 Shawn M. Crump
Shawn M. Crump Geoffrey W. Abbott
Geoffrey W. Abbott- Bioelectricity Laboratory, Department of Pharmacology, Department of Physiology and Biophysics, School of Medicine, University of California, Irvine, CA, USA
There are twenty-five known inherited cardiac arrhythmia susceptibility genes, all of which encode either ion channel pore-forming subunits or proteins that regulate aspects of ion channel biology such as function, trafficking, and localization. The human KCNE gene family comprises five potassium channel regulatory subunits, sequence variants in each of which are associated with cardiac arrhythmias. KCNE gene products exhibit promiscuous partnering and in some cases ubiquitous expression, hampering efforts to unequivocally correlate each gene to specific native potassium currents. Likewise, deducing the molecular etiology of cardiac arrhythmias in individuals harboring rare KCNE gene variants, or more common KCNE polymorphisms, can be challenging. In this review we provide an update on putative arrhythmia-causing KCNE gene variants, and discuss current thinking and future challenges in the study of molecular mechanisms of KCNE-associated cardiac rhythm disturbances.
Introduction
A quarter of a century ago, Takumi and colleagues discovered a fraction of rat kidney mRNA that generated an unusual, slow-activating K+-selective current when injected into Xenopus laevis oocytes (Takumi et al., 1988). The protein product required for this slow current has been variously termed “minimal potassium channel” (MinK), “IsK” (for slow potassium current), and more recently KCNE1—the gene name KCNE1 now being most commonly also used when referring to the protein product, for simplicity. We now know that KCNE1 is the founding member of a five-strong family of single transmembrane domain potassium channel ancillary (β) subunits (Figures 1, 2) that do not form currents alone but are essential for generation of some native K+ currents by virtue of formation of heteromeric ion channel complexes with voltage-gated potassium (Kv) channel pore-forming α subunits (Abbott and Goldstein, 1998). Because KCNE1 was relatively quickly found to be a molecular correlate of the slowly activating ventricular myocyte K+ current, IKs (Freeman and Kass, 1993), study of the KCNE family as a whole has historically been focused primarily on the heart. This is especially true for the study of the role of KCNE gene variants in human disease.
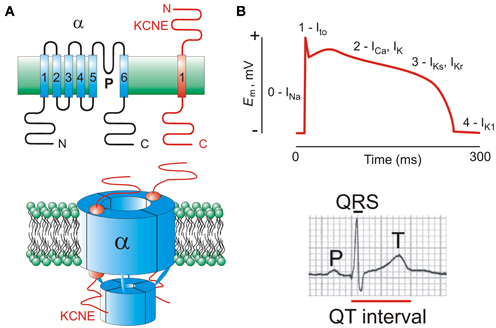
Figure 1. KCNE subunits and the ventricular myocyte action potential. (A) Upper, transmembrane topology of Kv α and KCNE subunits with transmembrane segments numbered; lower, one suggested stoichiometry of a KCNE-containing Kv channel complex. Extracellular side is uppermost in each case. (B) Upper, a ventricular action potential waveform indicating the major ionic currents that contribute to its morphology and duration; lower, a human surface ECG waveform showing the QT interval.

Figure 2. Human KCNE1-KCNE5 protein sequence alignments and gene variants. Image of aligned sequences generated using http://www.uniprot.org/align. Colors highlight inherited or sporadic non-synonymous mutations or polymorphisms resulting in single amino acid changes (changes involving >1 amino acid are omitted). In cases where an amino acid substitution is associated with LQTS in addition to another arrhythmia, only the latter is color-coded (see Table S1 for full information). The predicted transmembrane domain for each subunit is outlined in red.
Although in the new millennium the role of various KCNE subunits in epithelia has been extensively explored, this work has been largely conducted using mouse models (Arrighi et al., 2001; Dedek and Waldegger, 2001; Barriere et al., 2003; Rivas and Francis, 2005; Roepke et al., 2006, 2009, 2011a,b; Preston et al., 2010). The existing evidence from human genetics of the necessity for KCNE proteins in extracardiac tissue (Schulze-Bahr et al., 1997; Tyson et al., 1997) probably represents the tip of the iceberg in terms of the actual importance of human KCNE proteins to tissues outside the heart, including polarized epithelia. In contrast, more than sixty KCNE gene variants have been suggested to associate with human cardiac arrhythmias. In this mini-review we summarize current knowledge on arrhythmia-associated KCNE gene variants and discuss the difficulties in establishing causality and molecular etiology when dealing with rare diseases and promiscuous regulatory proteins.
Kv channels play a central role in active repolarization of all excitable cells, including cardiac myocytes. In human ventricles, three types of Kv channel in particular are important for timely myocyte repolarization, and also for the action potential morphology optimal for rhythmic contractions, incorporating a plateau phase followed by relatively steep phase 3 repolarization (Figure 1B). During an action potential, membrane depolarization primarily from Na+ influx through voltage-gated Na+ channels is counteracted by a transient outward K+ (Ito) current, producing the initial repolarization “notch.” Subsequently, slower delayed rectifier-generated outward K+ currents (IKr and IKs) counteract inward Ca2+ flux through voltage-gated Ca2+ channels, modulating the strength of contraction and duration of the action potential plateau (Sanguinetti and Jurkiewicz, 1990; Niwa and Nerbonne, 2010).
KCNE Regulation of hERG: the α Subunit Underlying Ventricular IKr
The “rapidly activating” K+ current (IKr) is the predominant human ventricular repolarization current under normal circumstances. IKr is generated by channels comprising a tetramer of hERG α subunits, encoded by the KCNH2 gene. KCNH2 gene mutations are (together with KCNQ1) one of the top two identified inherited causes of the cardiac arrhythmia Long QT Syndrome (LQTS), which results from delayed ventricular myocyte repolarization, manifests as a prolonged electrocardiogram QT interval (Figure 1B), and can cause lethal ventricular fibrillation (Curran et al., 1995; Sanguinetti et al., 1995). hERG channels exhibit unusual properties that influence both cardiac electrical function and arrhythmogenesis. First, the majority of pathologic KCNH2 gene mutations cause loss of function via protein maturation/trafficking defects rather than channel conduction or gating defects (Anderson et al., 2006). Second, upon membrane depolarization, hERG channels open and then rapidly inactivate. As the membrane begins to repolarize, hERG recovers rapidly from inactivation but deactivates slowly. This creates an atypical mode of inward rectification (classic inward rectifier K+ channels being generated instead by tetramers of two-transmembrane domain α subunits) (Smith et al., 1996). It ensures that hERG channels pass robust currents relatively late in the ventricular action potential, to speed phase 3 repolarization, without curtailing the preceding plateau phase. Third, hERG is highly susceptible to block by drugs from a wide range of chemical structures, making it the bane of pharmaceutical companies attempting to bring to market otherwise efficacious drugs that fail safety standards because they inhibit hERG and therefore are predicted (or demonstrated) to cause drug-induced LQTS (diLQTS) (Sanguinetti et al., 1995; Chen et al., 2002).
hERG channels are modulated by both KCNE1 and KCNE2 (originally named MinK-related Protein 1 or MiRP1) in vitro and potentially also in human heart (McDonald et al., 1997; Abbott et al., 1999). Currents generated by expression of hERG alone in heterologous expression systems recapitulate most of the functional properties of native IKr, including the unusual (for an S4 family α subunit) inward rectification and the exquisite drug sensitivity (Sanguinetti et al., 1995). However, KCNE1 forms heteromeric complexes with hERG and increases hERG currents by an as yet unknown mechanism when the two proteins are co-expressed in COS cells (McDonald et al., 1997). In addition, inherited KCNE1 mutants associated with human LQTS impair hERG function and/or trafficking (Table S1). These findings suggest that KCNE1 modulates hERG in vivo in at least some areas of the ventricle (Bianchi et al., 1999).
hERG also forms heteromeric complexes with KCNE2, when the two are co-expressed in Chinese Hamster Ovary (CHO) cells. When co-expressed in either Xenopus oocytes or CHO cells, KCNE2 alters hERG function, shifting the voltage dependence of activation, decreasing unitary conductance, and speeding deactivation. Importantly, inherited gene variants in human KCNE2 that are associated with LQTS impair hERG gating, which would be predicted to delay ventricular repolarization as is seen in LQTS (Abbott et al., 1999) (Table S1). More strikingly, KCNE2 gene variants associated with drug-induced LQTS (diLQTS) in some cases increase the sensitivity of hERG in vitro to block by the specific drug that precipitated the arrhythmic episode in vivo (Abbott et al., 1999; Sesti et al., 2000).
This is highly supportive of a role for KCNE2 in direct regulation of hERG channels in human ventricular myocardium. Indeed, KCNE2-rERG complexes have been isolated from rat heart and a plethora of other evidence suggests that KCNE2 regulates ERG in the hearts of several species (Jiang et al., 2004; McCrossan et al., 2009; Zhang et al., 2011). However, the debate that has surrounded the existence and necessity of hERG-KCNE2 complexes in human heart highlights the difficulties in nailing down the molecular correlates of multi-subunit channels. This problem is exacerbated when considering human heart, which compared to animal studies, involves more restrictions in tissue availability and less practicable experimental options. Temporal dynamism and spatial diversity in the makeup of these complexes (as almost certainly occurs with KCNE-containing channels) also stymies this research, as does the fact (as in the case of KCNE2-hERG) that the effects of KCNE2 on hERG are relatively subtle and may be expression system-dependent. Furthermore, when disease associations are relatively rare and the subunits involved exhibit promiscuous partnering, even human genetics does not automatically uncover the precise functional role of a regulatory subunit. Mouse models have been useful in discovering physiological and arrhythmogenic roles for KCNE subunits and their disruption (Temple et al., 2005; Roepke et al., 2008; Hu et al., 2013) but come with the caveat, especially for the heart, that there is a big divide between mouse and human heart in terms of physiology, the primary repolarizing currents, and their molecular underpinnings (Nerbonne et al., 2001).
KCNE Modulation of KCNQ1: the α Subunit Underlying Human Ventricular IKs
KCNQ1 is the pore-forming subunit of cardiac IKs, a slow-activating K+ current that, in human ventricles, may act primarily as a back-up for IKr when the latter is diminished by e.g., drug block, mutation, or during periods of increased heart-rate (Barhanin et al., 1996; Sanguinetti et al., 1996; Silva and Rudy, 2005). KCNQ1 is the endogenous Xenopus laevis oocyte α subunit that was awakened by injected KCNE1 in Takumi's original discovery of the KCNE family. It took a further 8 years before the cloning of human and Xenopus KCNQ1 (then termed KvLQT1) was reported (Barhanin et al., 1996; Sanguinetti et al., 1996), KCNQ1 was linked to LQTS (Wang et al., 1996), and the KCNE1 functionality and IKs molecular correlate conundrums solved.
Or were they? We know for certain that ventricular KCNQ1-KCNE1 complexes exist in the hearts of some large mammals, and almost certainly contribute to human ventricular myocyte repolarization. Loss-of-function gene variants in either gene reduce repolarizing force and delay ventricular repolarization, causing LQTS, particularly manifest (for KCNQ1 mutants) during periods of sympathetic stimulation such as while swimming (Wang et al., 1996; Tyson et al., 1997; Ackerman et al., 1999).
However, it is also highly likely that other KCNQ1-KCNE channels help to repolarize some ventricular myocytes in human heart, and that KCNE1 regulates other Kv α subunits in human heart as well. This means that KCNE1-associated LQTS (termed LQT5) could be much more complicated than just disruption of ventricular KCNQ1-KCNE1. Another fly in the ointment for those wishing to rationalize the genetics of ventricular arrhythmias is that KCNQ1-hERG complexes almost certainly exist in human heart and loss-of-function variants in either subunit may affect the function of the other (Ehrlich et al., 2004; Ehrlich, 2010; Ren et al., 2010). Add to this the notion that different KCNEs may participate in the same multi-subunit channel complex with KCNQ1 (Wu et al., 2006) and the riddle that is the molecular etiology of KCNE-associated arrhythmogenesis becomes ever more enigmatic. KCNQ1 can be modulated by all five known human KCNEs, with diverse functional outcomes (McCrossan and Abbott, 2004). The stoichiometry of KCNQ1-KCNE complexes is still under debate, but there are almost certainly four KCNQ1 α subunits in complex with 2–4 KCNE1 subunits (and possibly a variable number of KCNE1 subunits within these limits, depending upon expression levels) (Chen et al., 2003; Nakajo et al., 2010; Yu et al., 2013).
KCNE1 slows KCNQ1 activation 5–10-fold, eliminates its inactivation, increases unitary conductance and tweaks ion selectivity and pharmacology (Sesti and Goldstein, 1998). Strikingly, KCNE1 also strongly modulates KCNQ1 affinity for PIP2 (an important regulatory factor) (Loussouarn et al., 2003), and KCNE1 mediates protein kinase C-stimulated clathrin-mediated endocytosis of KCNQ1-KCNE1 (Kanda et al., 2011). KCNE1 also regulates KCNQ1 in the inner ear, which is why some individuals harboring loss-of-function mutants in KCNQ1 or KCNE1 in both alleles succumb to Jervell and Lange-Nielsen Syndrome (JLNS), comprising both LQTS and sensorineural deafness. Rather than mediating repolarization, in the inner ear KCNQ1-KCNE1 probably serves primarily to maintain a K+-rich environment in the endolymph (Wangemann et al., 1995; Vetter et al., 1996; Wangemann et al., 1996).
KCNE2 performs electrical alchemy with KCNQ1, converting it into a constitutively active channel that has lost much of its voltage dependence (Tinel et al., 2000). KCNQ1-KCNE2 is especially important in various polarized epithelia (Abbott, 2012) but could also be present in human heart. Some atrial fibrillation (AF)-associated KCNQ1 and KCNE2 human gene variants augment KCNQ1-KCNE2 currents (which are typically much smaller in terms of outward current even than KCNQ1 alone) and would therefore be predicted to shorten the atrial action potential, thought to predispose to AF (Yang et al., 2004) (Table S1). KCNE3, one variant of which also associates with AF (Zhang et al., 2005) has broadly similar effects on KCNQ1, but the resultant currents are much larger than those of KCNQ1-KCNE2 and important in different epithelial cells to those of KCNQ1-KCNE2 (Schroeder et al., 2000). Either heteromer type could contribute to background K+ currents in human cardiomyocytes but this has yet to be established. Similarly, KCNE2 and KCNE3 could each contribute to tripartite complexes with KCNQ1 and KCNE1, or to variegated macromolecular assemblies also including hERG, with potentially complex and dynamic functional attributes. Understanding the expression and regulation of such complexes in vivo is challenging but could lead to development of anti-arrhythmic drugs with, for example, improved spatial selectivity (Yu et al., 2013).
KCNE4 inhibits KCNQ1 and may potentially serve this function in vivo in human heart (Grunnet et al., 2002, 2005). KCNQ1-KCNE5 complexes generate currents superficially similar to those of KCNQ1-KCNE1, but with much more positive activation voltages (Angelo et al., 2002), suggesting either an inhibitory role for KCNE5 (originally termed KCNE1L), or perhaps an as-yet not understood role in more populous complexes with other KCNEs. KCNE4 and KCNE5 gene variants that dampened their inhibitory effects on KCNQ1 (gain-of-function mutants) also associate with AF (Ravn et al., 2008; Ohno et al., 2011) (Table S1).
KCNE Modulation of the Kv4 α Subunits Underlying Human Ventricular Ito
Human ventricular cardiomyocyte action potentials exhibit a sharp peak and a notch, as depolarization primarily by Na+ influx is curtailed abruptly by K+ exiting through rapidly activating Kv channels. The channels whose primary task is to stem phase 0 depolarization are, in human heart, the Kv4.2 and Kv4.3 channels, via generation of the transient outward Kv current (Ito). This current is transient because both channel types also inactivate rapidly. KCNE subunits regulate Kv4.2 and Kv4.3 when co-expressed in vitro, and it is thought that this type of regulation also occurs in human heart (Zhang et al., 2001; Niwa and Nerbonne, 2010).
Kv4 channels are also regulated by the cytoplasmic KChIP2 subunit in vivo, and KCNEs can regulate Kv4-KChIP2 complexes. KCNE1 and KCNE3-5 subunits each accelerate Kv4.3-KChIP2 inactivation, while KCNE2 slows inactivation and induces an overshoot of inactivation recovery in vitro in CHO cells, similar to that observed in human heart for Ito. KCNE2 augments Kv4.2 current by mechanisms including slowing of inactivation (Radicke et al., 2006); although also slowing Kv4.3 inactivation, KCNE2 reduces its peak current in vitro (Liu et al., 2006; Wu et al., 2010). KCNE subunits also modulate Kv4 channel pharmacology and temperature sensitivity (Radicke et al., 2008, 2009, 2013).
Inherited loss-of-function sequence variants in the cardiac Nav1.5 channel α subunit gene (SCN5A) are the most common identified genetic cause of Brugada Syndrome (BrS), a lethal ventricular arrhythmia (Brugada et al., 2006). More recently, BrS-associated KCNE gene variants have emerged that augment Kv4 currents, mimicking SCN5A loss of function (Delpon et al., 2008; Ohno et al., 2011; Nakajima et al., 2012) (Table S1). These are rare, and like the majority of KCNE arrhythmia-associated gene variants, are not backed by familial analyses or the statistical confidence that comes with prevalence in large cohorts. However, they have been given some credence in the field, largely because their effects in vitro are consistent with what would be predicted from work with SCN5A (which is strongly statistically linked to BrS), and they occur in BrS patients lacking SCN5A gene mutations.
Arrhythmogenic KCNE Gene Variants: Two Decades of Discovery
The first KCNE gene variant identified in the human population was the KCNE1 S38G polymorphism (Lai et al., 1994). The majority of people harbor one S and one G allele, with the 38GG genotype being next most common, and the 38SS being the least common (~10%, depending on the population studied). S38G genotype reportedly influences predisposition to both AF and LQTS (Fatini et al., 2006; Prystupa et al., 2006; Xu et al., 2008; Husser et al., 2009), and heart failure (Fatini et al., 2010), depending on factors including sex, age, BMI, diabetes, fibrinogen, hypercholesterolemia, hypertension, and another KCNE1 SNP (Friedlander et al., 2005).
The first KCNE gene variant to be identified as pathologic was D76N, a dominant-negative mutation that causes JLNS (Schulze-Bahr et al., 1997) and impairs KCNQ1-KCNE1 current by a combination of reduced unitary conductance and impaired gating. This was closely followed by discovery of S74L, which like D76N shifts the voltage dependence of activation and accelerates KCNQ1-KCNE1 channel closing (Splawski et al., 1997; Sesti and Goldstein, 1998).
The next KCNE gene to be associated with cardiac arrhythmia was KCNE2, with discovery of the rare M54T, I57T, A116V loss-of-function mutants, and the T8A and Q9E polymorphisms (Abbott et al., 1999; Sesti et al., 2000). T8A, harbored by >1% of US Caucasians, is pathogenic only in combination with drug interaction, as it has no apparent effects without drug but results in loss of a glycosylation site that shields hERG-KCNE2 channels from block by sulfamethoxazole (Sesti et al., 2000; Park et al., 2003). Q9E, represented in 1–2% of African Americans, impairs channel function slightly without drug, and also increases sensitivity of hERG-KCNE2 to block by macrolide antibiotics (Abbott et al., 1999; Ackerman et al., 2003).
Following these findings and the cloning of KCNE3-5, gene variants in all five KCNE genes have been associated with LQTS, BrS, and/or AF (Table S1). KCNE3 variants are implicated in AF, BrS6, and LQTS (Zhang et al., 2005; Delpon et al., 2008; Ohno et al., 2009). KCNE4 E45D augments KCNQ1-KCNE4 current and was discovered in a Chinese patient with AF (Zeng et al., 2007). KCNE5 gene variants include AF-associated L65F and BrS/idiopathic ventricular fibrillation-associated Y81H and D92E/E93X (Ohno et al., 2011). In contrast, the KCNE5-C97T polymorphism may be protective against AF (Ravn et al., 2005).
Reported KCNE sequence variants in arrhythmias offer as yet scant information on which to make concrete links between types/positions of sequence variants and classes of arrhythmia. However, some patterns emerge when contemplating the 49 variants that fall into the category of point mutants causing single amino acid changes (Figure 2), of the 59 reported KCNE variants for which we consider sufficient evidence is available that they at least be seriously considered as having a pathogenic role in the heart (Table S1). These are variants reported absent in control patients sequenced in the same study, and/or those for which cellular electrophysiology is consistent with disease association. For KCNE1 and KCNE2, pathogenic mutations in the transmembrane domain occur with a periodicity suggestive of the known α-helicity of this region, perhaps indicating disruption of the face most important to α subunit gating. BrS-associated KCNE variants cluster on the intracellular side; AF-associated variants in KCNE1-3 cluster in the extracellular region, whereas the opposite is true for KCNE4 and 5 (Figure 2).
To conclude, KCNE proteins are essential for normal cardiac function, and human genetics studies are essential in our understanding of this. However, human KCNE gene variants are mostly quite rare and typically lacking familial linkage, therefore some of the listed variants come into the category of genetic noise—variants that happen to be found in patients but do not contribute to the arrhythmia (Kapplinger et al., 2013). Also, in addition to their partnering promiscuity, all KCNE genes are expressed in multiple extracardiac tissues (McCrossan and Abbott, 2004). Therefore, KCNE sequence variants may manifest in multiple tissues, and these pathologies could indirectly impact cardiac function both electrically and structurally, further complicating our efforts to comprehend KCNE-related cardiac diseases. Comparatively little is known of the extracardiac effects of human KCNE gene disruption: the KCNE1-linked inner ear defect in JLNS, and genome-wide association studies showing statistical linkage to early-onset myocardial infarction and reduced lung capacity (Kathiresan et al., 2009; Soler Artigas et al., 2011). In contrast, mouse Kcne genes are known to be important in, e.g., the kidneys, adrenals, stomach, colon, airways, and thyroid (Arrighi et al., 2001; Dedek and Waldegger, 2001; Barriere et al., 2003; Rivas and Francis, 2005; Roepke et al., 2006, 2009, 2011a,b; Preston et al., 2010), and in some cases dysfunction of these tissues has been demonstrated to negatively impact the heart (Roepke et al., 2009; Hu et al., 2013, 2014). Kcne knockout mouse studies are providing invaluable inroads into the maze of KCNE physiology and disease, and constitute a substrate for future human genetics studies necessary for extrapolation of mouse data to human systems.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for financial support from the US National Institutes of Health (HL079275 to Geoffrey W. Abbott).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2014.00003/abstract
Abbreviations
AF, atrial fibrillation; BrS, Brugada syndrome; CHO, Chinese hamster ovary; diLQTS, drug-induced Long QT Syndrome; IKs, cardiac-delayed rectifier-like K+ current; JLNS, Jervell and Lange-Nielsen syndrome.
References
Abbott, G. W. (2012). KCNE2 and the K (+) channel: the tail wagging the dog. Channels (Austin) 6, 1–10. doi: 10.4161/chan.19126
Abbott, G. W., and Goldstein, S. A. (1998). A superfamily of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs). Q. Rev. Biophys. 31, 357–398. doi: 10.1017/S0033583599003467
Abbott, G. W., Sesti, F., Splawski, I., Buck, M. E., Lehmann, M. H., Timothy, K. W., et al. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187. doi: 10.1016/S0092-8674(00)80728-X
Ackerman, M. J., Tester, D. J., Jones, G. S., Will, M. L., Burrow, C. R., and Curran, M. E. (2003). Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin. Proc. 78, 1479–1487. doi: 10.4065/78.12.1479
Ackerman, M. J., Tester, D. J., and Porter, C. J. (1999). Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin. Proc. 74, 1088–1094. doi: 10.4065/74.11.1088
Anderson, C. L., Delisle, B. P., Anson, B. D., Kilby, J. A., Will, M. L., Tester, D. J., et al. (2006). Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113, 365–373. doi: 10.1161/CIRCULATIONAHA.105.570200
Angelo, K., Jespersen, T., Grunnet, M., Nielsen, M. S., Klaerke, D. A., and Olesen, S. P. (2002). KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys. J. 83, 1997–2006. doi: 10.1016/S0006-3495(02)73961-1
Arrighi, I., Bloch-Faure, M., Grahammer, F., Bleich, M., Warth, R., Mengual, R., et al. (2001). Altered potassium balance and aldosterone secretion in a mouse model of human congenital long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 98, 8792–8797. doi: 10.1073/pnas.141233398
Barhanin, J., Lesage, F., Guillemare, E., Fink, M., Lazdunski, M., and Romey, G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80. doi: 10.1038/384078a0
Barriere, H., Rubera, I., Belfodil, R., Tauc, M., Tonnerieux, N., Poujeol, C., et al. (2003). Swelling-activated chloride and potassium conductance in primary cultures of mouse proximal tubules. Implication of KCNE1 protein. J. Membr. Biol. 193, 153–170. doi: 10.1007/s00232-003-2014-z
Bianchi, L., Shen, Z., Dennis, A. T., Priori, S. G., Napolitano, C., Ronchetti, E., et al. (1999). Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 8, 1499–1507. doi: 10.1093/hmg/8.8.1499
Brugada, R., Brugada, P., and Brugada, J. (2006). Electrocardiogram interpretation and class I blocker challenge in Brugada syndrome. J. Electrocardiol. 39(4 Suppl.), S115–S118. doi: 10.1016/j.jelectrocard.2006.05.014
Chen, H., Kim, L. A., Rajan, S., Xu, S., and Goldstein, S. A. (2003). Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron 40, 15–23. doi: 10.1016/S0896-6273(03)00570-1
Chen, J., Seebohm, G., and Sanguinetti, M. C. (2002). Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. U.S.A. 99, 12461–12466. doi: 10.1073/pnas.192367299
Curran, M. E., Splawski, I., Timothy, K. W., Vincent, G. M., Green, E. D., and Keating, M. T. (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803. doi: 10.1016/0092-8674(95)90358-5
Dedek, K., and Waldegger, S. (2001). Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers Arch. 442, 896–902. doi: 10.1007/s004240100609
Delpon, E., Cordeiro, J. M., Nunez, L., Thomsen, P. E., Guerchicoff, A., Pollevick, G. D., et al. (2008). Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218. doi: 10.1161/CIRCEP.107.748103
Ehrlich, J. R. (2010). Cardiac delayed rectifiers–together as one? A patho-physiologically relevant interaction between IKr and IKs. Heart Rhythm 7, 981–982. doi: 10.1016/j.hrthm.2010.04.013
Ehrlich, J. R., Pourrier, M., Weerapura, M., Ethier, N., Marmabachi, A. M., Hebert, T. E., et al. (2004). KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha-subunit interaction between delayed rectifier currents. J. Biol. Chem. 279, 1233–1241. doi: 10.1074/jbc.M309087200
Fatini, C., Sticchi, E., Genuardi, M., Sofi, F., Gensini, F., Gori, A. M., et al. (2006). Analysis of minK and eNOS genes as candidate loci for predisposition to non-valvular atrial fibrillation. Eur. Heart J. 27, 1712–1718. doi: 10.1093/eurheartj/ehl087
Fatini, C., Sticchi, E., Marcucci, R., Verdiani, V., Nozzoli, C., Vassallo, C., et al. (2010). S38G single-nucleotide polymorphism at the KCNE1 locus is associated with heart failure. Heart Rhythm 7, 363–367. doi: 10.1016/j.hrthm.2009.11.032
Freeman, L. C., and Kass, R. S. (1993). Expression of a minimal K+ channel protein in mammalian cells and immunolocalization in guinea pig heart. Circ. Res. 73, 968–973. doi: 10.1161/01.RES.73.5.968
Friedlander, Y., Vatta, M., Sotoodehnia, N., Sinnreich, R., Li, H., Manor, O., et al. (2005). Possible association of the human KCNE1 (minK) gene and QT interval in healthy subjects: evidence from association and linkage analyses in Israeli families. Ann. Hum. Genet. 69(Pt 6), 645–656. doi: 10.1046/j.1529-8817.2005.00182.x
Grunnet, M., Jespersen, T., Rasmussen, H. B., Ljungstrom, T., Jorgensen, N. K., Olesen, S. P., et al. (2002). KCNE4 is an inhibitory subunit to the KCNQ1 channel. J. Physiol. 542(Pt 1), 119–130. doi: 10.1113/jphysiol.2002.017301
Grunnet, M., Olesen, S. P., Klaerke, D. A., and Jespersen, T. (2005). hKCNE4 inhibits the hKCNQ1 potassium current without affecting the activation kinetics. Biochem. Biophys. Res. Commun. 328, 1146–1153. doi: 10.1016/j.bbrc.2005.01.071
Hu, Z., Crump, S. M., Anand, M., Kant, R., Levi, R., and Abbott, G. W. (2013). Kcne3 deletion initiates extracardiac arrhythmogenesis in mice. FASEB J. doi: 10.1096/fj.13-241828. [Epub ahead of print].
Hu, Z., Kant, R., Anand, M., King, E. C., Krogh-Madsen, T., Christini, D. J., et al. (2014). Kcne2 Deletion Creates a Multisystem Syndrome Predisposing to Sudden Cardiac Death. Circ. Cardiovasc. Genet. doi: 10.1161/CIRCGENETICS.113.000315. [Epub ahead of print].
Husser, D., Stridh, M., Sornmo, L., Roden, D. M., Darbar, D., and Bollmann, A. (2009). A genotype-dependent intermediate ECG phenotype in patients with persistent lone atrial fibrillation genotype ECG-phenotype correlation in atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 24–28. doi: 10.1161/CIRCEP.108.799098
Jiang, M., Zhang, M., Tang, D. G., Clemo, H. F., Liu, J., Holwitt, D., et al. (2004). KCNE2 protein is expressed in ventricles of different species, and changes in its expression contribute to electrical remodeling in diseased hearts. Circulation 109, 1783–1788. doi: 10.1161/01.CIR.0000124225.43852.50
Kanda, V. A., Purtell, K., and Abbott, G. W. (2011). Protein kinase C downregulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm 8, 1641–1647. doi: 10.1016/j.hrthm.2011.04.034
Kapplinger, J., Tester, D. J., and Ackerman, M. J. (2013). The compounding dilemma of genetic noise: identification of the genetic noise rate in genes associated with cardiac channelopathies. Heart Rhythm 10, 1752–1753. doi: 10.1016/j.hrthm.2013.09.047
Kathiresan, S., Voight, B. F., Purcell, S., Musunuru, K., Ardissino, D., Mannucci, P. M., et al. (2009). Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 41, 334–341. doi: 10.1038/ng.327
Lai, L. P., Deng, C. L., Moss, A. J., Kass, R. S., and Liang, C. S. (1994). Polymorphism of the gene encoding a human minimal potassium ion channel (minK). Gene 151, 339–340. doi: 10.1016/0378-1119(94)90685-8
Liu, J., Deng, J. X., Pan, B. X., and Huang, Q. B. (2006). [KCNE2 modulates the function of Kv4.3 channel]. Nan Fang Yi Ke Da Xue Xue Bao 26, 1754–1756.
Loussouarn, G., Park, K. H., Bellocq, C., Baro, I., Charpentier, F., and Escande, D. (2003). Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 22, 5412–5421. doi: 10.1093/emboj/cdg526
McCrossan, Z. A., and Abbott, G. W. (2004). The MinK-related peptides. Neuropharmacology 47, 787–821. doi: 10.1016/j.neuropharm.2004.06.018
McCrossan, Z. A., Roepke, T. K., Lewis, A., Panaghie, G., and Abbott, G. W. (2009). Regulation of the Kv2.1 potassium channel by MinK and MiRP1. J. Membr. Biol. 228, 1–14. doi: 10.1007/s00232-009-9154-8
McDonald, T. V., Yu, Z., Ming, Z., Palma, E., Meyers, M. B., Wang, K. W., et al. (1997). A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 388, 289–292. doi: 10.1038/40882
Nakajima, T., Wu, J., Kaneko, Y., Ashihara, T., Ohno, S., Irie, T., et al. (2012). KCNE3 T4A as the genetic basis of Brugada-pattern electrocardiogram. Circ. J. 76, 2763–2772. doi: 10.1253/circj.CJ-12-0551
Nakajo, K., Ulbrich, M. H., Kubo, Y., and Isacoff, E. Y. (2010). Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc. Natl. Acad. Sci. U.S.A. 107, 18862–18867. doi: 10.1073/pnas.1010354107
Nerbonne, J. M., Nichols, C. G., Schwarz, T. L., and Escande, D. (2001). Genetic manipulation of cardiac K(+) channel function in mice: what have we learned, and where do we go from here? Circ. Res. 89, 944–956. doi: 10.1161/hh2301.100349
Niwa, N., and Nerbonne, J. M. (2010). Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J. Mol. Cell. Cardiol. 48, 12–25. doi: 10.1016/j.yjmcc.2009.07.013
Ohno, S., Toyoda, F., Zankov, D. P., Yoshida, H., Makiyama, T., Tsuji, K., et al. (2009). Novel KCNE3 mutation reduces repolarizing potassium current and associated with long QT syndrome. Hum. Mutat. 30, 557–563. doi: 10.1002/humu.20834
Ohno, S., Zankov, D. P., Ding, W. G., Itoh, H., Makiyama, T., Doi, T., et al. (2011). KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 4, 352–361. doi: 10.1161/CIRCEP.110.959619
Park, K. H., Kwok, S. M., Sharon, C., Baerga, R., and Sesti, F. (2003). N-Glycosylation-dependent block is a novel mechanism for drug-induced cardiac arrhythmia. FASEB J. 17, 2308–2309. doi: 10.1096/fj.03-0577fje
Preston, P., Wartosch, L., Gunzel, D., Fromm, M., Kongsuphol, P., Ousingsawat, J., et al. (2010). Disruption of the K+ channel beta-subunit KCNE3 reveals an important role in intestinal and tracheal Cl- transport. J. Biol. Chem. 285, 7165–7175. doi: 10.1074/jbc.M109.047829
Prystupa, A., Dzida, G., Myslinski, W., Malaj, G., and Lorenc, T. (2006). MinK gene polymorphism in the pathogenesis of lone atrial fibrillation. Kardiol. Pol. 64, 1205–1211; discussion 1212–1203.
Radicke, S., Cotella, D., Graf, E. M., Banse, U., Jost, N., Varro, A., et al. (2006). Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc. Res. 71, 695–703. doi: 10.1016/j.cardiores.2006.06.017
Radicke, S., Cotella, D., Sblattero, D., Ravens, U., Santoro, C., and Wettwer, E. (2009). The transmembrane beta-subunits KCNE1, KCNE2, and DPP6 modify pharmacological effects of the antiarrhythmic agent tedisamil on the transient outward current Ito. Naunyn Schmiedebergs Arch. Pharmacol. 379, 617–626. doi: 10.1007/s00210-008-0389-1
Radicke, S., Riedel, T., Cotella, D., Turnow, K., Ravens, U., Schaefer, M., et al. (2013). Accessory subunits alter the temperature sensitivity of Kv4.3 channel complexes. J. Mol. Cell. Cardiol. 56, 8–18. doi: 10.1016/j.yjmcc.2012.12.017
Radicke, S., Vaquero, M., Caballero, R., Gomez, R., Nunez, L., Tamargo, J., et al. (2008). Effects of MiRP1 and DPP6 beta-subunits on the blockade induced by flecainide of Kv4.3/KChIP2 channels. Br. J. Pharmacol. 154, 774–786. doi: 10.1038/bjp.2008.134
Ravn, L. S., Aizawa, Y., Pollevick, G. D., Hofman-Bang, J., Cordeiro, J. M., Dixen, U., et al. (2008). Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm 5, 427–435. doi: 10.1016/j.hrthm.2007.12.019
Ravn, L. S., Hofman-Bang, J., Dixen, U., Larsen, S. O., Jensen, G., Haunso, S., et al. (2005). Relation of 97T polymorphism in KCNE5 to risk of atrial fibrillation. Am. J. Cardiol. 96, 405–407. doi: 10.1016/j.amjcard.2005.03.086
Ren, X. Q., Liu, G. X., Organ-Darling, L. E., Zheng, R., Roder, K., Jindal, H. K., et al. (2010). Pore mutants of HERG and KvLQT1 downregulate the reciprocal currents in stable cell lines. Am. J. Physiol. Heart Circ. Physiol. 299, H1525–H1534. doi: 10.1152/ajpheart.00479.2009
Rivas, A., and Francis, H. W. (2005). Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: a model of Jervell and Lange-Nielsen syndrome. Otol. Neurotol. 26, 415–424. doi: 10.1097/01.mao.0000169764.00798.84
Roepke, T. K., Anantharam, A., Kirchhoff, P., Busque, S. M., Young, J. B., Geibel, J. P., et al. (2006). The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J. Biol. Chem. 281, 23740–23747. doi: 10.1074/jbc.M604155200
Roepke, T. K., Kanda, V. A., Purtell, K., King, E. C., Lerner, D. J., and Abbott, G. W. (2011a). KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J. 25, 4264–4273. doi: 10.1096/fj.11-187609
Roepke, T. K., King, E. C., Purtell, K., Kanda, V. A., Lerner, D. J., and Abbott, G. W. (2011b). Genetic dissection reveals unexpected influence of beta subunits on KCNQ1 K+ channel polarized trafficking in vivo. FASEB J. 25, 727–736. doi: 10.1096/fj.10-173682
Roepke, T. K., King, E. C., Reyna-Neyra, A., Paroder, M., Purtell, K., Koba, W., et al. (2009). Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat. Med. 15, 1186–1194. doi: 10.1038/nm.2029
Roepke, T. K., Kontogeorgis, A., Ovanez, C., Xu, X., Young, J. B., Purtell, K., et al. (2008). Targeted deletion of kcne2 impairs ventricular repolarization via disruption of I(K,slow1) and I(to,f). FASEB J. 22, 3648–3660. doi: 10.1096/fj.08-110171
Sanguinetti, M. C., Curran, M. E., Zou, A., Shen, J., Spector, P. S., Atkinson, D. L., et al. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83. doi: 10.1038/384080a0
Sanguinetti, M. C., Jiang, C., Curran, M. E., and Keating, M. T. (1995). A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307. doi: 10.1016/0092-8674(95)90340-2
Sanguinetti, M. C., and Jurkiewicz, N. K. (1990). Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96, 195–215. doi: 10.1085/jgp.96.1.195
Schroeder, B. C., Waldegger, S., Fehr, S., Bleich, M., Warth, R., Greger, R., et al. (2000). A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199. doi: 10.1038/35003200
Schulze-Bahr, E., Wang, Q., Wedekind, H., Haverkamp, W., Chen, Q., Sun, Y., et al. (1997). KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat. Genet. 17, 267–268. doi: 10.1038/ng1197-267
Sesti, F., Abbott, G. W., Wei, J., Murray, K. T., Saksena, S., Schwartz, P. J., et al. (2000). A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 97, 10613–10618. doi: 10.1073/pnas.180223197
Sesti, F., and Goldstein, S. A. (1998). Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112, 651–663. doi: 10.1085/jgp.112.6.651
Silva, J., and Rudy, Y. (2005). Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation 112, 1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306
Smith, P. L., Baukrowitz, T., and Yellen, G. (1996). The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379, 833–836. doi: 10.1038/379833a0
Soler Artigas, M., Loth, D. W., Wain, L. V., Gharib, S. A., Obeidat, M., Tang, W., et al. (2011). Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet. 43, 1082–1090. doi: 10.1038/ng.941
Splawski, I., Tristani-Firouzi, M., Lehmann, M. H., Sanguinetti, M. C., and Keating, M. T. (1997). Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 17, 338–340. doi: 10.1038/ng1197-338
Takumi, T., Ohkubo, H., and Nakanishi, S. (1988). Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science 242, 1042–1045. doi: 10.1126/science.3194754
Temple, J., Frias, P., Rottman, J., Yang, T., Wu, Y., Verheijck, E. E., et al. (2005). Atrial fibrillation in KCNE1-null mice. Circ. Res. 97, 62–69. doi: 10.1161/01.RES.0000173047.42236.88
Tinel, N., Diochot, S., Borsotto, M., Lazdunski, M., and Barhanin, J. (2000). KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19, 6326–6330. doi: 10.1093/emboj/19.23.6326
Tyson, J., Tranebjaerg, L., Bellman, S., Wren, C., Taylor, J. F., Bathen, J., et al. (1997). IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum. Mol. Genet. 6, 2179–2185. doi: 10.1093/hmg/6.12.2179
Vetter, D. E., Mann, J. R., Wangemann, P., Liu, J., McLaughlin, K. J., Lesage, F., et al. (1996). Inner ear defects induced by null mutation of the isk gene. Neuron 17, 1251–1264. doi: 10.1016/S0896-6273(00)80255-X
Wang, Q., Curran, M. E., Splawski, I., Burn, T. C., Millholland, J. M., VanRaay, T. J., et al. (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23. doi: 10.1038/ng0196-17
Wangemann, P., Liu, J., Shen, Z., Shipley, A., and Marcus, D. C. (1995). Hypo-osmotic challenge stimulates transepithelial K+ secretion and activates apical IsK channel in vestibular dark cells. J. Membr. Biol. 147, 263–273. doi: 10.1007/BF00234524
Wangemann, P., Shen, Z., and Liu, J. (1996). K(+)-induced stimulation of K+ secretion involves activation of the IsK channel in vestibular dark cells. Hear. Res. 100, 201–210. doi: 10.1016/0378-5955(96)00127-X
Wu, D. M., Jiang, M., Zhang, M., Liu, X. S., Korolkova, Y. V., and Tseng, G. N. (2006). KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of I(Ks) current amplitude in the heart. Heart Rhythm 3, 1469–1480. doi: 10.1016/j.hrthm.2006.08.019
Wu, J., Shimizu, W., Ding, W. G., Ohno, S., Toyoda, F., Itoh, H., et al. (2010). KCNE2 modulation of Kv4.3 current and its potential role in fatal rhythm disorders. Heart Rhythm 7, 199–205. doi: 10.1016/j.hrthm.2009.10.012
Xu, L. X., Yang, W. Y., Zhang, H. Q., Tao, Z. H., and Duan, C. C. (2008). [Study on the correlation between CETP TaqIB, KCNE1 S38G and eNOS T-786C gene polymorphisms for predisposition and non-valvular atrial fibrillation]. Zhonghua Liu Xing Bing Xue Za Zhi 29, 486–492.
Yang, Y., Xia, M., Jin, Q., Bendahhou, S., Shi, J., Chen, Y., et al. (2004). Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 75, 899–905. doi: 10.1086/425342
Yu, H., Lin, Z., Mattmann, M. E., Zou, B., Terrenoire, C., Zhang, H., et al. (2013). Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc. Natl. Acad. Sci. U.S.A. 110, 8732–8737. doi: 10.1073/pnas.1300684110
Zeng, Z., Tan, C., Teng, S., Chen, J., Su, S., Zhou, X., et al. (2007). The single nucleotide polymorphisms of I(Ks) potassium channel genes and their association with atrial fibrillation in a Chinese population. Cardiology 108, 97–103. doi: 10.1159/000095943
Zhang, D. F., Liang, B., Lin, J., Liu, B., Zhou, Q. S., and Yang, Y. Q. (2005). [KCNE3 R53H substitution in familial atrial fibrillation]. Chin. Med. J. (Engl) 118, 1735–1738.
Zhang, M., Jiang, M., and Tseng, G. N. (2001). minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ. Res. 88, 1012–1019. doi: 10.1161/hh1001.090839
Zhang, M., Wang, Y., Jiang, M., Zankov, D. P., Chowdhury, S., Kasirajan, V., et al. (2011). KCNE2 protein is more abundant in ventricles than in atria and can accelerate hERG protein degradation in a phosphorylation-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 302, H910–H922. doi: 10.1152/ajpheart.00691.2011
Keywords: MinK-related peptide, MiRP, Long QT Syndrome, atrial fibrillation, Brugada Syndrome
Citation: Crump SM and Abbott GW (2014) Arrhythmogenic KCNE gene variants: current knowledge and future challenges. Front. Genet. 5:3. doi: 10.3389/fgene.2014.00003
Received: 20 November 2013; Accepted: 04 January 2014;
Published online: 24 January 2014.
Edited by:
Junjie Xiao, Shanghai University, ChinaCopyright © 2014 Crump and Abbott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geoffrey W. Abbott, Bioelectricity Laboratory, Department of Pharmacology, Department of Physiology and Biophysics, School of Medicine, University of California, 360 Medical Surge II, Irvine, CA 92697, USA e-mail:YWJib3R0Z0B1Y2kuZWR1