 Brendan E. Russ
Brendan E. Russ Julia E. Prier
Julia E. Prier Sudha Rao
Sudha Rao Stephen J. Turner
Stephen J. Turner- 1Department of Microbiology and Immunology, The University of Melbourne, Parkville, VIC, Australia
- 2Department of Molecular and Cellular Biology, Canberra University, Canberra, ACT, Australia
Cellular differentiation is regulated by the strict spatial and temporal control of gene expression. This is achieved, in part, by regulating changes in histone post-translational modifications (PTMs) and DNA methylation that in turn, impact transcriptional activity. Further, histone PTMs and DNA methylation are often propagated faithfully at cell division (termed epigenetic propagation), and thus contribute to maintaining cellular identity in the absence of signals driving differentiation. Cardinal features of adaptive T cell immunity include the ability to differentiate in response to infection, resulting in acquisition of immune functions required for pathogen clearance; and the ability to maintain this functional capacity in the long-term, allowing more rapid and effective pathogen elimination following re-infection. These characteristics underpin vaccination strategies by effectively establishing a long-lived T cell population that contributes to an immunologically protective state (termed immunological memory). As we discuss in this review, epigenetic mechanisms provide attractive and powerful explanations for key aspects of T cell-mediated immunity – most obviously and notably, immunological memory, because of the capacity of epigenetic circuits to perpetuate cellular identities in the absence of the initial signals that drive differentiation. Indeed, T cell responses to infection are an ideal model system for studying how epigenetic factors shape cellular differentiation and development generally. This review will examine how epigenetic mechanisms regulate T cell function and differentiation, and how these model systems are providing general insights into the epigenetic regulation of gene transcription during cellular differentiation.
Introduction
Protection from the myriad of infectious pathogens we are exposed to on a daily basis largely results from the coordinated interaction of the cells and molecules of the mammalian immune system. Key cellular components of the adaptive immune system are white blood cells (lymphocytes) of which there are two types: B and T cells. B and T cells share features of adaptive immunity that include the ability to recognize pathogen components via clonal expression of a unique cell surface receptor; the ability to rapidly proliferate upon recognition of a pathogen, coincident with acquisition of cell lineage-specific immune functions; and finally, the ability to persist after the infection is cleared, combined with the capacity to “remember” the pathogen and respond more rapidly and vigorously upon re-infection (termed immunological memory).
T cells can be further divided into helper T (TH) cells and cytotoxic (killer) T cells. TH cells are distinguished by cell surface expression of CD4 (i.e. CD4+ T cells) and promote effective immunity by secreting molecules that promote effective antibody and cellular responses upon infection. Further, TH cells can differentiate into at least six subtypes, each characterized by expression of different immune molecules (termed effector molecules), which in turn, dictates that each TH subset can play a different role in immunity to infection. In contrast, killer T cells, distinguished by cell surface expression of CD8 (i.e., CD8+ T cells), are the “hit-men” of the immune system, typically locating and destroying virus-infected host cells, and thus limiting and contributing to the eventual clearance of infection. Killer T cells express a range of effector molecules that equip them to mediate this signature killing capacity.
A cardinal feature of T cell immunity is the ability of naïve T cells to undergo a program of proliferation and functional differentiation upon activation, resulting in a large pool of cells, all capable of recognizing a particular pathogen, and that have acquired the immune functions necessary to control and eventually clear infection (Kaech et al., 2002; van Stipdonk et al., 2003; Figure 1). Once an infection is cleared, the majority of the expanded effector T cell population dies, leaving behind a small pool of long-lived cells that can recognize the same pathogen that triggered their initial activation (termed memory T cells; Marshall et al., 2001; Kaech et al., 2002; La Gruta et al., 2004). Importantly, these memory T cells produce a broader array of immune molecules than naïve cells, and in larger quantities, and unlike naïve cells, can respond to infection without the need for further differentiation (Lalvani et al., 1997; Agarwal and Rao, 1998; Oehen and Brduscha-Riem, 1998; Veiga-Fernandes et al., 2000). These features, combined with persistence at a higher frequency, enable memory T cells to respond more rapidly upon secondary infection, enabling earlier control and clearance of infection (Figure 1), and together, these features of memory T cells provide the basis of T cell-mediated immunity. Importantly, our understanding of the molecular factors that shape cell fate decisions and drive acquisition of T cell effector function is limited, and questions remaining to be determined include how a T cell decides to be a memory versus an effector cell, and what are the molecular mechanisms that enable stable maintenance of rapid effector function within memory T cells in the long-term? In this review we describe what we think are some of the more interesting and important studies addressing these and similar questions, with the aim of demonstrating the utility of the immune system as a tool for studying epigenetics and cellular differentiation. We start by discussing the diversity of T cells phenotypes, before describing our current understanding of how epigenetic regulation influences how these distinct functional T cell populations arise and are maintained.
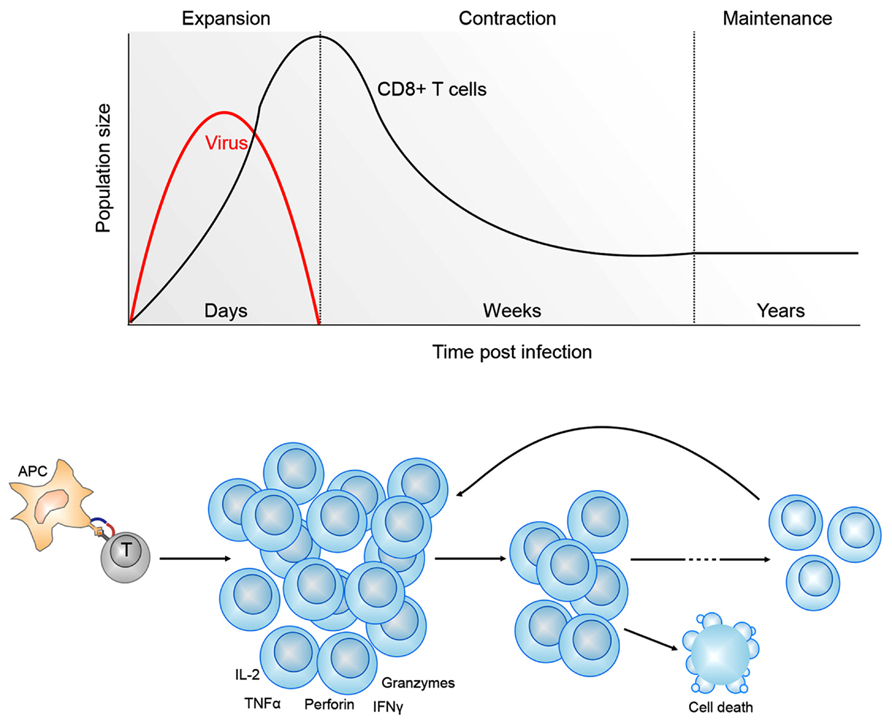
FIGURE 1. Kinetics of CD8+ T cell differentiation following viral infection. Shown is a typical CD8+ T cell response to a acute viral infection. Antigen presenting cells (APC) present viral antigens to CD8+ T cells. This initiates a program of clonal expansion and differentiation into effector CD8+ T cells capable of lineage-specific effector functions, including the ability to secrete pro-inflammatory (TNF-α, IFN-γ) and cytotoxic (perforin, granzyme) molecules. Following viral clearance, the CD8+ T cells undergo an extensive contraction phase, mediated by programed cell death. The remaining memory CD8+ T cells can persist in the host for years. In the event of a secondary exposure to the same virus, memory CD8+ T cells can rapidly expand and acquire effector functions.
Defining the Differing Roles of Distinct T Cell Subsets in Mediating Immunity
An important feature of T cell immunity is the enormous proliferative potential and functional plasticity of naïve T cells. Acquisition of lineage-specific T cell effector functions is clearly linked to an extended proliferative response, suggesting that T cell activation engages a differentiation program that facilitates effector gene expression (Gett and Hodgkin, 1998; Lawrence and Braciale, 2004; Jenkins et al., 2008). An example of T cell functional plasticity is found after activation of naïve TH cells that have the potential to differentiate into distinct T cell subsets, largely defined by the soluble effector molecules they secrete (Figure 2; Zhu et al., 2010). The best characterized of these are the TH1 and TH2 subsets, however, other subsets include TH17, Tregs (regulatory T cells), TFH (follicular TH cells) and the more recently described TH9 cells (Figure 2). TH1 and TH2 T cells are best characterized by their capacity to secrete interferon-gamma (IFN-γ) and interleukin (IL)-4, respectively. The tailoring of TH cell responses into distinct functional lineages is a consequence of integration of multiple signals that are present during initial T cell activation (Figure 2). For example, naïve TH cell activation in the presence of the pro-inflammatory molecules, IFN-γ and IL-12, induces TH1 differentiation while IL-4 is a potent inducer of TH2 differentiation (Zhu et al., 2010). Importantly, induction of transcription factor (TF) expression by extracellular signals received by activated TH cells drives T cell differentiation (Kanno et al., 2012); TH1 differentiation is dependent on STAT1 activation and expression of the T-box TF Tbx21 (T-bet; Djuretic et al., 2007). Conversely, IL-4 signals activate STAT6 resulting in up-regulation of the TF Gata3 (Ansel et al., 2003). TH17 differentiation is associated with IL-6/IL-21 induced expression of the RORγT TF (Dong, 2008) and Treg differentiation with FoxP3 (reviewed in Josefowicz et al., 2012). Such is the importance of these TFs in directing naïve TH cell commitment to a specific lineage that they are used as definitive markers of TH subset differentiation (Figure 2).
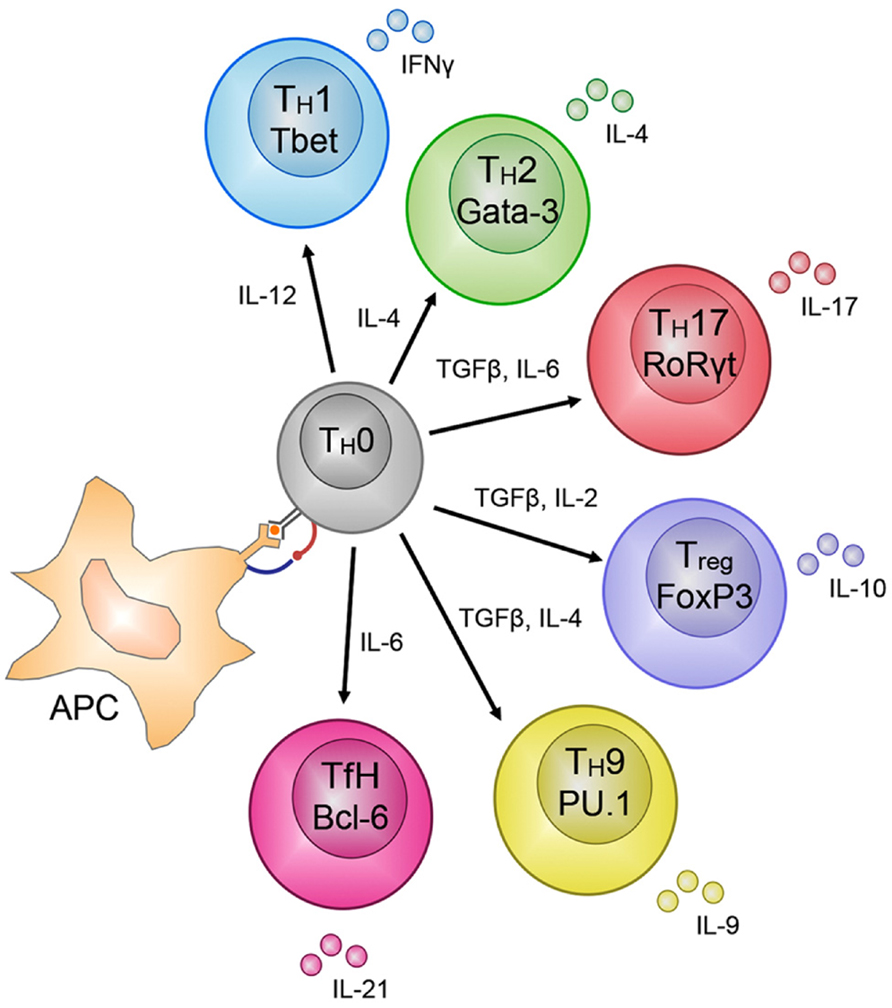
FIGURE 2. CD4+ TH – cell subset differentiation. CD4+ T cells show remarkable plasticity and are able to differentiate into many different subsets based on the soluble molecules secreted during priming of the subsets by antigen presenting cells (APC), e.g., IL-12 for TH1 cells. The different subsets can be distinguished by the transcription factors that regulate and maintain their lineage-specific effector functions, e.g., T-bet for TH1 cells. The molecules secreted by these subsets, e.g., IFN-γ for TH1 cells, are finely tuned to control the pathogen that mediated the release of the specific molecules by the APC during activation of the TH0 cells into the various subsets.
As we learn more about these TH subsets, it is clear that there is heterogeneity of effector function within a responding T cell population such that no one immune response is uniquely represented by a single TH subset. Rather, there is tailoring of the total T cell population such that a particular subset may be over-represented. For example, TH1 type cells dominate the response to extracellular bacterial infections, and in this case, expression of the TH1 cytokine, IFN-γ, is required to promote immune control of these particular pathogens. In this way, the immune system ensures that the most appropriate immune response is engaged to promote control of infection.
Killer T cells contribute to the control and eventual elimination of intracellular bacteria, viruses and tumor challenges via the coordinated interplay of varied effector mechanisms (Russ et al., 2012). This includes the production of pro-inflammatory cytokines such as IFN-γ and tumor necrosis factor alpha (TNF-α; La Gruta et al., 2004) and the expression of cytolytic effector molecules including perforin (Pfp; Kagi et al., 1994) and the granule enzymes (granzymes, Gzm) A, B, and K (Jenkins et al., 2007; Peixoto et al., 2007; Moffat et al., 2009). Whilst killer T cells are not typically associated with commitment to distinct lineages, it is clear that specific TFs are also important in regulating their differentiation and acquisition of effector function. For instance, two T-box TFs, T-bet (encoded by Tbx21) and Eomesodermin (encoded by Eomes; Intlekofer et al., 2005) play essential roles in effector CTL differentiation. Analogous to its role in TH1 T cells, T-bet is rapidly up-regulated upon naïve killer T cell activation and directly regulates the rapid acquisition of IFN-γ production (Cruz-Guilloty et al., 2009). Eomesodermin, a homolog of T-bet, was originally implicated in the regulation of CD8+ T cell granzyme B expression (Pearce et al., 2003), however, recent studies suggest that Eomesodermin is expressed later during CTL differentiation and contributes more to acquisition of perforin expression, while helping sustain the capacity to express IFN-γ (Cruz-Guilloty et al., 2009). IL-2 is a cytokine required for inducing proliferation and survival of activated T cells (Miyazaki et al., 1995). Importantly, high levels of IL-2 signaling at the time of killer T cell activation contribute to granzyme B and perforin expression via STAT5 activation (Janas et al., 2005; Pipkin et al., 2010). In this way, killer T cells integrate signals delivered by extrinsic inflammatory and survival signals during infection that promote effector T cell differentiation.
While the importance of these TFs in lineage determination is clear, exactly how they convey their effects on T cell differentiation is less well understood. As we describe below, at least some of these TFs (i.e., STAT6 and T-bet) exert their effects on T cell differentiation through the recruitment of chromatin modifying enzymes to the sites of TF binding (Lewis et al., 2007; Miller et al., 2010; Onodera et al., 2010). Further, such mechanisms of TF action are known from other systems, suggesting that this mechanism may be common. Thus it appears that TFs and chromatin modifying enzymes cooperate, with the former providing the DNA binding specificity, and the latter the catalytic activity. As described below, once modified, the chromatin can then serve as a substrate for yet other protein complexes that physically rearrange the chromatin, making it more or less permissive for transcription.
Epigenetic Regulation of Cellular Differentiation
Cellular differentiation is regulated by the strict spatial and temporal control of gene expression, which at the most fundamental level, is controlled by modulating access of the transcriptional machinery to gene regulatory regions, including promoters and enhancers. In eukaryotic cells, transcription occurs in the context of chromatin – a complex formed between the genome and histone protein octomers (termed nucleosomes), around which the DNA is wound. As the intimate nature of the nucleosome–DNA interaction can occlude binding of the transcriptional machinery, preventing transcription, this interaction must be tightly regulated to allow appropriate gene expression; this is achieved by controlling the positioning of nucleosomes, and by modulating their affinity for DNA. Histone post-translational modifications (PTMs) are key regulators of changes in chromatin structure that then influence gene expression. Importantly, these modifications are often propagated faithfully at cell division (termed epigenetic propagation), maintaining cellular identity in the absence of signals driving cellular differentiation.
Histone PTMs occur primarily at the solvent exposed N-termini, and can take a number of forms, including acetylation, methylation, and ubiquitination (Kouzarides, 2007). The transcriptional consequences of these modifications are then manifested either due to the direct biophysical consequences of the modification, or through the catalytic activities of proteins and protein complexes that recognize and bind modified histones. For instance, acetylation, which reduces the net positive charge on the nucleosome, results in decreased stability of histone associations with the negatively charged DNA, promoting transcription. Therefore, by balancing the expression and genomic localization of histone acetyltransferases (HATs) and histone deacetylases (HDACs), which add and remove acetyl groups, respectively, transcription can be activated or repressed (reviewed in Bannister and Kouzarides, 2011). Alternatively, it appears that the effects of histone methylation are conveyed indirectly, with methylated histones serving as a substrate for protein complexes that bind and reconfigure the chromatin. Importantly, histone methylation is associated with both active and repressed transcription, depending on the residue methylated. For example, trimethylation of lysine 4 of histone H3 (H3K4me3) is enriched at promoters of many actively transcribed genes, while trimethylation of lysine 27 of H3 (H3K27me3) is associated with transcriptionally repressed genes (Barski et al., 2007; Wang et al., 2008; Ernst et al., 2011).
Interestingly, activating and repressive modifications can co-localize, even occurring on the same nucleosome, and it appears that the combination and balance of these modifications serves to tune levels of transcription (Wang et al., 2008). Importantly in the context of cellular differentiation, co-localization of opposing PTMs is also employed to poise genes for rapid activation or repression (Bernstein et al., 2006).
As well as controlling access of the transcriptional machinery to the DNA template by modulating nucleosome positioning, transcription is controlled epigenetically by changing the structure of the DNA itself, through the addition and removal of bulky methyl groups. DNA methylation occurs predominantly at cytosine residues occurring in the context of cytosine–guanine di-nucleotides (termed CpG methylation), and results in transcriptional repression, both through steric hindrance of transcriptional activator binding (as described below for FoxP3), and through recruitment of methyl-CpG-binding domain proteins (MBD), that in turn, recruit HDACs. For instance, MBD2 has been shown to directly recruit HDAC1, resulting in histone deacetylation, and transcriptional repression (Ng and Bird, 1999). Thus, CpG methylation does not represent a separate system of epigenetic regulation to that described for histone PTMs, but rather is part of the same, inter-connected system.
Epigenetic Control of CD8+ T Cell Effector Function
The function of CD8+ killer T cells is defined largely by their capacity to produce effector molecules such as anti-viral cytokines and cytolytic molecules. As with naïve TH cells, the Ifng locus of naïve CD8+ killer T cells is heavily marked by the repressive H3K27me3, with little or none of the permissive H3K9Ac or H3K4me3 PTMs (Denton et al., 2011). Upon differentiation from naïve to effector killer T cells, transcriptional activation of Ifng is associated with removal of H3K27me3 and deposition of the permissive H3K9Ac and H3K4me3 PTMs (Denton et al., 2011). Further, in effector CD8+ killer T cells, the Ifng locus had reduced levels of total histone H3, indicating nucleosome evacuation from the region, presumably to allow the transcriptional machinery to access the promoter. Taken together, these data suggest that reconfiguration of the chromatin structure within naïve cells is necessary to enable Ifng transcription. Moreover, there appears to be conservation of chromatin restructuring and histone PTM modification with a similar pattern observed within other effector gene loci such as granzyme B (Gzmb; Juelich et al., 2009) and granzyme A (Gzma; Lauren Hatton, Michelle Nguyen, Brendan Russ, and Stephen Turner, data not shown).
As mentioned earlier, memory T cells maintain the capacity for rapid effector gene expression without the need for further differentiation. Strikingly, the permissive signature within the Ifng promoter of effector CD8+ killer T cells is maintained into long-term memory. Further, although memory CD8+ killer T cells exhibit little Ifng transcriptional activity prior to re-infection, RNA polymerase (RNAp) is docked at the Ifng promoter (Denton et al., 2011; Zediak et al., 2011). Taken together, these data suggest that the ability of memory cells to produce IFN-γ rapidly following re-infection is due to the promoter being maintained in a transcriptionally permissive state, and that the rate-limiting step in re-expression of IFN-γ is transcriptional initiation (Figures 3A,B). It remains to be determined whether transcriptional poising (as measured by RNAp docking) at other effector gene loci with low transcriptionally activity is evident within memory CD8+ killer T cells. Further, it would be of particular interest to determine the extent of transcriptional poising in memory T cells at a genome-wide level and compare this to naïve and effector cells. In this way, it could determined to what extent transcriptional poising underpins memory T cell characteristics. Moreover, given the direct effect of acetylation on nucleosome density, increased acetylation in memory cells (Araki et al., 2008; Denton et al., 2011) may explain their ability to produce more IFN-γ upon re-infection (La Gruta et al., 2004). In this way, memory T cells are reconfigured at the chromatin level to exhibit more potent effector function and this, in turn, helps ensure more effective and more rapid control of a secondary infection.
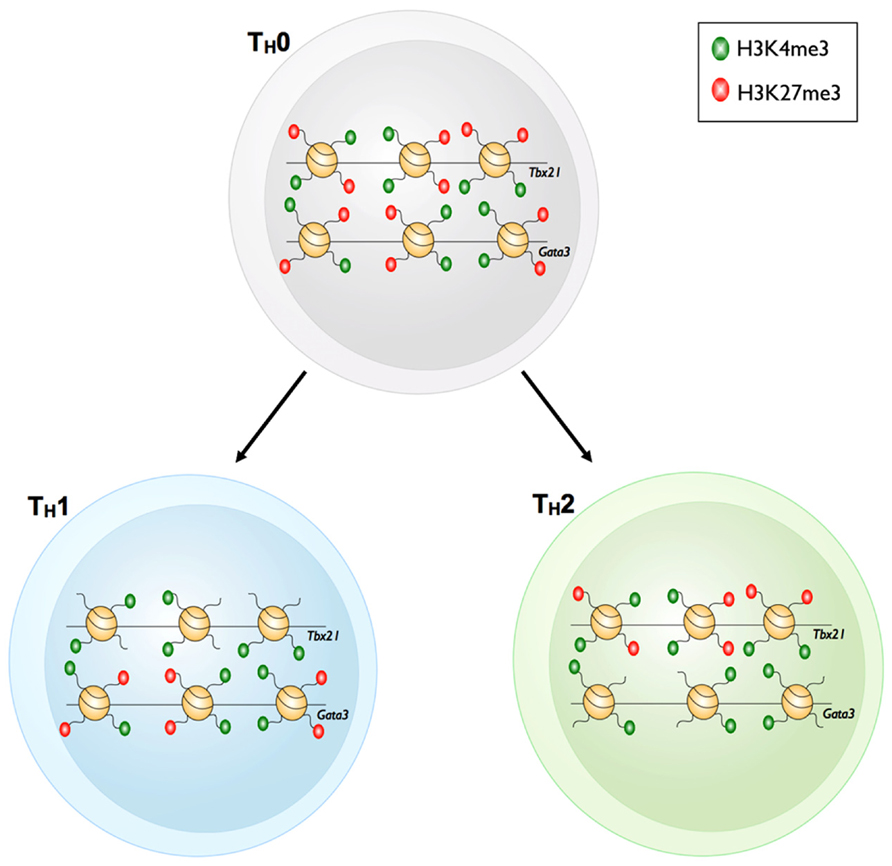
FIGURE 3. Bivalency of master regulator gene loci in TH1 and TH2 cells. Prior to differentiation of TH0 cells into the TH1 or TH2 cell subsets, the master regulator loci of each subset, Tbx21 and Gata3 respectively, have both active H3K4me3 and repressive H3K27me3 marks. The bivalent Tbx21 locus loses the repressive H3K27me3 mark upon differentiation into TH1 cells. However, the Gata3 locus remains bivalent. The reverse is true for TH2 cells whereby Gata3 loses the repressive H3K27me3 mark yet retains bivalency at the Tbx21 locus.
Recently, Scharer et al. (2013) applied global approaches to compare CpG methylation in naïve and effector CD8+ T cells. Combining immunoprecipitation of methylated genomic regions with high-throughput sequencing (MeDIP-seq), they identified ~650,000 regions that were differentially methylated between the two populations, indicating the likely importance of CpG methylation as a means of regulating CD8+ T cell differentiation. As expected, CpG methylation of gene promoters was inversely correlated with gene transcription, but interestingly, ~40% of genomic regions that differed in methylation state between naïve and effector occurred away from gene promoters. Further analysis showed that these promoter-distal regions largely overlapped candidate transcriptional enhancers identified in developing T cells in the thymus using next-generation sequencing and chromatin immunoprecipitation (ChIP-Seq) for enhancer-enriched histone PTMs (H3K27Ac and H3K4me1). Finally, when these putative enhancers were surveyed for over-represented TF binding sites, known and putative transcriptional regulators of CD8+ T cell differentiation were identified. Therefore, it seems likely that CpG methylation is employed to regulate CD8+ T cell differentiation, both by influencing protein–DNA interactions at gene promoters, and at transcriptional enhancers. Further, this study highlights the utility of such approaches in the identification of regulatory circuits controlling cellular differentiation.
CD4+ T Cell Differentiation: A Model for Understanding Epigenetic Regulation
The fact that distinct signals are capable of driving naïve TH cell differentiation in vitro into well-defined subsets makes CD4+ T cell activation a useful model for understanding how epigenetic regulation can influence cellular differentiation and fate determination. Comparison of the epigenetic profiles of signature effector gene loci within TH1 and TH2 cells has been particularly informative. In response to TH1 differentiation signals, the IFN-γ locus of naïve TH cells is remodeled to a permissive epigenetic signature that reinforces and heritably maintains IFN-γ gene expression in the long-term. At the same time, the IL-4 locus is remodeled to have a repressive epigenetic signature resulting in the shutdown of IL-4 gene expression.
Recent work using ChIP-Seq has been instrumental in providing genome-level insights into how epigenetic processes might regulate TH cell fate selection. For instance, genome-wide comparison of H3K4me3 and H3K27me3 distribution in naïve, TH1, TH2, and TH17 cells, combined with global transcriptional profiling demonstrated that the distribution of just two histone PTMs (H3K27me3 and H3K4me3) could provide a simple explanation for the differences in phenotypes observed amongst these different T cell subsets.
For example, upon differentiation from a naïve TH state into the various TH subsets, H3K4me3 deposition was observed at signature effector gene loci within distinct TH subsets (e.g., Ifng in TH1, Il4 in TH2, and Il17 in TH17). Moreover, H3K27me3 deposition was correlated with transcriptional shutdown of effector gene loci that are characteristic of other TH subsets (Wei et al., 2009; Table 1). One might have expected that changes in the epigenetic signatures within gene loci encoding lineage-defining TFs, would simply reflect those observed for lineage-specific effector gene loci. For example, the gene locus encoding the TH17 TF Rorc (retinoid-related orphan receptor-γ) was decorated with H3K27me3 in the naïve state, and only acquired H3K4me3, and losing H3K27me3 after TH17 differentiation. In contrast, the repressive H3K27me3 signature was reinforced under TH1 and TH2 differentiation conditions (Araki et al., 2009). However, this was not always the case. The Tbx21 (TH1) and Gata3 (TH2) gene loci in naïve TH cells were marked with both H3K4me3 and H3K27me3 (termed bivalent loci), and whilst these loci resolved to a permissive epigenetic signature (H3K4me3+/H3K27me3-) under TH1 and TH2 differentiation conditions, respectively, they did not acquire a repressive epigenetic signature when differentiated into opposing lineages, but rather maintained a bivalent state (Figure 4). Similarly, the Tbx21 locus within TH17 cells was also maintained in a bivalent state. In the case of TH17 cells, re-stimulation of TH17 cells in the presence of IL-12 resulted in expression of IFN-γ and conversion to a TH1 phenotype. This was associated with acquisition of permissive epigenetic signatures at the IFN-γ locus and IL-12-dependent STAT-4 and Tbx21-dependent epigenetic silencing of the TH17 associated Rorc locus (Mukasa et al., 2010). Given that epigenetic bivalency is considered a mechanism for poising gene loci for rapid activation or repression, these data suggest that CD4+ TH subsets can maintain some level of functional plasticity despite lineage commitment. It is tempting to speculate that this provides the immune system with inherent flexibility, allowing the redirection of pathogen-specific TH responses. In the case of TH17 cells, it may represent a mechanism that enables switching from a potent inflammatory TH17 response to a less damaging, more controlled effector response. It also suggests that targeted interventions that drive epigenetic reprograming of TH responses involved in autoimmune diseases (such as TH17 in the context of multiple sclerosis) might represent novel immunotherapeutic targets that could lead to decreased pathology.
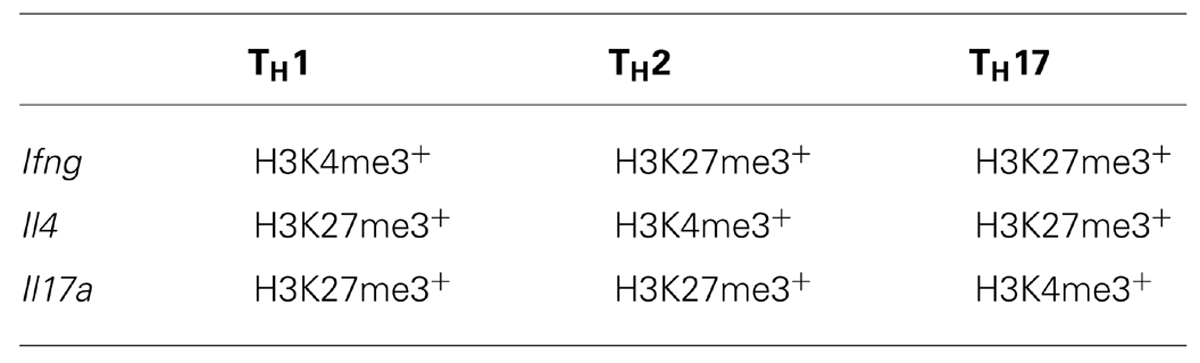
TABLE 1. Major histone methylation patterns at lineage-specific effector gene loci in differentiated CD4+ TH populations.
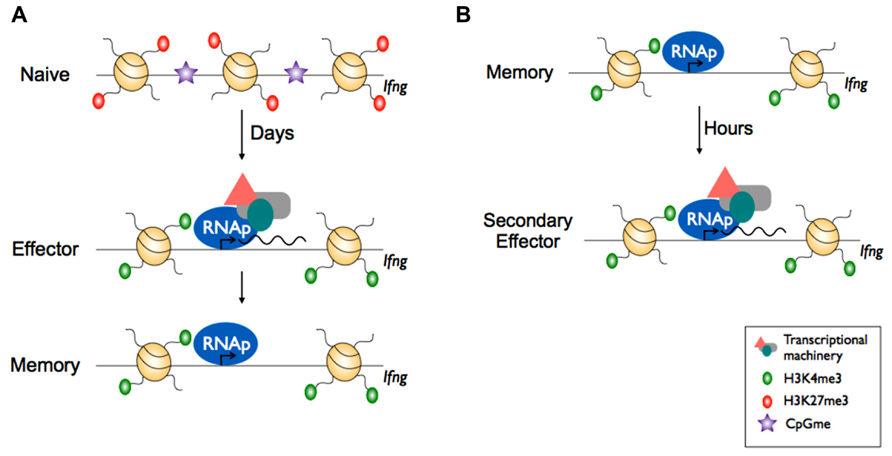
FIGURE 4. Epigenetic reprogramming within effector gene loci of CD8+ memory T cells enables rapid effector function. (A) In naïve CD8+ T cells, effector loci such as Ifng display repressive epigenetic marks e.g., H3K27me3 and is inaccessible to transcriptional machinery due to the heterochromatin structure. Upon activation, the chromatin is remodelled whereby it acquires active epigenetic marks e.g., H3K4me3 at key effector loci and nucleosome exit to make the loci accessible by transcriptional machinery and RNA polymerase II (RNAp), allowing transcription. Upon differentiation to memory CD8+ T cells, the chromatin retains the permissive H3K4me3 mark and RNAp remains docked. (B) Upon re-infection, the effector loci in memory CD8+ T cells is poised and can undergo rapid transcription.
A number of studies have also defined roles for CpG methylation in the differentiation of CD4+ T cells. For instance, regulated deposition of CpG methylation is important for maintenance of CD4+ T cells that have differentiated to become Tregs. Zheng et al. (2010) showed that mice that had a conserved non-coding sequence (CNS2) within the Foxp3 locus deleted, had wild-type levels of Tregs in young mice, but greatly reduced numbers in older mice. Further, this was due to a loss of FoxP3 expression in the peripheral Tregs, indicating a role for this TF, not just in Treg differentiation, as described previously (reviewed in Josefowicz et al., 2012), but also in the maintenance of the Treg phenotype. Finally, they were able to show that FoxP3 binds to the CNS2 in Tregs, but not in naïve CD4+ T cells, and that FoxP3 binding was dependent on differentiation-induced demethylation of CpG sites within this region. Thus, FoxP3 binding to CNS2, enabled by differentiation-dependent CpG demethylation, results in a feed-forward signal that enforces Treg fate.
Enzymes Modulating Histone Modification During T Cell Differentiation
Whilst there is a growing understanding of how changes in histone PTMs correlate with dynamic changes in T cell effector functions, it is less clear how the factors that write or erase these histone PTMs are involved in directing T cell differentiation during an immune response. Using the CD4 TH1 versus TH2 model system, Allan et al. (2012) examined the role of the histone methyltransferase, Suv39H1, in epigenetic regulation of TH2 differentiation. Suv39H1 specifically trimethylates H3K9 – a PTM typically associated with transcriptional silencing of gene loci that is in turn recognized by heterochromatin protein 1α (HP1α; Lachner et al., 2001; Peters et al., 2001). Docking of HP1α onto H3K9me3+ gene loci in turn recruits HDAC1 and 2, and the transcriptional repressor MBD1 (Fujita et al., 2003). In this way, H3K9 acetylation, a PTM associated with transcriptional activation, is limited. Thus, Suv39H1-mediated trimethylation of H3K9 is an initial step that triggers histone deacetylation and binding of transcriptional repressor protein complexes that stably silence targeted loci.
While it was possible to skew naïve TH cells from Suv39H1 gene-deficient mice into the TH2 lineage in vitro, these cells could be reprogrammed to secrete IFN-γ after re-culture in TH1-inducing conditions. Thus, a lack of Suv39H1 resulted in an inability to stably repress TH1 effector gene expression. This appeared largely due to an inability of Suv39H1 gene-deficient TH2 cells to stably silence the transcriptional potential of the key TH1 TF, T-bet (encoded by Tbx21). Consistent with this, Suv39H1-deficient TH2 cells exhibited increased levels of histone acetylation at the Tbx21 locus. Of particular interest was the fact that TH1 cells from Suv39H1 gene-deficient mice stably repressed expression of TH2 effector genes after re-culture in TH2-inducing conditions. This suggests that histone PTMs, other than H3K9me3, are used to heritably silence TH2 effector gene expression during TH1 differentiation, or alternatively, other H3K9 methyltransferases (such as GP9a, SETDB1/2, or Suv39H2) are utilized by TH1 cells to establish H3K9me3 repression at TH2 gene loci. Such a hypothesis would require selective targeting of H3K9 methyltransferases to specific gene loci and this could potentially be facilitated via interactions with specific TFs that bind to specific regulatory regions within target gene loci. Such a precedent has been observed with the demonstration that members of the T-box family of TFs serve to recruit histone methyltransferases to signature effector gene loci within TH1 cells to promote gene transcription (Lewis et al., 2007). Thus, this mechanism could potentially be a way of ensuring that only certain gene loci are targeted for silencing within either TH1 or TH2 cell subsets, thereby ensuring appropriate gene expression, and appropriate immune function.
Taken together, these data demonstrate that Suv39H1 acts to specifically promote TH2 lineage commitment via epigenetic silencing (via H3K9me3 deposition) of gene loci that drive TH1 fate commitment (Figure 5). One interesting observation was the fact that despite TH2 cells exhibiting an overall repressive signature within the Tbx21 locus, there is still evidence of H3K4me3 deposition at the promoter. Thus, pharmacological interventions that block Suv39H1 activity could serve to promote Tbx21 transcription and subsequent TH1 gene expression. The clinical relevance was made apparent when treatment of mice with a Suv39H1 inhibitor, was able to ameliorate TH2 cell driven tissue damage in a model of allergic asthma. Treatment of mice resulted in higher numbers of TH1 T cells, and redirected the immune response toward a less pathogenic state. This study highlights the potential for manipulating epigenetic programing of effector T cell responses using small molecule inhibitors to either promote immunity, in the case of vaccination, or suppress the damage caused by inappropriate immune responses, as is found in autoimmune disease or allergy.
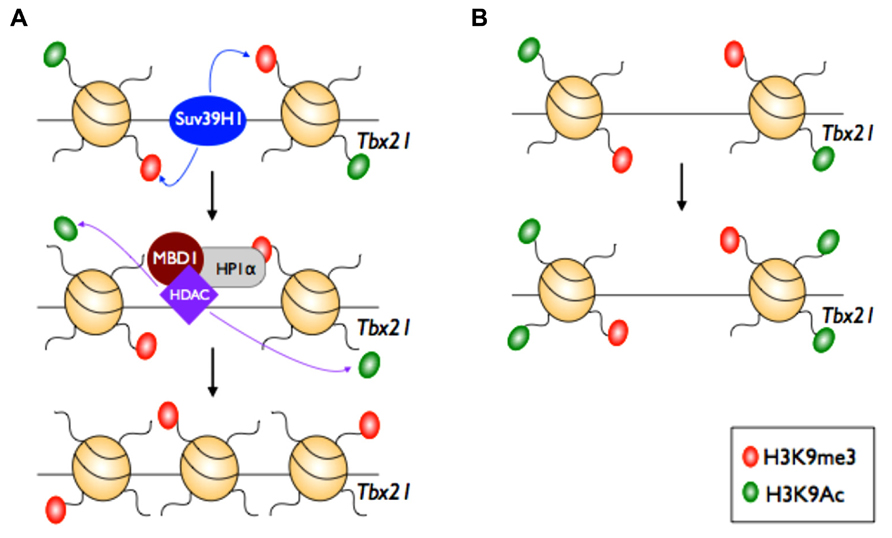
FIGURE 5. Epigenetic maintenance of TH2 lineage commitment. In the TH2 cell subset, the master regulator of TH1 cells (Tbx21) is silenced. The histone methylase Suv39H1 adds the repressive H3K9me3 mark at the Tbx21 locus. This initiates recruitment and docking of heterochromatin protein 1 alpha (HP1α), histone deacetylase (HDAC) 1 and 2, and methyl-binding domain protein (MBD1). HDACs then remove the active H3K9ac mark to maintain silencing, mediated by H3K9me3, at the Tbx21 locus.
Epigenetic Control of T Cell Development
Mature, immunologically naïve CD4+ and CD8+ T cells develop in the thymus from multipotent hematopoietic progenitors. Within the thymus, these progenitors progress through at least ten phenotypically distinct stages of development, before exiting the thymus as mature, naïve CD4+ or CD8+ T cells (reviewed in Rothenberg et al., 2010). Hematopoietic progenitor cells enter the thymus expressing neither CD4 nor CD8, and are hence termed double-negative (DN). They then progress through five phenotypically distinct stages of maturation (DN1, DN2a, DN2b, DN3a, and DN3b) before up-regulating both CD4 and CD8 (termed double-positive, DP), and following further differentiation, permanently down-regulate either CD4 or CD8 (becoming single-positive, SP), before migrating from the thymus. Importantly, events occurring in the thymus not only determine lineage commitment (CD4+ versus CD8+), but also the potential fates of mature T cells; commitment to the CD8+ lineage results in cells with specialized cytotoxic potential, while commitment to the CD4+ lineage results in naïve cells with much broader differentiation potential. Thus an interesting question is when is fate potential programed, and what is the contribution of epigenetic mechanisms?
Rothenberg’s group recently studied the molecular signatures that underpin lineage commitment and differentiation occurring in the early phases (DN1–DP) of thymic development in mice (Zhang et al., 2012). Combining ChIP-Seq and RNA-Seq, they determined the global distribution and dynamics of three histone PTMs, and the transcriptional signatures of immature thymocytes, at each stage of differentiation. Specifically, they studied the distribution of H3K9/14Ac (Gett and Hodgkin, 1998; Djuretic et al., 2007) and H3K27me3, which is enriched within the promoters and enhancers of actively transcribed and repressed genes, respectively, and H3K4me2, which defines active enhancer elements, and is often associated with transcriptionally poised gene promoters.
Aside from highlighting the extraordinary complexity of the mechanisms regulating T cell differentiation, this study provided novel insights into the mechanisms controlling cellular differentiation. A key finding of the paper was that the repressive H3K27me3 PTM is often deposited at genes after transcription has already been shutdown, indicating that the likely role of this modification is not to directly regulate transcription, as is generally accepted, but rather to stabilize repression. Further, there appeared to be multiple mechanisms of transcriptional repression, since only approximately a third of genes that were developmentally repressed during thymic differentiation were associated with H3K27me3.
In contrast, histone acetylation was strongly and temporally correlated with mRNA levels, indicating that this modification may be added just prior to transcription, and as such, likely represents a rate-limiting step in the activation of gene transcription. Further histone deacetylation may be a key means of gene repression during T cell differentiation since this observation also implies that acetylation is either rapidly removed from promoters following transcriptional repression, or is a direct cause of transcriptional repression. Finally, H3K4me2 deposition often preceded transcription. This finding is consistent with a previous study showing that H3K4me2 marks lineage-specific hematopoietic genes in multipotent progenitor cells, in the absence of transcription (Orford et al., 2008). As many H3K4me2-marked genes lost this modification as differentiation preceded (toward an erythroid fate), it appears that H3K4me2 poises genes for a rapid response to differentiation signals, whereby, following the receipt of signals, non-lineage-specific genes lose H3K4me2 and are not expressed, while at lineage-specific genes, H3K4me2 is converted to H3K4me3 – a positive correlate of transcription.
Taken together, these studies suggest that different histone PTMs play distinct roles in transcriptional regulation; acetylated histones appear to rate-limit transcription, probably by directly regulating promoter accessibility, while H3K27me3 appears to operate “after the fact” – stabilizing transcriptional repression rather than directly repressing transcription. Finally, H3K4me2 apparently functions as an intermediate between unmethylated H3K4, and the activating trimethylated state at gene promoters, thus allowing rapid transcriptional change following differentiation signals.
In the context of T cell development, CpG methylation plays important roles both during thymic development, and in later (peripheral) fate decisions (described above). For instance, by deleting DNA methyltransferase 1 (DNMT1) at the DN stage of thymic development, Lee et al. (2001) showed an ~90% reduction in the numbers of DP T cells, as well as large decreases in mature peripheral T cells of both CD4+ and CD8+ lineages. Further, the T cells that did develop had greatly reduced survival relative to the wild-type. However, when DNMT1 was deleted at the (later) DP stage, peripheral T cell numbers and composition were normal, but when either (Dnmt1-/-) CD4+ or CD8+ T cells where stimulated in vitro, they had aberrant cytokine production profiles, in that they produced IL-2, IL-3, and IFN-γ more rapidly than wild-type cells. This latter observation is consistent with the demonstration that demethylation of regions controlling the transcription of Ifng and Il2 in effector CD8+ T cells (Kersh et al., 2006; Northrop et al., 2006) and Il2 in effector CD4+ T cells (Thomas et al., 2005) coincides with their demethylation. Further, it suggests that methylation might be a safeguard against inappropriate expression of these genes, which might otherwise lead to immune pathology. Taken together, these results indicated a central role for DNMT1, and CpG methylation, both during thymic and post-thymic development and differentiation of T cells of both CD4+ and CD8+ lineages.
Summary
Both current effective vaccine strategies, and the design of novel vaccine strategies that specifically target adaptive T cell immunity, rely on acquisition and maintenance of T cell functional potential to establish protective immunity. Conversely, these same characteristics of adaptive T cell immunity are also at play during adverse immune reactions where priming of T cells to either environmental or self-antigens, can manifest as T cell hypersensitivities or T cell-mediated autoimmune diseases, respectively. Thus, a greater understanding of the molecular mechanisms, and specifically epigenetic mechanisms, that shape acquisition and maintenance of lineage-specific T cell function, will be key if we are to make advances in novel therapeutic strategies for a variety of disease contexts. We have tried to highlight what we think are some of the key findings and general themes emerging from the studies of T cell differentiation, as well as the utility of the immune system as a tool for studying differentiation and development. By comparison with studies performed on stem cells, it appears that conclusions made from studies of T cells are broadly relevant to differentiation in other cell types and tissues. In particular, the concepts of transcriptional poising and promoter bivalency as mechanisms that regulate fate decisions are pertinent during the differentiation of stem cells and less primitive tissues. The studies of Rothenberg et al. (2010), in particular, highlight the value of the immune system as a tool for studying differentiation – because of the detailed ontogenies and the ability to resolve different stages of T cell development based on characteristic and defined cell surface phenotypes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council of Australia (program grant #5671222 awarded to Stephen J. Turner; project grant #APP1003131 awarded to Stephen J. Turner and Sudha Rao). Stephen J. Turner is supported by an Australian Research Council Future Fellowship; Julia E. Prier is supported by an Australian Government Postgraduate Award.
References
Agarwal, S., and Rao, A. (1998). Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 9, 765–775. doi: S1074-7613(00)80642-1
Allan, R. S., Zueva, E., Cammas, F., Schreiber, H. A., Masson, V., Belz, G. T., et al. (2012). An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 487, 249–253. doi: 10.1038/nature11173
Ansel, K. M., Lee, D. U., and Rao, A. (2003). An epigenetic view of helper T cell differentiation. Nat. Immunol. 4, 616–623. doi: 10.1038/ni0703-616
Araki, Y., Fann, M., Wersto, R., and Weng, N. P. (2008). Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J. Immunol. 180, 8102–8108. doi: 180/12/8102
Araki, Y., Wang, Z., Zang, C., Wood, W. H. III, Schones, D., Cui, K., et al. (2009). Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 30, 912–925. doi: 10.1016/j.immuni.2009.05.006
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. doi: 10.1016/j.cell.2006.02.041
Cruz-Guilloty, F., Pipkin, M. E., Djuretic, I. M., Levanon, D., Lotem, J., Lichtenheld, M. G., et al. (2009). Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 206, 51–59. doi: 10.1084/jem.20081242
Denton, A. E., Russ, B. E., Doherty, P. C., Rao, S., and Turner, S. J. (2011). Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. U.S.A. 108, 15306–15311. doi: 10.1073/pnas.1112520108
Djuretic, I. M., Levanon, D., Negreanu, V., Groner, Y., Rao, A., and Ansel, K. M. (2007). Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat. Immunol. 8, 145–153. doi: 10.1038/ni1424
Dong, C. (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 8, 337–348. doi: 10.1038/nri2295
Ernst, J., Kheradpour, P., Mikkelsen, T. S., Shoresh, N., Ward, L. D., Epstein, C. B., et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49. doi: 10.1038/nature09906
Fujita, N., Watanabe, S., Ichimura, T., Tsuruzoe, S., Shinkai, Y., Tachibana, M., et al. (2003). Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 278, 24132–24138. doi: 10.1074/jbc.M302283200
Gett, A. V., and Hodgkin, P. D. (1998). Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc. Natl. Acad. Sci. U.S.A. 95, 9488–9493. doi: 10.1073/pnas.95.16.9488
Intlekofer, A. M., Takemoto, N., Wherry, E. J., Longworth, S. A., Northrup, J. T., Palanivel, V. R., et al. (2005). Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, 1236–1244. doi: 10.1038/ni1268
Janas, M. L., Groves, P., Kienzle, N., and Kelso, A. (2005). IL-2 regulates perforin and granzyme gene expression in CD8+ T cells independently of its effects on survival and proliferation. J. Immunol. 175, 8003–8010.
Jenkins, M. R., Kedzierska, K., Doherty, P. C., and Turner, S. J. (2007). Heterogeneity of effector phenotype for acute phase and memory influenza A virus-specific CTL. J. Immunol. 179, 64–70.
Jenkins, M. R., Mintern, J., La Gruta, N. L., Kedzierska, K., Doherty, P. C., and Turner, S. J. (2008). Cell cycle-related acquisition of cytotoxic mediators defines the progressive differentiation to effector status for virus-specific CD8+ T cells. J. Immunol. 181, 3818–3822.
Josefowicz, S. Z., Lu, L. F., and Rudensky, A. Y. (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564. doi: 10.1146/annurev.immunol.25.022106.141623
Juelich, T., Sutcliffe, E. L., Denton, A., He, Y., Doherty, P. C., Parish, C. R., et al. (2009). Interplay between chromatin remodeling and epigenetic changes during lineage-specific commitment to granzyme B expression. J. Immunol. 183, 7063–7072. doi: 10.4049/jimmunol.0901522
Kaech, S. M., Hemby, S., Kersh, E., and Ahmed, R. (2002). Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851. doi: 10.1016/S0092-8674(02)01139-X
Kagi, D., Ledermann, B., Burki, K., Seiler, P., Odermatt, B., Olsen, K. J., et al. (1994). Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369, 31–37. doi: 10.1038/369031a0
Kanno, Y., Vahedi, G., Hirahara, K., Singleton, K., and O’Shea, J. J. (2012). Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu. Rev. Immunol. 30, 707–731. doi: 10.1146/annurev-immunol-020711-075058
Kersh, E. N., Fitzpatrick, D. R., Murali-Krishna, K., Shires, J., Speck, S. H., Boss, J. M., et al. (2006). Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J. Immunol. 176, 4083–4093.
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Lachner, M., O’Carroll, D., Rea, S., Mechtler, K., and Jenuwein, T. (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. doi: 10.1038/35065132
La Gruta, N. L., Turner, S. J., and Doherty, P. C. (2004). Hierarchies in cytokine expression profiles for acute and resolving influenza virus-specific CD8+ T cell responses: correlation of cytokine profile and TCR avidity. J. Immunol. 172, 5553–5560.
Lalvani, A., Brookes, R., Hambleton, S., Britton, W. J., Hill, A. V., and McMichael, A. J. (1997). Rapid effector function in CD8+ memory T cells. J. Exp. Med. 186, 859–865. doi: 10.1084/jem.186.6.859
Lawrence, C. W., and Braciale, T. J. (2004). Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J. Immunol. 173, 1209–1218.
Lee, P. P., Fitzpatrick, D. R., Beard, C., Jessup, H. K., Lehar, S., Makar, K. W., et al. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774. doi: 10.1016/S1074-7613(01)00227-8
Lewis, M. D., Miller, S. A., Miazgowicz, M. M., Beima, K. M., and Weinmann, A. S. (2007). T-bet’s ability to regulate individual target genes requires the conserved T-box domain to recruit histone methyltransferase activity and a separate family member-specific transactivation domain. Mol. Cell. Biol. 27, 8510–8521. doi: 10.1128/MCB.01615-07
Marshall, D. R., Turner, S. J., Belz, G. T., Wingo, S., Andreansky, S., Sangster, M. Y., et al. (2001). Measuring the diaspora for virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. U.S.A. 98, 6313–6318. doi: 10.1073/pnas.101132698
Miller, S. A., Mohn, S. E., and Weinmann, A. S. (2010). Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol. Cell 40, 594–605. doi: 10.1016/j.molcel.2010.10.028
Miyazaki, T., Liu, Z. J., Kawahara, A., Minami, Y., Yamada, K., Tsujimoto, Y., et al. (1995). Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic cell proliferation. Cell 81, 223–231. doi: 10.1016/0092-8674(95)90332-1
Moffat, J. M., Gebhardt, T., Doherty, P. C., Turner, S. J., and Mintern, J. D. (2009). Granzyme A expression reveals distinct cytolytic CTL subsets following influenza A virus infection. Eur. J. Immunol. 39, 1203–1210. doi: 10.1002/eji.200839183
Mukasa, R., Balasubramani, A., Lee, Y. K., Whitley, S. K., Weaver, B. T., Shibata, Y., et al. (2010). Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32, 616–627. doi: 10.1016/j.immuni.2010.04.016
Ng, H. H., and Bird, A. (1999). DNA methylation and chromatin modification. Curr. Opin. Genet. Dev. 9, 158–163. doi: 10.1016/S0959-437X(99)80024-0
Northrop, J. K., Thomas, R. M., Wells, A. D., and Shen, H. (2006). Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J. Immunol. 177, 1062–1069.
Oehen, S., and Brduscha-Riem, K. (1998). Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J. Immunol. 161, 5338–5346.
Onodera, A., Yamashita, M., Endo, Y., Kuwahara, M., Tofukuji, S., Hosokawa, H., et al. (2010). STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in T helper type 2 cells. J. Exp. Med. 207, 2493–2506. doi: 10.1084/jem.20100760
Orford, K., Kharchenko, P., Lai, W., Dao, M. C., Worhunsky, D. J., Ferro, A., et al. (2008). Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev. Cell 14, 798–809. doi: 10.1016/j.devcel.2008.04.002
Pearce, E. L., Mullen, A. C., Martins, G. A., Krawczyk, C. M., Hutchins, A. S., Zediak, V. P., et al. (2003). Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043. doi: 10.1126/science.1090148
Peixoto, A., Evaristo, C., Munitic, I., Monteiro, M., Charbit, A., Rocha, B., et al. (2007). CD8 single-cell gene coexpression reveals three different effector types present at distinct phases of the immune response. J. Exp. Med. 204, 1193–1205. doi: 10.1084/jem.20062349
Peters, A. H., O’Carroll, D., Scherthan, H., Mechtler, K., Sauer, S., Schofer, C., et al. (2001). Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337. doi: S0092-8674(01)00542-6
Pipkin, M. E., Sacks, J. A., Cruz-Guilloty, F., Lichtenheld, M. G., Bevan, M. J., and Rao, A. (2010). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90. doi: 10.1016/j.immuni.2009.11.012
Rothenberg, E. V., Zhang, J., and Li, L. (2010). Multilayered specification of the T-cell lineage fate. Immunol. Rev. 238, 150–168. doi: 10.1111/j.1600-065X.2010.00964.x
Russ, B. E., Denton, A. E., Hatton, L., Croom, H., Olson, M. R., and Turner, S. J. (2012). Defining the molecular blueprint that drives CD8(+) T cell differentiation in response to infection. Front. Immunol. 3:371. doi: 10.3389/fimmu.2012.00371
Scharer, C. D., Barwick, B. G., Youngblood, B. A., Ahmed, R., and Boss, J. M. (2013). Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 91, 3419–3429.doi: 10.4049/jimmunol.1301395
Thomas, R. M., Gao, L., and Wells, A. D. (2005). Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J. Immunol. 174, 4639–4646.
van Stipdonk, M. J., Hardenberg, G., Bijker, M. S., Lemmens, E. E., Droin, N. M., Green, D. R., et al. (2003). Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4, 361–365. doi: 10.1038/ni912
Veiga-Fernandes, H., Walter, U., Bourgeois, C., McLean, A., and Rocha, B. (2000). Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat. Immunol. 1, 47–53. doi: 10.1038/76907
Wang, Z., Zang, C., Rosenfeld, J. A., Schones, D. E., Barski, A., Cuddapah, S., et al. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903. doi: 10.1038/ng.154
Wei, G., Wei, L., Zhu, J., Zang, C., Hu-Li, J., Yao, Z., et al. (2009). Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167. doi: 10.1016/j.immuni.2008.12.009
Zediak, V. P., Johnnidis, J. B., Wherry, E. J., and Berger, S. L. (2011). Cutting edge: persistently open chromatin at effector gene loci in resting memory CD8+ T cells independent of transcriptional status. J. Immunol. 186, 2705–2709. doi: 10.4049/jimmunol.1003741
Zhang, J. A., Mortazavi, A., Williams, B. A., Wold, B. J., and Rothenberg, E. V. (2012). Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149, 467–482. doi: 10.1016/j.cell.2012.01.056
Zheng, Y., Josefowicz, S., Chaudhry, A., Peng, X. P., Forbush, K., and Rudensky, A. Y. (2010). Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812. doi: 10.1038/nature08750
Keywords: epigenetics, T cell Immunity, T cell memory, viral immunity, T cell differentiation
Citation: Russ BE, Prier JE, Rao S and Turner SJ (2013) T cell immunity as a tool for studying epigenetic regulation of cellular differentiation. Front. Genet. 4:218. doi: 10.3389/fgene.2013.00218
Received: 02 August 2013; Accepted: 08 October 2013;
Published online: 12 November 2013.
Edited by:
Silvia Carolina Galvan, Universidad Nacional Autónoma de, MéxicoReviewed by:
Ichiro Taniuchi, Research Center for Allergy and Immunology, JapanSteven Zvi Josefowicz, Rockefeller University, USA
Copyright © 2013 Russ, Prier, Rao and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen J. Turner, Department of Microbiology and Immunology, The University of Melbourne, Royal Parade, Parkville, VIC 3010, Australia e-mail:c2p0dXJuQHVuaW1lbGIuZWR1LmF1