 Ridgely Fisk Green1*
Ridgely Fisk Green1* Julianne Byrne2
Julianne Byrne2 Krista S. Crider3
Krista S. Crider3 Margaret Gallagher4
Margaret Gallagher4 Deborah Koontz4
Deborah Koontz4 Robert J. Berry3
Robert J. Berry3- 1Carter Consulting, Inc. and Division of Birth Defects and Developmental Disabilities, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, Atlanta, GA, USA
- 2Boyne Research Institute, Drogheda, Ireland
- 3Division of Birth Defects and Developmental Disabilities, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, Atlanta, GA, USA
- 4Newborn Screening and Molecular Biology Branch, Division of Laboratory Science, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta, GA, USA
Periconceptional folic acid use can often prevent neural tube defects (NTDs). Variants of genes involved in folate metabolism in mothers and children have been associated with occurrence of NTDs. We identified Irish families with individuals affected by neural tube defects. In these families, we observed that neural tube defects and birth defects overall occurred at a higher rate in the maternal lineage compared with the paternal lineage. The goal of this study was to look for evidence for genetic effects that could explain the discrepancy in the occurrence of these birth defects in the maternal vs. paternal lineage. We genotyped blood samples from 322 individuals from NTD-affected Irish families, identified through their membership in spina bifida associations. We looked for differences in distribution in maternal vs. paternal lineages of five genetic polymorphisms: the DHFR 19 bp deletion, MTHFD1 1958G>A, MTHFR 1298A>C, MTHFR 677C>T, and SLC19A1 80A>G. In addition to looking at genotypes individually, we determined the number of genotypes associated with decreased folate metabolism in each relative (“risk genotypes”) and compared the distribution of these genotypes in maternal vs. paternal relatives. Overall, maternal relatives had a higher number of genotypes associated with lower folate metabolism than paternal relatives (p = 0.017). We expected that relatives would share the same risk genotype as the individuals with NTDs and/or their mothers. However, we observed that maternal relatives had an over-abundance of any risk genotype, rather than one specific genotype. The observed genetic effects suggest an epigenetic mechanism in which decreased folate metabolism results in epigenetic alterations related to the increased rate of NTDs and other birth defects seen in the maternal lineage. Future studies on the etiology of NTDs and other birth defects could benefit from including multigenerational extended families, in order to explore potential epigenetic mechanisms.
Introduction
Neural tube defects (NTDs) are congenital anomalies of the brain and spinal cord that cause significant morbidity and mortality (Sutton et al., 2008). Historically, Ireland has had high rates of NTDs (Coffey, 1983), with rates decreasing significantly in the last 50 years (Botto et al., 2006). Both environmental and genetic factors have been implicated in the etiology of NTDs. The evidence for environmental factors lies in the protective effect of periconceptional folic acid intake (Czeizel and Dudas, 1992). The evidence for genetic factors lies in the increased recurrence risk in siblings of individuals with NTDs, the increased risk for NTDs among other relatives, and the increased risk for birth defects overall among both close and distant relatives (MRC Vitamin Study Research Group, 1991; Deak et al., 2008). Maternal relatives have higher NTD and overall birth defect rates than paternal relatives, consistent with a combination of genetic and epigenetic mechanisms contributing to NTD and overall birth defects risk (Byrne et al., 1996; Byrne, 2008, 2010, 2011; Deak et al., 2008).
The genetic mechanisms associated with NTDs have been addressed through studies on associations between NTD risk and variants in genes involved in folate metabolism, chosen because of the protective effect of folic acid on NTDs (Molloy et al., 2009; Shaw et al., 2009). Methylene tetrahydrofolate reductase (MTHFR) plays an important role in folate metabolism. The 677 T allele results in reduced enzyme activity and decreased folate metabolism (Frosst et al., 1995). The MTHFR 677 TT genotype in infants and mothers has been associated with increased risk for NTDs [reviewed in Botto and Yang (2000)]. Studies in Irish populations found significantly more individuals with NTDs with the TT and CT genotypes and more parents of affected individuals with the TT genotype (Whitehead et al., 1995; Shields et al., 1999; Kirke et al., 2004; Carter et al., 2011). The MTHFR 1298A>C variant also results in reduced enzyme activity, although its role in NTD risk is less clear (van der Put et al., 1998; Botto and Yang, 2000; Wang et al., 2012a). Parle-McDermott et al. (2003) did not find an association between the MTHFR 1298A>C variant and NTDs in an Irish population. Methylene tetrahydrofolate dehydrogenase 1 (MTHFD1) acts in folate metabolism and DNA synthesis, and the 1958G>A variant results in decreased enzyme stability (Christensen et al., 2009). Studies in Irish populations (Brody et al., 2002; Parle-McDermott et al., 2006) identified a significant excess of MTHFD1 AA homozygotes among mothers of NTD cases compared to controls. A recent larger scale study in an Irish population also identified the MTHFD1 A allele as a risk factor for NTDs (Pangilinan et al., 2012). Dihydrofolate reductase (DHFR) also acts in folate metabolism. Studies have yielded conflicting results on the effect of the 19 bp deletion at intron 1 on red blood cell folate concentrations (Kalmbach et al., 2008; Stanislawska-Sachadyn et al., 2008). Researchers from Trinity College in Dublin observed a slight protective effect of the homozygous DHFR deletion genotype in mothers (Parle-McDermott et al., 2007). SLC19A1 plays an important role in folate transport, and some previous studies have reported associations between the 80A>G variant and NTD risk (Chango et al., 2000; De Marco et al., 2003; Wang et al., 2012b). O'Leary et al. (2006) did not find a difference in the frequency of the SLC19A1 80A>G reduced folate carrier in mothers, fathers and affected individuals compared with controls in an Irish population.
Epigenetic mechanisms related to NTDs have not been well explored. The higher rates of NTDs in maternal relatives compared with paternal relatives suggest the presence of epigenetic effects. Folate metabolism genes are of interest because folate metabolism is critical to the production of one-carbon subunits needed for epigenetic modification (Crider et al., 2012). Decreased folate metabolism, seen with certain gene variants, might hinder proper establishment or maintenance of epigenetic modifications. The epigenetic changes of interest would occur in the maternal genome in utero, that is, during the grandmother's pregnancy (Crider et al., 2012). Thus, in contrast to genetic mechanisms, in which the mothers' and affected individuals' genotypes would be most relevant, for epigenetic mechanisms, the maternal and paternal grandparents' genotypes would be important. In the absence of information on grandparents' genotypes, aunts, uncles, and first cousins provide an indirect measure of the grandparents' genotypes.
In this hypothesis-generating study, we investigated possible mechanisms for the excess risk for NTDs and birth defects overall in the maternal lineage of Irish individuals with NTDs. To do this, we evaluated five polymorphisms in genes involved in folate metabolism in the maternal vs. paternal lineage, both individually and with low folate-metabolizing genotypes grouped together. To our knowledge, this study is the first to look for genomic associations in extended relatives in NTD families and the effect of maternal vs. paternal lineage risk. We chose five polymorphic markers for evaluation, which were among the most intensely studied so far: the 19 bp deletion in DHFR, MTHFD1 1958G>A, MTHFR 1298A>C, MTHFR 677C>T, and SLC19A1 80A>G.
Materials and Methods
Study Design and Participants
The participants in this cross-sectional study were members of Irish families with NTDs. These families had previously participated in epidemiologic studies of the Boyne Research Institute [for instance, (Byrne, 2011)]. The families were identified initially through their membership in the Louth-Meath (northeastern region of the Republic of Ireland) branch of the Irish Association for Spina Bifida and Hydrocephalus, and the Ballymena (Northern Ireland) branch of the (UK) Association for Spina Bifida and Hydrocephalus. Families were also recruited through word of mouth and through radio announcements in the Louth-Meath area.
To be eligible for the study, male and female participants had to be 15 years of age or older, reside in the northeast region of the Republic of Ireland or Northern Ireland, and be either the individual with an NTD (only one NTD in sibship) or a blood relative of the affected individual. In terms of relationship, relatives (defined according to their relationship to the affected individual) had to be one of the following: parents, siblings, nieces, nephews, offspring (of the affected individual), grandparents, uncles, aunts, first cousins and first cousins once removed (the offspring of the first cousins).
Family members were interviewed in phases. Phase I evaluated pregnancy histories and the health of siblings in the original nuclear families (1995–2002). Phase II subjects (uncles and aunts of the affected individuals) were identified by their siblings during Phase I and interviewed between 2000 and 2002. Phase III consisted of interviews with first cousins of the affected individuals (the children of the uncles and aunts) between 2002 and 2009. Between 2005 and 2006, affected individuals and their siblings were interviewed.
For the present study, conducted in 2007, the response rate among eligible, locatable relatives was 49.5%, or 331 participants. From these, 322 gave a blood sample. Of these with a blood sample, 34 were relatives in families in which the individual with an NTD had anencephaly, 41 in which the affected individual had spina bifida occulta, 240 in which the affected individual had open spina bifida and 7 in which the affected individual had encephalocele.
This study was conducted according to the guidelines from the Declaration of Helsinki and all procedures involving human subjects were approved by the Ethics Board of the Boyne Research Institute and the Ethics Committee of the Health Service Executive of the Irish Government's Department of Health and Children. Written informed consent was obtained from all subjects.
Statistical Analysis
Data were managed using Microsoft EXCEL©, and statistical analysis was conducted using SAS 9.1 (SAS Institute, Cary, NC, USA), STATA©, and IBM® SPSS® v19. Measures of association were evaluated with t-tests for continuous variables and chi-square and Fisher's exact tests for categorical variables, with the level of statistical significance set at 0.05. First-degree relatives were parents, siblings, and children; second-degree relatives were aunts, uncle, nieces, nephews, and grandparents; and third-degree and higher relatives were cousins and first cousins once removed. Genotypes were analyzed using an additive model, consistent with previous studies. For logistic regression analyses, genotypes were dichotomized using a recessive model, consistent with previous studies. For each individual, we calculated the number of “risk genotypes” present. “Risk genotypes” were those associated with lower folate metabolism in previous studies (DHFR WT/WT, MTHFD1 1958 AA, MTHFR 1298 CC, MTHFR 677 TT, and SLC19A1 80 GG).
DNA Methods
DNA was extracted from blood clots using Gentra Puregene Blood kit and Clotspin Baskets per manufacturer's protocol (Qiagen, Valencia, CA). Quantitation was performed by spectrophotometric reading at 260nm and all samples normalized to 10 ng/μl. For PCR reactions, 2 μl genomic DNA (20 ng) was used. Pyrosequencing technology (Qiagen, Valencia, CA) was used to genotype DNA samples for DHFR 19 bp deletion (rs70991108), MTHFD1 1958G>A (rs2236225), MTHFR 1298A>C (rs1801131), MTHFR 677C>T (rs1801133), and SLC19A1 80A>G (rs1051266). PCR and pyrosequencing primers (Table 1) were designed using PSQ Assay Design Software Version 1.0.6. After amplification, the pyrosequencing primer was used to extend the PCR product by addition of nucleotides in a specific sequential order based on the known sequence. The resulting pyrogram of signal peaks was used to determine the genotype. PCR amplification for the DHFR 19 bp deletion was performed using the GC Rich PCR System (Roche, Indianapolis, IN) as previously described (Parle-McDermott et al., 2007). PCR for all other variants was performed in a 20 μl reaction mix containing 0.5 μM each forward and reverse primer and Applied Biosystems GeneAmp® Fast PCR Master Mix. Amplification was performed using the 9800 Fast PCR System according to manufacturer's suggested protocol (Applied Biosystems, Foster City, CA). Pyrosequencing was carried out as described by manufacturer's protocol (Qiagen, Valencia, CA).
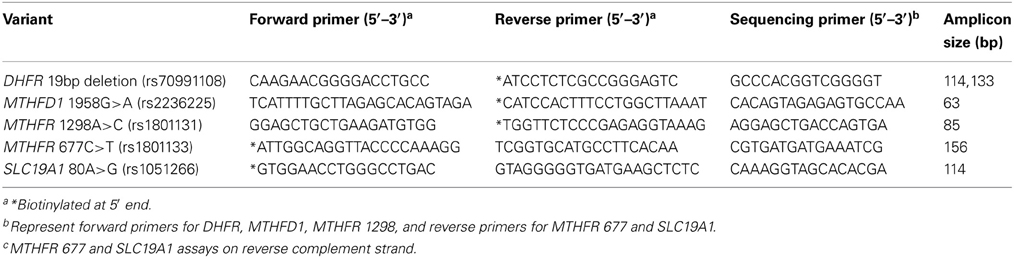
Table 1. Primer sequences and PCR amplicon size for DHFR, MTHFD1, MTHFR, and SLC19A1 variant analysis.
Quality Control
Five percent of the samples were selected at random for repeat testing of the five polymorphisms, and the concordance was checked. We also tested each variant for any significant departure from Hardy-Weinberg equilibrium (HWE) by a chi-square goodness-of-fit test to verify data quality. In addition, Mendelian inheritance for all variants was assessed in 13 families with full trios using the PedCheck program (O'Connell and Weeks, 1998). Water and genotype controls (Coriell Cell Repositories, Camden, NJ) were included on each assay plate. The tests for HWE indicated that none of the variants deviated significantly from HWE, supporting high quality genotype data. All replica test results were 100% concordant.
Results
We obtained blood samples from all categories of relatives, including individuals with NTDs (n = 13), offspring (N = 2), fathers (N = 29), mothers (N = 37), siblings (N = 59), nieces and nephews (N = 2), uncles and aunts (N = 66), first cousins (N = 105), and grandparents (N = 2) from 47 families. Paternal relatives (N = 65) and maternal relatives (N = 115) comprised uncles and aunts, first cousins, first cousins once removed, and grandparents, who were related to the affected individual through the father and mother, respectively. The average age of the study participants was 41 years. The overall distribution of each of the five polymorphisms is shown in Table 2. The genotype distributions for the five polymorphisms in our study were similar to those from other studies, with a few exceptions (Table 3).
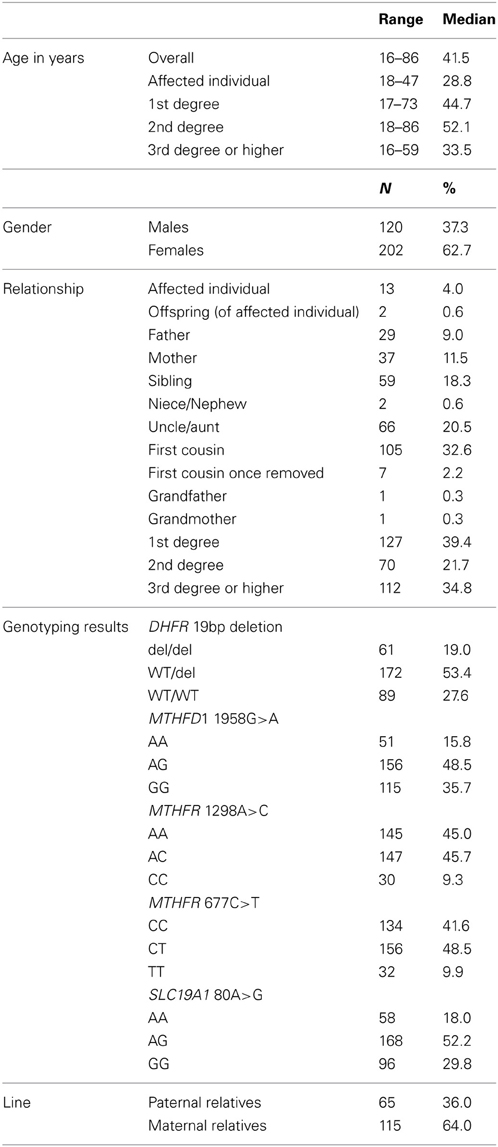
Table 2. Characteristics of the participants, northeast region of the Republic of Ireland and Northern Ireland, 1995–2009.
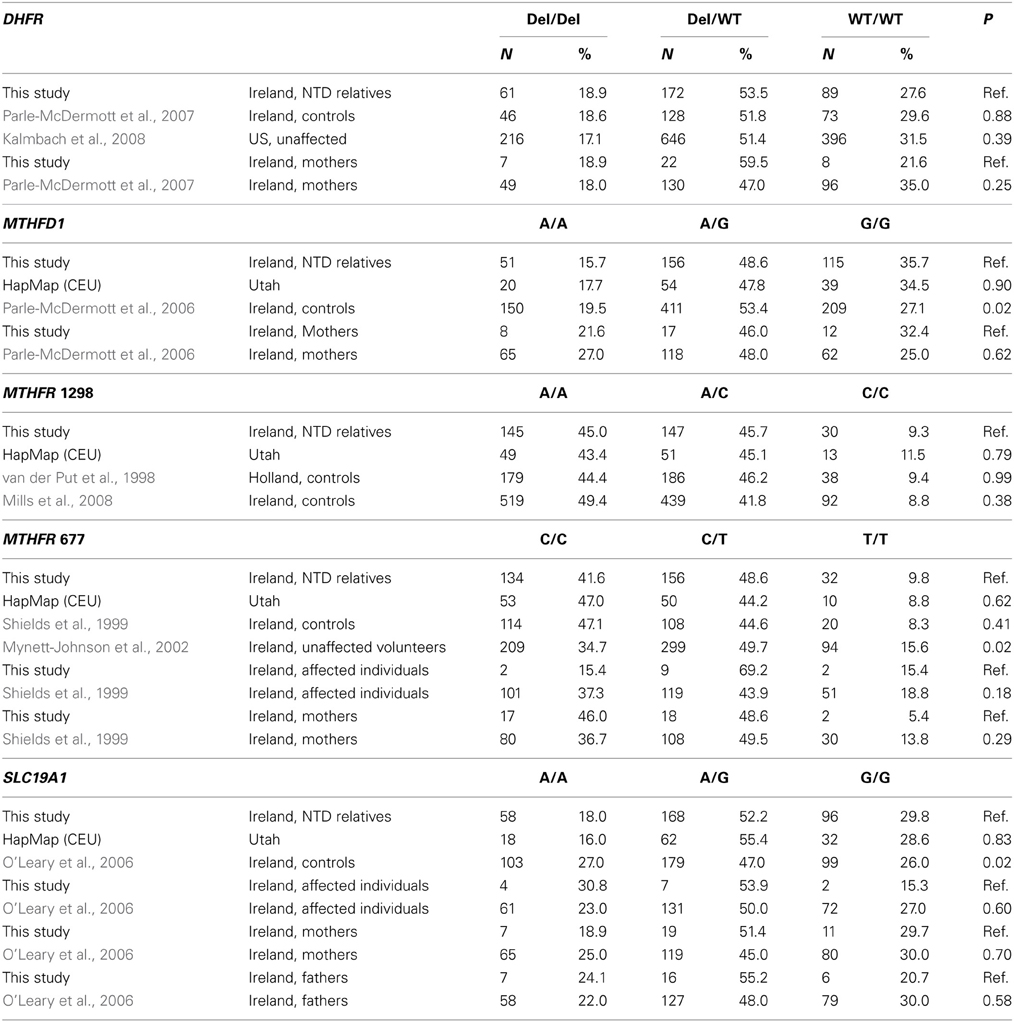
Table 3. Distribution of genotypes and comparison with other studies.
Analysis of Individual Polymorphisms
The distributions of each genotype and allele by relationship to the individual with an NTD, degree of relationship to the affected individual, and maternal vs. paternal lineage are shown in Table 4. We did not observe any significant associations between individual genotypes and the affected individuals or mothers, although the higher folate-metabolizing MTHFR 677 CC genotype tended to be found less often in affected individuals (15.4%) than in all other relatives (42.7%) (Fisher's exact test, p = 0.08) (Table 4 and data not shown). We also did not see any significant associations between individual genotypes and maternal vs. paternal lineage (Table 4).
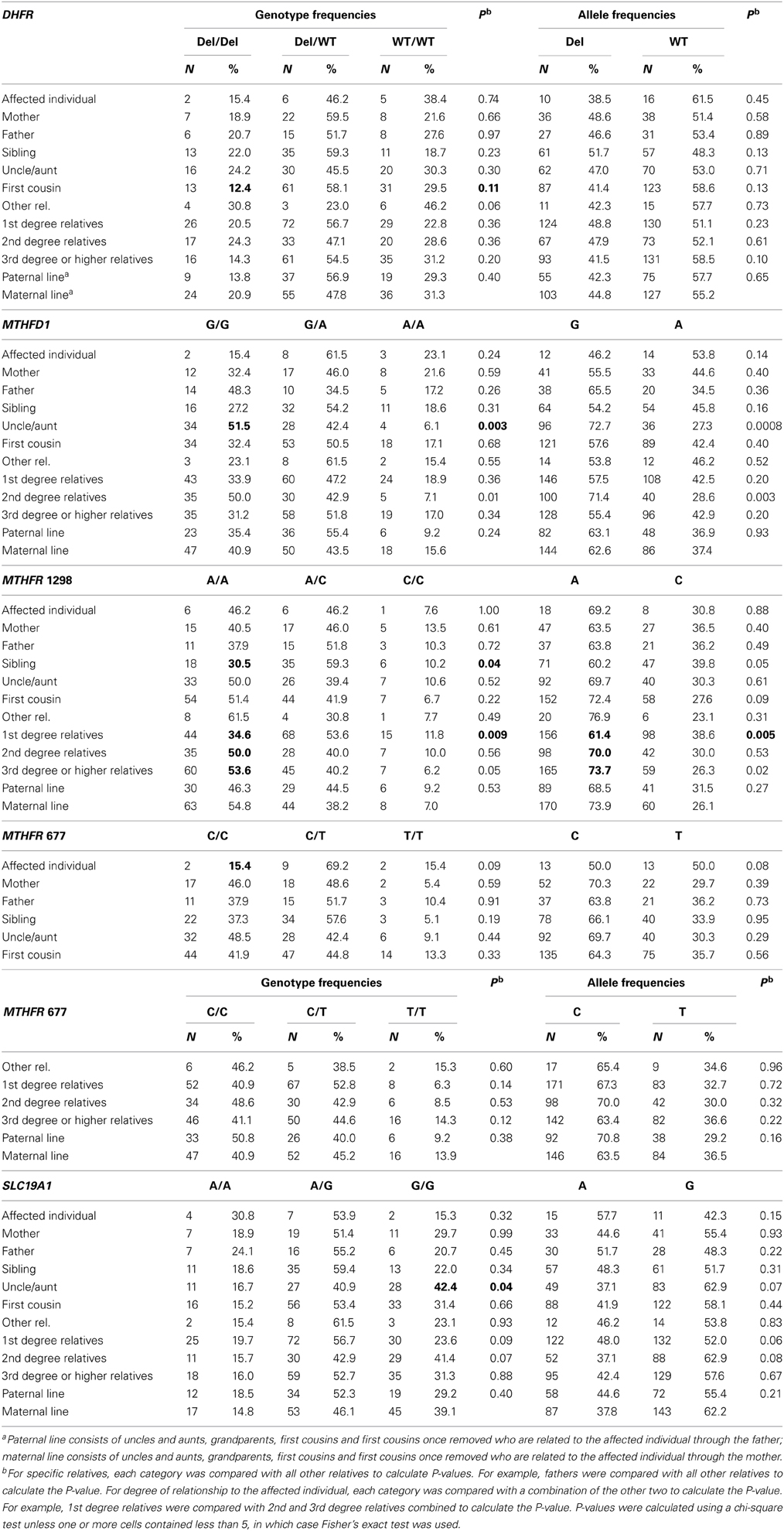
Table 4. Distribution of genotypes and alleles according to relationship within NTD-affected families and lineage, northeast region of the Republic of Ireland and Northern Ireland, 1995–2009.
Relatives of individuals with NTDs might be expected to have genotypes associated with increased or decreased risk for NTDs, and the prevalence of these genotypes could increase or decrease with degree of closeness to the affected individual. For MTHFR 1298A>C, a lower percentage of first-degree relatives (34.6%) had the higher folate-metabolizing AA genotype compared with second-degree (50.0%) relatives and third-degree and higher (53.6%) relatives (p = 0.009). Similarly, the A allele was less common in first-degree relatives (61.4%) than in second-degree relatives (70.0%) and third-degree and higher (73.7%) relatives (p = 0.005). When the genotype (AA vs. A/C, C/C) and relatives (first-degree relatives vs. all other relatives) were dichotomized, the odds ratio for the association of the AA genotype with first-degree relatives was 0.49 (0.30–0.78, p = 0.002) (data not shown). Consistent with this, a lower percentage of siblings, 30.5%, had the AA genotype compared to 48.3% among other relatives (p = 0.04, Table 4). We did not observe a consistent trend of genotypes being more or less common in relatives more closely related to the affected individual for any of the other genotypes (Table 4).
We observed some associations with individual categories of relatives. For example, fewer first cousins had the higher folate-metabolizing DHFR del/del genotype compared with all other relatives (12.4 vs. 22.1%, p = 0.11, Table 4 and data not shown), the higher folate-metabolizing MTHFD1 GG genotype was higher in uncles and aunts compared with all other relatives (51.5 vs. 31.6%, p = 0.003) (Table 4), and the lower folate-metabolizing SLC19A1 GG genotype was more common in uncles and aunts than in other relatives (42.4 vs. 26.6%, p = 0.04) (Table 4). However, the meaning of these isolated associations is unclear.
Number of Risk Genotypes
While the frequencies of individual genotypes did not differ significantly between maternal and paternal relatives (Table 4), the number of risk genotypes, defined as the number of homozygous genotypes associated with lower folate metabolism, did show significant variation between the maternal and paternal lines (p = 0.02) (Table 5). A lower percentage of maternal relatives (20.9%) had no (0) risk genotypes compared with paternal relatives (41.5%), while more maternal relatives (54.8%) than paternal relatives (35.4%) had 1 risk genotype. When the risk genotypes were dichotomized (0, 1–3), the odds ratio for association with the maternal line compared with the paternal line was 2.69 (1.38–5.25, p = 0.004).
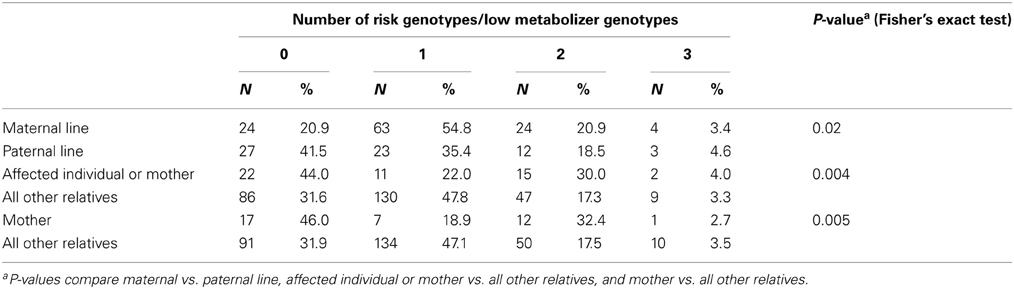
Table 5. Number of risk genotypes by relationship to affected individuals with NTDs and lineage, northeast region of the Republic of Ireland and Northern Ireland, 1995–2009.
The numbers of risk genotypes in affected individuals and mothers (p = 0.004) or mothers alone (p = 0.005), compared with all other relatives were also statistically significant (Table 5). A higher percentage of affected individuals and mothers (30.0 vs. 17.3%) and mothers alone (32.4 vs. 17.5%) had 2 risk genotypes compared with all other relatives, while a lower percentage of affected individuals and mothers (22.0 vs. 47.8%) and mothers alone (18.9 vs. 47.1%) had 1 risk genotype. A higher percentage of affected individuals and mothers (44.0 vs. 31.6%) and mothers alone (46.0 vs. 31.9%) had 0 risk genotypes.
Discussion
In the Irish families in our study, we found that maternal relatives were more likely than paternal relatives to have one or more genotypes associated with decreased folate metabolism. As expected from previous studies, mothers and affected individuals had differential distributions of risk genotypes compared with all other relatives.
The observed increased number of affected relatives in the maternal lineage of families could be due to genetic and/or epigenetic mechanisms. A simple genetic explanation would be that more maternal relatives have alleles associated with increased risk for NTDs simply due to chance, so the likelihood of inheriting these alleles from the maternal side would be increased. With a simple genetic model, we would expect that relatives would share the same risk genotype as the affected individuals and/or mothers. However, we did not observe this, but instead found that maternal relatives had an over-abundance of any risk genotype, rather than one specific genotype. We found that more maternal relatives had one risk genotype, while more paternal relatives had no risk genotypes. However, if this were the main explanation behind the increased number of affected individuals on the maternal side, we would expect that relatives more closely related to the affected individuals would share more genetic risk factors, and thus the prevalence of higher risk genotypes would be greater in first-degree relatives than in more distant ones. However, with the exception of MTHFR 1298, we did not observe consistent patterns of increasing numbers of risk genotypes with closer degree of relationship to the affected individual.
The mother's genotype during pregnancy plays a significant role in NTD risk, best studied in MTHFR (Yan et al., 2012), and the inheritance of these alleles would necessarily come from the maternal line. In this case, mothers and possibly affected individuals might be expected to have an increased prevalence of genotypes associated with greater NTD risk. In our study, fewer affected individuals had the higher folate-metabolizing MTHFR 677 CC genotype, consistent with earlier reports that the homozygous TT genotype is a risk factor for NTDs (Shields et al., 1999; Botto and Yang, 2000). Likewise, affected individuals and mothers, as well as mothers only, were more likely to have 2 risk genotypes. Our study of these associations was limited by our small sample size and was not the main objective of this study. While our findings were consistent with previous literature, these associations alone were not strong enough to explain the increased number of affected relatives on the mother's side, suggesting that other mechanisms are also at work.
Whenever there is preferential inheritance of a condition from the maternal vs. paternal line, epigenetic effects are strongly suspected. The genes included in our study are not located in known imprinted regions. However, folate metabolism is critical to the production of one-carbon subunits needed for epigenetic modification. Certain variants of folate metabolism genes (possibly including those in our study), which are associated with NTD risk, might affect proper imprinting and establishment or maintenance of other epigenetic changes. These epigenetic effects could occur in the maternal genome and would have been initiated when the mother was a fetus herself, but may also have taken place in the genomes of the mother's parents (Crider et al., 2012). If this mechanism were at work, the maternal and paternal grandparents' genotypes would be of importance. Unfortunately, we were unable to obtain samples from many grandparents. However, aunts, uncles, and first cousins provide an indirect measure of the grandparents' genotypes, and significant associations for these relatives were seen for DHFR, MTHFD1, and SLC19A1. The increased number of risk genotypes in maternal vs. paternal relatives is consistent with an epigenetic mechanism.
Our study is the first to look at the genotypes of second- and third-degree or more relatives in families affected by NTDs and to begin to explore the genetic and epigenetic mechanisms behind the increased NTD and overall birth defects risks in these families. However, our study is preliminary and has a number of limitations. The limitations of our study include a relatively small sample size, lack of triads (both parents and offspring) among distant relatives to allow for heritability testing, and inadequate number of samples from grandparents to evaluate epigenetic effects. Additionally, more maternal than paternal relatives participated in the study. However, while this might have affected the stability of the point estimate for the association of risk genotypes with the maternal vs. paternal line, it did not affect the magnitude or direction. The response rate was relatively low for our study, and the participation of families and of relatives within families could be biased and not reflect the true underlying distribution of families with NTDs in Ireland. However, selection for and participation in the study was independent of genotype and thus participants can be considered close to a random sample of relatives. The candidate gene approach used here evaluated a small number of polymorphisms and would have missed associations with other variants in folate metabolism pathway genes. An important limitation of our study (and others) is that we were not able to measure the mother's intake of folate and folic acid during pregnancy. The genes included in this study function in folate metabolism, so that their effect depends on the levels of folate present in the body. For example, high intake of folic acid and folate could mask genetic effects that would be seen at lower intake levels. Lastly, the number of multiple comparisons evaluated in our study likely produced some spurious findings.
Our results are consistent with both genetic and epigenetic mechanisms contributing to the increased NTD and overall birth defects risk in the maternal lineage. The diversity of genetic risk seen in these families suggests that etiology of NTDs is multi-faceted. Future studies will benefit from including other relatives, especially grandparents, in addition to mothers, fathers, and affected individuals, especially if epigenetic mechanisms are to be addressed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the families who chose to participate in our studies, to Rebecca Lawler, Sharon McGinty, and the volunteers who helped to conduct this study, to Claymon Laboratories for biochemical tests, to Stanimila Nikolova for technical assistance and to Mary E. Cogswell, Quanhe Yang, and Ramal Moonesinghe for their insightful comments. The Joseph E. and Marjorie B. Jones Foundation of Washington, DC provided partial support to Julianne Byrne. No additional external funding was received for this study.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
Botto, L. D., Lisi, A., Bower, C., Canfield, M. A., Dattani, N., De Vigan, C., et al. (2006). Trends of selected malformations in relation to folic acid recommendations and fortification: an international assessment. Birth Defects Res. A. Clin. Mol. Teratol. 76, 693–705. doi: 10.1002/bdra.20307
Botto, L. D., and Yang, Q. (2000). 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am. J. Epidemiol. 151, 862–877. doi: 10.1093/oxfordjournals.aje.a010290
Brody, L. C., Conley, M., Cox, C., Kirke, P. N., McKeever, M. P., Mills, J. L., et al. (2002). A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am. J. Hum. Genet. 71, 1207–1215. doi: 10.1086/344213
Byrne, J. (2008). Birth defects in uncles and aunts from Irish families with neural tube defects. Birth Defects Res. A. Clin. Mol. Teratol. 82, 8–15. doi: 10.1002/bdra.20406
Byrne, J. (2010). Birth defects among maternal first cousins in Irish families with a neural tube defect. Ir. J. Med. Sci. 179, 375–380. doi: 10.1007/s11845-009-0381-x
Byrne, J. (2011). Three generations of matrilineal excess of birth defects in Irish families with neural tube defects. Ir. J. Med. Sci. 180, 69–72. doi: 10.1007/s11845-010-0632-x
Byrne, J., Cama, A., Reilly, M., Vigliarolo, M., Levato, L., Boni, L., et al. (1996). Multigeneration maternal transmission in Italian families with neural tube defects. Am. J. Med. Genet. 66, 303–310. doi: 10.1002/(SICI)1096-8628(19961218)66:3<303::AID-AJMG13>3.3.CO;2-2
Carter, T. C., Pangilinan, F., Troendle, J. F., Molloy, A. M., VanderMeer, J., Mitchell, A., et al. (2011). Evaluation of 64 candidate single nucleotide polymorphisms as risk factors for neural tube defects in a large Irish study population. Am. J. Med. Genet. A 155, 14–21. doi: 10.1002/ajmg.a.33755
Chango, A., Emery-Fillon, N., de Courcy, G. P., Lambert, D., Pfister, M., Rosenblatt, D. S., et al. (2000). A polymorphism (80G–>A) in the reduced folate carrier gene and its associations with folate status and homocysteinemia. Mol. Genet. Metab. 70, 310–315. doi: 10.1006/mgme.2000.3034
Christensen, K. E., Rohlicek, C. V., Andelfinger, G. U., Michaud, J., Bigras, J. L., Richter, A., et al. (2009). The MTHFD1 p.Arg653Gln variant alters enzyme function and increases risk for congenital heart defects. Hum. Mutat. 30, 212–220. doi: 10.1002/humu.20830
Coffey, V. P. (1983). Neural tube defects in Dublin 1953–1954 and 1961–1982. Ir. Med. J. 76, 411–413.
Crider, K. S., Yang, T. P., Berry, R. J., and Bailey, L. B. (2012). Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv. Nutr. 3, 21–38. doi: 10.3945/an.111.000992
Czeizel, A. E., and Dudas, I. (1992). Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 327, 1832–1835. doi: 10.1056/NEJM199212243272602
Deak, K. L., Siegel, D. G., George, T. M., Gregory, S., Ashley-Koch, A., Speer, M. C., et al. (2008). Further evidence for a maternal genetic effect and a sex-influenced effect contributing to risk for human neural tube defects. Birth Defects Res. A. Clin. Mol. Teratol. 82, 662–669. doi: 10.1002/bdra.20511
De Marco, P., Calevo, M. G., Moroni, A., Merello, E., Raso, A., Finnell, R. H., et al. (2003). Reduced folate carrier polymorphism (80A–>G) and neural tube defects. Eur. J. Hum. Genet. 11, 245–252. doi: 10.1038/sj.ejhg.5200946
Frosst, P., Blom, H. J., Milos, R., Goyette, P., Sheppard, C. A., Matthews, R. G., et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 10, 111–113. doi: 10.1038/ng0595-111
Kalmbach, R. D., Choumenkovitch, S. F., Troen, A. P., Jacques, P. F., D'Agostino, R., and Selhub, J. (2008). A 19-base pair deletion polymorphism in dihydrofolate reductase is associated with increased unmetabolized folic acid in plasma and decreased red blood cell folate. J. Nutr. 138, 2323–2327. doi: 10.3945/jn.108.096404
Kirke, P. N., Mills, J. L., Molloy, A. M., Brody, L. C., O'Leary, V. B., Daly, L., et al. (2004). Impact of the MTHFR C677T polymorphism on risk of neural tube defects: case-control study. BMJ 328, 1535–1536. doi: 10.1136/bmj.38036.646030.EE
Mills, J. L., Molloy, A. M., Parle-McDermott, A., Troendle, J. F., Brody, L. C., Conley, M. R., et al. (2008). Folate-related gene polymorphisms as risk factors for cleft lip and cleft palate. Birth Defects Res. A. Clin. Mol. Teratol. 82, 636–643. doi: 10.1002/bdra.20491
Molloy, A. M., Brody, L. C., Mills, J. L., Scott, J. M., and Kirke, P. N. (2009). The search for genetic polymorphisms in the homocysteine/folate pathway that contribute to the etiology of human neural tube defects. Birth Defects Res. A. Clin. Mol. Teratol. 85, 285–294. doi: 10.1002/bdra.20566
MRC Vitamin Study Research Group. (1991). Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 338, 131–137. doi: 10.1016/0140-6736(91)90133-A
Mynett-Johnson, L. A., Keenan, C., Black, I. L., Livingstone, W. J., Lawler, M., Roche, H. M., et al. (2002). Thermolabile methylenetetrahydrofolate reductase (C677T): frequency in the Irish population. Ir. J. Med. Sci. 171, 37–39. doi: 10.1007/BF03168940
O'Connell, J. R., and Weeks, D. E. (1998). PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 63, 259–266. doi: 10.1086/301904
O'Leary, V. B., Pangilinan, F., Cox, C., Parle-McDermott, A., Conley, M., Molloy, A. M., et al. (2006). Reduced folate carrier polymorphisms and neural tube defect risk. Mol. Genet. Metab. 87, 364–369. doi: 10.1016/j.ymgme.2005.09.024
Pangilinan, F., Molloy, A. M., Mills, J. L., Troendle, J. F., Parle-McDermott, A., Signore, C., et al. (2012). Evaluation of common genetic variants in 82 candidate genes as risk factors for neural tube defects. BMC Med. Genet. 13:62. doi: 10.1186/1471-2350-13-62
Parle-McDermott, A., Kirke, P. N., Mills, J. L., Molloy, A. M., Cox, C., O'Leary, V. B., et al. (2006). Confirmation of the R653Q polymorphism of the trifunctional C1-synthase enzyme as a maternal risk for neural tube defects in the Irish population. Eur. J. Hum. Genet. 14, 768–772. doi: 10.1038/sj.ejhg.5201603
Parle-McDermott, A., Mills, J. L., Kirke, P. N., O'Leary, V. B., Swanson, D. A., Pangilinan, F., et al. (2003). Analysis of the MTHFR 1298A–>C and 677C–>T polymorphisms as risk factors for neural tube defects. J. Hum. Genet. 48, 190–193. doi: 10.1007/s10038-003-0008-4
Parle-McDermott, A., Pangilinan, F., Mills, J. L., Kirke, P. N., Gibney, E. R., Troendle, J., et al. (2007). The 19-bp deletion polymorphism in intron-1 of dihydrofolate reductase (DHFR) may decrease rather than increase risk for spina bifida in the Irish population. Am. J. Med. Genet. A 143A, 1174–1180. doi: 10.1002/ajmg.a.31725
Shaw, G. M., Lu, W., Zhu, H., Yang, W., Briggs, F. B., Carmichael, S. L., et al. (2009). 118 SNPs of folate-related genes and risks of spina bifida and conotruncal heart defects. BMC Med. Genet. 10:49. doi: 10.1186/1471-2350-10-49
Shields, D. C., Kirke, P. N., Mills, J. L., Ramsbottom, D., Molloy, A. M., Burke, H., et al. (1999). The “thermolabile” variant of methylenetetrahydrofolate reductase and neural tube defects: an evaluation of genetic risk and the relative importance of the genotypes of the embryo and the mother. Am. J. Hum. Genet. 64, 1045–1055. doi: 10.1086/302310
Stanislawska-Sachadyn, A., Brown, K. S., Mitchell, L. E., Woodside, J. V., Young, I. S., Scott, J. M., et al. (2008). An insertion/deletion polymorphism of the dihydrofolate reductase (DHFR) gene is associated with serum and red blood cell folate concentrations in women. Hum. Genet. 123, 289–295. doi: 10.1007/s00439-008-0475-y
Sutton, M., Daly, L. E., and Kirke, P. N. (2008). Survival and disability in a cohort of neural tube defect births in Dublin, Ireland. Birth Defects Res. A. Clin. Mol. Teratol. 82, 701–709. doi: 10.1002/bdra.20498
van der Put, N. M., Gabreels, F., Stevens, E. M., Smeitink, J. A., Trijbels, F. J., Eskes, T. K., et al. (1998). A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am. J. Hum. Genet. 62, 1044–1051. doi: 10.1086/301825
Wang, X. W., Luo, Y. L., Wang, W., Zhang, Y., Chen, Q., and Cheng, Y. L. (2012a). Association between MTHFR A1298C polymorphism and neural tube defect susceptibility: a metaanalysis. Am. J. Obstet. Gynecol. 206, 251.e1–7. doi: 10.1016/j.ajog.2011.12.021
Wang, H. G., Wang, J. L., Zhang, J., Zhao, L. X., Zhai, G. X., Xiang, Y. Z., et al. (2012b). Reduced folate carrier A80G polymorphism and susceptibility to neural tube defects: a meta-analysis. Gene 510, 180–184. doi: 10.1016/j.gene.2012.02.020
Whitehead, A. S., Gallagher, P., Mills, J. L., Kirke, P. N., Burke, H., Molloy, A. M., et al. (1995). A genetic defect in 5,10 methylenetetrahydrofolate reductase in neural tube defects. QJM 88, 763–766.
Keywords: neural tube defects, folate metabolism, DHFR 19bp deletion, MTHFD1 1958G>A, MTHFR 1298A>C, MTHFR 677C>T, SLC19A1 80A>G, maternal inheritance
Citation: Fisk Green R, Byrne J, Crider KS, Gallagher M, Koontz D and Berry RJ (2013) Folate-related gene variants in Irish families affected by neural tube defects. Front. Genet. 4:223. doi: 10.3389/fgene.2013.00223
Received: 09 July 2013; Accepted: 15 October 2013;
Published online: 06 November 2013.
Edited by:
Zdenka Pausova, The Hospital for Sick Children, CanadaReviewed by:
Alice Hudder, Lake Erie College of Osteopathic Medicine, USAChris A. Murgatroyd, Manchester Metropolitan University, UK
Eva Reischl, Helmholtz Zentrum München - German Research Centre for Environmental Health GmbH, Germany
Copyright © 2013 Fisk Green, Byrne, Crider, Gallagher, Koontz and Berry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ridgely Fisk Green, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, 1600 Clifton Rd., MS E–86, Atlanta, GA 30333, USA e-mail:Z3JmMUBjZGMuZ292