L. L., and Thorley-Lawson, D. A. (2005). Terminal differentiation into plasma cells initiates the replicative cycle of Epstein–Barr virus in vivo. J. Virol. 79, 1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005
Landeira, D., Sauer, S., Poot, R., Dvorkina, M., Mazzarella, L., Jorgensen, H. F., et al. (2010). Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA polymerase II to developmental regulators. Nat. Cell Biol. 12, 618–624. doi: 10.1038/ncb2065
Leidal, A. M., Pringle, E. S., and McCormick, C. (2012). Evasion of oncogene-induced senescence by gammaherpesviruses. Curr. Opin. Virol. 2, 748–754. doi: 10.1016/j.coviro.2012.09.009
Lowe, S. W., Cepero, E., and Evan, G. (2004). Intrinsic tumour suppression. Nature 432, 307–315. doi: 10.1038/nature03098
Margueron, R., and Reinberg, D. (2011). The polycomb complex PRC2 and its mark in life. Nature 469, 343–349. doi: 10.1038/nature09784
Maruo, S., Johannsen, E., Illanes, D., Cooper, A., and Kieff, E. (2003). Epstein–Barr virus nuclear protein EBNA3A is critical for maintaining lymphoblastoid cell line growth. J. Virol. 77, 10437–10447. doi: 10.1128/JVI.77.19.10437-10447.2003
Maruo, S., Wu, Y., Ishikawa, S., Kanda, T., Iwakiri, D., and Takada, K. (2006). Epstein–Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc. Natl. Acad. Sci. U.S.A. 103, 19500–19505. doi: 10.1073/pnas.0604919104
Maruo, S., Zhao, B., Johannsen, E., Kieff, E., Zou, J., and Takada, K. (2011). Epstein–Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc. Natl. Acad. Sci. U.S.A. 108, 1919–1924. doi: 10.1073/pnas.1019599108
McClellan, M. J., Khasnis, S., Wood, C. D., Palermo, R. D., Schlick, S. N., Kanhere, A. S., et al. (2012). Downregulation of integrin receptor-signaling genes by Epstein–Barr virus EBNA 3C via promoter-proximal and -distal binding elements. J. Virol. 86, 5165–5178. doi: 10.1128/JVI.07161-11
Murzina, N. V., Pei, X. Y., Zhang, W., Sparkes, M., Vicente-Garcia, J., Pratap, J. V., et al. (2008). Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure 16, 1077–1085. doi: 10.1016/j.str.2008.05.006
Nikitin, P. A., and Luftig, M. A. (2012). The DNA damage response in viral-induced cellular transformation. Br. J. Cancer 106, 429–435. doi: 10.1038/bjc.2011.612
Nikitin, P. A., Yan, C. M., Forte, E., Bocedi, A., Tourigny, J. P., White, R. E., et al. (2010). An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein–Barr virus transformation of primary human B cells. Cell Host Microbe 8, 510–522. doi: 10.1016/j.chom.2010.11.004
Parker, G. A., Crook, T., Bain, M., Sara, E. A., Farrell, P. J., and Allday, M. J. (1996). Epstein–Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 13, 2541–2549.
Paschos, K., Parker, G. A., Watanatanasup, E., White, R. E., and Allday, M. J. (2012). BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 40, 7233–7246. doi: 10.1093/nar/gks391
Paschos, K., Smith, P., Anderton, E., Middeldorp, J. M., White, R. E., and Allday, M. J. (2009). Epstein–Barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 5:e1000492. doi: 10.1371/journal.ppat.1000492
Popov, N., and Gil, J. (2010). Epigenetic regulation of the INK4b-ARF-INK4a locus: in sickness and in health. Epigenetics 5, 685–690. doi: 10.4161/epi.5.8.12996
Radkov, S. A., Touitou, R., Brehm, A., Rowe, M., West, M., Kouzarides, T., et al. (1999). Epstein–Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 73, 5688–5697.
Roughan, J. E., and Thorley-Lawson, D. A. (2009). The intersection of Epstein–Barr virus with the germinal center. J. Virol. 83, 3968–3976. doi: 10.1128/JVI.02609-08
Rowe, M., Kelly, G. L., Bell, A. I., and Rickinson, A. B. (2009). Burkitt’s lymphoma: the rosetta stone deciphering Epstein–Barr virus biology. Semin. Cancer Biol. 19, 377–388. doi: 10.1016/j.semcancer.2009.07.004
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. doi: 10.1016/S0092-8674(00)81902-9
Sherr, C. J. (1998). Tumor surveillance via the ARF-p53 pathway. Genes Dev. 12, 2984–2991. doi: 10.1101/gad.12.19.2984
Sherr, C. J. (2012). Ink4-Arf locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 1, 731–741. doi: 10.1002/wdev.40
Shirata, N., Kudoh, A., Daikoku, T., Tatsumi, Y., Fujita, M., and Kiyono, T. (2005). Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 280, 30336–30341. doi: 10.1074/jbc.M500976200
Simon, J. A., and Kingston, R. E. (2013). Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824. doi: 10.1016/j.molcel.2013.02.013
Sinclair, A. J., Palmero, I., Peters, G., and Farrell, P. J. (1994). EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein–Barr virus. EMBO J. 13, 3321–3328.
Skalska, L., White, R. E., Franz, M., Ruhmann, M., and Allday, M. J. (2010). Epigenetic repression of p16(INK4A) by latent Epstein–Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 6:e1000951. doi: 10.1371/journal.ppat.1000951
Skalska, L., White, R. E., Parker, G. A., Sinclair, A. J., Paschos, K., and Allday, M. J. (2013). Induction of p16(INK4a) is the major barrier to proliferation when Epstein–Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 9:e1003187. doi: 10.1371/journal.ppat.1003187
Spender, L. C., Cannell, E. J., Hollyoake, M., Wensing, B., Gawn, J. M., Brimmell, M., et al. (1999). Control of cell cycle entry and apoptosis in B lymphocytes infected by Epstein–Barr virus. J. Virol. 73, 4678–4688.
Strasser, A. (2005). The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5, 189–200. doi: 10.1038/nri1568
Tarakanova, V. L., Leung-Pineda, V., Hwang, S., Yang, C. W., Matatall, K., Basson, M., et al. (2007). Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1, 275–286. doi: 10.1016/j.chom.2007.05.008
Thorley-Lawson, D. A., and Allday, M. J. (2008). The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat. Rev. Microbiol. 6, 913–924. doi: 10.1038/nrmicro2015
Thorley-Lawson, D. A., and Gross, A. (2004). Persistence of the Epstein–Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 350, 1328–1337. doi: 10.1056/NEJMra032015
Thorley-Lawson, D. A., Hawkins, J. B., Tracy, S. I., and Shapiro, M. (2013). The pathogenesis of Epstein–Barr virus persistent infection. Curr. Opin. Virol. 3, 227–232. doi: 10.1016/j.coviro.2013.04.005
Tomkinson, B., and Kieff, E. (1992). Use of second-site homologous recombination to demonstrate that Epstein–Barr virus nuclear protein 3B is not important for lymphocyte infection or growth transformation in vitro. J. Virol. 6, 2893–2903.
Tomkinson, B., Robertson, E., and Kieff, E. (1993). Epstein–Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 67, 2014–2025.
Touitou, R., Hickabottom, M., Parker, G., Crook, T., and Allday, M. J. (2001). Physical and functional interactions between the corepressor CtBP and the Epstein–Barr virus nuclear antigen EBNA3C. J. Virol. 75, 7749–7755. doi: 10.1128/JVI.75.16.7749-7755.2001
Voigt, P., Tee, W. W., and Reinberg, D. (2013). A double take on bivalent promoters. Genes Dev. 27, 1318–1338. doi: 10.1101/gad.219626.113
Vousden, K. H., and Prives, C. (2009). Blinded by the light: the growing complexity of p53. Cell 137, 413–431. doi: 10.1016/j.cell.2009.04.037
Wade, M., and Wahl, G. M. (2006). c-Myc, genome instability, and tumorigenesis: the devil is in the details. Curr. Top. Microbiol. Immunol 302, 169–203. doi: 10.1007/3-540-32952-8_7
White, R. E., Groves, I. J., Turro, E., Yee, J., Kremmer, E., and Allday, M. J. (2010). Extensive co-operation between the Epstein–Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS ONE 5:e13979. doi: 10.1371/journal.pone.0013979
White, R. E., Ramer, P. C., Naresh, K. N., Meixlsperger, S., Pinaud, L., Rooney, C., et al. (2012). EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Invest. 122, 1487–1502. doi: 10.1172/JCI58092
Widschwendter, M., Fiegl, H., Egle, D., Mueller-Holzner, E., Spizzo, G., Marth, C., et al. (2007). Epigenetic stem cell signature in cancer. Nat. Genet. 39, 157–158. doi: 10.1038/ng1941
Williams, H., and Crawford, D. H. (2006). Epstein–Barr virus: the impact of scientific advances on clinical practice. Blood 107, 862–869. doi: 10.1182/blood-2005-07-2702
Young, L. S., and Rickinson, A. B. (2004). Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768. doi: 10.1038/nrc1452
Keywords: Epstein–Barr virus, PcG, epigenetic, oncogenic stress response, oncogene-induced senescence, p16INK4a, BIM, B cell transformation
Citation: Allday MJ (2013) EBV finds a polycomb-mediated, epigenetic solution to the problem of oncogenic stress responses triggered by infection. Front. Genet. 4:212. doi: 10.3389/fgene.2013.00212
Received: 25 June 2013; Accepted: 01 October 2013;
Published online: 24 October 2013.
Edited by:
Félix Recillas-Targa, Universidad Nacional Autónoma de México, MexicoReviewed by:
Ian C. G. Weaver, Dalhousie University, CanadaSteven G. Gray, St James Hospital/Trinity College Dublin, Ireland
Copyright © 2013 Allday. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin J. Allday, Section of Virology, Department of Medicine, Imperial College London, St Mary’s Campus, Norfolk Place, London W2 1PG, UK e-mail:bS5hbGxkYXlAaW1wZXJpYWwuYWMudWs=