 Yasuko Kikuchi1,2
Yasuko Kikuchi1,2 Eiichi Tsuji2Koichi Yagi1Keisuke Matsusaka3
Eiichi Tsuji2Koichi Yagi1Keisuke Matsusaka3 Shingo Tsuji1
Shingo Tsuji1 Junichi Kurebayashi4Toshihisa Ogawa2Hiroyuki Aburatani1
Junichi Kurebayashi4Toshihisa Ogawa2Hiroyuki Aburatani1 Atsushi Kaneda1,5,6*
Atsushi Kaneda1,5,6*- 1Genome Science Division, Research Center for Advanced Science and Technology, The University of Tokyo, Tokyo, Japan
- 2Department of Metabolic Care and Endocrine Surgery, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 3Department of Pathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 4Department of Breast and Endocrine Surgery, Kawasaki Medical University, Okayama, Japan
- 5Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Chiba, Japan
- 6CREST, Japan Science and Technology Agency, Saitama, Japan
Cancer arises through accumulation of epigenetic and genetic alteration. Aberrant promoter methylation is a common epigenetic mechanism of gene silencing in cancer cells. We here performed genome-wide analysis of DNA methylation of promoter regions by Infinium HumanMethylation27 BeadChip, using 14 clinical papillary thyroid cancer samples and 10 normal thyroid samples. Among the 14 papillary cancer cases, 11 showed frequent aberrant methylation, but the other three cases showed no aberrant methylation at all. Distribution of the hypermethylation among cancer samples was non-random, which implied existence of a subset of preferentially methylated papillary thyroid cancer. Among 25 frequently methylated genes, methylation status of six genes (HIST1H3J, POU4F2, SHOX2, PHKG2, TLX3, HOXA7) was validated quantitatively by pyrosequencing. Epigenetic silencing of these genes in methylated papillary thyroid cancer cell lines was confirmed by gene re-expression following treatment with 5-aza-2′-deoxycytidine and trichostatin A, and detected by real-time RT-PCR. Methylation of these six genes was validated by analysis of additional 20 papillary thyroid cancer and 10 normal samples. Among the 34 cancer samples in total, 26 cancer samples with preferential methylation were significantly associated with mutation of BRAF/RAS oncogene (P = 0.04, Fisher's exact test). Thus, we identified new genes with frequent epigenetic hypermethylation in papillary thyroid cancer, two subsets of either preferentially methylated or hardly methylated papillary thyroid cancer, with a concomitant occurrence of oncogene mutation and gene methylation. These hypermethylated genes may constitute potential biomarkers for papillary thyroid cancer.
Introduction
Papillary thyroid cancer is the most common cancer derived from follicular cells. It is estimated that approximately 23,500 cases of differentiated thyroid cancer occur per year in the United States (Jemal et al., 2005), and 19,000 papillary thyroid cancer cases per year in the European Union (http://globocan.iarc.fr/). In Japan, about 8000 patients suffer from thyroid cancer every year, 80% of which are papillary cancer. While prognosis for papillary thyroid cancer is generally good, with a 10-year survival rate above 90%, some patients die of distant metastases and/or repeated recurrence (Ezaki et al., 1992; Yamashita et al., 1998).
RET/PTC (Rearranged in Transformation/Papillary Thyroid Carcinoma) re-arrangement, BRAF (V-Raf murine sarcoma viral oncogene homolog B) and RAS (Rat Sarcoma viral oncogene homolog) point mutations are frequently observed in papillary thyroid cancer (Mitsutake et al., 2006; Knauf and Fagin, 2009). Mutation of T to A at 1799 in the exon 15 of the BRAF gene has been reported in 28–69% of papillary thyroid cancer cases, while point mutations of RAS genes are detected in 5–20% (Cohen et al., 2003; Kimura et al., 2003; Namba et al., 2003; Kondo et al., 2007). Papillary thyroid cancer with poor prognosis is associated with BRAF mutation (Xing et al., 2005; Lee et al., 2012), whereas concomitantly, lengthy disease-free interval is not (Ulisse et al., 2012).
Patients with papillary thyroid cancer are generally treated by surgery. But it is difficult to decide whether total thyroidectomy, hemithyroidectomy, prophylactic central neck dissection or no dissection, should be performed in patients without preoperative or intraoperative evidence of metastatic lymph nodes (Xing et al., 2013). Association of BRAF mutation with occult central neck lymph node metastases (Joo et al., 2012) might support use of BRAF mutation as an indication for prophylactic central neck dissection for patients with conventionally low- to intermediate-risk papillary thyroid cancer. But precise diagnosis to define prognosis and suitable therapy is currently impossible. Molecular biomarkers would therefore simplify disease management (McLeod et al., 2013).
Along with genetic alterations, accumulation of epigenetic alterations is known to affect cancer development (Baylin and Ohm, 2006; Feinberg et al., 2006; Esteller, 2007). Aberrant DNA methylation at promoter regions is a major epigenetic alteration to silence tumor suppressor genes in many cancer types. RASSF1A (Ras association (RalGDS/AF-6) domain family member 1) is methylated in 20% of papillary thyroid cancer, leading to activation of the RAS-MAPK (Mitogen-Activated Protein Kinase) signal (Xing et al., 2004). Papillary thyroid cancer is also reported to involve methylation of other genes, including RARB (Retinoic Acid Receptor, Beta), p16INK4A (CDKN2A, Cyclin-Dependent Kinase Inhibitor 2A), TSHR (Thyroid Stimulating Hormone Receptor), CDH1 (Cadherin 1, type 1, E-cadherin), DAPK (Death-Associated Protein Kinase 1), and MLH1 (mutL Homolog 1) (Hoque et al., 2005; Guan et al., 2008; Mohammadi-asl et al., 2011). While a few genes known to be aberrantly methylated in other cancers were analyzed in these studies, methylation frequencies ranged from 15 to 33%. Involvement of genes in aberrant DNA methylation, however, has not been well-clarified in papillary thyroid cancer. Whether any subset of papillary thyroid cancer shows preferential aberrant methylation, and whether such methylation and other clinicopathological factors are associated are also unclear.
We here analyzed DNA methylation status of promoter regions on a genome-wide scale, using the Illumina Infinium HumanMethylation27 BeadChip technique on 14 clinical papillary thyroid cancer samples and 10 normal thyroid samples. For genes frequently hypermethylated in cancer, methylation status was validated quantitatively by pyrosequencing, using 20 additional clinical cancer samples and 10 additional normal samples. Methylation-associated gene silencing was confirmed by gene re-expression following 5-aza-2′-deoxycytidine and trichostatin A treatment, and by quantitative reverse transcription-polymerase chain reaction (RT-PCR) on thyroid cancer cell lines. We found a number of genes with frequent aberrant methylation and silencing in papillary thyroid cancer, and a subset of cancer with preferential aberrant methylation.
Materials and Methods
Clinical Samples and Cell Lines
We obtained 79 primary papillary thyroid cancer samples from patients who underwent thyroidectomy at The University of Tokyo, with written informed consent. These samples were immediately frozen with liquid nitrogen and stored at −80°C. The frozen materials were microscopically examined for cancer cell contents by pathologists and were dissected to enrich cancer cells when necessary. Thirty-four cancer samples containing more than 40% of cancer cells were selected and used for further analysis. DNA was extracted by QIAamp DNA Micro Kit (QIAGEN, Valencia, CA). Thyroid cancer cell line BHT-101 was obtained from DSMZ (Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures), TPC-1 was provided from Dr. Mitsutake, University of Nagasaki (Ishizaka et al., 1989) and KTC-1 and KTC-3 cell lines were provided from Dr. Kurebayashi, Kawasaki Medical University (Kurebayashi et al., 2000, 2006). These cell lines were maintained in RPMI 1640 (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum, streptomycin sulfate (100 mg/L), and penicillin G sodium (100 mg/L). Peripheral blood cell samples were purchased from The Coriell Cell Repositories. This research was certified by the Ethics Committee of The University of Tokyo and Chiba University.
Infinium Assays
High-resolution methylation analysis was conducted on the Illumina Infinium HumanMethylation27 microarray platform. This BeadChip assay measures methylation, given as a β-value, at more than 27,000 CpG loci covering 15,000 genes. For each CpG site, the β-value is the ratio of the fluorescence signal from the methylated probe over the sum of methylated and unmethylated probe signals. The β-value, ranging from 0.00 to 1.00, reflects the methylation level of the individual CpG site. Bisulfite conversion, whole-genome amplification, labeling, hybridization, and scanning were carried out according to the manufacturer's protocols. According to the previously proposed classification (Weber et al., 2007), Infinium probes were classified into three categories: high-CpG, intermediate-CpG, and low-CpG probes, on the basis of CpG ratio (the ratio of observed CpG count over expected CpG count) and GC contents within 500 bp region around the probe site (Matsusaka et al., 2011). Genes in X and Y chromosomes were excluded to avoid gender differences. Infinium enables us to analyze DNA methylation levels systematically for more than 14,000 genes, but methylation level of a single CpG site detected by Infinium may not precisely represent methylation status of promoter CpG island. Some Infinium probes might be less quantitative; in analysis of control samples with known levels of methylation (0, 25, 50, 75, 100%), the observed β-values generally correlated with the expected β-values, while some probes showed lower β-values (0.0–0.3) for 75% control or higher β-values (0.7–1.0) for 25% control (Nagae et al., 2011).
Pyrosequencing Analysis
Quantitative validation for methylation status was carried out by pyrosequencing as previously reported (Matsusaka et al., 2011). Primers were designed to include no or only one CpG site in a primer sequence using Pyro Q-CpG Software (QIAGEN), to amplify bisulfite-treated DNA regions containing several CpG sites. For C of CpG site within a primer sequence, a nucleotide which does not anneal to C or U was chosen, e.g., adenosine (A). Briefly, the biotinylated PCR product was bound to streptavidin sepharose beads HP (Amersham Biosciences, Sweden), washed, and denatured using a 0.2 mol/L NaOH solution. After addition of 0.3 μmol/L sequencing primer to the purified, single-stranded PCR product, pyrosequencing was carried out using PyroMark Q96 ID System (QIAGEN) according to the manufacturer's instructions. Primer sequences and conditions for HIST1H3J (Histone cluster 1, H3j), POU4F2 (POU class 4 homeobox 2), SHOX2 (Short stature homeobox 2), PHKG2 (Phosphorylase Kinase, Gamma 2), TLX3 (T-cell Leukemia homeobox 3), and HOXA7 (Homeobox A7), are shown in Table 1. Control samples with known levels of methylation (0, 25, 50, 75, 100%) were prepared as previously described (Yagi et al., 2010). Pyrosequencing is not systematic, but highly quantitative (Matsusaka et al., 2011), and enables us to precisely validate the methylation level at one CpG site, as determined by the Infinium assay, as well as at surrounding CpG sites. Mean methylation levels of these CpG sites were calculated to represent methylation status of each gene promoter, and were displayed in figures by heatmap or dot chart.

Table 1. Primer sequences for pyrosequencing of six potential methylation biomarkers.
Mutation analysis
Mutations at BRAF 1799, KRAS (Kirsten Rat Sarcoma viral oncogene homolog) 34, 35, 38, NRAS (Neuroblastoma RAS viral oncogene homolog) 181, 182, 183, HRAS (Harvey Rat Sarcoma viral oncogene homolog) 35, 37, 181, 182, 183, were analyzed by genotyping assays on MassARRAY platform, by detecting mass difference of the extended nucleotide using matrix assisted laser desorption ionization-time of flight-mass spectrometry (MALDI-TOF-MS) (Yagi et al., 2012). First, PCR amplification primers and a post-PCR extension primer were designed using MassARRAY Assay Design 3.0 software (Sequenom), and listed in Table 2. Those mutations were analyzed in a single reaction by multiplex PCR. PCR amplification was performed in 5-μL volume containing 0.5 unit of Taq polymerase, 5 ng of genomic DNA, 0.5 pmol of PCR primer and 2.5 nmol of dNTPs. PCR reactions were cycled at 94°C for 15 min, followed by 45 cycles of 94°C for 20 s, 56°C for 30 s and 72°C for 1 min. Shrimp alkaline phosphatase treatment was performed at 37°C for 20 min and 85°C for 5 min. Post-PCR primer extension was carried out using 5.6 pmol of extension primer. Extension reaction was cycled at 94°C for 30 s, followed by 40 cycles of 94°C for 5 s, 5 cycles of 52°C for 5 s and 80°C for 5 s, and 72°C for 3 min. Reaction products were transferred to a SpectroCHIP (Sequenom) and mass difference was analyzed using MALDI-TOF-MS. Irradiation of the matrix-oligonucleotide-cocrystal with a laser beam ultimately results in desorption and ionization of the oligonucleotides, which then can be accelerated in an electrical field into the TOF device. The TOF device separates the accelerated analyte ions of different mass-to-charge (m/z) ratios by providing a field-free drift tube of defined length. After passing through the tube, ions are detected; every signal is assigned to a specific molecular mass calculated from the TOF. The extended bases at possible mutation sites are determined from the difference of nucleotide molecular masses (Vogel et al., 2009).
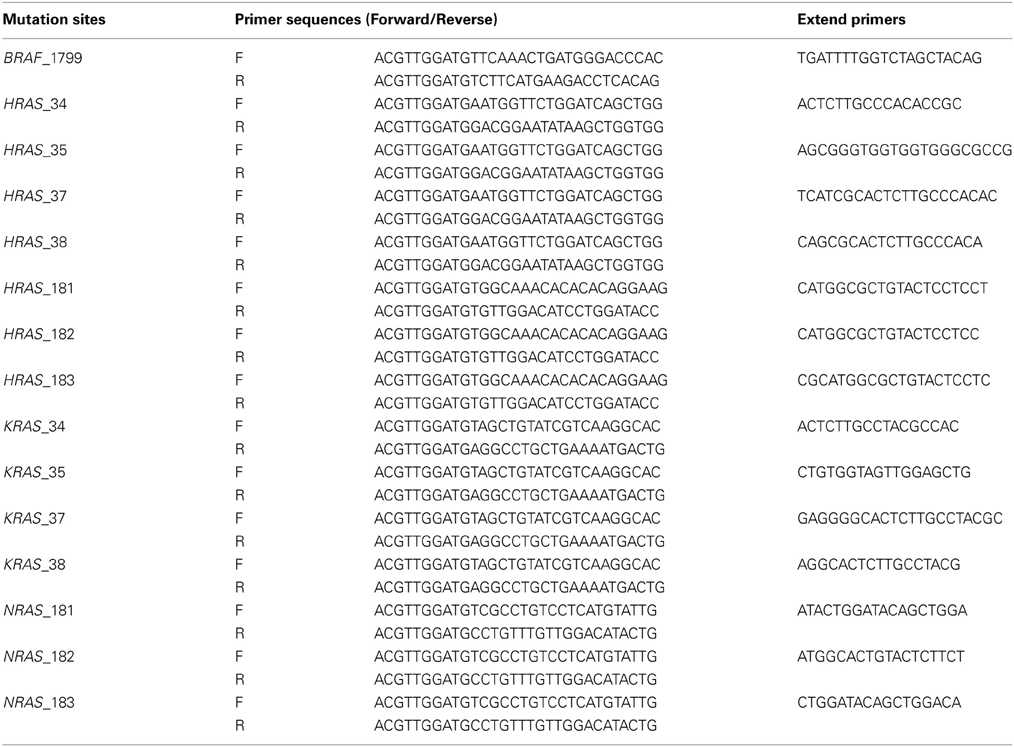
Table 2. Primer sequences used for mutation analysis (MALDI-TOF-MS assays).
5-Aza-2′-Deoxycytidine and Trichostatin A Treatment
Thyroid cancer cell lines were cultured at a density of 3 × 105 cells/10-cm dish on Day 0. Cells were exposed to 3 μM 5-aza-2′-deoxycytidine (Sigma-Aldrich, St. Louis, MO) on Days 1, 2, and 3, and to 300 nM trichostatin A (Sigma-Aldrich) on the Day 3. 5-Aza-2′-deoxycytidine induces hypomethylation of DNA by inhibiting DNA methyltransferase, and re-expression of silenced genes by 5-aza-2′-deoxycytidine treatment is synergistically enhanced by trichostatin A, a histone deacetylase inhibitor (Suzuki et al., 2002). 5-Aza-2′-deoxycytidine is unstable in aqueous solution, and thus a 20 mM solution in dimethyl sulfoxide (DMSO) was freshly prepared, and diluted in medium to 3 μM every day immediately before medium change. The medium was changed every 24 h, and the cells were harvested on Day 4.
Quantitative PCR Analysis
RT-PCR was performed using CFX96 Touch TM Real-Time PCR Detection System (BIORAD Laboratories). cDNA was synthesized from 1 μg of total RNA treated with DNase I with a Superscript III kit (Invitrogen, Life Technologies). The quantity of cDNA of each gene in a sample was measured by comparing it with standard samples that contained 101 to 106 copies of the genes, and normalized to that of PPIA (Peptidylprolyl Isomerase A). Primer sequences are shown in Table 3.

Table 3. Primer sequences for real-time RT-PCR in gene re-expression analysis.
Statistical Analysis
P-values were calculated to compare methylation(+) cancer and methylation(−) cancer and to analyze the correlation of the methylation status to clinicopathological features. Fisher's exact test was used for analysis of binary features such as sex, distant metastasis, recurrence, and mutation of BRAF/RAS oncogenes (with simple choice between male and female, occurrence and no occurrence); t-test was used for more descriptive features that do not imply a choice, such as age, tumor size, number of lymph nodes with metastasis, and thyroglobulin. When P < 0.05, the correlation was considered statistically significant. P-values were also calculated by t-test to compare distribution of methylation ratios between cancer and normal samples. When P < 0.05, the difference of the methylation ratios between cancer and normal samples was considered statistically significant. The dot chart and heatmap were drawn using Excel software and Java TreeView software (http://jtreeview.sourceforge.net/).
Results
Oncogene Mutation Status in Papillary Thyroid Cancer
We analyzed mutation status of BRAF and RAS (HRAS, NRAS, and KRAS) oncogenes in 34 papillary thyroid cancer samples using MALDI-TOF-MS (Figure 1). BRAF mutation was detected in 67% (23/34) of the 34 samples, whereas HRAS, NRAS, and KRAS mutations were detected less frequently, in 3% (1/34), 3% (1/34), and 0% (0/34) sample, respectively. Each oncogene mutation was mutually exclusive; 25 among the 34 samples (75%) were oncogene-mutation(+) cancer.
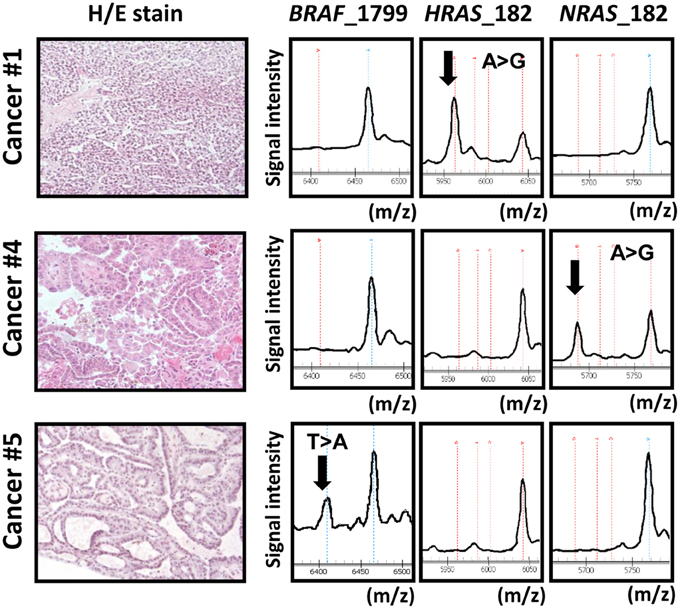
Figure 1. Representative data obtained from MALDI-TOF-MS imaging for three cancer samples. Cancer samples #1, #4, and #5 correspond to papillary cancer samples #1, #4, and #5 in Figure 3. First column: photomicrograph of a tumor section with hematoxylin/eosin (H/E) staining. Second, third, and fourth columns: MALDI-TOF-MS profile with detection of BRAF/RAS mutations. X-axis indicates mass-to-charge ratio (m/z) to distinguish wild-type and mutant allele, and Y-axis indicates signal intensity.
DNA Methylation Analysis using Illumina Infinium Beadarray
Among 34 papillary thyroid cancer and 24 normal thyroid samples, 14 and 10 samples, respectively, were analyzed using Infinium 27K BeadArray. Methylation data of other cancer types (80 head and neck squamous cell cancers, 50 gastric cancers, 80 colorectal cancers, 80 prostate cancers, 24 chronic myeloid leukemias, 50 glioblastomas), and normal samples of corresponding tissues were collected from National Center for Biotechnology Information, Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/gds: GSE25089 for head and neck squamous cell carcinoma, GSE31789 for gastric cancer, GSE27130 for colorectal cancer, GSE26126 for prostate cancer, GSE31600 for chronic myeloid leukemia, and GSE22867 for glioblastoma). To analyze aberrantly methylated genes in cancer samples, probes with β-value < 0.17 in all the normal samples and with standard deviation of β-values in cancer samples >0.15 were selected, and shown in Figure 2. Each cancer type including papillary thyroid cancer showed a unique pattern of aberrant promoter methylation.
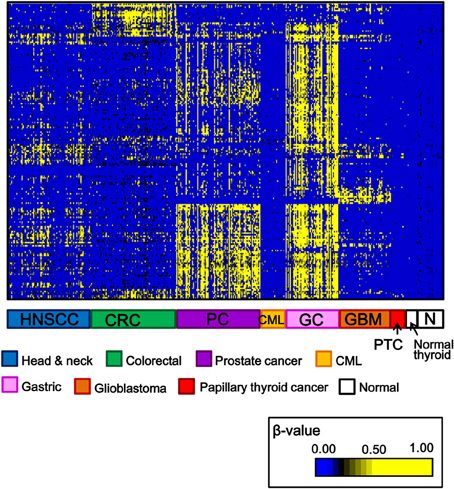
Figure 2. Heatmap for methylation status of several cancer types. Genes with β-value <0.17 in normal samples and with standard deviation of β-value in cancer samples >0.15, were selected and their β-values were shown by heatmap. Each cancer type showed different methylation profile, and papillary thyroid cancer displayed fewer aberrantly methylated genes than other cancer types. HNSCC, head and neck squamous cell carcinoma; CRC, colorectal cancer; PC, prostate cancer; CML, chronic myeloid leukemia; GC, gastric cancer; GBM, glioblastoma; PTC, papillary thyroid cancer; N, normal samples corresponding to these cancer types.
Aberrantly Methylated Genes in Papillary Thyroid Cancer
While the number of aberrantly methylated genes was relatively small in papillary thyroid cancer (Figure 2), 25 genes showed frequent hypermethylation (β > 0.25) in three or more samples among the 14 papillary thyroid cancer samples, and no methylation (β < 0.2) in all the 10 normal samples (Figure 3). To check that the hypermethylation status was not due to contaminated blood cells, the methylation status of these genes in peripheral blood cells was also analyzed to see that none of them were methylated in blood (Figure 3).
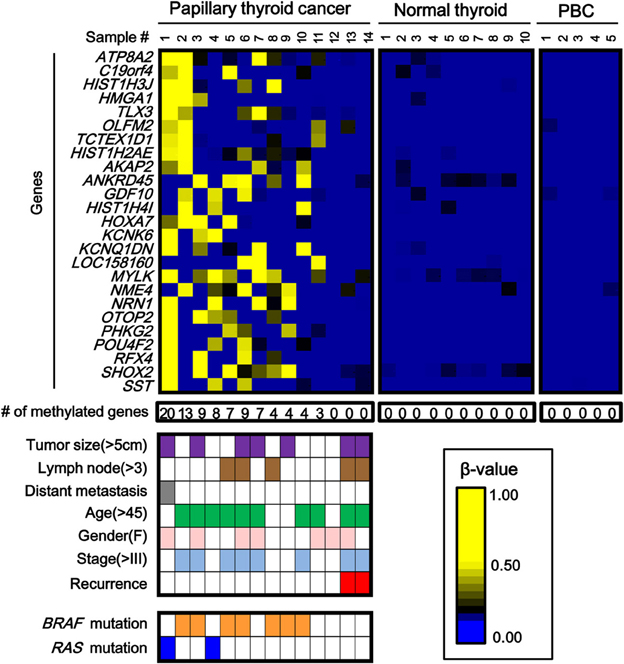
Figure 3. Aberrantly methylated genes frequently observed in papillary thyroid cancer. PBC, peripheral blood cells. We detected 25 genes that were hypermethylated (β > 0.25) in at least three of 14 papillary thyroid cancer samples, but not methylated (β < 0.2) in 10 normal samples. Among the 14 papillary cancer samples, 11 showed aberrant methylation in three or more genes, whereas the other three samples had no aberrant methylation. Purple, tumor size; brown, number of metastatic lymph nodes >3; gray, distant metastasis(+); green, age over 45 years old; pink, female; pale blue, stage III or IV; red, recurrence (+); orange, BRAF mutation(+); blue, RAS mutation (+). The two samples with recurrence (red) had no aberrant methylation (P = 0.03, Fisher's exact test). BRAF/RAS oncogene mutations were all observed in methylation(+) samples (P = 0.03, Fisher's exact test).
Among 14 papillary cancer samples, 11 samples showed aberrant methylation in three or more genes, whereas the other three samples showed no aberrant methylation at all (Figure 3). When methylation status was compared with clinicopathological factors, the two cancer cases with recurrence were both methylation-negative (P = 0.03, Fisher's exact test) (Figure 3). Nine of the 11 frequently methylated samples showed mutation of BRAF/RAS oncogenes, whereas none of the three methylation-negative samples showed oncogene mutation (P = 0.03, Fisher's exact test). Other clinicopathological factors, including tumor size, lymph node metastasis, distant metastasis, tumor stage, age, or sex, did not show significant difference.
To validate the methylation status of these genes, six out of the 25 genes, HIST1H3J, POU4F2, SHOX2, PHKG2, TLX3, and HOXA7, were randomly chosen and analyzed by pyrosequencing, a highly quantitative method (Figure 4). Although one normal sample showed high methylation in POU4F2, frequent hypermethylation of these genes in papillary cancer samples was confirmed, while normal thyroid samples were generally unmethylated.
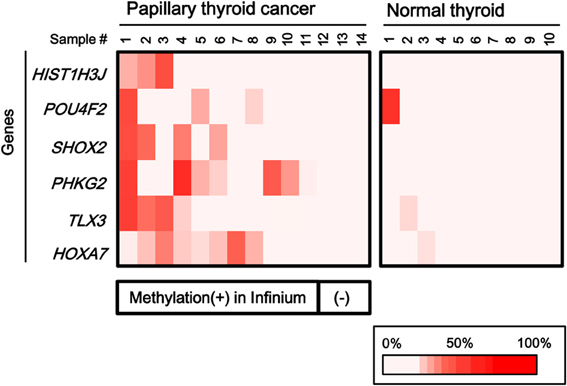
Figure 4. Confirmation of aberrant methylation by pyrosequencing. Six genes were randomly chosen among the 25 frequently methylated genes, and their methylation status was quantitatively validated by pyrosequencing, using the 14 papillary thyroid cancer samples and 10 normal thyroid samples that were analyzed by Infinium. Although one normal sample showed high methylation in POU4F2, aberrant methylation of the six genes in the papillary cancer samples was confirmed. Bottom, the corresponding data for aberrant methylation obtained by Infinium analysis (Figure 3).
Evaluation of Gene Silencing
The analyzed tissue samples include a part of non-tumor cells (see Materials and Methods). To evaluate whether these aberrantly methylated genes were silenced in cancer cells, we analyzed methylation status of these six genes in papillary thyroid cancer cell lines (TPC1, KTC1, and KTC3) and anaplastic thyroid cancer cell line BHT-101 (Figure 5A). All the genes except SHOX2 showed dense methylation in at least one papillary thyroid cancer cell line, confirming that hypermethylation detected in cancer tissue samples is due to hypermethylation in cancer cells.
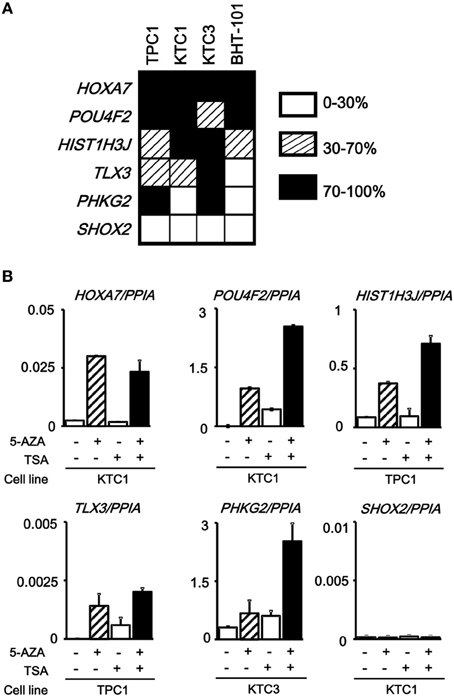
Figure 5. Silencing of aberrantly methylated genes. (A) Methylation status of the HOXA7, POU4F2, HIST1H3J, TLX3, PHKG2, SHOX2 genes was analyzed in papillary thyroid cancer cell lines TPC1, KTC1, and KTC3 and anaplastic thyroid cancer cell line BHT-101 by pyrosequencing. Open box, 0–30% methylation, presumably no allele methylation. Hatched box, 30–70% methylation, presumably hemi-allelic or partial methylation. Closed box, 70–100% methylation, presumably bi-allelic and dense methylation. All the genes except SHOX2 showed dense methylation in at least one papillary thyroid cancer cell line. (B) Real-time RT-PCR analysis of the methylated genes. Cells were treated with 5-aza-2′-deoxycytidine (5-AZA) and/or trichostatin A (TSA). Gene expression levels were normalized to that of PPIA (Peptidylprolyl Isomerase A, or cyclophilin A). All the genes except SHOX2 showed no or very low expression in the analyzed cancer cell line, and showed re-expression in cells treated with 5-AZA/TSA.
We next performed real-time RT-PCR for the six genes. All the genes except SHOX2 showed no or very low expression in the analyzed, methylated cancer cell line, and showed re-expression in cells treated with 5-aza-2′-deoxycytidine and/or trichostatin A (Figure 5B). SHOX2 was neither expressed, nor methylated in KTC1 (Figure 5A). Consequently, its expression was not reversed by the deoxycytidine/trichostatin treatment (Figure 5B). This is presumably because SHOX2 was silenced in KTC1 by mechanisms other than promoter methylation, e.g., by depletion of appropriate transcription factors.
Methylation Analysis of the Six Genes in Additional Samples
To statistically extend the validation of aberrant methylation of the six genes, we analyzed the methylation status in 20 additional papillary thyroid cancer samples and 10 additional normal thyroid samples by pyrosequencing (Figure 6). A similar fraction of cancer samples showed high methylation in each gene (2/20 for HIST1H3J, 8/20 for POU4F2, 4/20 for SHOX2, 5/20 for PHKG2, 6/20 for TLX3, and 4/20 for HOXA7).
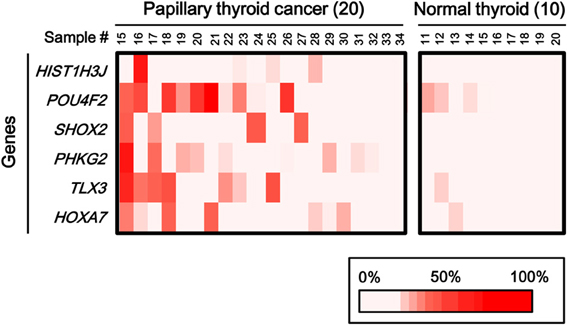
Figure 6. Aberrant methylation of HOXA7, POU4F2, HIST1H3J, TLX3, PHKG2, SHOX2 genes in additional samples: 20 cancer samples and 10 normal ones. Methylation status of the six genes was confirmed by pyrosequencing. Bottom, color scale with known methylation degrees (0–100%).
When methylation ratios were compared between 34 cancer samples in total and 20 normal samples in total, five genes (HIST1H3J, SHOX2, PHKG2, TLX3, and HOXA7) showed significantly higher methylation in cancer (P < 0.05, ranging from 0.0001 to 0.004, t-test), and POU4F2 tended to show higher methylation in cancer (P = 0.07, t-test) (Figure 7A). Among the 34 cancer samples, 26 showed aberrant methylation in at least one gene, but eight showed no aberrant methylation at all (Figure 7B). When clinicopathological features were compared between methylation(+) cancer and methylation(−) cancer, mutations of BRAF/RAS oncogenes significantly correlated to methylation(+) groups (P = 0.04, Fisher's exact test) (Table 4). Although it was not statistically significant, methylation(+) cancer tended to show larger size of tumor (P = 0.06, t-test) and higher levels of thyroglobulin (P = 0.08, t-test).
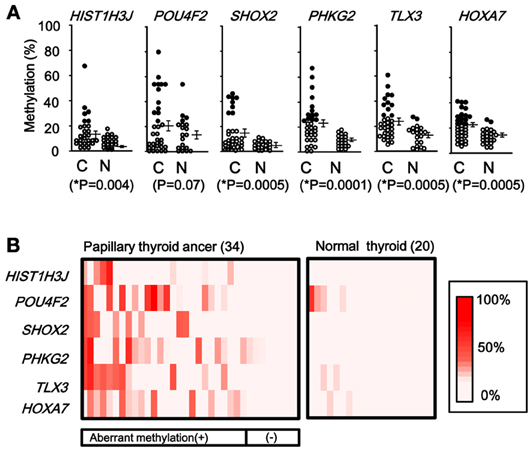
Figure 7. Methylation status of the six genes in 54 samples analyzed by pyrosequencing. (A) Each gene was methylated to a different extent in each sample. C, 34 cancer samples. N, 20 normal samples. Circle, methylation ratio of each sample. Filled circles indicate that the gene is methylated with the methylation ratio >25%. Bars, the mean and standard error of methylation ratios. P-values were calculated by t-test to compare distribution of methylation ratios between cancer (C) and normal (N) samples. Five genes (HIST1H3J, SHOX2, PHKG2, TLX3, and HOXA7) showed significantly higher methylation in cancer (P < 0.05, t-test). POU4F2 tended to show higher methylation in cancer (P = 0.07, t-test). (B) Heatmap for methylation status of the 54 analyzed samples. Among the 34 papillary thyroid cancer samples, 26 were aberrant methylation(+) in at least one of six genes, while eight had no methylation. In Table 4, the number of cancer samples with aberrant methylation (n = 26) and the number of samples without aberrant methylation (n = 8) refer to these data.
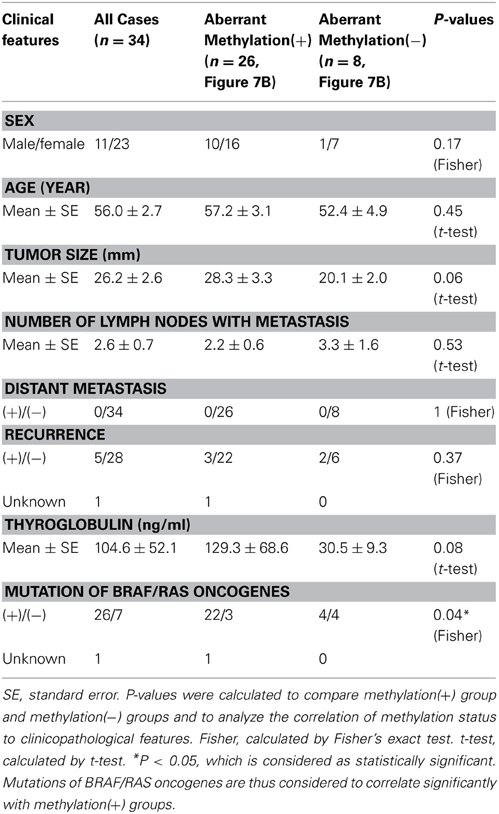
Table 4. Aberrant methylation and clinicopathological features.
Discussion
In this study, we performed genome-wide DNA methylation analysis in 14 human papillary thyroid cancer samples and 10 normal samples. Although papillary thyroid cancer apparently involves fewer aberrantly methylated genes than other types of cancers, we detected 25 genes frequently hypermethylated in papillary thyroid cancer. Methylation status was quantitatively validated in six out of the 25 genes by pyrosequencing, using the genome-widely analyzed samples and additional samples. Gene silencing in papillary thyroid cancer cell lines was confirmed by real-time RT-PCR. While a subset of cancer cases had no aberrant methylation at all, cancer with preferential methylation tended to have oncogene mutation and to be larger tumor.
Papillary thyroid cancer displayed fewer aberrantly methylated genes, compared with other cancer types (Figure 2). For genes previously reported to be methylated in thyroid cancer, such as TSHR (Xing et al., 2003), or in other cancer types, such as RASSF1A, RAR-β2, p16, CDH1, DAPK, and MLH1, the methylation frequency in papillary thyroid cancer ranges from 15 to 33% (Hoque et al., 2005; Guan et al., 2008; Mohammadi-asl et al., 2011). In these reports, no or few normal samples were analyzed (Guan et al., 2008; Mohammadi-asl et al., 2011), methylation was also detected in normal samples (Hoque et al., 2005), or a non-quantitative method, i.e., standard methylation-specific PCR, was used (Guan et al., 2008). Standard methylation-specific PCR (Herman et al., 1996) can amplify and detect minor fraction of methylated alleles, but its high sensitivity can lead to overestimation of methylation frequency. Our analysis did not select these genes as frequently methylated ones in the first 14 cancer samples, because normal thyroid tissues also showed high methylation levels or because methylation frequencies in papillary thyroid cancer samples were low (≤2 of 14 cancer samples). Instead, we detected 25 novel genes that were frequently aberrantly methylated (β > 0.25) in at least three of the 14 cancer samples, and not methylated in any of the 10 normal thyroid samples (β < 0.2).
Interestingly, three of the 14 papillary thyroid cancer samples showed no aberrant methylation in the 25 genes, but the other 11 cancer samples showed hypermethylation in at least three of the 25 genes. No cancer sample showed aberrant methylation in just one or two genes. This unusual distribution of aberrant methylation is similar to the CpG island methylator phenotype (CIMP), which was first proposed in colorectal cancer (Toyota et al., 1999). As Toyota et al. demonstrated in colorectal cancer (Toyota et al., 1999), we calculated probability of methylation distribution in papillary thyroid cancer using these 25 genes. The fraction of methylated tumors in each gene was 3/14 for ATP8A2, 3/14 for C19orf4, …, 5/14 for ANKRD45, …, 6/14 for MYLK, …, 4/14 for NRN1, …, 5/14 for SHOX2, and 3/14 for SST (Figure 3). Assuming that methylation of these genes is random, the probability that none of the 25 genes would be methylated in three cancer samples is P = 1.2 × 10−8. This was calculated using the following formula:
where x indicates number of samples which do not have methylated genes (x = 3 in the present case), N indicates number of cancer samples (N = 14 here), g indicates one of the 25 genes, and f(g) indicates the number of samples in which g is methylated. Similarly, the probability that at least three of the 25 genes are methylated in 11 cancer samples would be P = 0.0028. In each case, a random event is highly unlikely. We rather observe that the associated methylation or un-methylation events both occur at relatively high frequencies. Our data thus suggest that there are two distinct classes of papillary thyroid cancer. One is a subset of hardly methylated cancer. The second one is a subset of preferentially methylated cancer, prone to transcriptional silencing and with the potential to inactivate several genes simultaneously, as CIMP has been proposed in colorectal cancer and other cancers (Toyota et al., 1999; Kaneda et al., 2002; Noushmehr et al., 2010).
Although the number of analyzed samples was not large, preferentially methylated papillary thyroid cancer showed mutation of BRAF/RAS oncogenes more frequently than methylation(−) cancer (P = 0.04, Fisher's exact test). In previous studies of papillary thyroid cancer, although methylation of RASSF1A and BRAF mutation were detected in a mutually exclusive manner (Xing et al., 2004; Hoque et al., 2005), methylation of RAR-β2 or MLH1 significantly correlated to BRAF mutation (Hoque et al., 2005; Guan et al., 2008). Correlation of aberrant methylation and oncogene mutation are also reported in colorectal cancer; high-methylation and intermediate-methylation epigenotypes strongly correlated to BRAF mutation and KRAS mutation, respectively, and low-methylation epigenotype strongly correlated to lack of oncogene mutation (Shen et al., 2007; Yagi et al., 2010; Hinoue et al., 2012). The mechanism of these correlations is still unknown, but oncogene mutation may somehow induce aberrant methylation, or aberrant methylation may help escape from senescence by disrupting factors critical in RAF/RAS-induced senescence (Kaneda and Yagi, 2011).
Preferentially methylated cancer also tended to have larger tumors and higher thyroglobulin levels, which might relate to cancer progression (Piccardo et al., 2013). Although 90% of papillary thyroid cancers are considered to be at low risk with a mortality rate of 1–2%, the mortality rate of the high risk group is 50–75% (Hay et al., 1993; Noguchi et al., 1994; Shaha et al., 1996; Dean and Hay, 2000). The tumor-node-metastasis (TNM) classification is a tool for cancer prognosis; each variable used in TNM staging (age, tumor size, extent of primary tumor, and presence of nodal or distant metastases) shows significant association with observed end points of cancer recurrence or death. Cancer recurrence and mortality ratios are significantly lower in stage I (15.4% and 1.7%, respectively) compared with more advanced tumors (22% and 15.8% in stage II, 46.4% and 30% in stage III, and 66.7% and 60.9% in stage IV, respectively) (Loh et al., 1997). Molecular diagnostic markers are still not used, although their development is anticipated (McLeod et al., 2013). Although aberrant methylation was not significantly associated with lymph node metastasis, distant metastasis, or recurrence in analysis of the 34 cancer samples in this study, further study should be performed using larger set of clinical samples for comparison of aberrant methylation, gene mutation status, and prognosis.
As for detected genes, TLX3 (HOX11L2) is a transcription factor highly expressed in T-cell leukemia (Baak et al., 2008), and its aberrant methylation was observed in cisplatin-resistant bladder cancer (Tada et al., 2011). SHOX2 is a member of the homeobox gene family, and is reported to relate to a short-stature phenotype of Turner syndrome (Clement-Jones et al., 2000). DNA methylation of SHOX2 was suggested to be a biomarker for diagnosis of lung cancer based on bronchial aspirates (Schmidt et al., 2010). HOXA7 is also a transcription factor belonging to the homeobox gene family that regulates gene expression, morphogenesis, and differentiation (La Celle and Polakowska, 2001). POU4F2 is one of POU family genes with Pit-Oct-Unc domain, and is a transcription factor with a role in cell identity and regulation of nerve cell or retinal development (Weishaupt et al., 2005). PHKG2 is the gamma subunit of phosphorylase kinase, containing the active site of the enzyme. Phosphorylase kinase-deficient liver glycogenosis can be caused by mutations of phosphorylase kinase subunits, PHKA2, PHKB, or PHKG2, but PHKG2 mutation was reported to cause a severe phenotype of this disease (Burwinkel et al., 2003). HIST1H3J encodes a member of histone H3 family, and is found in the small histone gene cluster on chromosome 6p22-p21.3 (NCBI gene data bank). If the role of histone modifications is known to affect the regulation of gene expression, less is known about the possible direct involvement of histones, an H3 variant in the present case, in thyroid tumorigenesis. Further investigation is necessary to clarify tumorigenic roles of these genes and their methylation, in papillary thyroid cancer and other types of cancer (Schmidt et al., 2010; Tada et al., 2011).
In summary, 25 new genes were found to be frequently methylated in papillary thyroid cancer. There might be subsets of papillary thyroid cancer hardly methylated and preferentially methylated, and aberrant methylation of these genes correlates a priori to BRAF/RAS oncogene mutation in papillary thyroid cancer.
Author Contributions
Yasuko Kikuchi, Koichi Yagi, and Keisuke Matsusaka performed the experiments. Eiichi Tsuji and Toshihisa Ogawa prepared clinical samples and information. Junichi Kurebayashi established and supplied cell lines. Yasuko Kikuchi, Shingo Tsuji and Atsushi Kaneda analyzed and interpreted the data. Yasuko Kikuchi and Atsushi Kaneda wrote the manuscript. Toshihisa Ogawa, Hiroyuki Aburatani, and Atsushi Kaneda supervised the study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Mitsutake, University of Nagasaki, for kindly providing cancer cell line TPC-1, and Hiroko Meguro, Kyoko Fujinaka and Kaori Nakano for their technical help. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by JST CREST program.
References
Baak, U., Gokbuget, N., Orawa, H., Schwartz, S., Hoelzer, D., Thiel, E., et al. (2008). Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22, 1154–1160. doi: 10.1038/leu.2008.52
Baylin, S. B., and Ohm, J. E. (2006). Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction. Nat. Rev. Cancer 6, 107–116. doi: 10.1038/nrc1799
Burwinkel, B., Rootwelt, T., Kvittingen, E. A., Chakraborty, P. K., and Kilimann, M. W. (2003). Severe phenotype of phosphorylase kinase-deficient liver glycogenosis with mutations in the PHKG2 gene. Pediatr. Res. 54, 834–839. doi: 10.1203/01.PDR.0000088069.09275.10
Clement-Jones, M., Schiller, S., Rao, E., Blaschke, R. J., Zuniga, A., Zeller, R., et al. (2000). The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum. Mol. Genet. 9, 695–702. doi: 10.1093/hmg/9.5.695
Cohen, Y., Xing, M., Mambo, E., Guo, Z., Wu, G., Trink, B., et al. (2003). BRAF mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 95, 625–627. doi: 10.1093/jnci/95.8.625
Dean, D. S., and Hay, I. D. (2000). Prognostic indicators in differentiated thyroid carcinoma. Cancer Control 7, 229–239.
Esteller, M. (2007). Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 8, 286–298. doi: 10.1038/nrg2005
Ezaki, H., Ebihara, S., Fujimoto, Y., Iida, F., Ito, K., Kuma, K., et al. (1992). Analysis of thyroid carcinoma based on material registered in Japan during 1977-1986 with special reference to predominance of papillary type. Cancer 70, 808–814. doi: 10.1002/1097-0142(19920815)70:4<808::AID-CNCR2820700415>3.0.CO;2-L
Feinberg, A. P., Ohlsson, R., and Henikoff, S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33. doi: 10.1038/nrg1748
Guan, H., Ji, M., Hou, P., Liu, Z., Wang, C., Shan, Z., et al. (2008). Hypermethylation of the DNA mismatch repair gene hMLH1 and its association with lymph node metastasis and T1799A BRAF mutation in patients with papillary thyroid cancer. Cancer 113, 247–255. doi: 10.1002/cncr.23548
Hay, I. D., Bergstralh, E. J., Goellner, J. R., Ebersold, J. R., and Grant, C. S. (1993). Predicting outcome in papillary thyroid carcinoma: development of a reliable prognostic scoring system in a cohort of 1779 patients surgically treated at one institution during 1940 through 1989. Surgery 114, 1050–1057. discussion: 7–8.
Herman, J. G., Graff, J. R., Myohanen, S., Nelkin, B. D., and Baylin, S. B. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826. doi: 10.1073/pnas.93.18.9821
Hinoue, T., Weisenberger, D. J., Lange, C. P., Shen, H., Byun, H. M., Van Den Berg, D., et al. (2012). Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 22, 271–282. doi: 10.1101/gr.117523.110
Hoque, M. O., Rosenbaum, E., Westra, W. H., Xing, M., Ladenson, P., Zeiger, M. A., et al. (2005). Quantitative assessment of promoter methylation profiles in thyroid neoplasms. J. Clin. Endocrinol. Metab. 90, 4011–4018. doi: 10.1210/jc.2005-0313
Ishizaka, Y., Itoh, F., Tahira, T., Ikeda, I., Ogura, T., Sugimura, T., et al. (1989). Presence of aberrant transcripts of ret proto-oncogene in a human papillary thyroid carcinoma cell line. Jpn. J. Cancer Res. 80, 1149–1152. doi: 10.1111/j.1349-7006.1989.tb01645.x
Jemal, A., Murray, T., Ward, E., Samuels, A., Tiwari, R. C., Ghafoor, A., et al. (2005). Cancer statistics, 2005. CA. Cancer J. Clin. 55, 10–30. doi: 10.3322/canjclin.55.1.10
Joo, J. Y., Park, J. Y., Yoon, Y. H., Choi, B., Kim, J. M., Jo, Y. S., et al. (2012). Prediction of occult central lymph node metastasis in papillary thyroid carcinoma by preoperative BRAF analysis using fine-needle aspiration biopsy: a prospective study. J. Clin. Endocrinol. Metab. 97, 3996–4003. doi: 10.1210/jc.2012-2444
Kaneda, A., Kaminishi, M., Yanagihara, K., Sugimura, T., and Ushijima, T. (2002). Identification of silencing of nine genes in human gastric cancers. Cancer Res. 62, 6645–6650.
Kaneda, A., and Yagi, K. (2011). Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci. 102, 18–24. doi: 10.1111/j.1349-7006.2010.01712.x
Kimura, E. T., Nikiforova, M. N., Zhu, Z., Knauf, J. A., Nikiforov, Y. E., and Fagin, J. A. (2003). High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 63, 1454–1457.
Knauf, J. A., and Fagin, J. A. (2009). Role of MAPK pathway oncoproteins in thyroid cancer pathogenesis and as drug targets. Curr. Opin. Cell Biol. 21, 296–303. doi: 10.1016/j.ceb.2009.01.013
Kondo, T., Nakazawa, T., Murata, S., Kurebayashi, J., Ezzat, S., Asa, S. L., et al. (2007). Enhanced B-Raf protein expression is independent of V600E mutant status in thyroid carcinomas. Hum. Pathol. 38, 1810–1818. doi: 10.1016/j.humpath.2007.04.014
Kurebayashi, J., Okubo, S., Yamamoto, Y., Ikeda, M., Tanaka, K., Otsuki, T., et al. (2006). Additive antitumor effects of gefitinib and imatinib on anaplastic thyroid cancer cells. Cancer Chemother. Pharmacol. 58, 460–470. doi: 10.1007/s00280-006-0185-x
Kurebayashi, J., Tanaka, K., Otsuki, T., Moriya, T., Kunisue, H., Uno, M., et al. (2000). All-trans-retinoic acid modulates expression levels of thyroglobulin and cytokines in a new human poorly differentiated papillary thyroid carcinoma cell line, KTC-1. J. Clin. Endocrinol. Metab. 85, 2889–2896. doi: 10.1210/jc.85.8.2889
La Celle, P. T., and Polakowska, R. R. (2001). Human homeobox HOXA7 regulates keratinocyte transglutaminase type 1 and inhibits differentiation. J. Biol. Chem. 276, 32844–32853. doi: 10.1074/jbc.M104598200
Lee, K. C., Li, C., Schneider, E. B., Wang, Y., Somervell, H., Krafft, M., et al. (2012). Is BRAF mutation associated with lymph node metastasis in patients with papillary thyroid cancer. Surgery 152, 977–983. doi: 10.1016/j.surg.2012.08.019
Loh, K. C., Greenspan, F. S., Gee, L., Miller, T. R., and Yeo, P. P. (1997). Pathological tumor-node-metastasis (pTNM) staging for papillary and follicular thyroid carcinomas: a retrospective analysis of 700 patients. J. Clin. Endocrinol. Metab. 82, 3553–3562. doi: 10.1210/jc.82.11.3553
Matsusaka, K., Kaneda, A., Nagae, G., Ushiku, T., Kikuchi, Y., Hino, R., et al. (2011). Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 71, 7187–7197. doi: 10.1158/0008-5472.CAN-11-1349
McLeod, D. S., Sawka, A. M., and Cooper, D. S. (2013). Controversies in primary treatment of low-risk papillary thyroid cancer. Lancet 381, 1046–1057. doi: 10.1016/S0140-6736(12)62205-3
Mitsutake, N., Miyagishi, M., Mitsutake, S., Akeno, N., Mesa, C. Jr., Knauf, J. A., et al. (2006). BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology 147, 1014–1019. doi: 10.1210/en.2005-0280
Mohammadi-asl, J., Larijani, B., Khorgami, Z., Tavangar, S. M., Haghpanah, V., Kheirollahi, M., et al. (2011). Qualitative and quantitative promoter hypermethylation patterns of the P16, TSHR, RASSF1A and RARbeta2 genes in papillary thyroid carcinoma. Med. Oncol. 28, 1123–1128. doi: 10.1007/s12032-010-9587-z
Nagae, G., Isagawa, T., Shiraki, N., Fujita, T., Yamamoto, S., Tsutsumi, S., et al. (2011). Tissue-specific demethylation in CpG-poor promoters during cellular differentiation. Hum. Mol. Genet. 20, 2710–2721. doi: 10.1093/hmg/ddr170
Namba, H., Nakashima, M., Hayashi, T., Hayashida, N., Maeda, S., Rogounovitch, T. I., et al. (2003). Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J. Clin. Endocrinol. Metab. 88, 4393–4397. doi: 10.1210/jc.2003-030305
Noguchi, S., Murakami, N., and Kawamoto, H. (1994). Classification of papillary cancer of the thyroid based on prognosis. World J. Surg. 18, 552–7. discussion: 8. doi: 10.1007/BF00353763
Noushmehr, H., Weisenberger, D. J., Diefes, K., Phillips, H. S., Pujara, K., Berman, B. P., et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522. doi: 10.1016/j.ccr.2010.03.017
Piccardo, A., Arecco, F., Puntoni, M., Foppiani, L., Cabria, M., Corvisieri, S., et al. (2013). Focus on high-risk DTC patients: high postoperative serum thyroglobulin level is a strong predictor of disease persistence and is associated to progression-free survival and overall survival. Clin. Nucl. Med. 38, 18–24. doi: 10.1097/RLU.0b013e318266d4d8
Schmidt, B., Liebenberg, V., Dietrich, D., Schlegel, T., Kneip, C., Seegebarth, A., et al. (2010). SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer 10:600. doi: 10.1186/1471-2407-10-600
Shaha, A. R., Shah, J. P., and Loree, T. R. (1996). Risk group stratification and prognostic factors in papillary carcinoma of thyroid. Ann. Surg. Oncol. 3, 534–538. doi: 10.1007/BF02306085
Shen, L., Toyota, M., Kondo, Y., Lin, E., Zhang, L., Guo, Y., et al. (2007). Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 18654–18659. doi: 10.1073/pnas.0704652104
Suzuki, H., Gabrielson, E., Chen, W., Anbazhagan, R., van Engeland, M., Weijenberg, M. P., et al. (2002). A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 31, 141–149. doi: 10.1038/ng892
Tada, Y., Yokomizo, A., Shiota, M., Tsunoda, T., Plass, C., and Naito, S. (2011). Aberrant DNA methylation of T-cell leukemia, homeobox 3 modulates cisplatin sensitivity in bladder cancer. Int. J. Oncol. 39, 727–733. doi: 10.3892/ijo.2011.1049
Toyota, M., Ahuja, N., Ohe-Toyota, M., Herman, J. G., Baylin, S. B., and Issa, J. P. (1999). CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 96, 8681–8686. doi: 10.1073/pnas.96.15.8681
Ulisse, S., Baldini, E., Sorrenti, S., Barollo, S., Prinzi, N., Catania, A., et al. (2012). In papillary thyroid carcinoma BRAFV600E is associated with increased expression of the urokinase plasminogen activator and its cognate receptor, but not with disease-free interval. Clin. Endocrinol. (Oxf.) 77, 780–786. doi: 10.1111/j.1365-2265.2012.04465.x
Vogel, N., Schiebel, K., and Humeny, A. (2009). Technologies in the whole-genome age: MALDI-TOF-based genotyping. Transfus. Med. Hemother. 36, 253–262. doi: 10.1159/000225089
Weber, M., Hellmann, I., Stadler, M. B., Ramos, L., Paabo, S., Rebhan, M., et al. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466. doi: 10.1038/ng1990
Weishaupt, J. H., Klocker, N., and Bahr, M. (2005). Axotomy-induced early down-regulation of POU-IV class transcription factors Brn-3a and Brn-3b in retinal ganglion cells. J. Mol. Neurosci. 26, 17–25. doi: 10.1385/JMN:26:1:017
Xing, M., Cohen, Y., Mambo, E., Tallini, G., Udelsman, R., Ladenson, P. W., et al. (2004). Early occurrence of RASSF1A hypermethylation and its mutual exclusion with BRAF mutation in thyroid tumorigenesis. Cancer Res. 64, 1664–1668. doi: 10.1158/0008-5472.CAN-03-3242
Xing, M., Haugen, B. R., and Schlumberger, M. (2013). Progress in molecular-based management of differentiated thyroid cancer. Lancet 381, 1058–1069. doi: 10.1016/S0140-6736(13)60109-9
Xing, M., Usadel, H., Cohen, Y., Tokumaru, Y., Guo, Z., Westra, W. B., et al. (2003). Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res. 63, 2316–2321.
Xing, M., Westra, W. H., Tufano, R. P., Cohen, Y., Rosenbaum, E., Rhoden, K. J., et al. (2005). BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 90, 6373–6379. doi: 10.1210/jc.2005-0987
Yagi, K., Akagi, K., Hayashi, H., Nagae, G., Tsuji, S., Isagawa, T., et al. (2010). Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res. 16, 21–33. doi: 10.1158/1078-0432.CCR-09-2006
Yagi, K., Takahashi, H., Akagi, K., Matsusaka, K., Seto, Y., Aburatani, H., et al. (2012). Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am. J. Pathol. 180, 616–625. doi: 10.1016/j.ajpath.2011.10.010
Keywords: DNA methylation, thyroid cancer, CIMP (CpG island methylator phenotype), BRAF, RAS, oncogene mutation
Citation: Kikuchi Y, Tsuji E, Yagi K, Matsusaka K, Tsuji S, Kurebayashi J, Ogawa T, Aburatani H and Kaneda A (2013) Aberrantly methylated genes in human papillary thyroid cancer and their association with BRAF/RAS mutation. Front. Genet. 4:271. doi: 10.3389/fgene.2013.00271
Received: 21 September 2013; Accepted: 18 November 2013;
Published online: 05 December 2013.
Edited by:
Yoshimasa Saito, Keio University Faculty of Pharmacy, JapanReviewed by:
Craig A. Cooney, Central Arkansas Veterans Healthcare System, USAMichèle Amouyal, Centre National de la Recherche Scientifique, France
Copyright © 2013 Kikuchi, Tsuji, Yagi, Matsusaka, Tsuji, Kurebayashi, Ogawa, Aburatani and Kaneda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Kaneda, Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Inohana 1-8-1, Chuo-ku, Chiba-City 260-8670, Japan e-mail:a2FuZWRhQGNoaWJhLXUuanA=