 Kousuke Watanabe1
Kousuke Watanabe1 Daiya Takai2*
Daiya Takai2*- 1Department of Respiratory Medicine, The University of Tokyo Hospital, Bunkyo-ku, Tokyo, Japan
- 2Department of Clinical Laboratory, The University of Tokyo Hospital, Bunkyo-ku, Tokyo, Japan
Two decades have passed since the discovery of microRNA (miRNA), which determines cell fate in nematodes. About one decade ago, the conservation of miRNA in humans was also discovered. At present, the loss of certain miRNAs and the overexpression of miRNAs have been demonstrated in many types of diseases, especially cancer. A key miRNA in lung cancer was reported soon after the initial discovery of a tumor-suppressive miRNA in a hematological malignancy. Various causes of miRNA disruption are known, including deletions, mutations, and epigenetic suppression as well as coding genes. The recent accumulation of knowledge regarding epigenetic transcriptional suppression has revealed the suppression of several miRNAs in lung cancer in response to epigenetic changes, such as H3K9 methylation prior to DNA methylation and H3K27 methylation independent of DNA methylation. In this review, recent knowledge of miRNA disruption in lung cancer as a result of epigenetic changes is discussed. Additionally, emerging cancer-specific changes in RNA editing and their impact on miRNA function are described.
MicroRNAs and Cancer: a Historical Perspective
MicroRNAs (miRNAs) are small non-coding RNA molecules (approximately 22 nucleotides) that function as versatile regulators of gene expression. miRNAs negatively regulate the expression of thousands of genes through the destabilization and/or translational suppression of mRNAs by binding to complementary sequences in the 3′ untranslated regions (3′UTRs) of target mRNAs (Lee et al., 1993; Wightman et al., 1993).
The first miRNA to be discovered, lin-4, was determined to be an essential regulator of development in the nematode Caenorhabditis elegans (Lee et al., 1993; Wightman et al., 1993). This short non-coding RNA was considered to be a peculiar constituent specific to worms. Seven years passed before a second miRNA, let-7, was discovered in nematodes (Reinhart et al., 2000). Shortly thereafter, let-7 was found to be broadly conserved across many species, including humans (Pasquinelli et al., 2000). In 2001, a large number of such genes were identified, and the term “microRNA” was coined (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). Currently, more than 2,000 mature miRNAs have been documented in the miRNA registry (Sanger miRBase release 20; http://www.mirbase.org).
MicroRNAs are involved in many biological processes such as cell cycle control, cell differentiation, and apoptosis. Alterations in miRNA expression have been increasingly recognized as playing important roles in the pathogenesis of human cancers. For example, the first tumor-suppressive miRNAs mir-15 and mir-16 located at 13q14 are frequently deleted and downregulated in chronic lymphocytic leukemia (Calin et al., 2002). In lung cancer, a reduction in let-7 expression is significantly associated with a shorter postoperative survival (Takamizawa et al., 2004). The let-7 miRNAs target important oncogenes such as the Ras family (Johnson et al., 2005) and HMGA2 (Mayr et al., 2007). The mir-17-92 miRNA cluster, which was the first oncogenic miRNA to be reported, is amplified and over expressed in B cell lymphoma (He et al., 2005). Moreover, the mir-17-92 miRNA cluster is also amplified and overexpressed in small-cell lung cancer (SCLC) and enhances the proliferation of cancer cells (Hayashita et al., 2005).
MicroRNAs can be used as biomarkers for the diagnosis and prognosis of malignancies. In general, miRNA expression is downregulated in tumors, compared with normal tissues, and analyzes of the expression of 217 miRNAs in various human cancers reflect the developmental lineage and differentiation of the tumor (Lu et al., 2005). Furthermore, certain miRNAs can aid in classifying the histological subtype (adenocarcinoma or squamous cell carcinoma) of lung cancer (Bishop et al., 2010). The miRNA signature can also predict the survival and relapse of patients with lung cancer (Yu et al., 2008).
Despite growing evidence of the involvement of miRNAs in human carcinogenesis, limited information is available regarding how miRNA expressions are deregulated in cancer. In this article, we review the mechanisms responsible for the changes in miRNA expression in lung cancer, focusing particularly on epigenetic mechanisms, such as DNA methylation and histone modifications.
Mechanisms of Deregulated miRNA Expression in Cancer
In animals, miRNAs are generally transcribed by RNA polymerase II (Lee et al., 2004) to form primary transcripts (pri-miRNAs). Pri-miRNAs form hairpin structures in the nucleus and are processed by the Drosha/DGCR8 complex to form approximately 60 nt precursor miRNAs (pre-miRNAs; Gregory et al., 2004). Pre-miRNAs are transported to the cytoplasm through the RAN GTP-dependent transporter exportin-5 (Lund et al., 2004) and are cleaved by Dicer into mature miRNAs (Hutvagner et al., 2001; Figure 1).
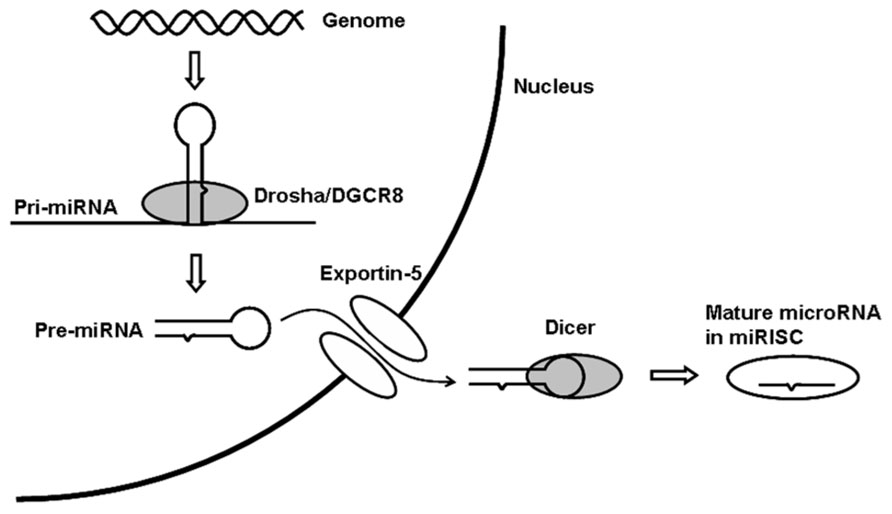
FIGURE 1. miRNA biogenesis pathway. miRISC, microRNA-induced silencing complex.
miRNA are frequently located at fragile sites as well as minimal regions of loss of heterozygosity, minimal regions of amplification, or common breakpoint regions in cancer (Calin et al., 2004). In addition to such genomic changes, any alteration in the miRNA biogenesis pathway described above can affect miRNA expression in cancer. The currently known mechanisms responsible for changes in miRNA expression in cancer include genomic deletions or amplifications, chromosomal translocations, epigenetic silencing by DNA methylation, and impairments of the miRNA biogenesis pathway, such as the frameshift mutation of exportin-5 (Melo et al., 2010), the downregulation of Dicer (Karube et al., 2005; Merritt et al., 2008), and the frameshift mutation of TARBP2 (Melo et al., 2009).
As mentioned above, mir-15 and mir-16, located at 13q14, are deleted in more than half of all cases of chronic lymphocytic leukemia (Calin et al., 2002), and amplification of the mir-17-92 miRNA cluster located at 13q31 is observed in B cell lymphoma and SCLC (Hayashita et al., 2005; He et al., 2005). In addition, mir-125b-1 is a target of the chromosomal translocation t(11,14) in B-cell precursor acute lymphoblastic leukemia, and this translocation results in mir-125b overexpression that is controlled by an immunoglobulin heavy-chain gene regulatory element (Bousquet et al., 2008; Chapiro et al., 2010). However, with regard to lung cancer, no chromosomal translocations involving miRNAs have been previously reported.
Kumar et al. (2007) reported that the impairment of the miRNA biogenesis pathway in cancer results in the global downregulation of miRNA and the enhancement of cellular transformation and tumorigenesis. They demonstrated that the knockdown of DGCR8, Drosha, or Dicer in cancer cells resulted in a pronounced transformed phenotype and that the conditional deletion of Dicer enhanced tumor development in a K-ras-induced mouse model of lung cancer. Importantly, a reduction in Dicer expression is associated with a poor prognosis in patients with lung cancer (Karube et al., 2005) and ovarian cancer (Merritt et al., 2008). Interestingly, a germline mutation of Dicer1 has been discovered in pleuropulmonary blastoma, a rare pediatric lung tumor (Hill et al., 2009). These findings clearly demonstrate that the miRNA biogenesis pathway plays a crucial role in normal lung development and lung carcinogenesis. Frameshift mutations of exportin-5 and TARBP2 have been reported in colorectal cancer with microsatellite instability, but not in lung cancer (Melo et al., 2009, 2010).
Epigenetic changes in cancer genomes, such as DNA methylation and histone modifications, cause the silencing of tumor suppressor genes and contribute to human carcinogenesis (Jones and Takai, 2001; Jones and Baylin, 2002; Herman and Baylin, 2003). Recently, DNA methylation in cancerous tissue has been shown to silence miRNAs in cancer (Saito et al., 2006; Lujambio et al., 2007). Saito et al. reported that the expression of mir-127, which is embedded in a CpG island, was induced by treatment with the chromatin-modifying drugs 5-aza-2’-deoxycytidine and 4-phenylbutyric acid in a bladder cancer cell line. Lujambio et al. analyzed the miRNA expression profile of a colon cancer cell line, which was genetically deficient for the DNA methyltransferase (DNMT) enzymes DNMT1 and DNMT3b, and identified the epigenetic silencing of mir-124a in various types of cancer, including colon, breast, and lung cancers as well as leukemia and lymphoma. As the epigenetic silencing of key tumor suppressor genes, such as APC and p16INK4A, is a common event in lung carcinogenesis (Takai et al., 2001; Sano et al., 2007; Brock et al., 2008; Kusakabe et al., 2010) and miRNA expression is altered in lung cancer (Yanaihara et al., 2006), some miRNAs are thought to be silenced by DNA methylation or histone modification in lung cancer. In fact, growing evidence demonstrates that epigenetic changes contribute to miRNA silencing in lung cancer (Liu et al., 2013).
DNA Methylation and miRNA Expression
The earliest papers on miRNA expression profiling in lung cancer were published in 2006 (Volinia et al., 2006; Yanaihara et al., 2006). These studies used miRNA microarrays and found that a high level of mir-155 expression and a low level of let-7a-2 expression were significantly correlated with a poor survival of lung cancer patients. The number of miRNA profiling studies in lung cancer has grown rapidly in recent years, and these studies have led to the discovery of the role of miRNAs in lung carcinogenesis and their potential as diagnostic, prognostic, or predictive markers (Rothschild, 2013). Vosa et al. performed a meta-analysis of 20 published miRNA expression profiling studies in lung cancer and identified a meta-signature of seven up-regulated (mir-21, mir-210, mir-182, mir-31, mir-200b, mir-205, and mir-183) and eight down-regulated (mir-126-3p, mir-30a, mir-30d, mir-486-5p, mir-451a, mir-126-5p, mir-143, and mir-145) miRNAs (Vosa et al., 2013). Guan et al. (2012) also reported a meta-analysis of 14 published miRNA expression profiling studies and their results agreed well with those published by Vosa et al.
One approach to identifying epigenetically silenced miRNAs is to analyze the miRNA expression profile of cancer cells before and after treatment with chromatin-modifying drugs. Lujambio et al. analyzed the miRNA expression profile of three metastatic cancer cell lines with and without the DNMT inhibitor 5-aza-2’-deoxycytidine and reported that the DNA methylation of three miRNAs (mir-9, mir-34b/c, and mir-148a) was associated with the metastasis of human cancers including lung cancer (Lujambio et al., 2008). Heller et al. analyzed the miRNA expression profile changes in A549 lung cancer cells treated with 5-aza-2’-deoxycytidine and the histone deacetylase inhibitor tricostatin A and identified mir-9-3 and mir-193a as targets for DNA methylation in non-small cell lung cancer (NSCLC; Heller et al., 2012). Our research team analyzed the expressions of 55 in silico selected candidate miRNAs treated with or without 5-aza-2’-deoxycytidine and found that mir-34b/c and mir-126 are silenced by DNA methylation in NSCLC (Watanabe et al., 2012). We also revealed that the DNA methylation of mir-9-3, -124-2, and -124-3 was individually associated with an advanced T factor, and that the methylation of multiple miRNA loci was associated with a poor prognosis (Kitano et al., 2011). The correlation between miRNA methylation and the T factor suggested that the DNA methylation of these miRNA loci accumulates during tumor progression. A list of miRNAs that are silenced by DNA methylation in lung cancer is shown in Table 1.

TABLE 1. miRNAs silenced by DNA methylation in lung cancer.
The mir-34 family is comprised of three miRNAs (mir-34a, mir-34b, and mir-34c) that are derived from two transcripts (mir-34a on chromosome 1 and mir-34b/c on chromosome 11). In mice, mir-34a is ubiquitously expressed, with the highest expression being in the brain, whereas mir-34b/c is mainly expressed in the lung (Bommer et al., 2007). The mir-34 genes induce cell cycle arrest, cellular senescence, and apoptosis when ectopically expressed (Bommer et al., 2007; He et al., 2007; Welch et al., 2007) through the downregulation of multiple target genes such as Bcl-2, Cyclin D1, Cyclin E2, CDK4, CDK6, c-Myc, and c-Met (Hermeking, 2010). Moreover, mir-34s have been identified as direct targets of the p53 transcription factor (Bommer et al., 2007; Corney et al., 2007; He et al., 2007), and their expression is induced in response to DNA damage or oncogenic stress. These results indicate that mir-34s play a critical role in the tumor-suppressive program governed by p53. Interestingly, the chromosomal locus 1p36, where mir-34a is located, has been proposed to harbor a tumor suppressor gene, since a homozygous deletion at this locus has been reported in neuroblastoma, and mir-34a has been identified as a candidate tumor suppressor at this locus (Cole et al., 2008).
In lung cancer, mir-34a and mir-34b/c are targets of epigenetic silencing by DNA methylation (Lodygin et al., 2008; Gallardo et al., 2009; Wang et al., 2011; Watanabe et al., 2012). In primary NSCLC, a low mir-34a expression level is significantly associated with a high probability of relapse after surgery (Gallardo et al., 2009). We previously reported that mir-34b/c is methylated in 41% of primary NSCLC cases and that mir-34b/c methylation is associated with lymphatic invasion (Watanabe et al., 2012). The DNA methylation of mir-34b/c is associated with a poorer prognosis in patients with NSCLC (Wang et al., 2011). In addition, the mir-34s are silenced by DNA methylation in SCLC (Tanaka et al., 2012). In primary SCLC, mir-34a and mir-34b/c were methylated in 15% and 67% of the cases, respectively. The CpG island methylation of mir-34b/c has also been reported in colorectal cancer (Toyota et al., 2008), oral squamous cell cancer (Kozaki et al., 2008), melanoma, and breast cancer (Lujambio et al., 2008). Thus, the epigenetic inactivation of mi-34s is a common event in human cancer.
mir-34a and mir-34b/c are intergenic miRNAs, and their expressions are regulated by the DNA methylation of their own promoters. Importantly, many miRNA encoding sequences are located within the introns of protein coding genes, and intronic miRNAs are usually expressed in a coordinate manner along with their host gene mRNAs (Baskerville and Bartel, 2005). We previously reported that mir-126, which is located within the intron of EGFL7, is silenced by the DNA methylation of its host gene in NSCLC (Watanabe et al., 2012). mir-126 functions as a tumor-suppressive miRNA, suppressing metastasis in breast cancer (Tavazoie et al., 2008) and inhibiting the invasion of NSCLC cell lines by targeting Crk (Crawford et al., 2008). Moreover, mir-126 was recently reported to be an essential regulator of angiogenesis. Vascular endothelial growth factor (VEGF)-A is a target of mir-126, and the downregulation of mir-126 increases the activity of VEGF-A in lung cancer (Liu et al., 2009). A reduction in mir-126 expression is significantly associated with increased microvessel density in oral squamous cell cancer (Sasahira et al., 2012) and NSCLC (Jusufovic et al., 2012), suggesting a negative regulatory role of mir-126 in tumor angiogenesis. In addition, decreased mir-126 expression is significantly associated with a shorter survival period in patients with NSCLC (Jusufovic et al., 2012). In contrast, Donnem et al. demonstrated that an elevated level of mir-126 expression is associated with a shorter survival period in patients with NSCLC and that an elevated level of mir-126 expression was associated with an increase in VEGF-A expression in NSCLC (Donnem et al., 2011). The targeted deletion of mir-126 in mice impaired normal angiogenesis, suggesting a positive regulatory role of mir-126 in angiogenesis (Wang et al., 2008). Additional research is required to elucidate the relation between mir-126, tumor angiogenesis, and tumor progression. Furthermore, we reported that the DNA methylation of EGFL7 (a host gene of mir-126) was only observed in 7% of the clinical samples that were evaluated (Watanabe et al., 2012), which cannot completely explain the frequent downregulation of this miRNA in NSCLC. In fact, our analyzes of DNA methylation of the coding genes (Sano et al., 2007; Kusakabe et al., 2010) and miRNAs (Kitano et al., 2011; Watanabe et al., 2012) revealed that the ratio of DNA methylation is often quite low in primary NSCLCs. This low ratio of DNA methylation may be a result of insufficient DNA methylation following changes in histone modification, rather than the result of the coexistence of non-cancerous tissues, and may be responsible for the low frequency of DNA methylation of mir-126 in primary NSCLCs if the method used to detect DNA methylation is not sufficiently sensitive.
Cigarette smoking is the most important risk factor for lung cancer. Accumulating evidence suggests that tobacco induces the epigenetic silencing of certain miRNAs in lung carcinogenesis (Tellez et al., 2011; Xi et al., 2013). Tellez et al. reported that the exposure of human bronchial epithelial cells (HBECs) to tobacco carcinogens decreased the expressions of mir-200b, -200c, and -205 and induced the epithelial-to-mesenchymal transition (EMT). The mir-200 family and mir-205 function as key negative regulators of the EMT through the direct targeting of ZEB1 and ZEB2 (Gregory et al., 2008; Park et al., 2008). In HBECs, tobacco carcinogens initially induced an increase in H3K27me3 (inactive closed chromatin) and subsequently induced the DNA methylation of sequences encoding these miRNAs. The loss of mir-200c expression as a result of DNA methylation has been shown to induce an aggressive, invasive, and chemoresistant phenotype of NSCLC (Ceppi et al., 2010). In addition, Shien et al. (2013) demonstrated that a lung cancer cell line with acquired resistance to an EGFR tyrosine kinase inhibitor exhibited EMT features and the downregulation of mir-200c as a result of DNA methylation. Xi et al. reported that tobacco carcinogens induced the epigenetic silencing of mir-487b and that mir-487b functions as a tumor suppressive miRNA in NSCLC by targeting SUZ12, BMI1, WNT5A, MYC, and K-ras. These studies highlight the potential of epigenetic drugs to reverse tobacco-induced reprogramming in lung cancer cells.
H3K27me3 and miRNAs
Epigenetic silencing in mammalian cells is mediated by at least two distinct histone modifications: histone H3 trimethylation at lysine 27 (H3K27me3) and histone H3 dimethylation and trimethylation at lysine 9 (H3K9me2 and H3K9me3). A recent genome-wide study of histone modifications in prostate cancer cells revealed H3K27me3 as a mechanism of tumor-suppressor gene silencing in cancer that occurs independently of promoter DNA methylation (Kondo et al., 2008). A polycomb group protein, EZH2, exhibits histone methyltransferase activity with substrate specificity for H3K27 (Cao and Zhang, 2004). EZH2 overexpression is associated with a poor prognosis in lung cancer, and the knockdown of EZH2 expression decreases the growth and invasion of lung cancer cells (Huqun et al., 2012). These findings suggest that aberrant H3K27me3 contributes to tumor-suppressor gene silencing in lung cancer, but genome-wide analyzes of H3K27me3 in lung cancer have not been reported.
Recently, Au et al. (2012) analyzed the changes in miRNA expression profiles induced by EZH2 knockdown and found that some tumor-suppressive miRNAs (mir-139, -125b, -101, -200b, and let-7c) are silenced by H3K27me3 in hepatocellular carcinoma. In lung cancer, mir-212 is silenced by histone modifications rather than DNA methylation (Incoronato et al., 2011). Incoronato et al. showed that increases in H3K27me3 and H3K9me2 are observed in the mir-212 promoter region in the lung cancer cell line Calu-1, which has a low mir-212 expression level. EZH2 may exert its oncogenic function, at least in part, by silencing tumor-suppressive miRNAs, and further investigation is required to verify the association between H3K27me3 and miRNA expression in lung cancer.
miRNAs that Target Epigenetic Machinery
While miRNA expression is regulated by DNA methylation and histone modifications, genes encoding the epigenetic machinery are also targeted by miRNAs. The mir-29 family is the prototype of such miRNAs (Fabbri et al., 2007). The mir-29 family is comprised of three miRNAs (mir-29a, mir-29b, and mir-29c) that are derived from two transcripts (mir-29b-1/29a on chromosome 7 and mir-29b-2/29c on chromosome 1). The mir-29 family is highly expressed in normal tissues and is downregulated in many types of human cancers including lung cancer (Yanaihara et al., 2006; Xu et al., 2009). mir-29a reportedly functions as an anti-metastatic and anti-proliferative miRNA in lung cancer (Muniyappa et al., 2009). mir-29b has also been reported to function as an anti-metastatic miRNA in lung cancer through the regulation of the Src-ID1 pathway (Rothschild et al., 2012).
Recently, the mir-29 family was shown to directly target DNMT3A and DNMT3B, two enzymes involved in de novo DNA methylation (Fabbri et al., 2007). The expression of mir-29s is inversely correlated with that of DNMT3A and DNMT3B in lung cancer tissue, and the enforced expression of mir-29s in lung cancer cell lines restores the normal pattern of DNA methylation, induces the re-expression of methylation-silenced tumor suppressor genes (such as FHIT and WWOX), and inhibits tumorigenicity both in vitro and in vivo. mir-29b also induces PTEN expression through the downregulation of DNA methyltransferases (DNMTs) and the subsequent hypomethylation of the PTEN promoter in a lung cancer xenograft model (Li et al., 2012). Samakoglu et al. (2012) also report that a combination therapy consisting of an EGFR antibody with cisplatin and gemcitabine induces mir-29b expression, the downregulation of DNMT3b, and the hypomethylation of tumor-suppressor genes in a lung cancer xenograft model. mir-29b has also been shown to down-regulate DNMT1, an enzyme involved in the maintenance of DNA methylation, indirectly by targeting Sp1, a transactivator of the DNMT1 gene in leukemia (Garzon et al., 2009).
In addition to DNMTs, miRNAs can also target histone modification enzymes. mir-449a/b is downregulated and directly targets HDAC1, a histone deacetylase in lung cancer (Jeon et al., 2012). mir-101 reportedly targets EZH2, and the genomic loss of mir-101 leads to the overexpression of EZH2 in prostate cancer cells (Varambally et al., 2008). These results clearly show a strong interplay between miRNA and the epigenetic machinery, providing new insights into the molecular mechanism of aberrant DNA methylation and histone modifications in cancer.
RNA Editing of miRNAs
The most prevalent type of RNA editing in humans is the deamination of adenosine to inosine in double-stranded RNA (A-to-I editing). This process is catalyzed by two family members of adenosine deaminases acting on RNA (ADAR): ADAR1 and ADAR2. All known A-to-I edited sites have been attributed to ADAR1 or ADAR2 (Zinshteyn and Nishikura, 2009). Inosine is present in mRNA at tissue-specific levels that are correlated with ADAR expression. Analyzes of the amount of inosine in various mammalian tissues has revealed that inosine is most abundant in the brain, where one inosine molecule is present for every 17,000 ribonucleotides of mRNA; the second highest frequency of inosine has been observed in the lung, where one inosine molecule is present for every 33,000 ribonucleotides of mRNA (Paul and Bass, 1998).
Recently, certain pri-miRNAs have been reported to be subject to A-to-I editing. Kawahara et al. examined 209 pri-miRNAs and identified 47 pri-miRNAs as the targets of A-to-I editing in the human brain (Kawahara et al., 2008), suggesting that miRNA editing could have a considerable impact on miRNA-mediated gene silencing. Most A-to-I editing of pri-miRNAs results in altered miRNA processing by Drosha and Dicer (Kawahara et al., 2008). However, in rare cases, such as mir-376, RNA editing causes base substitution in the seed sequence and generates edited mature miRNAs with unique target genes and functions different from those of the unedited miRNAs (Kawahara et al., 2007).
Emerging lines of evidence suggest a link between A-to-I editing and cancer. Anomalous ADAR activity in cancer may lead to alterations in the efficiency of A-to-I editing. For example, the glutamate receptor subunit B (GluR-B) is nearly 100% edited at one position (Q/R site) in the normal brain. In primary glioblastoma, this position is substantially under-edited, compared with normal tissues, because of the decreased activity of ADAR2, which is responsible for the A-to-I editing of GluR-B (Maas et al., 2001). Recently, the efficiency of A-to-I editing of mir-376a* was found to be significantly attenuated in glioblastoma cells (Choudhury et al., 2012). Unedited mir-376a* promotes the migration and invasion of glioma cells, whereas edited mir-376a* suppresses these features. These results suggest that the attenuation of A-to-I editing of mir-376a* promotes the invasiveness of glioblastoma. Considering the relatively high prevalence of inosine in lung mRNA and the frequent A-to-I editing of miRNAs in the brain, it would be tempting to conduct a large-scale survey to evaluate the A-to-I editing of pri-miRNAs in normal lung and to analyze the alteration of miRNA editing and ADAR activity in lung cancer.
Conclusions and Future Perspectives
The two major challenges in studying the role of miRNA in cancer are (i) the identification of target genes, and (ii) the elucidation of the mechanisms that regulate miRNA expression in both normal and cancer cells. Limited knowledge is available regarding miRNA transcription, primarily because of inadequate information on the precise locations of the promoters and transcriptional start sites of the miRNAs. Approximately half of all miRNAs are intragenic sequences that are located within the exons, introns, or 3′UTRs of protein-coding genes. These intragenic miRNAs share promoters with their host genes and are co-regulated with their host genes, as in the case of mir-126. The remaining 50% of miRNAs are intergenic miRNAs with their own promoters, which have not been experimentally validated in most cases. Comprehensive analyzes of the miRNA transcription unit will help to elucidate the transcription factors or epigenetic changes responsible for alterations in miRNA expression in cancer.
The impairment of the miRNA biogenesis pathway and the attenuation of A-to-I editing add to the growing complexity of miRNA deregulation in cancer. Moreover, 3′UTRs of certain mRNAs are progressively shortened in cancer cells as a result of changes in alternative cleavage and polyadenylation (APA), a phenomenon that alters the 3′UTR length. Progressive 3′UTR shortening in cancer cells may lead to the disruption of miRNA-mediated gene silencing (Mayr and Bartel, 2009). Understanding these complexities as well as those of miRNA transcriptional regulation may lead to the identification of novel biomarkers and should help to unravel the impact of miRNA in lung carcinogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by KAKENHI (25830098, 24659267, 23390147).
References
Au, S. L., Wong, C. C., Lee, J. M., Fan, D. N., Tsang, F. H., Ng, I. O., et al. (2012). Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 56, 622–631. doi: 10.1002/hep.25679
Baskerville, S., and Bartel, D. P. (2005). Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11, 241–247. doi: 10.1261/rna.7240905
Bishop, J. A., Benjamin, H., Cholakh, H., Chajut, A., Clark, D. P., and Westra, W. H. (2010). Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin. Cancer Res. 16, 610–619. doi: 10.1158/1078-0432.CCR-09-2638
Bommer, G. T., Gerin, I., Feng, Y., Kaczorowski, A. J., Kuick, R., Love, R. E., et al. (2007). p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 17, 1298–1307. doi: 10.1016/j.cub.2007.06.068
Bousquet, M., Quelen, C., Rosati, R., Mansat-De Mas, V., La Starza, R., Bastard, C., et al. (2008). Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J. Exp. Med. 205, 2499–2506. doi: 10.1084/jem.20080285
Brock, M. V., Hooker, C. M., Ota-Machida, E., Han, Y., Guo, M., Ames, S., et al. (2008). DNA methylation markers and early recurrence in stage I lung cancer. N. Engl. J. Med. 358, 1118–1128. doi: 10.1056/NEJMoa0706550
Calin, G. A., Dumitru, C. D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., et al. (2002). Frequent deletions and down-regulation of micro – RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 99, 15524–15529. doi: 10.1073/pnas.242606799
Calin, G. A., Sevignani, C., Dumitru, C. D., Hyslop, T., Noch, E., Yendamuri, S., et al. (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. U.S.A. 101, 2999–3004. doi: 10.1073/pnas.0307323101
Cao, R., and Zhang, Y. (2004). The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 14, 155–164. doi: 10.1016/j.gde.2004.02.001
Ceppi, P., Mudduluru, G., Kumarswamy, R., Rapa, I., Scagliotti, G. V., Papotti, M., et al. (2010). Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 8, 1207–1216. doi: 10.1158/1541-7786.MCR-10-0052
Chapiro, E., Russell, L. J., Struski, S., Cave, H., Radford-Weiss, I., Valle, V. D., et al. (2010). A new recurrent translocation t(11;14)(q24;q32) involving IGH@ and miR-125b-1 in B-cell progenitor acute lymphoblastic leukemia. Leukemia 24, 1362–1364. doi: 10.1038/leu.2010.93
Chen, Y., Min, L., Zhang, X., Hu, S., Wang, B., Liu, W., et al. (2013). Decreased miRNA-148a is associated with lymph node metastasis and poor clinical outcomes and functions as a suppressor of tumor metastasis in non-small cell lung cancer. Oncol. Rep. 30, 1832–1840. doi: 10.3892/or.2013.2611.
Choudhury, Y., Tay, F. C., Lam, D. H., Sandanaraj, E., Tang, C., Ang, B. T., et al. (2012). Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Invest. 122, 4059–4076. doi: 10.1172/JCI62925
Cole, K. A., Attiyeh, E. F., Mosse, Y. P., Laquaglia, M. J., Diskin, S. J., Brodeur, G. M., et al. (2008). A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 6, 735–742. doi: 10.1158/1541-7786.MCR-07-2102
Corney, D. C., Flesken-Nikitin, A., Godwin, A. K., Wang, W., and Nikitin, A. Y. (2007). MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 67, 8433–8438. doi: 10.1158/0008-5472.CAN-07-1585
Crawford, M., Brawner, E., Batte, K., Yu, L., Hunter, M. G., Otterson, G. A., et al. (2008). MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines. Biochem. Biophys. Res. Commun. 373, 607–612. doi: 10.1016/j.bbrc.2008.06.090
Donnem, T., Lonvik, K., Eklo, K., Berg, T., Sorbye, S. W., Al-Shibli, K., et al. (2011). Independent and tissue-specific prognostic impact of miR-126 in nonsmall cell lung cancer: coexpression with vascular endothelial growth factor-A predicts poor survival. Cancer 117, 3193–3200. doi: 10.1002/cncr.25907
Fabbri, M., Garzon, R., Cimmino, A., Liu, Z., Zanesi, N., Callegari, E., et al. (2007). MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. U.S.A. 104, 15805–15810. doi: 10.1073/pnas.0707628104
Gallardo, E., Navarro, A., Vinolas, N., Marrades, R. M., Diaz, T., Gel, B., et al. (2009). miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis 30, 1903–1909. doi: 10.1093/carcin/bgp219
Garzon, R., Liu, S., Fabbri, M., Liu, Z., Heaphy, C. E., Callegari, E., et al. (2009). MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113, 6411–6418. doi: 10.1182/blood-2008-07-170589
Gregory, P. A., Bert, A. G., Paterson, E. L., Barry, S. C., Tsykin, A., Farshid, G., et al. (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601. doi: 10.1038/ncb1722
Gregory, R. I., Yan, K. P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., et al. (2004). The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. doi: 10.1038/nature03120
Guan, P., Yin, Z., Li, X., Wu, W., and Zhou, B. (2012). Meta-analysis of human lung cancer microRNA expression profiling studies comparing cancer tissues with normal tissues. J. Exp. Clin. Cancer Res. 31, 54. doi: 10.1186/1756-9966-31-54
Hayashita, Y., Osada, H., Tatematsu, Y., Yamada, H., Yanagisawa, K., Tomida, S., et al. (2005). A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 65, 9628–9632. doi: 10.1158/0008-5472.CAN-05-2352
He, L., He, X., Lim, L. P., De Stanchina, E., Xuan, Z., Liang, Y., et al. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134. doi: 10.1038/nature05939
He, L., Thomson, J. M., Hemann, M. T., Hernando-Monge, E., Mu, D., Goodson, S., et al. (2005). A microRNA polycistron as a potential human oncogene. Nature 435, 828–833. doi: 10.1038/nature03552
Heller, G., Weinzierl, M., Noll, C., Babinsky, V., Ziegler, B., Altenberger, C., et al. (2012). Genome-wide miRNA expression profiling identifies miR-9-3 and miR-193a as targets for DNA methylation in non-small cell lung cancers. Clin. Cancer Res. 18, 1619–1629. doi: 10.1158/1078-0432.CCR-11-2450
Herman, J. G., and Baylin, S. B. (2003). Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 349, 2042–2054. doi: 10.1056/NEJMra023075
Hermeking, H. (2010). The miR-34 family in cancer and apoptosis. Cell Death Differ. 17, 193–199. doi: 10.1038/cdd.2009.56
Hill, D. A., Ivanovich, J., Priest, J. R., Gurnett, C. A., Dehner, L. P., Desruisseau, D., et al. (2009). DICER1 mutations in familial pleuropulmonary blastoma. Science 325, 965. doi: 10.1126/science.1174334
Hutvagner, G., Mclachlan, J., Pasquinelli, A. E., Balint, E., Tuschl, T., and Zamore, P. D. (2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838. doi: 10.1126/science.1062961
Huqun, Ishikawa, R., Zhang, J., Miyazawa, H., Goto, Y., Shimizu, Y., et al. (2012). Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer 118, 1599–1606. doi: 10.1002/cncr.26441.
Incoronato, M., Urso, L., Portela, A., Laukkanen, M. O., Soini, Y., Quintavalle, C., et al. (2011). Epigenetic regulation of miR-212 expression in lung cancer. PLoS ONE 6:e27722. doi: 10.1371/journal.pone.0027722
Jeon, H. S., Lee, S. Y., Lee, E. J., Yun, S. C., Cha, E. J., Choi, E., et al. (2012). Combining microRNA-449a/b with a HDAC inhibitor has a synergistic effect on growth arrest in lung cancer. Lung Cancer 76, 171–176. doi: 10.1016/j.lungcan.2011.10.012
Johnson, S. M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., et al. (2005). RAS is regulated by the let-7 microRNA family. Cell 120, 635–647. doi: 10.1016/j.cell.2005.01.014
Jones, P. A., and Baylin, S. B. (2002). The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3, 415–428. doi: 10.1038/nrg816
Jones, P. A., and Takai, D. (2001). The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070.
Jusufovic, E., Rijavec, M., Keser, D., Korosec, P., Sodja, E., Iljazovic, E., et al. (2012). let-7b and miR-126 are down-regulated in tumor tissue and correlate with microvessel density and survival outcomes in non-small-cell lung cancer. PLoS ONE 7:e45577. doi: 10.1371/journal.pone.0045577
Karube, Y., Tanaka, H., Osada, H., Tomida, S., Tatematsu, Y., Yanagisawa, K., et al. (2005). Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 96, 111–115.
Kawahara, Y., Megraw, M., Kreider, E., Iizasa, H., Valente, L., Hatzigeorgiou, A. G., et al. (2008). Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 36, 5270–5280. doi: 10.1111/j.1349-7006.2005.00015.x
Kawahara, Y., Zinshteyn, B., Sethupathy, P., Iizasa, H., Hatzigeorgiou, A. G., and Nishikura, K. (2007). Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315, 1137–1140. doi: 10.1093/nar/gkn479
Kitano, K., Watanabe, K., Emoto, N., Kage, H., Hamano, E., Nagase, T., et al. (2011). CpG island methylation of microRNAs is associated with tumor size and recurrence of non-small-cell lung cancer. Cancer Sci. 102, 2126–2131. doi: 10.1126/science.1138050
Kondo, Y., Shen, L., Cheng, A. S., Ahmed, S., Boumber, Y., Charo, C., et al. (2008). Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 40, 741–750. doi: 10.1111/j.1349-7006.2011.02101.x
Kozaki, K., Imoto, I., Mogi, S., Omura, K., and Inazawa, J. (2008). Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 68, 2094–2105. doi: 10.1038/ng.159
Kumar, M. S., Lu, J., Mercer, K. L., Golub, T. R., and Jacks, T. (2007). Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 39, 673–677. doi: 10.1158/0008-5472.CAN-07-5194
Kusakabe, M., Kutomi, T., Watanabe, K., Emoto, N., Aki, N., Kage, H., et al. (2010). Identification of G0S2 as a gene frequently methylated in squamous lung cancer by combination of in silico and experimental approaches. Int. J. Cancer 126, 1895–1902. doi: 10.1038/ng2003
Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. (2001). Identification of novel genes coding for small expressed RNAs. Science 294, 853–858. doi: 10.1126/science.1064921
Lau, N. C., Lim, L. P., Weinstein, E. G., and Bartel, D. P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862. doi: 10.1126/science.1065062
Lee, R. C., and Ambros, V. (2001). An extensive class of small RNAs in Caenorhabditis elegans. Science 294, 862–864. doi: 10.1126/science.1065329
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Lee, Y., Kim, M., Han, J., Yeom, K. H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi: 10.1038/sj.emboj.7600385
Li, G., Zhao, J., Peng, X., Liang, J., Deng, X., and Chen, Y. (2012). The mechanism involved in the loss of PTEN expression in NSCLC tumor cells. Biochem. Biophys. Res. Commun. 418, 547–552. doi: 10.1016/j.bbrc.2012.01.065
Liu, S. V., Fabbri, M., Gitlitz, B. J., and Laird-Offringa, I. A. (2013). Epigenetic therapy in lung cancer. Front. Oncol. 3:135. doi: 10.3389/fonc.2013.00135
Liu, B., Peng, X. C., Zheng, X. L., Wang, J., and Qin, Y. W. (2009). MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 66, 169–175. doi: 10.1016/j.lungcan.2009.01.010
Lodygin, D., Tarasov, V., Epanchintsev, A., Berking, C., Knyazeva, T., Korner, H., et al. (2008). Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7, 2591–2600. doi: 10.4161/cc.7.16.6533
Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., et al. (2005). MicroRNA expression profiles classify human cancers. Nature 435, 834–838. doi: 10.1038/nature03702
Lujambio, A., Calin, G. A., Villanueva, A., Ropero, S., Sanchez-Cespedes, M., Blanco, D., et al. (2008). A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 105, 13556–13561. doi: 10.1073/pnas.0803055105
Lujambio, A., Ropero, S., Ballestar, E., Fraga, M. F., Cerrato, C., Setien, F., et al. (2007). Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 67, 1424–1429. doi: 10.1158/0008-5472.CAN-06-4218
Lund, E., Guttinger, S., Calado, A., Dahlberg, J. E., and Kutay, U. (2004). Nuclear export of microRNA precursors. Science 303, 95–98. doi: 10.1126/science.1090599
Maas, S., Patt, S., Schrey, M., and Rich, A. (2001). Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. U.S.A. 98, 14687–14692. doi: 10.1073/pnas.251531398
Mayr, C., and Bartel, D. P. (2009). Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. doi: 10.1016/j.cell.2009.06.016
Mayr, C., Hemann, M. T., and Bartel, D. P. (2007). Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315, 1576–1579. doi: 10.1126/science.1137999
Melo, S. A., Moutinho, C., Ropero, S., Calin, G. A., Rossi, S., Spizzo, R., et al. (2010). A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 18, 303–315. doi: 10.1016/j.ccr.2010.09.007
Melo, S. A., Ropero, S., Moutinho, C., Aaltonen, L. A., Yamamoto, H., Calin, G. A., et al. (2009). A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat. Genet. 41, 365–370. doi: 10.1038/ng.317
Merritt, W. M., Lin, Y. G., Han, L. Y., Kamat, A. A., Spannuth, W. A., Schmandt, R., et al. (2008). Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 359, 2641–2650. doi: 10.1056/NEJMoa0803785
Muniyappa, M. K., Dowling, P., Henry, M., Meleady, P., Doolan, P., Gammell, P., et al. (2009). MiRNA-29a regulates the expression of numerous proteins and reduces the invasiveness and proliferation of human carcinoma cell lines. Eur. J. Cancer 45, 3104–3118. doi: 10.1016/j.ejca.2009.09.014
Park, S. M., Gaur, A. B., Lengyel, E., and Peter, M. E. (2008). The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 22, 894–907. doi: 10.1101/gad.1640608
Pasquinelli, A. E., Reinhart, B. J., Slack, F., Martindale, M. Q., Kuroda, M. I., Maller, B., et al. (2000). Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408, 86–89. doi: 10.1038/35040556
Paul, M. S., and Bass, B. L. (1998). Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 17, 1120–1127. doi: 10.1093/emboj/17.4.1120
Reinhart, B. J., Slack, F. J., Basson, M., Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E., et al. (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906. doi: 10.1038/35002607
Rothschild, S. I. (2013). Epigenetic therapy in lung cancer – role of microRNAs. Front. Oncol. 3:158. doi: 10.3389/fonc.2013.00158
Rothschild, S. I., Tschan, M. P., Federzoni, E. A., Jaggi, R., Fey, M. F., Gugger, M., et al. (2012). MicroRNA-29b is involved in the Src-ID1 signaling pathway and is dysregulated in human lung adenocarcinoma. Oncogene 31, 4221–4232. doi: 10.1038/onc.2011.578
Saito, Y., Liang, G., Egger, G., Friedman, J. M., Chuang, J. C., Coetzee, G. A., et al. (2006). Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9, 435–443. doi: 10.1016/j.ccr.2006.04.020
Samakoglu, S., Deevi, D. S., Li, H., Wang, S., Murphy, M., Bao, C., et al. (2012). Preclinical rationale for combining an EGFR antibody with cisplatin/gemcitabine for the treatment of NSCLC. Cancer Genomics Proteomics 9, 77–92.
Sano, A., Kage, H., Sugimoto, K., Kitagawa, H., Aki, N., Goto, A., et al. (2007). A second-generation profiling system for quantitative methylation analysis of multiple gene promoters: application to lung cancer. Oncogene 26, 6518–6525. doi: 10.1038/sj.onc.1210483
Sasahira, T., Kurihara, M., Bhawal, U. K., Ueda, N., Shimomoto, T., Yamamoto, K., et al. (2012). Downregulation of miR-126 induces angiogenesis and lymphangiogenesis by activation of VEGF-A in oral cancer. Br. J. Cancer 107, 700–706. doi: 10.1038/bjc.2012.330
Shien, K., Toyooka, S., Yamamoto, H., Soh, J., Jida, M., Thu, K. L., et al. (2013). Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 73, 3051–3061. doi: 10.1158/0008-5472.CAN-12-4136
Takai, D., Yagi, Y., Wakazono, K., Ohishi, N., Morita, Y., Sugimura, T., et al. (2001). Silencing of HTR1B and reduced expression of EDN1 in human lung cancers, revealed by methylation-sensitive representational difference analysis. Oncogene 20, 7505–7513. doi: 10.1038/sj.onc.1204940
Takamizawa, J., Konishi, H., Yanagisawa, K., Tomida, S., Osada, H., Endoh, H., et al. (2004). Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 64, 3753–3756. doi: 10.1158/0008-5472.CAN-04-0637
Tanaka, N., Toyooka, S., Soh, J., Kubo, T., Yamamoto, H., Maki, Y., et al. (2012). Frequent methylation and oncogenic role of microRNA-34b/c in small-cell lung cancer. Lung Cancer 76, 32–38. doi: 10.1016/j.lungcan.2011.10.002
Tavazoie, S. F., Alarcon, C., Oskarsson, T., Padua, D., Wang, Q., Bos, P. D., et al. (2008). Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152. doi: 10.1038/nature06487
Tellez, C. S., Juri, D. E., Do, K., Bernauer, A. M., Thomas, C. L., Damiani, L. A., et al. (2011). EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 71, 3087–3097. doi: 10.1158/0008-5472.CAN-10-3035
Toyota, M., Suzuki, H., Sasaki, Y., Maruyama, R., Imai, K., Shinomura, Y., et al. (2008). Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 68, 4123–4132. doi: 10.1158/0008-5472.CAN-08-0325
Varambally, S., Cao, Q., Mani, R. S., Shankar, S., Wang, X., Ateeq, B., et al. (2008). Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695–1699. doi: 10.1126/science.1165395
Volinia, S., Calin, G. A., Liu, C. G., Ambs, S., Cimmino, A., Petrocca, F., et al. (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261. doi: 10.1073/pnas.0510565103
Vosa, U., Vooder, T., Kolde, R., Vilo, J., Metspalu, A., and Annilo, T. (2013). Meta-analysis of microRNA expression in lung cancer. Int. J. Cancer 132, 2884–2893. doi: 10.1002/ijc.27981
Wang, S., Aurora, A. B., Johnson, B. A., Qi, X., Mcanally, J., Hill, J. A., et al. (2008). The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15, 261–271. doi: 10.1016/j.devcel.2008.07.002
Wang, Z., Chen, Z., Gao, Y., Li, N., Li, B., Tan, F., et al. (2011). DNA hypermethylation of microRNA-34b/c has prognostic value for stage non-small cell lung cancer. Cancer Biol. Ther. 11, 490–496. doi: 10.4161/cbt.11.5.14550
Watanabe, K., Emoto, N., Hamano, E., Sunohara, M., Kawakami, M., Kage, H., et al. (2012). Genome structure-based screening identified epigenetically silenced microRNA associated with invasiveness in non-small-cell lung cancer. Int. J. Cancer 130, 2580–2590. doi: 10.1002/ijc.26254
Welch, C., Chen, Y., and Stallings, R. L. (2007). MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 26, 5017–5022. doi: 10.1038/sj.onc.1210293
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. doi: 10.1016/0092-8674(93)90530-4
Xi, S., Xu, H., Shan, J., Tao, Y., Hong, J. A., Inchauste, S., et al. (2013). Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J. Clin. Invest. 123, 1241–1261. doi: 10.1172/JCI61271
Xu, H., Cheung, I. Y., Guo, H. F., and Cheung, N. K. (2009). MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: potential implications for immune based therapy of human solid tumors. Cancer Res. 69, 6275–6281. doi: 10.1158/0008-5472.CAN-08-4517
Yanaihara, N., Caplen, N., Bowman, E., Seike, M., Kumamoto, K., Yi, M., et al. (2006). Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189–198. doi: 10.1016/j.ccr.2006.01.025
Yu, S. L., Chen, H. Y., Chang, G. C., Chen, C. Y., Chen, H. W., Singh, S., et al. (2008). MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 13, 48–57. doi: 10.1016/j.ccr.2007.12.008
Keywords: microRNA, lung cancer, DNA methylation, histone modification, RNA editing
Citation: Watanabe K and Takai D (2013) Disruption of the expression and function of microRNAs in lung cancer as a result of epigenetic changes. Front. Genet. 4:275. doi: 10.3389/fgene.2013.00275
Received: 18 September 2013; Accepted: 19 November 2013;
Published online: 03 December 2013.
Edited by:
Yoshimasa Saito, Keio University, JapanReviewed by:
Nejat Dalay, Istanbul University Oncology Institute, TurkeyAlexander Kouzmenko, Alfaisal University, Saudi Arabia
Copyright © 2013 Watanabe and Takai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daiya Takai, Department of Clinical Laboratory, The University of Tokyo Hospital, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan e-mail:ZHRha2FpLWluZEB1bWluLmFjLmpw