 Scott J. Webster1
Scott J. Webster1 Adam D. Bachstetter1
Adam D. Bachstetter1 Peter T. Nelson1,2
Peter T. Nelson1,2 Frederick A. Schmitt1,3
Frederick A. Schmitt1,3 Linda J. Van Eldik1,4*
Linda J. Van Eldik1,4*- 1Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA
- 2Division of Neuropathology, Department of Pathology and Laboratory Medicine, University of Kentucky, Lexington, KY, USA
- 3Department of Neurology, University of Kentucky, Lexington, KY, USA
- 4Department of Anatomy and Neurobiology, University of Kentucky, Lexington, KY, USA
The goal of this review is to discuss how behavioral tests in mice relate to the pathological and neuropsychological features seen in human Alzheimer's disease (AD), and present a comprehensive analysis of the temporal progression of behavioral impairments in commonly used AD mouse models that contain mutations in amyloid precursor protein (APP). We begin with a brief overview of the neuropathological changes seen in the AD brain and an outline of some of the clinical neuropsychological assessments used to measure cognitive deficits associated with the disease. This is followed by a critical assessment of behavioral tasks that are used in AD mice to model the cognitive changes seen in the human disease. Behavioral tests discussed include spatial memory tests [Morris water maze (MWM), radial arm water maze (RAWM), Barnes maze], associative learning tasks (passive avoidance, fear conditioning), alternation tasks (Y-Maze/T-Maze), recognition memory tasks (Novel Object Recognition), attentional tasks (3 and 5 choice serial reaction time), set-shifting tasks, and reversal learning tasks. We discuss the strengths and weaknesses of each of these behavioral tasks, and how they may correlate with clinical assessments in humans. Finally, the temporal progression of both cognitive and non-cognitive deficits in 10 AD mouse models (PDAPP, TG2576, APP23, TgCRND8, J20, APP/PS1, TG2576 + PS1 (M146L), APP/PS1 KI, 5×FAD, and 3×Tg-AD) are discussed in detail. Mouse models of AD and the behavioral tasks used in conjunction with those models are immensely important in contributing to our knowledge of disease progression and are a useful tool to study AD pathophysiology and the resulting cognitive deficits. However, investigators need to be aware of the potential weaknesses of the available preclinical models in terms of their ability to model cognitive changes observed in human AD. It is our hope that this review will assist investigators in selecting an appropriate mouse model, and accompanying behavioral paradigms to investigate different aspects of AD pathology and disease progression.
Introduction
Alzheimer's disease (AD) is characterized by a progressive decline in cognitive function, usually starting with memory complaints and eventually progressing to involve multiple cognitive, neuropsychological and behavioral domains. The definitive diagnosis of AD comes from postmortem analysis of the neuropathological changes in the brain. Analyses of both clinical and pathological features, i.e., clinicopathological correlation studies, have provided important insights into how the pathology correlates with cognitive status. Complementing these studies in humans has been the development of preclinical model systems of AD pathology. These preclinical animal models, especially mouse models, have been extremely useful to test mechanistic hypotheses about AD pathophysiology and to predict outcomes from pharmacological interventions. However, no animal model recapitulates the entirety of AD in humans, and therefore it is important to understand both the utility and limitations of particular animal models. With this in mind, we will present an overview of the neuropathological changes seen in the AD patient population as individuals transition from normal cognitive aging to dementia, review the clinical neuropsychological assessments used in the AD field, review the mouse behavioral tasks commonly used in preclinical testing and discuss how they relate to these clinical neuropsychological assessments, and outline the temporal progression of cognitive and non-cognitive deficits seen in the commonly used mouse models of AD.
Overview of Neuropathological Changes in AD
In 2012, new consensus guidelines for neuropathologic evaluation of AD were adopted (Hyman et al., 2012; Montine et al., 2012). The AD neuropathologic change is now ranked on three parameters (Amyloid, Braak, CERAD) to obtain an “ABC” score: histopathologic assessments of beta-amyloid (Aβ)-containing amyloid plaques (A), Braak staging of neurofibrillary tangles (B), and scoring of neuritic amyloid plaques (C). Standard approaches for the workup of cases, preferred staining methods, reporting of results and clinicopathological correlations are also recommended. Unlike the prior AD neuropathologic criteria (Hyman and Trojanowski, 1997) that required a history of dementia, the current guidelines recognize that AD neuropathologic changes can be present in the brain in the apparent absence of cognitive impairment. The updated guidelines thus emphasize the continuum of neuropathologic changes that underlie AD. For a disease process that is known to occur over a decade or more (Blennow and Zetterberg, 2013; Rosen and Zetterberg, 2013; Rosen et al., 2013), and encompasses the age range where people are likely to die of other causes, it is inevitable that many people will die in a prodromal or “preclinical” stage of AD. This consideration points to the complexity that clinicopathological studies face when examining AD pathological contributions to cognitive deficits (Nelson et al., 2009b).
There have been numerous clinicopathological studies attempting to correlate amyloid plaques with the cognitive deficits seen in AD (Blessed et al., 1968; Tomlinson et al., 1970; Duyckaerts et al., 1990, 1998; Berg et al., 1998; Gold et al., 2000; Mungas et al., 2001; Tiraboschi et al., 2002, 2004; Guillozet et al., 2003; Kraybill et al., 2005; Holtzer et al., 2006; Markesbery et al., 2006; Nelson et al., 2007a, 2009a; Beach et al., 2009; Sabbagh et al., 2010; Robinson et al., 2011). Apparent inconsistencies in the conclusions of these studies are due to differences in study cohorts, methodology used to classify plaque subcategories, plaque-counting techniques, and metrics used to assess cognitive deficits. Nevertheless, several important concepts have emerged pertaining to plaque pathology and cognition. First, the strongest correlation between amyloid plaques and cognition is in the early stages of the disease and this association weakens as NFTs and gross neocortical neurodegeneration become more widespread (Thal et al., 2002; Nelson et al., 2009b, 2012). As the disease progresses into the later stages, there is little evidence to support a continued contribution by amyloid plaques to the late-stage AD cognitive decline (Nelson et al., 2009b, 2012). Second, it appears that density of neuritic plaques correlates more strongly with the cognitive deficits than do “diffuse” amyloid plaques (Mckee et al., 1991; Crystal et al., 1993; Tiraboschi et al., 2004; Nelson et al., 2007a; Braak et al., 2011).
In contrast to the literature concerning amyloid plaques, a large number of studies have arrived at a common finding, namely, there is a strong link between neocortical NFTs and cognitive decline (Tomlinson et al., 1970; Duyckaerts et al., 1990, 1997, 1998; Mckee et al., 1991; Arriagada et al., 1992; Bierer et al., 1995; Davis et al., 1995; Dickson et al., 1995; Nagy et al., 1995, 1999; Cummings et al., 1996; Berg et al., 1998; Grober et al., 1999; Sabbagh et al., 1999; Gold et al., 2000; Mungas et al., 2001; Riley et al., 2002; Silver et al., 2002; Tiraboschi et al., 2002; Guillozet et al., 2003; Bennett et al., 2004; Kraybill et al., 2005; Holtzer et al., 2006; Markesbery et al., 2006; Koepsell et al., 2008; Whitwell et al., 2008; Beach et al., 2009; Brayne et al., 2009; Giannakopoulos et al., 2009; Sabbagh et al., 2010; Robinson et al., 2011). It should be noted that outside of frontotemporal lobar degeneration (FTLD), one does not see widespread cortical NFTs without abundant plaque pathology. In the earliest stages of AD (Braak stage I-II), NFTs are restricted to the entorhinal cortex (Braak and Del Tredici, 2011; Braak et al., 2011). NFTs spread to the limbic and medial temporal lobe (Braak stage III-IV), and this stage correlates with early AD symptoms related to memory (Schmitt et al., 2000; Riley et al., 2011). During the late stages (Braak stage V–VI), NFTs increase in number and manifest in neocortical areas responsible for higher cognitive domains such as executive function, visuospatial capacities, and speech in synchrony with observed AD-related cognitive deficits in these respective cognitive domains. Not all AD cases fall within the standard NFT distribution described by the Braak staging (Hof et al., 1997; Abner et al., 2011; Murray et al., 2011). Some cases classified as “high level” of AD neuropathological changes may show subtle or undetectable cognitive impairment, yet all cases with quantifiably “end stage” neocortical NFT pathology show cognitive impairment (Dickson et al., 1995; Berlau et al., 2007; Nelson et al., 2009a, 2012; Abner et al., 2011; Santacruz et al., 2011; Jicha et al., 2012). In sum, the correlations noted in human material support the hypothesis that plaques and tangles correlate with cognitive status. The data also support the “Amyloid Cascade Hypothesis”(Karran et al., 2011), a deceptively complex hypothesis which posits that beta-amyloid/plaque pathology kindles widespread tau/NFT pathology, with the tau/NFT pathology constituting the more direct cause of the cell loss and synapse elimination underlying clinical disease (Nelson et al., 2009b, 2012).
Cognitive Neuropsychological Assessments used in the AD Field
Neuropsychological assessment is the most reliable means to clinically evaluate the cognitive deficits seen in humans. Many neuropsychological tests have been developed which are highly sensitive to the cognitive behavioral symptoms seen in AD, and these tests are extensively used as clinical diagnostic tools (Schmitt, 1994) as well as to track the progression of the disease (Flicker et al., 1984; Morris et al., 1989; Storandt and Hill, 1989; Storandt, 1991; Welsh et al., 1991, 1992; Locascio et al., 1995; Albert, 1996; Storandt et al., 1998; Schmitt et al., 2000; Salmon and Bondi, 2009). Current neuropsychological assessments (from the National Institute on Aging workgroups on diagnostic guidelines for AD) aim to detect disruptions in cognitive domains such as episodic memory, semantic memory, working memory, and attention, as well as dysfunction in language, praxis, and executive functioning (Flicker et al., 1984; Baddeley et al., 1986, 1991, 2001; Huff et al., 1987; Knopman and Ryberg, 1989; Hodges et al., 1992; Parasuraman and Nestor, 1993; Hodges and Patterson, 1995; Perry and Hodges, 1999; Salmon et al., 1999; Perry et al., 2000; Backman et al., 2001; Lambon Ralph et al., 2003). In the following section, we will cover several of the most common neuropsychological tests used clinically to assess the mental status and memory disruptions in AD (for more in depth reviews see Perry and Hodges, 1999; Budson and Price, 2005; Bondi et al., 2008; Weintraub et al., 2012). Table 1 provides an overview of four mental status examinations and two brief memory tests commonly used clinically.
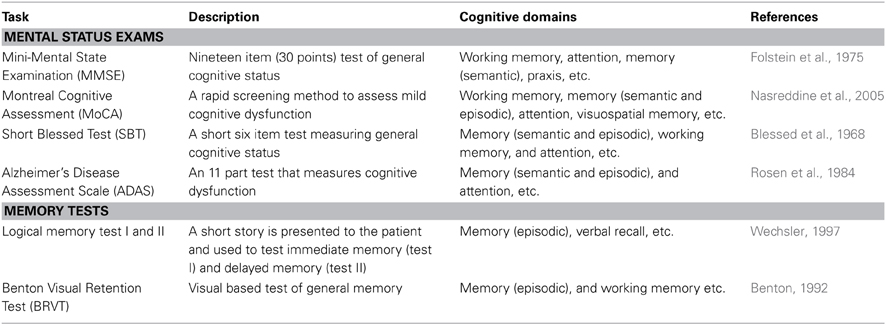
Table 1. Common neuropsychological assessment tasks seen clinically.
Mental Status Exams
Mental status examinations assess multiple mental functions and cognitive abilities across multiple domains (see Table 1), and are generally more encompassing than specific verbal and visual memory tests (examples also seen in Table 1). Both categories of tests are clinically useful in assessing the cognitive progression of AD. Most mental status examinations assess mental functions and cognitive ability across a wide range of areas such as: language skills, arithmetic ability, visuospatial ability, attention, memory, and orientation to time and place.
The Mini-Mental Status Examination (MMSE) is one of the most commonly used neuropsychological screening tools for cognitive impairments seen in AD (Simard and Van Reekum, 1999; Snyderman and Rovner, 2009). The MMSE is a brief questionnaire that can both diagnose cognitive impairment and track the severity of this cognitive impairment throughout the pathogenesis of the disease. The MMSE covers multiple areas such as: attention, memory (semantic and episodic), orientation to time and place, and working memory. The scoring system ranges from 0 to 30 points. In general, a score of 27 or greater reflects normal cognition, a score of 19–24 represents mild impairment, a score of 10–18 represents moderate impairment, and a score below 9 indicates severe cognitive impairment. The MMSE has excellent reproducibility that lends itself well to longitudinal use in tracking the progression of the cognitive impairments associated with AD (Jacqmin-Gadda et al., 1997; Aevarsson and Skoog, 2000; Chatfield et al., 2007).
The Montreal Cognitive Assessment (MoCA) is a more recent mental status examination used for cognitive dysfunction seen in AD. This test battery takes approximately 10 min to administer and covers many similar cognitive domains to that of the MMSE. The total possible score is 30 points, with a score of 26 or above considered within the normal range. An example of a task included in the MoCA is the forward and backward digit span test. In the forward digit span test, a sequence of five numbers is read at a rate of one number per second and the test taker is required to repeat a set of numbers in exactly the same sequence as they were presented. In the backward digit span task, the test taker is required to repeat a three number sequence in the reverse order in which it was presented. One point is awarded for each of the digit span tests in which the test taker made no errors. The MoCA also includes a delayed recall memory test. Relatively near the beginning of the test, the examiner presents a short word list for the patient to remember. At the end of the test, the patient is again prompted to recall the word list and for each of the words correctly remembered one point is awarded.
Another commonly used test battery is that of the Short Blessed Test (SBT) which consists of a six-item test designed to identify the cognitive dysfunction seen in AD (Katzman et al., 1983). An appealing advantage of the SBT is the ease and speed with which it can be administered (often taking only a few minutes). In the SBT, errors are scored for incorrect answers and the scoring range falls between 0 and 28, with a score of 0–4 representing normal cognition, a score of 5–9 representing early impairment, and a score of 10 or more representing impaired cognition. Despite the simplicity and brevity of the SBT, the results that it produces have demonstrated excellent reliability (Fuld, 1978). Similarly, the SBT was the first mental status examination to be correlated with amyloid plaque burden at autopsy (Carpenter et al., 2011).
The Alzheimer's Disease Assessment Scale (ADAS) was specifically developed to measure the severity of symptoms commonly seen in AD (Rosen et al., 1984). Initially, it was developed in two parts (sub-scales): one for cognitive symptoms and one for non-cognitive symptoms. The cognitive sub-scale of the ADAS is commonly referred to as the ADAS-cog, and has become one of the most common neuropsychological tests used to assess AD progression. The scoring for the ADAS-cog ranges from 0 to 70, with a low score representing a cognitively intact person and a high score representing someone with cognitive impairment. Because the ADAS-cog has an excellent test-retest reliability and is considered to be one of the most sensitive scales for assessing cognitive changes related to AD (Emilien et al., 2004), this test is extensively used in AD clinical trials as an outcome measure of cognitive change (Schmitt and Wichems, 2006; Connor and Sabbagh, 2008).
Memory Tests
Individual memory tests are generally shorter than mental status examinations and focus solely on assessing memory deficits. Both verbal and visual memory tests are commonly used clinically as stand alone tests or incorporated into a more comprehensive mental status examination. Examples of such memory tests are the Logical Memory Test I and II (LM-I and LM-II) and the Benton Visual Retention Test (BVRT) (Benton, 1992; Wechsler, 1997).
The LM-I and LM-II were originally developed as subtests to the Wechsler memory scale, but are commonly used as stand-alone memory tests. Both are verbal based memory tests that involve a short story read to the patient. In the LM-I, the patient answers immediate questions related to the narrative, whereas in the LM-II there is a delay between the presentation of the story and the questions. Thus, these memory tests are used to assess immediate recall (LM-I) and delayed recall (LM-II).
In the BVRT, the patient is shown 10 different visual designs, one at a time, and is then asked to reproduce each one from memory exactly as it appeared. While scoring the BVRT, errors of omissions, distortions, perseverations, rotations, misplacements, and size are all looked for and can give some insight into the progression of the disease. For example, if the patient has a high number of perseveration errors it is likely that the AD pathology has manifest itself in neocortical areas responsible for higher cognitive domains such as executive function, and visuospatial capacities (Braak Stage V–VI).
Cognitive Neuropsychological Assessment Summary
Each of the tests described above is aimed at assessing deficits in different cognitive domains. Each of these domains has been shown to be impaired at some point in the spectrum of human AD. However, they are not uniformly affected throughout the course of the disease. Deficits in some domains occur relatively early, while deficits in others occur much later in the progression of the disease. Figure 1A depicts an overview of the time course of affected cognitive domains in human AD. It has become increasingly clear that identifying and targeting the cognitive deficits that occur early in the course of the disease is critical to producing the maximum impact of treatment on cognitive function and quality of life (Salmon et al., 2002). Thus, great efforts have been made to better understand the profile of cognitive deficits associated with early AD, and have resulted in earlier and more reliable clinical diagnosis (Bondi et al., 1995, 1999; Jacobson et al., 2002; Lange et al., 2002; Mickes et al., 2007).
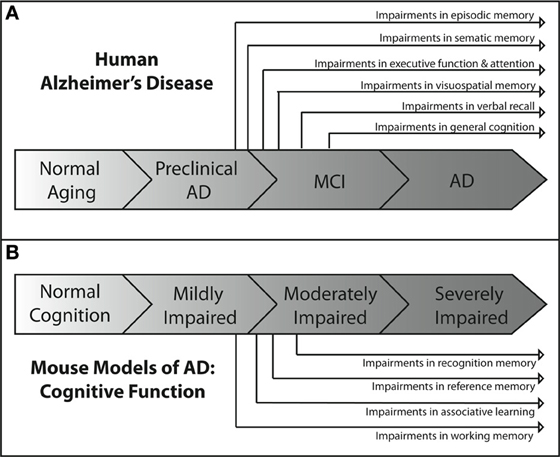
Figure 1. Overview of the progression of cognitive deficits in human AD and in mouse models of AD. (A) In the human disease, the earliest AD-related cognitive deficits present themselves as episodic memory impairment during the late preclinical phase of the disease (Backman et al., 2005; Twamley et al., 2006; Bondi et al., 2008). Semantic memory deficits are the next to develop (Tuokko et al., 2005; Storandt et al., 2006), followed by impairments in executive functioning (Bondi et al., 2008), attention (Bondi et al., 2008), and visuospatial memory (Twamley et al., 2006) near the beginning of the MCI phase of the disease. As MCI progresses, deficits in verbal recall (Kryscio et al., 2006; Bondi et al., 2008) develop and impairments in general cognition (Bondi et al., 2008) become apparent. As the patient transitions into AD, all cognitive domains become affected. (B) The development of cognitive deficits in APP mouse models of AD shows similar patterns of progression. Consistently, the earliest observable impairments are in spatial working memory (Webster et al., 2013), as assessed through the use of water maze based tasks. These impairments are generally followed temporally by impairments in associative learning and reference memory, as assessed by maze alternation (Lalonde et al., 2012) and fear conditioning tasks (Kobayashi and Chen, 2005). Deficits in recognition memory usually present later in the spectrum of cognitive impairment than deficits in other domains (Eriksen and Janus, 2007; Hall and Roberson, 2012; Webster et al., 2013).
Some of the earliest neuropathological changes in AD are in the hippocampus and entorhinal cortex, followed by changes in the medial temporal lobe. Consistent with this progression of pathology, the earliest detectable deficits in cognition are seen in medial temporal lobe-dependent episodic memory (Bondi et al., 1999; Collie and Maruff, 2000; Schmitt et al., 2000; Smith et al., 2007). These early deficits in episodic memory are followed closely by deficits in semantic memory, and both are developed before other deficits in cognitive domains such as attention, visuospatial memory, or executive function (Bondi et al., 2008). This suggests that cognitive functions such as episodic and semantic memory that depend heavily on the neural circuitry of the medial and lateral temporal lobes may be impaired earlier than cognitive abilities that depend on the circuitry of other brain regions. Further support for this idea comes from the time course of the frontal lobe dependent executive function deficits observed in patients. Slight deficits in executive functioning are first detectable near the end of the preclinical phase of AD but after the observed deficits in episodic and semantic memory (Storandt et al., 2006; Twamley et al., 2006). As the patient moves from the preclinical phase of AD into MCI, more cognitive domains begin to be affected. Most studies of MCI patients show consistent impairments in verbal recall (Larrieu et al., 2002; Tuokko et al., 2005; Kryscio et al., 2006) and a decline in general memory functioning (Tuokko et al., 2005; Bondi et al., 2008). Once the AD patient progresses past MCI and into dementia, general cognition continues to decline with deficits appearing in all respective cognitive domains (Huff et al., 1987; Locascio et al., 1995; Lambon Ralph et al., 2003; Mckhann et al., 2011).
The importance of neuropsychological testing cannot be overstated, as it is the only measure that provides information about a patient's current cognitive status and remains the most reliable means to clinically diagnose probable AD. Neuropsychological testing provides information on both general cognitive status and specific information on different cognitive domains affected in AD. Composite scores encompassing multiple neuropsychological tests are often used and can provide some of the most reliable assessments of global cognitive status relating to AD as well as serving as efficacy outcome measures in clinical trials (Bernick et al., 2012).
How Commonly used Preclinical Mouse Behavioral Tasks Relate to the Clinical Neuropsychological Assessment Tests in Human AD?
Ideally, preclinical rodent cognitive testing would assess identical cognitive domains to those examined through neuropsychological testing in human AD. Indeed, many rodent behavioral tasks have been specifically designed with this in mind, and while each task varies with respect to face, construct, and predictive validity, they all attempt to model different aspects of the cognition disrupted in AD and targeted by the human neuropsychological assessments listed in the previous section. Some cognitive domains disrupted in AD have been extensively modeled (reference memory, working memory and executive function), some less so (attention), and some nearly not at all (episodic memory). Reference memory, while not used clinically to describe human cognition, refers to learned knowledge for an aspect of a task that remains constant throughout the behavioral task and most closely correlates to human semantic memory. Working memory refers to a mental processing system used to hold transitory information for a limited time where it can be manipulated and operated on and used to guide behavior. Recognition memory refers to the ability to recognize previously encountered events, objects, or individuals and is classified as part of long-term declarative memory. Other cognitive domains impaired in human AD such as those involving language (i.e., verbal acuity tasks and verbal recall tasks) simply cannot be modeled in rodent models. Table 2 provides a short description of various behavioral tasks used to assess AD-like cognitive deficits in mice, and summarizes the respective cognitive domains measured by each task.
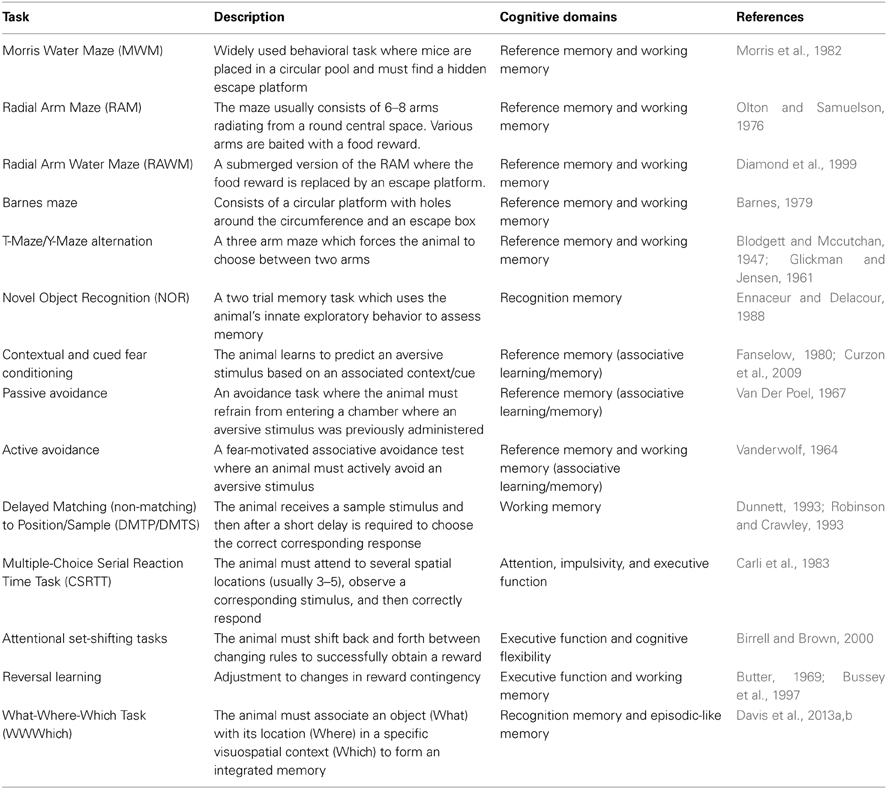
Table 2. Commonly used mouse behavioral tasks.
Modeling Working Memory
Working memory is perhaps one of the most well modeled aspects of the memory deficits in AD. Clinically, many of the neuropsychological tests that assess working memory rely heavily on the use of verbal tasks (Kaplan et al., 1978; Benedict et al., 1998; Spreen and Strauss, 1998; Delis et al., 2000), employing language as a core construct and thus are not feasible to model using mice. Instead, spatial based working memory tasks are heavily employed in murine working memory testing and likely are more depictive of the visuospatial working memory tasks used clinically (Benton, 1992; Benedict and Groninger, 1994).
The most widely used paradigms for working memory in mice are maze type tasks which require spatial working memory to solve. The earliest variants of these are the T-maze and Y-maze alternation tasks, which are relatively simple tests consisting of three arms with a single intersection. These tasks rely on the natural exploratory behavior (tendency to choose an alternative arm over an arm which has been previously explored) of rodents and are considered the most rudimentary tasks to assess spatial working memory (Dudchenko, 2004). A more complex maze type task used to test murine spatial working memory is that of the Radial Arm Maze (RAM), consisting of several arms (usually 6–8) radiating outwards from a central platform (Olton and Samuelson, 1976; Olton et al., 1979). In the RAM, the animal is started in the center area and then some of the arms or all of the arms can be baited with a food reward. Depending on the baiting paradigm, unimpaired rats and mice will quickly learn where the food reward is and which arms have previously been visited, and will avoid re-entering a previously entered arm. Perhaps the most widely employed spatial working memory task is that of the Morris Water Maze (MWM) (Morris et al., 1982). The MWM consists of a large open pool with a hidden (submerged) escape platform located somewhere within the pool. Animals must learn where the platform is, remember the platform's location, and then use spatial cues on subsequent trials to navigate back to the hidden platform. Large numbers of AD mouse models have been tested in the MWM and most show AD related cognitive deficits (Webster et al., 2013). Other common tasks of murine spatial working memory are the Radial Arm Water Maze (RAWM) and the Barnes Maze (Barnes, 1979; Diamond et al., 1999; Alamed et al., 2006).
It is important to note that while many of the previously described behavioral tasks are considered tasks of working memory they can also be modified to test reference memory depending on the testing protocol used. Similarly, not all models of murine working memory are spatial working memory tasks. For example, there exist versions of both the RAM and RAWM that are considered non-spatial working memory tasks (Olton and Feustle, 1981; Crusio et al., 1993; Hyde et al., 2000). Other examples of non-spatial working memory tasks are operant tasks such as the Delayed Match to Sample (DMTS), Delayed Non-Match to Sample (DNMTS), and Delayed Stimulus Discrimination Task (DSDT) (Dudchenko, 2004; Buccafusco et al., 2008). In these non-spatial working memory tasks the animal is required to remember a stimulus (over a delay period) that is paired with a particular type of response (generally a lever press or a nose poke) and a correct response is rewarded. In many of the non-spatial operant working memory tasks each animal can perform many trials per day and thus can serve as its own control. This lends itself nicely to pharmacological based studies assessing potential therapeutic compounds for the treatment of the cognitive deficits seen in AD (Buccafusco et al., 2008).
Modeling Executive Function
Executive function refers to a broad range of higher cognitive processes such as: reasoning, planning, cognitive flexibility, sequencing, response inhibition, and abstract concept formation. The current mouse models of executive function most closely replicate the human aspects of cognitive flexibility and response inhibition in executive function. Attentional set-shifting tasks are one of the main behavioral tasks used to assess executive function in the mouse. In many ways, set-shifting tasks are similar to the Wisconsin Card Sorting Task in that they form the gold standard for assessing executive function (Drewe, 1974; Robinson et al., 1980; Arnett et al., 1994). In both the Wisconsin Card Sorting Task in humans and set-shifting tasks in mice, the dorsolateral and orbital prefrontal cortex is critical for successful performance (Weinberger et al., 1986; Berman et al., 1995; Brigman et al., 2005; Bissonette et al., 2008). In the most common version of the murine set-shifting task, mice are required to select a bowl in which to dig for a food reward. Bowls can be discriminated from each other according to different stimulus dimensions such as texture and odor. Successful completion of the task requires the animal to shift between stimuli dimensions to successfully retrieve the food reward. The ability to extract knowledge from different stimuli dimensions suggests that the mouse is capable of using at least some aspects of higher-order cognitive functions seen in human executive functioning (Chudasama, 2011). Numerous different transgenic mouse models of AD have shown deficits in set-shifting tasks (Zhuo et al., 2007, 2008; Marchese et al., 2013).
Reversal learning is another way that aspects of executive function are modeled in the mouse. While less complex than attentional set shifting, reversal learning does require both cognitive flexibility and impulse control, thus tapping into components of human executive function (Chudasama, 2011; Stopford et al., 2012). There are many different variations of reversal learning tasks in mouse behavior, but they all work on the same principle. The animal first learns that a particular response to a stimulus will be rewarded, while a response to a different stimulus will be unrewarded. Then the stimulus-reward is switched so that the previously unrewarded stimulus becomes the rewarded stimulus. The animal must learn to reverse responses in order to receive the reward. Wild type control mice are able to quickly adjust their response in order to obtain the reward. However, animals with prefrontal cortex lesions display profound deficits in reversal learning (Chudasama, 2011; Izquierdo and Jentsch, 2012). Similarly, many different AD mouse models have shown impairment in reversal learning (Angelo et al., 2003; Dong et al., 2005; Filali et al., 2012; Cheng et al., 2013; Musilli et al., 2013; Papadopoulos et al., 2013).
Another aspect of executive function that is modeled in mice is response inhibition. Response inhibition is required for the appropriate control of an individual's behavioral actions in response to a stimulus (Robbins, 1996; Humby et al., 1999; Perry and Hodges, 1999; Romberg et al., 2013a). The five choice serial reaction time task (5-CSRTT) is a behavioral task that measures the response inhibition component of executive function (5-CSRTT is also used to model aspects of attention, see below section) in mice (Robbins, 2002; Bari et al., 2008; Chudasama, 2011; Romberg et al., 2013a). The 5-CSRTT can test two different aspects of response inhibition: (1) a failure to withhold the impulsive urge to respond while anticipating correct response (premature responses) and (2) a failure to disengage from repeating past correct responses (perseveration responses) (Chudasama, 2011). Several mouse models of AD have shown deficits in response inhibition using the 5-CSRTT (Romberg et al., 2011, 2013b).
Modeling Attention
Several behavioral tasks have been developed for modeling attention in mice that provide reliable measures comparable to the neuropsychological assessments used in AD. The most widely used of these tasks is the 5-CSRTT (Muir, 1996; Humby et al., 1999; Robbins, 2002; De Bruin et al., 2006; Gibson et al., 2006; Lambourne et al., 2007; Pattij et al., 2007). This task employs an operant box with nose poke holes on the front wall of the chamber. Animals are trained to respond to brief flashes of light corresponding to five different spatial locations on this front side of the chamber and correct responses are rewarded with a food pellet released to a feeder box at the rear of the chamber. Touchscreen versions of this task are also available in which the nose poke holes and stimulus lights are replaced with an LCD screen (Romberg et al., 2011; Bussey et al., 2012). For both standard and touchscreen versions of the task, multiple trials are run each day and both the duration of the stimulus itself or the interval between the stimulus and response can be manipulated to increase the attention demands placed on the animal. Sustained attention is measured by examining when the animal responds to a different (incorrect) hole than where the stimulus light appeared (called errors of commission), fails to respond within the allotted time to the stimulus (errors of omission), and the speed with which the animal responds (reaction time). Aspects of selective attention can also be modeled with the 5-CSRTT by introducing brief bursts of white noise that the animal must ignore while still detecting the visual stimulus as it is presented (Robbins, 2002; Bari et al., 2008). The rodent 5-CSRTT is analogous to Leonard's 5-CSRTT used in humans (Wilkinson, 1963). Both tasks require subjects (mouse and human respectively) to utilize sustained attention divided among multiple spatial locations across which a large number of trials and errors of commission, omission, and reaction time are scored. Another task that can be considered somewhat analogous to the 5-CSRTT is the human Continuous Performance Tests (CPT) of sustained attention (Beck et al., 1956). In this task, the subjects are asked to respond to signal and non-signal events across numerous trials, and scores of hits, misses, rejections, and false alarms are recorded. Errors of commission in the 5-CSRTT are thought to be analogous to CPT false alarms rates. Similarly, errors of omission in the 5-CSRTT are thought to be analogous to CPT misses. AD mice such as the 3×Tg-AD mice have been shown to have deficits in sustained attention using the 5-CSRTT (Romberg et al., 2011). However, the homology between mouse and human versions of these tasks is far from perfect, and caution should be used when drawing conclusions from the rodent 5-CSRTT and applying them to human attention (Young et al., 2009).
Modeling Episodic Memory
Episodic memory refers to the ability to encode and recall personal past events and experiences. Episodic memory has also been referred to as the “what, when, and where” aspect of a particular experience. Modeling AD-related deficits in episodic memory in mice is a less well-explored area than that of other aspects of working memory, executive function, or attention. Historically, episodic memory was thought to be unique to humans (Tulving and Markowitsch, 1998). However, work over the past few decades on episodic-like memory across a number of animal species has suggested otherwise, and several mouse behavioral tasks designed at assessing episodic-like memory have been developed (Clayton and Dickinson, 1998; Clayton et al., 2001; Griffiths and Clayton, 2001; Morris, 2001; Davis et al., 2013a,b). One such task is the What-Where-Which Task (WWWhich). This task is an adaptation of the NOR task. While the NOR task itself is too simplistic a task to be considered a true episodic memory task (rather it is considered a task of recognition memory), the WWWhich task is able to model episodic-like memory. In the WWWhich task, the animal must integrate the location of a particular object with specific contextual cues to form an episodic-like memory (Davis et al., 2013a,b). Several studies employing the WWWhich task have observed performance deficits related to the aging process and to AD disease state in several transgenic mouse lines (Davis et al., 2013a,b). While the WWWhich task models episodic memory in the mouse, it is not very comparable to any of the episodic memory tasks commonly used in neuropsychological testing for AD. This is largely because the human tasks rely on language as a foundational construct for assessment. Obviously, there exists no such component in the WWWhich task for mice. Therefore, caution should be used when attempting to correlate any preclinical finding concerning episodic memory in mice to that of human cognition.
Summary of Mouse Behavioral Tests
All of the rodent behavioral tasks discussed in this section have been specifically developed to assess deficits in cognitive domains related to what is seen in human AD. Just as multiple neuropsychological tests assessing different cognitive domains are often used clinically to provide a global cognitive profile, so multiple behavioral tasks assessing different cognitive domains should ideally be used when characterizing the profile of AD-related cognitive impairment in a particular mouse model of AD.
Temporal Progression of the Cognitive Deficits Seen in the Commonly used AD Mouse Models
Cognitive decline is a defining feature of AD, and many mouse models have been developed that recapitulate aspects of the cognitive impairments seen in AD (Elder et al., 2010; Hochgrafe et al., 2013; Platt et al., 2013). Although no one animal model fully replicates the progression of cognitive impairments seen in the human disease, AD mouse models have been invaluable in advancing our knowledge of the disease. It should be noted that most of the AD mouse models are representative of the familial form of AD (FAD) which accounts for only a small percentage of the total AD cases each year (Campion et al., 1999). In addition, the contributions of both background strain and likely overexpression of mutant human APP genes on brain development and function must always be considered with regard to observed cognitive deficits in the various AD mouse models. Each transgenic mouse model of AD provides different insights into aspects of AD pathogenesis and the cognitive deficits associated with the disease. A generalized time course of the development of cognitive deficits across the various mouse models is depicted in Figure 1B. For each specific mouse model the temporal time course and progression of cognitive deficits in each cognitive domain can be different. In addition, in some models, cognitive deficits can be detected prior to the appearance of significant neuropathology. Careful forethought is therefore required in the selection of an optimal model displaying the AD related cognitive deficits desired based on the specific research interests of the investigator. An overview of the progressive cognitive deficits and the time of appearance of amyloid pathology is presented in Table 3 for five mouse models that contain amyloid precursor protein (APP) mutations and in Table 4 for five other common mouse models that contain APP and presenilin (PS1) mutations, or APP/PS1/Tau mutations. These tables are by no means an all-encompassing list of mouse models; rather they are simply meant to be examples of some of the commonly used mouse models of AD that are characterized by APP mutations. For recent reviews of additional AD mouse strains not included here, see (Ashe and Zahs, 2010; Elder et al., 2010; Epis et al., 2010; Hall and Roberson, 2012; Platt et al., 2013).
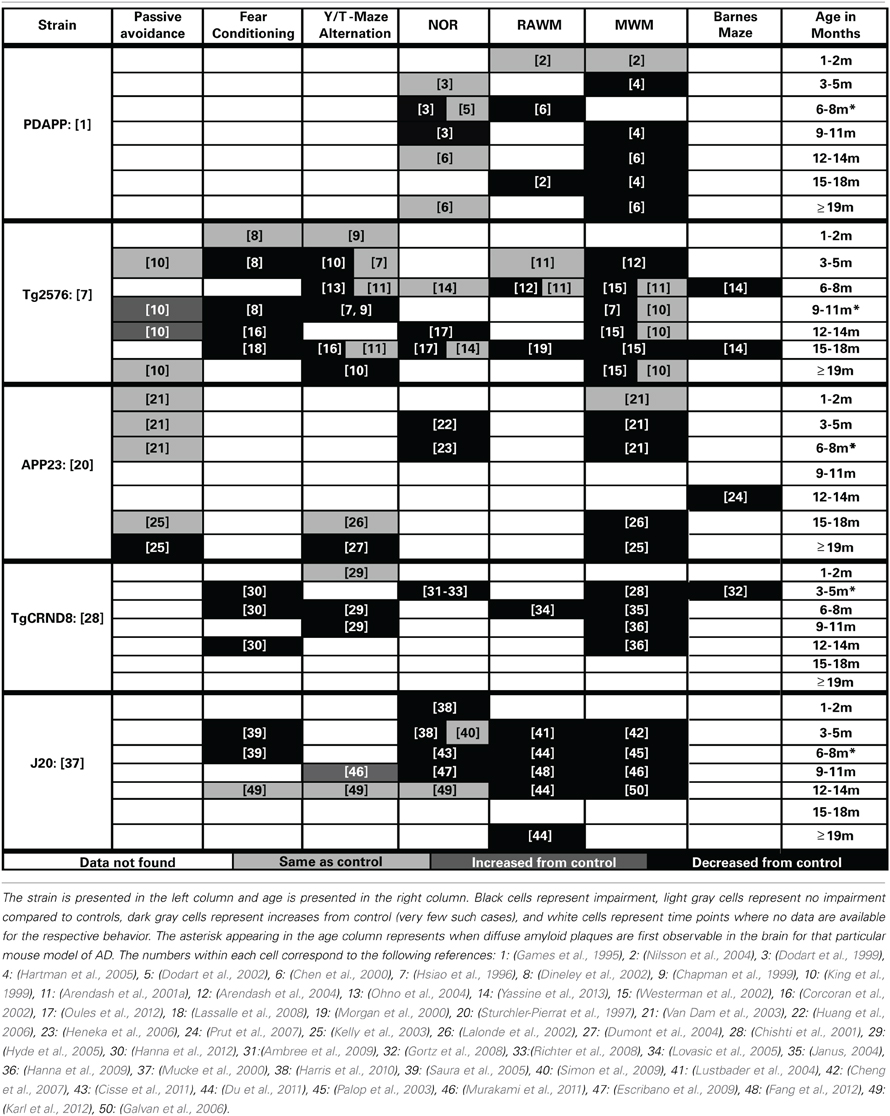
Table 3. Progression of cognitive deficits in APP mouse models of AD.
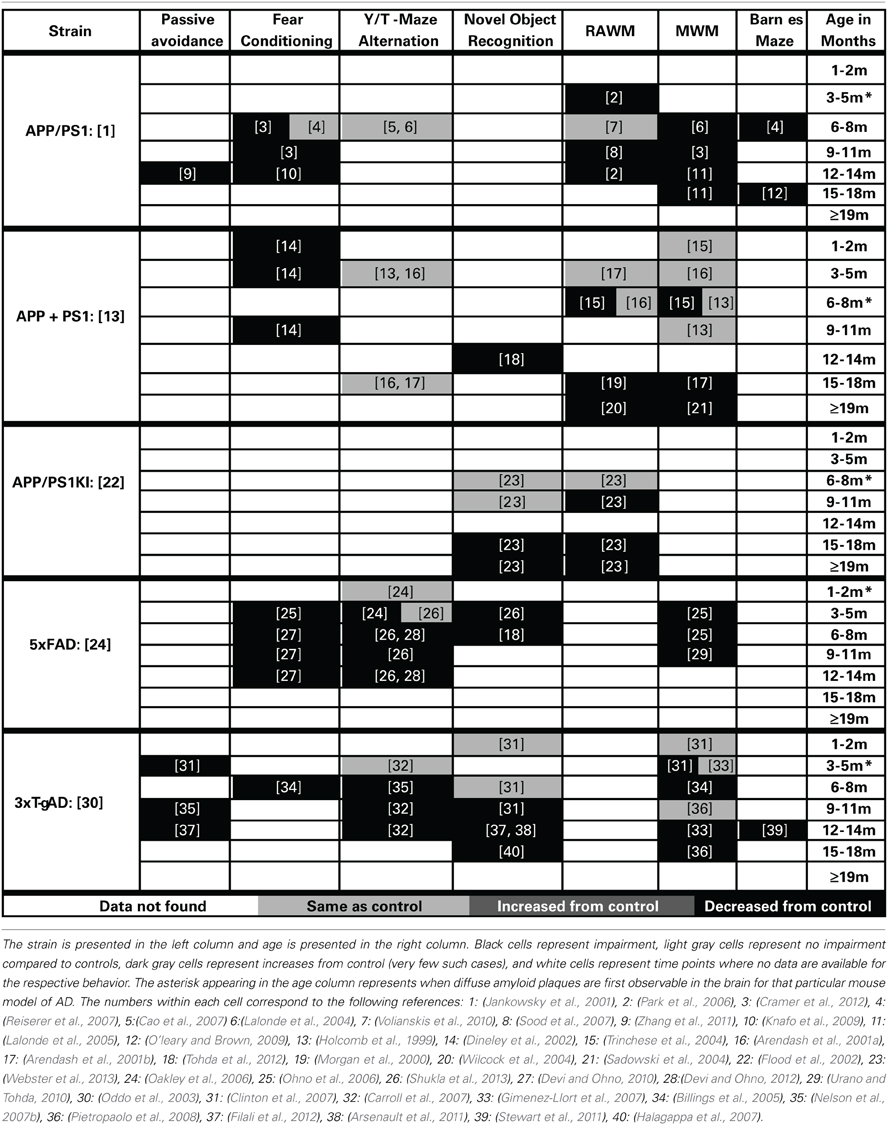
Table 4. Progression of memory deficits in other mouse models of AD (APP + PS1/Tau).
PDAPP
(Promoter: Platelet-Derived (PDGF) Promoter, Symbol: Tg (APPV717F) 109Ili, MGI ID: 2151935)
The PDAPP mouse was first described by Games in 1995 and is considered one of the earliest mouse models of AD (Chartier-Harlin et al., 1991; Games et al., 1995). In this model, the cognitive deficits first present themselves in spatial working memory at 4 months of age when assessed by MWM testing (Hartman et al., 2005). These deficits in working memory in the MWM are present throughout the rest of the life span for this model (Chen et al., 2000; Daumas et al., 2008). Deficits in recognition memory appear to develop after the working memory deficits in this model, as the first reported deficits in the NOR task are at 6 months old (Dodart et al., 1999). The cognitive deficits in recognition memory do not appear as robust as those in working memory in this model as there are inconsistent reports in the literature (Dodart et al., 1999, 2002; Chen et al., 2000). The cognitive defects in this model appear to present themselves before the appearance of plaque deposition (first appear at approximately 6 months) or other gross amyloid pathologies (Games et al., 1995; Hsiao et al., 1996; Schenk et al., 1999; Chen et al., 2000; Morgan, 2003).
TG2576
(Promoter: Hamster PrP Promoter, Symbol: Tg (APPSWE) 2576Kha, MGI ID: 2385631)
In the Tg2576 mouse (Hsiao et al., 1996), the first presentation of cognitive deficits is seen at 5 months of age in spatial working memory (Arendash et al., 2004). These spatial working memory deficits are generally accepted to be present across the rest of the life span for this model (Hsiao et al., 1996; Westerman et al., 2002; Arendash et al., 2004). However, methodology used to assess spatial memory appears to be very important, as several different reports have failed to observe these same deficits at various ages (Hsiao et al., 1996; Arendash et al., 2001a; King and Arendash, 2002). Non-spatial working memory tasks show a similar time course progression, first appearing at 3–5 months of age and persisting across the lifespan (Hsiao et al., 1996; Chapman et al., 1999; King and Arendash, 2002; Lalonde et al., 2003; Ohno et al., 2004). Deficits in recognition memory do not appear until much later, first appearing at 12 months of age in the NOR task (Oules et al., 2012; Yassine et al., 2013).
APP23
(Promoter: Thy-1, Symbol: Tg (Thy1-APP) 3Somm, MGI ID: 2447146)
The APP23 mouse model was reported in 1997 (Sturchler-Pierrat et al., 1997). In this model, the cognitive deficits begin to first appear in both recognition memory and spatial working memory at 3 months of age. The deficits appear to be progressive with age, and at 12 months old the animals also show cognitive deficits in a reference memory version of the Barnes maze (Prut et al., 2007). This model develops non-spatial working memory deficits very late in the progression of the disease (only after 19 months of age) (Lalonde et al., 2002; Dumont et al., 2004). Interestingly, cognitive performance in passive avoidance memory tasks follows the same progression as non-spatial working memory deficits in this model, unimpaired at 15 months of age and then developing deficits between 19 and 20 months of age (Kelly et al., 2003).
TgCRND8
(Promoter: PrP, Symbol: Tg (PRNP-APPSweInd) 8Dwst, MGI ID: 3589475)
The TgCRND8 model, described by Chishti et al. (2001), exhibits early cognitive impairment that spans across multiple cognitive domains (Chishti et al., 2001). TgCRND8 are impaired on spatial working memory tasks starting at 3 months of age. These deficits are seen in the MWM and progress with age of the animal (Janus et al., 2000; Chishti et al., 2001; Gortz et al., 2008; Richter et al., 2008; Ambree et al., 2009). Reference memory deficits via Barnes maze testing are also present at 3 months of age (Gortz et al., 2008; Richter et al., 2008; Ambree et al., 2009). Similar to this observed temporal time course of spatial working memory and reference memory deficits are the development of deficits in both recognition memory and fear conditioning (Gortz et al., 2008; Richter et al., 2008; Ambree et al., 2009; Hanna et al., 2012). Deficits in alternation tasks develop by 6 and 9 months in Y-maze and T-maze alternation tasks, respectively (Hyde et al., 2005).
J20
(Promoter: Platelet-Derived (PDGF), Symbol: Tg (PDGFB-APPSwInd) 20Lms, MGI ID: 3057148)
The J20 mouse model was developed by (Mucke et al., 2000). This model is unique in that the first presented cognitive deficits are observed very early (at 1–2 months of age) in recognition memory (Harris et al., 2010). These deficits in recognition memory are present when assessed at several other time points (Escribano et al., 2009; Simon et al., 2009; Cisse et al., 2011). However, they do not appear to progress with the age of the animal and there has even been a report of no recognition memory deficits in old animals in advanced stages of the disease (Karl et al., 2012). Early memory deficits can also be observed in spatial working memory at 3 months of age when assessed by the MWM and the RAWM tasks (Lustbader et al., 2004; Cheng et al., 2007; Meilandt et al., 2008). These spatial working memory deficits are present across the rest of this model's lifespan (Palop et al., 2003; Galvan et al., 2006; Cisse et al., 2011; Du et al., 2011; Murakami et al., 2011; Fang et al., 2012). Fear conditioning deficits appear consistent with the presentation of spatial working memory and recognition memory deficits in this model (Saura et al., 2005). Interestingly, this model does not appear to display working memory deficits on tasks of alternation such as the Y-maze (Murakami et al., 2011; Karl et al., 2012).
APP/PS1
(Promoter: PrP, Symbol: Tg (APPswe, PSEN1dE9) 85Dbo, MGI ID: 3524957)
The cognitive deficits in the APP/PS1 mouse model, first described by Jankowsky et al. (2001), have been well characterized. Cognitive deficits are first seen at 3 months of age in the RAWM spatial working memory task and are also reported by 6 months of age in the MWM (Cao et al., 2007; Ding et al., 2008). Further, these deficits have been well characterized across the lifespan of this mouse model in water based spatial working memory tasks (Lalonde et al., 2005; Park et al., 2006; Cao et al., 2007; Sood et al., 2007; Ding et al., 2008; Volianskis et al., 2010; Zhang et al., 2011; Cramer et al., 2012; Ma et al., 2012). Impairments in reference memory develop by 6 months and persist through the rest of the life of this model (Reiserer et al., 2007; Bernardo et al., 2009; O'leary and Brown, 2009). Deficits in associative learning have also been described in fear conditioning tasks starting at 6–8 months of age (Knafo et al., 2009; Cramer et al., 2012). Similarly, passive avoidance deficits have also been described at 12 months of age (Zhang et al., 2011). No deficits were seen in alternation tasks of working memory for this model (Lalonde et al., 2004; Cao et al., 2007).
APP + PS1
(Promoter: Hamster PrP Promoter, Symbol: Tg (APPSWE) 2576Kha, MGI ID: 2385631) × (Platelet-Derived (PDGF), Symbol: Tg (PDGFB-PSEN1M146L) 2Jhd, MGI ID: 2447326)
Holcomb described a mouse model in 1998 that has been widely used to study cognitive deficits related to AD (Holcomb et al., 1998). The first observable deficits in this model are shown in associative learning and present themselves between 4 and 5 months of age (Dineley et al., 2002). The progression of the spatial working memory impairment in this model is relatively slow compared to most other models. The first reported impairment in spatial working memory was observed using 6-month-old animals (Trinchese et al., 2004). However, these cognitive deficits are not robust at this age, as others have observed no such deficit (Holcomb et al., 1999; Arendash et al., 2001a). By 15 months of age the spatial working memory is consistently impaired throughout the rest of the life span (Morgan et al., 2000; Arendash et al., 2001b; Gordon et al., 2001; Sadowski et al., 2004; Wilcock et al., 2004). Similarly, deficits in recognition memory occur later in this model, first observed at 12 months of age (Mori et al., 2013). No deficits were observed in alternation tasks of working memory (Holcomb et al., 1998; Arendash et al., 2001a).
APP/PS1 KI
(Promoter: Endogenous, Symbol: Apptm1.1Cep, MGI ID: 2652346) × (Promoter: Endogenous, Symbol: Psen1tm1Dgf, MGI ID: 3608968)
The APP/PS1 knock-in mouse model (first described in Flood et al., 2002) uses endogenous promoters to drive the expression of humanized amyloid beta sequence, and AD-like pathology and cognitive deficits develop in the absence of APP or PS1 overexpression (Flood et al., 2002). The earliest reports of cognitive deficits are reported at 7 months in this model (Bruce-Keller et al., 2011). However, the majority of cognitive deficits appear later. We have shown previously that the cognitive deficits in spatial working memory (assessed by RAWM testing) first appear at 9 months of age (Webster et al., 2013). These deficits are followed by impairments in associative memory (appearing by 14 months of age Thibault et al., 2012) and in recognition memory (not developing until 15 months of age Webster et al., 2013).
5×FAD
(Promoter: Thy-1, Symbol: Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799Vas, MGI ID: 3693208)
The 5×FAD model, first described by Oakley et al. (2006), develops progressive cognitive deficits with age. This model develops cognitive deficits by 3 months of age in spatial working memory (Ohno et al., 2006; Urano and Tohda, 2010). These working memory deficits are followed temporally with the development of associative learning impairment in fear conditioning (Ohno et al., 2006; Devi and Ohno, 2010) as well as the development of deficits in a working memory version of the Y-maze (Oakley et al., 2006; Devi and Ohno, 2012; Shukla et al., 2013). As with several of the other models (PDAPP, Tg2576, APP/PS1, and APP/PS1 KI) this model develops deficits in recognition memory later than the observed deficits in spatial working memory (Tohda et al., 2012).
3×Tg-AD
(Promoter: Thy-1, Symbol: Tg (APPSwe,tauP301L) 1Lfa, MGI ID: 2672831) × (Promoter: Endogenous, Symbol: Psen1tm1Mpm, MGI ID: 1930937)
The 3×Tg-AD mouse model, developed by Oddo et al. (2003), shows progressive cognitive impairments starting at a young age. The first deficits observed in this model are those of associative learning deficits, which begin between 3 and 5 months of age. These are then followed by deficits in spatial working memory at 6 months of age in the MWM task. Both Y-maze alternation and contextual fear conditioning impairment follow a similar temporal time course. Then deficits in recognition memory present themselves between 9 and 11 months of age. Finally, reference memory impairment in the Barnes maze task is observed at 12 months of age.
Temporal Progression of AD-Like Non-Cognitive Behavioral Abnormalities Seen in the Commonly Used Mouse Models
While most AD research has focused on the neurobiological mechanisms underlying the cognitive deficits seen in AD pathogenesis, there is a wide range of non-cognitive neuropsychiatric symptoms also associated with the disease. Indeed, these non-cognitive symptoms are seen as a very important concern among the family members of the patients and caregivers alike (Tan et al., 2005). These non-cognitive symptoms are often more difficult to deal with, as they compose important sources of distress and psychological burden on the family members/caregivers and can drastically affect the quality of life of patients by leading to institutionalization (Hope et al., 1998; Shin et al., 2005). Non-cognitive neuropsychological symptoms associated with AD include activity disturbances, affective disturbances, aggression, stereotypic behavior, circadian rhythm disturbances, and anxiety (Ancoli-Israel et al., 1989; Okawa et al., 1991; Vitiello et al., 1992; Bliwise, 1994; Satlin et al., 1995; Van Someren et al., 1996; Hope et al., 1998; Harper et al., 2004; Shin et al., 2005; Tan et al., 2005). While there has been less emphasis placed on the modeling of these non-cognitive neuropsychological disturbances in murine models of AD, several of the commonly used mouse models of AD do show a number of these disruptions. Non-cognitive symptoms associated with AD shown in the mouse include increased locomotor activity (Dodart et al., 1999; King et al., 1999; Arendash et al., 2001b; Dumont et al., 2004; Hyde et al., 2005; Cheng et al., 2007; Gil-Bea et al., 2007; Pietropaolo et al., 2008; Sanchez-Mejia et al., 2008; Ambree et al., 2009; Cisse et al., 2011; Mori et al., 2013), anxiety (Moechars et al., 1996, 1999; Lalonde et al., 2003, 2004; Gil-Bea et al., 2007; Reiserer et al., 2007; Lassalle et al., 2008; Espana et al., 2010; Bedrosian et al., 2011; Cisse et al., 2011; Murakami et al., 2011; Filali et al., 2012), circadian disturbances (Huitron-Resendiz et al., 2002; Vloeberghs et al., 2004; Wisor et al., 2005; Ambree et al., 2006; Sterniczuk et al., 2010; Bedrosian et al., 2011), and increased aggression (Moechars et al., 1996, 1998; Van Dorpe et al., 2000; Ambree et al., 2006; Vloeberghs et al., 2006; Pugh et al., 2007; Alexander et al., 2011).
Locomotor Activity
Most of the commonly used AD mouse models exhibit increased locomotor activity (Dodart et al., 1999; King et al., 1999; Arendash et al., 2001b; Dumont et al., 2004; Hyde et al., 2005; Cheng et al., 2007; Gil-Bea et al., 2007; Pietropaolo et al., 2008; Sanchez-Mejia et al., 2008; Ambree et al., 2009; Cisse et al., 2011; Mori et al., 2013). These disturbances include hyperactivity, stereotypic behaviors, and home cage activity disturbances (Dodart et al., 1999; Janus and Westaway, 2001; Auld et al., 2002; Dumont et al., 2004; Hyde et al., 2005; Cheng et al., 2007; Gil-Bea et al., 2007; Gimenez-Llort et al., 2007; Pietropaolo et al., 2008; Cisse et al., 2011) and have been linked to altered APP metabolism, amyloid levels, and disease progression (Van Someren et al., 1996; Harper et al., 2004). These activity disturbances do not seem to be constant, but rather present themselves with more severity at different times of the day. For example, in the TgCRND8 mice, the deficits seem most severe near the end of their wake cycle and less severe at other times of the day (Ambree et al., 2006). Further, these disturbances increase with the age of the animals and with the severity of the disease. Likewise, in the APP23 mice there have been reports of increased activity in the second half of the nocturnal phase (end of the active phase of the wake cycle) (Vloeberghs et al., 2004). These increased activity disturbances at the end of the activity cycle have been suggested to be similar to the exacerbation of activity behavioral symptoms observed in human AD patients late in the afternoon and evening time termed sundowning syndrome (Vitiello et al., 1992; Bliwise, 1994). Other AD mouse models that show activity disturbances are the PDAPP, TG2576, J20, APP + PS1 [Tg2576 + PS1 (M146L)], and 3×Tg-AD mouse models of AD. The onset of the disturbances is different for each model, with the J20 and the TgCRND8 mice developing disruptions earliest (approximately 1 month of age), followed by PDAPP and TG2576 (approximately 3 months of age), and the PS1 [Tg2576 + PS1 (M146L)], 3×TgAD, and APP23 mice developing last (approximately 6–9 months of age) (Dodart et al., 1999; King et al., 1999; Arendash et al., 2001b; Dumont et al., 2004; Hyde et al., 2005; Cheng et al., 2007; Gil-Bea et al., 2007; Gimenez-Llort et al., 2007; Pietropaolo et al., 2008; Sanchez-Mejia et al., 2008; Ambree et al., 2009; Harris et al., 2010; Cisse et al., 2011; Mori et al., 2013).
Circadian Rhythm and Sleep Disruptions
Similar to the reported activity disturbances, circadian rhythm disruptions are also observed in many of the AD mouse models (Huitron-Resendiz et al., 2002; Vloeberghs et al., 2004; Wisor et al., 2005; Ambree et al., 2006; Sterniczuk et al., 2010; Bedrosian et al., 2011). Circadian rhythm disturbances have also been well described in human AD patients (Ancoli-Israel et al., 1989; Okawa et al., 1991; Vitiello et al., 1992; Van Someren et al., 1996; Auld et al., 2002; Harper et al., 2004). These disturbances are characterized by the AD patient's propensity to frequently awaken during the nighttime and to increase the amount of time slept during the day (Ancoli-Israel et al., 1989; Okawa et al., 1991). While this behavior is in itself a non-cognitive behavior, it has been reported to possibly have important effects on patient cognition (Smith, 1985; Graves et al., 2001). Several mouse models of AD also display sleep disruption. Other similarities between the sleep disturbances seen clinically and those seen in the AD mouse models are: (1) as the severity of the disease progresses, the worse the sleep disturbances become (Smith, 1985; Graves et al., 2001; Huitron-Resendiz et al., 2002, 2005) and (2) the greater the sleep disruptions are, the more severe the cognitive decline seems to be (Huitron-Resendiz et al., 2002, 2005).
Anxiety Disturbances
Anxiety disturbances have been reported in many of the AD mouse models (Moechars et al., 1996, 1999; Lalonde et al., 2003, 2004; Gil-Bea et al., 2007; Reiserer et al., 2007; Lassalle et al., 2008; Espana et al., 2010; Bedrosian et al., 2011; Cisse et al., 2011; Murakami et al., 2011; Filali et al., 2012). The prevailing thought is that these anxiolytic-like behaviors stem from disinhibitory tendencies resulting from the underlying AD pathology (Lalonde et al., 2003; Ognibene et al., 2005). Both APP function (as these anxiolytic behaviors are more common in APP transgenic mice) (Moechars et al., 1996, 1999; Lalonde et al., 2003, 2004; Lassalle et al., 2008; Murakami et al., 2011; Filali et al., 2012) and disruption of the cholinergic system (Apelt et al., 2002; Klingner et al., 2003; Luth et al., 2003) because of its well-known role in behavioral inhibition (and disruption in several of the AD mouse lines) have been proposed as underlying causes of this behavioral abnormality. The temporal time course of these anxiety-like disturbances can vary depending on the mouse model. For example, in some models the behavioral disturbances start early at 1–2 months of age or 3–6 months of age for the J20 and the APP/PS1 models, respectively (Lalonde et al., 2004; Reiserer et al., 2007; Harris et al., 2010). Other models such as the TG2576 develop the anxiety-like disturbances later at the age of 9–11 months of age (Gil-Bea et al., 2007). It is also important to note that not all murine AD models exhibit anxiety disturbances (Arendash et al., 2001b; Lalonde et al., 2002; Webster et al., 2013). Still, the majority of models do display these disturbances and the prevailing thought is that this behavioral phenotype of disinhibition may be akin to the disinhibition seen in AD patients (exemplified by unacceptable behavior and inappropriate euphoria) (Daffner et al., 1992; Chung and Cummings, 2000).
Aggressive Behaviors
Increased aggressive behaviors are another common behavioral symptom of AD and present themselves in as much as 65% of AD patients (Burns et al., 1990b). As with other non-cognitive behavioral symptoms of AD, increased aggression can be emotionally stressful to both the patient and caregivers (Murman et al., 2002a,b). The exact mechanism that underlies these increased aggressive behaviors is not known but proposed mechanisms deal with dysregulation of different neurotransmitter systems such as serotonin, norepinephrine, dopamine, and GABA (Arsland, 1995; Meltzer et al., 1998). Many AD mouse models also display increased aggressive behavior (Moechars et al., 1996, 1998; Van Dorpe et al., 2000; Ambree et al., 2006; Vloeberghs et al., 2006; Pugh et al., 2007; Alexander et al., 2011). For example, in the TG2576 mouse increased aggressive behaviors display themselves in both the frequency of attacks on other home cage mice as well as on the latency to first attack when interacting with a novel mouse (Alexander et al., 2011). The APP23 mouse model of AD also shows increased aggressive behaviors (Vloeberghs et al., 2006). These aggressive disruptions appear to develop later than the onset of cognitive deficits in this model (Kelly et al., 2003; Van Dam et al., 2003; Vloeberghs et al., 2006). The aggression alterations in the APP23 model appear by 6 months of age (after amyloid pathology and behavioral deficits) and seem to remain relatively constant throughout the rest of the course of disease (Vloeberghs et al., 2006). This suggests that perhaps aggressive deficits correlate best with moderate to severe stages of the disease, which is also what is observed clinically in human AD (Senanarong et al., 2004).
Depressive Symptoms
Depressive symptoms/behaviors are a very common comorbidity with AD. The exact prevalence of this comorbidity is not known but is believed to range from as low as 2% to as high as 85% (variability likely due to methods of assessment, diagnostic criteria, stage of AD participants, and other factors) (Mendez et al., 1990; Burns et al., 1990a; Migliorelli et al., 1995a,b; Devanand et al., 1996; Cummings, 2000; Apostolova and Cummings, 2008). Numerous meta-analysis studies have linked depression and AD (Chen et al., 1999; Charlson and Peterson, 2002; Ownby et al., 2006; Lenoir et al., 2011; Diniz et al., 2013), and several even consider late-life depression a significant risk factor for future development of AD (Butters et al., 2000, 2008; Diniz et al., 2013). Generally, depressive symptoms precede the onset of AD (Devanand et al., 1996) and usually worsen with the progression of the disease (Mega et al., 1996). Similarly, patients with a history of depression prior to a diagnosis of AD are much more likely to experience depressive episodes in the course of AD (Pearlson et al., 1990; Strauss and Ogrocki, 1996). Despite this well documented connection between depression and AD and having a wide range of potentially applicable tools to study depression in animal models (Seligman et al., 1975; Jolly et al., 1999; Song and Leonard, 2005; Flint and Shifman, 2008; Nestler and Hyman, 2010), very little work has been devoted to determining the range of depressive behavioral symptoms in the commonly used mouse models of AD. Depressive-like behaviors have been reported in at least one mouse model of AD (Filali et al., 2009), and likely exist in many of the other commonly used models.
Conclusions
No one animal model fully replicates the pathogenesis of AD, but rather only model different aspects of the disease. Consequently, no one model recapitulates all of the cognitive deficits observed in human AD. Further, the anatomical makeup and cognitive ability of mice make it difficult to model all of the intricacies of higher-order cognitive function exclusive to humans. Instead, each mouse model allows us insight into different aspects of cognition related to AD. Several important points should be taken away from the preceding discussion of the temporal development of cognitive deficits in the various mouse models of AD. First, the temporal time course and progression of cognitive deficits in a specific cognitive domain/behavioral task can be quite different among the different mouse models. Investigators should use careful forethought in selection of an optimal model and planning experiments based on the progression of that model's specific deficits. Secondly, most models display deficits in spatial working memory earlier than the deficits in other cognitive domains. Similarly, most of the studies using mouse models of AD have focused on understanding/correcting the cognitive deficits associated with the disease. However, AD is not just a memory disorder, rather it is a complex disease with many different non-cognitive neuropsychiatric symptoms which are an important source of distress and a psychological burden on family members and caregivers alike. These non-cognitive symptoms are present across many of the different mouse models of AD and more emphasis should be placed on understanding/correcting these deficits, as well as the cognitive aspects of the disease. It is our hope that this comprehensive review of the spectrum of behavioral deficits present in commonly used AD mouse models and how well they model human cognitive and non-cognitive symptoms will assist investigators in selecting an appropriate mouse model to investigate different aspects of AD pathology and disease progression.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in our laboratories is supported by funds from the National Institutes of Health (National Institute on Aging, National Institute of Neurological Disorders and Stroke, National Institute of Child Health and Human Development, and National Institute of Nursing Research).
References
Abner, E. L., Kryscio, R. J., Schmitt, F. A., Santacruz, K. S., Jicha, G. A., Lin, Y., et al. (2011). “End-stage” neurofibrillary tangle pathology in preclinical Alzheimer's disease: fact or fiction? J. Alzheimers Dis. 25, 445–453. doi: 10.3233/JAD-2011-101980
Aevarsson, O., and Skoog, I. (2000). A longitudinal population study of the mini-mental state examination in the very old: relation to dementia and education. Dement. Geriatr. Cogn. Disord. 11, 166–175. doi: 10.1159/000017231
Alamed, J., Wilcock, D. M., Diamond, D. M., Gordon, M. N., and Morgan, D. (2006). Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc. 1, 1671–1679. doi: 10.1038/nprot.2006.275
Albert, M. S. (1996). Cognitive and neurobiologic markers of early Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 93, 13547–13551. doi: 10.1073/pnas.93.24.13547
Alexander, G., Hanna, A., Serna, V., Younkin, L., Younkin, S., and Janus, C. (2011). Increased aggression in males in transgenic Tg2576 mouse model of Alzheimer's disease. Behav. Brain Res. 216, 77–83. doi: 10.1016/j.bbr.2010.07.016
Ambree, O., Richter, H., Sachser, N., Lewejohann, L., Dere, E., De Souza Silva, M. A., et al. (2009). Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer's disease. Neurobiol. Aging 30, 1192–1204. doi: 10.1016/j.neurobiolaging.2007.11.010
Ambree, O., Touma, C., Gortz, N., Keyvani, K., Paulus, W., Palme, R., et al. (2006). Activity changes and marked stereotypic behavior precede Abeta pathology in TgCRND8 Alzheimer mice. Neurobiol. Aging 27, 955–964. doi: 10.1016/j.neurobiolaging.2005.05.009
Ancoli-Israel, S., Parker, L., Sinaee, R., Fell, R. L., and Kripke, D. F. (1989). Sleep fragmentation in patients from a nursing home. J. Gerontol. 44, M18–M21. doi: 10.1093/geronj/44.1.M18
Angelo, M., Plattner, F., Irvine, E. E., and Giese, K. P. (2003). Improved reversal learning and altered fear conditioning in transgenic mice with regionally restricted p25 expression. Eur. J. Neurosci. 18, 423–431. doi: 10.1046/j.1460-9568.2003.02746.x
Apelt, J., Kumar, A., and Schliebs, R. (2002). Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res. 953, 17–30. doi: 10.1016/S0006-8993(02)03262-6
Apostolova, L. G., and Cummings, J. L. (2008). Neuropsychiatric manifestations in mild cognitive impairment: a systematic review of the literature. Dement. Geriatr. Cogn. Disord. 25, 115–126. doi: 10.1159/000112509
Arendash, G. W., Gordon, M. N., Diamond, D. M., Austin, L. A., Hatcher, J. M., Jantzen, P., et al. (2001a). Behavioral assessment of Alzheimer's transgenic mice following long-term Abeta vaccination: task specificity and correlations between Abeta deposition and spatial memory. DNA Cell Biol. 20, 737–744. doi: 10.1089/10445490152717604
Arendash, G. W., King, D. L., Gordon, M. N., Morgan, D., Hatcher, J. M., Hope, C. E., et al. (2001b). Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 891, 42–53. doi: 10.1016/S0006-8993(00)03186-3
Arendash, G. W., Lewis, J., Leighty, R. E., Mcgowan, E., Cracchiolo, J. R., Hutton, M., et al. (2004). Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer's disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 1012, 29–41. doi: 10.1016/j.brainres.2004.02.081
Arnett, P. A., Rao, S. M., Bernardin, L., Grafman, J., Yetkin, F. Z., and Lobeck, L. (1994). Relationship between frontal lobe lesions and Wisconsin Card Sorting Test performance in patients with multiple sclerosis. Neurology 44, 420–425. doi: 10.1212/WNL.44.3_Part_1.420
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42, 631–639. doi: 10.1212/WNL.42.3.631
Arsenault, D., Julien, C., Tremblay, C., and Calon, F. (2011). DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS ONE 6:e17397. doi: 10.1371/journal.pone.0017397
Arsland, D. (1995). Aggressive-behavior in dementia and its pharmacological treatment - a review. Nord. J. Psychiatry 49, 111–118. doi: 10.3109/08039489509011892
Ashe, K. H., and Zahs, K. R. (2010). Probing the biology of Alzheimer's disease in mice. Neuron 66, 631–645. doi: 10.1016/j.neuron.2010.04.031
Auld, D. S., Kornecook, T. J., Bastianetto, S., and Quirion, R. (2002). Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68, 209–245. doi: 10.1016/S0301-0082(02)00079-5
Backman, L., Jones, S., Berger, A. K., Laukka, E. J., and Small, B. J. (2005). Cognitive impairment in preclinical Alzheimer's disease: a meta-analysis. Neuropsychology 19, 520–531. doi: 10.1037/0894-4105.19.4.520
Backman, L., Small, B. J., and Fratiglioni, L. (2001). Stability of the preclinical episodic memory deficit in Alzheimer's disease. Brain 124, 96–102. doi: 10.1093/brain/124.1.96
Baddeley, A., Logie, R., Bressi, S., Della Sala, S., and Spinnler, H. (1986). Dementia and working memory. Q. J. Exp. Psychol. A 38, 603–618. doi: 10.1080/14640748608401616
Baddeley, A. D., Baddeley, H. A., Bucks, R. S., and Wilcock, G. K. (2001). Attentional control in Alzheimer's disease. Brain 124, 1492–1508. doi: 10.1093/brain/124.8.1492
Baddeley, A. D., Bressi, S., Della Sala, S., Logie, R., and Spinnler, H. (1991). The decline of working memory in Alzheimer's disease. A longitudinal study. Brain 114(Pt 6), 2521–2542. doi: 10.1093/brain/114.6.2521
Bari, A., Dalley, J. W., and Robbins, T. W. (2008). The application of the 5-choice serial reaction time task for the assessment of visual attentional processes and impulse control in rats. Nat. Protoc. 3, 759–767. doi: 10.1038/nprot.2008.41
Barnes, C. A. (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 93, 74–104. doi: 10.1037/h0077579
Beach, T. G., Adler, C. H., Lue, L., Sue, L. I., Bachalakuri, J., Henry-Watson, J., et al. (2009). Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 117, 613–634. doi: 10.1007/s00401-009-0538-8
Beck, L. H., Bransome, E. D. Jr., Mirsky, A. F., Rosvold, H. E., and Sarason, I. (1956). A continuous performance test of brain damage. J. Consult. Psychol. 20, 343–350. doi: 10.1037/h0043220
Bedrosian, T. A., Herring, K. L., Weil, Z. M., and Nelson, R. J. (2011). Altered temporal patterns of anxiety in aged and amyloid precursor protein (APP) transgenic mice. Proc. Natl. Acad. Sci. USA. 108, 11686–11691. doi: 10.1073/pnas.1103098108
Benedict, R. H. B., and Groninger, L. (1994). Preliminary standardization and validation of a multiple-form test for the assessment of visuospatial memory. Arch. Clin. Neuropsychol. 9, 109–110.
Benedict, R. H. B., Schretlen, D., Groninger, L., and Brandt, J. (1998). Hopkins verbal learning test revised: normative data and analysis of inter-form and test-retest reliability. Clin. Neuropsychol. 12, 43–55. doi: 10.1076/clin.12.1.43.1726
Bennett, D. A., Schneider, J. A., Wilson, R. S., Bienias, J. L., and Arnold, S. E. (2004). Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch. Neurol. 61, 378–384. doi: 10.1001/archneur.61.3.378
Benton, A. L. (1992). Benton Visual Retention Test, 5th Edn. San Antonio, TX: The Psychological Corporation.
Berg, L., Mckeel, D. W. Jr., Miller, J. P., Storandt, M., Rubin, E. H., Morris, J. C., et al. (1998). Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch. Neurol. 55, 326–335. doi: 10.1001/archneur.55.3.326
Berlau, D. J., Kahle-Wrobleski, K., Head, E., Goodus, M., Kim, R., and Kawas, C. (2007). Dissociation of neuropathologic findings and cognition: case report of an apolipoprotein E epsilon2/epsilon2 genotype. Arch. Neurol. 64, 1193–1196. doi: 10.1001/archneur.64.8.1193
Berman, K. F., Ostrem, J. L., Randolph, C., Gold, J., Goldberg, T. E., Coppola, R., et al. (1995). Physiological activation of a cortical network during performance of the Wisconsin Card Sorting Test: a positron emission tomography study. Neuropsychologia 33, 1027–1046. doi: 10.1016/0028-3932(95)00035-2
Bernardo, A., Harrison, F. E., Mccord, M., Zhao, J., Bruchey, A., Davies, S. S., et al. (2009). Elimination of GD3 synthase improves memory and reduces amyloid-beta plaque load in transgenic mice. Neurobiol. Aging 30, 1777–1791. doi: 10.1016/j.neurobiolaging.2007.12.022
Bernick, C., Cummings, J., Raman, R., Sun, X., and Aisen, P. (2012). Age and rate of cognitive decline in Alzheimer disease: implications for clinical trials. Arch. Neurol. 69, 901–905. doi: 10.1001/archneurol.2011.3758
Bierer, L. M., Hof, P. R., Purohit, D. P., Carlin, L., Schmeidler, J., Davis, K. L., et al. (1995). Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch. Neurol. 52, 81–88. doi: 10.1001/archneur.1995.00540250089017
Billings, L. M., Oddo, S., Green, K. N., Mcgaugh, J. L., and Laferla, F. M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. doi: 10.1016/j.neuron.2005.01.040
Birrell, J. M., and Brown, V. J. (2000). Medial frontal cortex mediates perceptual attentional set shifting in the rat. J. Neurosci. 20, 4320–4324. Available online at: http://www.jneurosci.org/content/20/11/4320.long
Bissonette, G. B., Martins, G. J., Franz, T. M., Harper, E. S., Schoenbaum, G., and Powell, E. M. (2008). Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J. Neurosci. 28, 11124–11130. doi: 10.1523/JNEUROSCI.2820-08.2008
Blennow, K., and Zetterberg, H. (2013). The application of cerebrospinal fluid biomarkers in early diagnosis of Alzheimer disease. Med. Clin. North Am. 97, 369–376. doi: 10.1016/j.mcna.2012.12.012
Blessed, G., Tomlinson, B. E., and Roth, M. (1968). The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br. J. Psychiatry 114, 797–811. doi: 10.1192/bjp.114.512.797
Blodgett, H. C., and Mccutchan, K. (1947). Place versus response learning in the simple T-maze. J. Exp. Psychol. 37, 412–422. doi: 10.1037/h0059305
Bondi, M. W., Jak, A. J., Delano-Wood, L., Jacobson, M. W., Delis, D. C., and Salmon, D. P. (2008). Neuropsychological contributions to the early identification of Alzheimer's disease. Neuropsychol. Rev. 18, 73–90. doi: 10.1007/s11065-008-9054-1
Bondi, M. W., Salmon, D. P., Galasko, D., Thomas, R. G., and Thal, L. J. (1999). Neuropsychological function and apolipoprotein E genotype in the preclinical detection of Alzheimer's disease. Psychol. Aging 14, 295–303. doi: 10.1037/0882-7974.14.2.295
Bondi, M. W., Salmon, D. P., Monsch, A. U., Galasko, D., Butters, N., Klauber, M. R., et al. (1995). Episodic memory changes are associated with the APOE-epsilon 4 allele in nondemented older adults. Neurology 45, 2203–2206. doi: 10.1212/WNL.45.12.2203
Braak, H., and Del Tredici, K. (2011). The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 121, 171–181. doi: 10.1007/s00401-010-0789-4
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Brayne, C., Richardson, K., Matthews, F. E., Fleming, J., Hunter, S., Xuereb, J. H., et al. (2009). Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J. Alzheimers Dis. 18, 645–658. doi: 10.3233/JAD-2009-1182
Brigman, J. L., Bussey, T. J., Saksida, L. M., and Rothblat, L. A. (2005). Discrimination of multidimensional visual stimuli by mice: intra- and extradimensional shifts. Behav. Neurosci. 119, 839–842. doi: 10.1037/0735-7044.119.3.839
Bruce-Keller, A. J., Gupta, S., Knight, A. G., Beckett, T. L., Mcmullen, J. M., Davis, P. R., et al. (2011). Cognitive impairment in humanized APPxPS1 mice is linked to Abeta (1-42) and NOX activation. Neurobiol. Dis. 44, 317–326. doi: 10.1016/j.nbd.2011.07.012
Buccafusco, J. J., Terry, A. V. Jr., Webster, S. J., Martin, D., Hohnadel, E. J., Bouchard, K. A., et al. (2008). The scopolamine-reversal paradigm in rats and monkeys: the importance of computer-assisted operant-conditioning memory tasks for screening drug candidates. Psychopharmacology (Berl.) 199, 481–494. doi: 10.1007/s00213-007-0887-8
Budson, A. E., and Price, B. H. (2005). Memory dysfunction. N. Engl. J. Med. 352, 692–699. doi: 10.1056/NEJMra041071
Burns, A., Jacoby, R., and Levy, R. (1990a). Psychiatric phenomena in Alzheimer's disease. II: Disorders of perception. Br. J. Psychiatry 157, 76–81, 92–74.
Burns, A., Jacoby, R., and Levy, R. (1990b). Psychiatric phenomena in Alzheimer's disease. IV: Disorders of behaviour. Br. J. Psychiatry 157, 86–94. doi: 10.1192/bjp.157.1.86
Bussey, T. J., Holmes, A., Lyon, L., Mar, A. C., Mcallister, K. A., Nithianantharajah, J., et al. (2012). New translational assays for preclinical modelling of cognition in schizophrenia: the touchscreen testing method for mice and rats. Neuropharmacology 62, 1191–1203. doi: 10.1016/j.neuropharm.2011.04.011
Bussey, T. J., Muir, J. L., Everitt, B. J., and Robbins, T. W. (1997). Triple dissociation of anterior cingulate, posterior cingulate, and medial frontal cortices on visual discrimination tasks using a touchscreen testing procedure for the rat. Behav. Neurosci. 111, 920–936. doi: 10.1037/0735-7044.111.5.920
Butter, C. M. (1969). Perseveration in extinction and in discrimination reversal tasks following selective frontal ablations in Macaca mulatta. Physiol. Behav. 4, 163–171. doi: 10.1016/0031-9384(69)90075-4
Butters, M. A., Becker, J. T., Nebes, R. D., Zmuda, M. D., Mulsant, B. H., Pollock, B. G. 3rd., et al. (2000). Changes in cognitive functioning following treatment of late-life depression. Am. J. Psychiatry 157, 1949–1954. doi: 10.1176/appi.ajp.157.12.1949
Butters, M. A., Young, J. B., Lopez, O., Aizenstein, H. J., Mulsant, B. H., Reynolds, C. F., et al. (2008). Pathways linking late-life depression to persistent cognitive impairment and dementia. Dialogues Clin. Neurosci. 10, 345–357.
Campion, D., Dumanchin, C., Hannequin, D., Dubois, B., Belliard, S., Puel, M., et al. (1999). Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 65, 664–670. doi: 10.1086/302553
Cao, D., Lu, H., Lewis, T. L., and Li, L. (2007). Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 282, 36275–36282. doi: 10.1074/jbc.M703561200
Carli, M., Robbins, T. W., Evenden, J. L., and Everitt, B. J. (1983). Effects of lesions to ascending noradrenergic neurones on performance of a 5-choice serial reaction task in rats; implications for theories of dorsal noradrenergic bundle function based on selective attention and arousal. Behav. Brain Res. 9, 361–380. doi: 10.1016/0166-4328(83)90138-9
Carpenter, C. R., Bassett, E. R., Fischer, G. M., Shirshekan, J., Galvin, J. E., and Morris, J. C. (2011). Four sensitive screening tools to detect cognitive dysfunction in geriatric emergency department patients: brief Alzheimer's Screen, Short Blessed Test, Ottawa 3DY, and the caregiver-completed AD8. Acad. Emerg. Med. 18, 374–384. doi: 10.1111/j.1553-2712.2011.01040.x
Carroll, J. C., Rosario, E. R., Chang, L., Stanczyk, F. Z., Oddo, S., Laferla, F. M., et al. (2007). Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 27, 13357–13365. doi: 10.1523/JNEUROSCI.2718-07.2007
Chapman, P. F., White, G. L., Jones, M. W., Cooper-Blacketer, D., Marshall, V. J., Irizarry, M., et al. (1999). Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 2, 271–276. doi: 10.1038/6374
Charlson, M., and Peterson, J. C. (2002). Medical comorbidity and late life depression: what is known and what are the unmet needs? Biol. Psychiatry 52, 226–235. doi: 10.1016/S0006-3223(02)01422-1
Chartier-Harlin, M. C., Crawford, F., Houlden, H., Warren, A., Hughes, D., Fidani, L., et al. (1991). Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844–846. doi: 10.1038/353844a0
Chatfield, M., Matthews, F. E., Brayne, C., Medical Research Council Cognitive, F., and Ageing, S. (2007). Using the Mini-Mental State Examination for tracking cognition in the older population based on longitudinal data. J. Am. Geriatr. Soc. 55, 1066–1071. doi: 10.1111/j.1532-5415.2007.01216.x
Chen, G., Chen, K. S., Knox, J., Inglis, J., Bernard, A., Martin, S. J., et al. (2000). A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature 408, 975–979. doi: 10.1038/35046031
Chen, P., Ganguli, M., Mulsant, B. H., and Dekosky, S. T. (1999). The temporal relationship between depressive symptoms and dementia: a community-based prospective study. Arch. Gen. Psychiatry 56, 261–266. doi: 10.1001/archpsyc.56.3.261
Cheng, D., Low, J. K., Logge, W., Garner, B., and Karl, T. (2013). Novel behavioural characteristics of female APP/PS1DeltaE9 double transgenic mice. Behav. Brain Res. 260, 111–118. doi: 10.1016/j.bbr.2013.11.046
Cheng, I. H., Scearce-Levie, K., Legleiter, J., Palop, J. J., Gerstein, H., Bien-Ly, N., et al. (2007). Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 282, 23818–23828. doi: 10.1074/jbc.M701078200
Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. doi: 10.1074/jbc.M100710200
Chudasama, Y. (2011). Animal models of prefrontal-executive function. Behav. Neurosci. 125, 327–343. doi: 10.1037/a0023766
Chung, J. A., and Cummings, J. L. (2000). Neurobehavioral and neuropsychiatric symptoms in Alzheimer's disease: characteristics and treatment. Neurol. Clin. 18, 829–846. doi: 10.1016/S0733-8619(05)70228-0
Cisse, M., Sanchez, P. E., Kim, D. H., Ho, K., Yu, G. Q., and Mucke, L. (2011). Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 31, 10427–10431. doi: 10.1523/JNEUROSCI.1459-11.2011
Clayton, N. S., and Dickinson, A. (1998). Episodic-like memory during cache recovery by scrub jays. Nature 395, 272–274. doi: 10.1038/26216
Clayton, N. S., Griffiths, D. P., Emery, N. J., and Dickinson, A. (2001). Elements of episodic-like memory in animals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 1483–1491. doi: 10.1098/rstb.2001.0947
Clinton, L. K., Billings, L. M., Green, K. N., Caccamo, A., Ngo, J., Oddo, S., et al. (2007). Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol. Dis. 28, 76–82. doi: 10.1016/j.nbd.2007.06.013
Collie, A., and Maruff, P. (2000). The neuropsychology of preclinical Alzheimer's disease and mild cognitive impairment. Neurosci. Biobehav. Rev. 24, 365–374. doi: 10.1016/S0149-7634(00)00012-9
Connor, D. J., and Sabbagh, M. N. (2008). Administration and scoring variance on the ADAS-Cog. J. Alzheimers Dis. 15, 461–464.
Corcoran, K. A., Lu, Y., Turner, R. S., and Maren, S. (2002). Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer's disease. Learn. Mem. 9, 243–252. doi: 10.1101/lm.51002
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Crusio, W. E., Schwegler, H., and Brust, I. (1993). Covariations between hippocampal mossy fibres and working and reference memory in spatial and non-spatial radial maze tasks in mice. Eur. J. Neurosci. 5, 1413–1420. doi: 10.1111/j.1460-9568.1993.tb00927.x
Crystal, H. A., Dickson, D. W., Sliwinski, M. J., Lipton, R. B., Grober, E., Marks-Nelson, H., et al. (1993). Pathological markers associated with normal aging and dementia in the elderly. Ann. Neurol. 34, 566–573. doi: 10.1002/ana.410340410
Cummings, B. J., Pike, C. J., Shankle, R., and Cotman, C. W. (1996). Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol. Aging 17, 921–933. doi: 10.1016/S0197-4580(96)00170-4
Cummings, J. L. (2000). Cognitive and behavioral heterogeneity in Alzheimer's disease: seeking the neurobiological basis. Neurobiol. Aging 21, 845–861. doi: 10.1016/S0197-4580(00)00183-4
Curzon, P., Rustay, N. R., and Browman, K. E. (2009). “Cued and Contextual Fear Conditioning for Rodents,” in Methods of Behavior Analysis in Neuroscience, Chapter 2, 2nd Edn. ed J. J. Buccafusco (Boca Raton, FL: CRC Press), 26–49.
Daffner, K. R., Scinto, L. F., Weintraub, S., Guinessey, J. E., and Mesulam, M. M. (1992). Diminished curiosity in patients with probable Alzheimer's disease as measured by exploratory eye movements. Neurology 42, 320–328. doi: 10.1212/WNL.42.2.320
Daumas, S., Sandin, J., Chen, K. S., Kobayashi, D., Tulloch, J., Martin, S. J., et al. (2008). Faster forgetting contributes to impaired spatial memory in the PDAPP mouse: deficit in memory retrieval associated with increased sensitivity to interference? Learn. Mem. 15, 625–632. doi: 10.1101/lm.990208
Davis, K. E., Eacott, M. J., Easton, A., and Gigg, J. (2013a). Episodic-like memory is sensitive to both Alzheimer's-like pathological accumulation and normal ageing processes in mice. Behav. Brain Res. 254, 73–82. doi: 10.1016/j.bbr.2013.03.009
Davis, K. E., Easton, A., Eacott, M. J., and Gigg, J. (2013b). Episodic-like memory for what-where-which occasion is selectively impaired in the 3xTgAD mouse model of Alzheimer's disease. J. Alzheimers Dis. 33, 681–698. doi: 10.3233/JAD-2012-121543
Davis, P. C., Gearing, M., Gray, L., Mirra, S. S., Morris, J. C., Edland, S. D., et al. (1995). The CERAD experience, Part VIII: neuroimaging-neuropathology correlates of temporal lobe changes in Alzheimer's disease. Neurology 45, 178–179. doi: 10.1212/WNL.45.1.178
De Bruin, N. M., Fransen, F., Duytschaever, H., Grantham, C., and Megens, A. A. (2006). Attentional performance of (C57BL/6Jx129Sv) F2 mice in the five-choice serial reaction time task. Physiol. Behav. 89, 692–703. doi: 10.1016/j.physbeh.2006.08.009
Delis, D. C., Kaplan, E., Kramer, J. H., and Ober, B. A. (2000). California Verbal Learning Test - Second Edition (CVLT - II) Manual. San Antonio, TX: Psychological Corperation.
Devanand, D. P., Sano, M., Tang, M. X., Taylor, S., Gurland, B. J., Wilder, D., et al. (1996). Depressed mood and the incidence of Alzheimer's disease in the elderly living in the community. Arch. Gen. Psychiatry 53, 175–182. doi: 10.1001/archpsyc.1996.01830020093011
Devi, L., and Ohno, M. (2010). Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer's disease model mice. Eur. J. Neurosci. 31, 110–118. doi: 10.1111/j.1460-9568.2009.07031.x
Devi, L., and Ohno, M. (2012). Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice. Neurobiol. Dis. 45, 417–424. doi: 10.1016/j.nbd.2011.09.001
Diamond, D. M., Park, C. R., Heman, K. L., and Rose, G. M. (1999). Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus 9, 542–552.
Dickson, D. W., Crystal, H. A., Bevona, C., Honer, W., Vincent, I., and Davies, P. (1995). Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 16, 285–298. discussion: 298–304. doi: 10.1016/0197-4580(95)00013-5
Dineley, K. T., Xia, X., Bui, D., Sweatt, J. D., and Zheng, H. (2002). Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J. Biol. Chem. 277, 22768–22780. doi: 10.1074/jbc.M200164200
Ding, Y., Qiao, A., Wang, Z., Goodwin, J. S., Lee, E. S., Block, M. L., et al. (2008). Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. J. Neurosci. 28, 11622–11634. doi: 10.1523/JNEUROSCI.3153-08.2008
Diniz, B. S., Butters, M. A., Albert, S. M., Dew, M. A., and Reynolds, C. F. 3rd. (2013). Late-life depression and risk of vascular dementia and Alzheimer's disease: systematic review and meta-analysis of community-based cohort studies. Br. J. Psychiatry 202, 329–335. doi: 10.1192/bjp.bp.112.118307
Dodart, J. C., Bales, K. R., Gannon, K. S., Greene, S. J., Demattos, R. B., Mathis, C., et al. (2002). Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat. Neurosci. 5, 452–457. doi: 10.1038/nn842
Dodart, J. C., Meziane, H., Mathis, C., Bales, K. R., Paul, S. M., and Ungerer, A. (1999). Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav. Neurosci. 113, 982–990. doi: 10.1037/0735-7044.113.5.982
Dong, H., Csernansky, C. A., Martin, M. V., Bertchume, A., Vallera, D., and Csernansky, J. G. (2005). Acetylcholinesterase inhibitors ameliorate behavioral deficits in the Tg2576 mouse model of Alzheimer's disease. Psychopharmacology (Berl.) 181, 145–152. doi: 10.1007/s00213-005-2230-6
Drewe, E. A. (1974). The effect of type and area of brain lesion on Wisconsin card sorting test performance. Cortex 10, 159–170. doi: 10.1016/S0010-9452(74)80006-7
Du, H., Guo, L., Zhang, W., Rydzewska, M., and Yan, S. (2011). Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 32, 398–406. doi: 10.1016/j.neurobiolaging.2009.03.003
Dudchenko, P. A. (2004). An overview of the tasks used to test working memory in rodents. Neurosci. Biobehav. Rev. 28, 699–709. doi: 10.1016/j.neubiorev.2004.09.002
Dumont, M., Strazielle, C., Staufenbiel, M., and Lalonde, R. (2004). Spatial learning and exploration of environmental stimuli in 24-month-old female APP23 transgenic mice with the Swedish mutation. Brain Res. 1024, 113–121. doi: 10.1016/j.brainres.2004.07.052
Dunnett, S. B. (1993). The role and repair of forebrain cholinergic systems in short-term memory. Studies using the delayed matching-to-position task in rats. Adv. Neurol. 59, 53–65.
Duyckaerts, C., Bennecib, M., Grignon, Y., Uchihara, T., He, Y., Piette, F., et al. (1997). Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol. Aging 18, 267–273. doi: 10.1016/S0197-4580(97)80306-5
Duyckaerts, C., Colle, M. A., Dessi, F., Piette, F., and Hauw, J. J. (1998). Progression of Alzheimer histopathological changes. Acta Neurol. Belg. 98, 180–185.
Duyckaerts, C., Delaere, P., Hauw, J. J., Abbamondi-Pinto, A. L., Sorbi, S., Allen, I., et al. (1990). Rating of the lesions in senile dementia of the Alzheimer type: concordance between laboratories. A European multicenter study under the auspices of EURAGE. J. Neurol. Sci. 97, 295–323. doi: 10.1016/0022-510X(90)90226-D
Elder, G. A., Gama Sosa, M. A., and De Gasperi, R. (2010). Transgenic mouse models of Alzheimer's disease. Mt. Sinai J. Med. 77, 69–81. doi: 10.1002/msj.20159
Emilien, G., Durlach, C., Minaker, K. L., Winblad, B., Gauthier, S., and Maloteaux, J.-M. (2004). Alzheimer Disease: Neuropsychology and Pharmacology. Basel: Birkhauser Verlag.
Ennaceur, A., and Delacour, J. (1988). A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 31, 47–59. doi: 10.1016/0166-4328(88)90157-X
Epis, R., Gardoni, F., Marcello, E., Genazzani, A., Canonico, P. L., and Di Luca, M. (2010). Searching for new animal models of Alzheimer's disease. Eur. J. Pharmacol. 626, 57–63. doi: 10.1016/j.ejphar.2009.10.020
Eriksen, J. L., and Janus, C. G. (2007). Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav. Genet. 37, 79–100. doi: 10.1007/s10519-006-9118-z
Escribano, L., Simon, A. M., Perez-Mediavilla, A., Salazar-Colocho, P., Del Rio, J., and Frechilla, D. (2009). Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer's disease mouse model. Biochem. Biophys. Res. Commun. 379, 406–410. doi: 10.1016/j.bbrc.2008.12.071
Espana, J., Gimenez-Llort, L., Valero, J., Minano, A., Rabano, A., Rodriguez-Alvarez, J., et al. (2010). Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer's disease transgenic mice. Biol. Psychiatry 67, 513–521. doi: 10.1016/j.biopsych.2009.06.015
Fang, F., Chen, X., Huang, T., Lue, L. F., Luddy, J. S., and Yan, S. S. (2012). Multi-faced neuroprotective effects of Ginsenoside Rg1 in an Alzheimer mouse model. Biochim. Biophys. Acta 1822, 286–292. doi: 10.1016/j.bbadis.2011.10.004
Fanselow, M. S. (1980). Conditioned and unconditional components of post-shock freezing. Pavlov. J. Biol. Sci. 15, 177–182.
Filali, M., Lalonde, R., and Rivest, S. (2009). Cognitive and non-cognitive behaviors in an APPswe/PS1 bigenic model of Alzheimer's disease. Genes Brain Behav. 8, 143–148. doi: 10.1111/j.1601-183X.2008.00453.x
Filali, M., Lalonde, R., Theriault, P., Julien, C., Calon, F., and Planel, E. (2012). Cognitive and non-cognitive behaviors in the triple transgenic mouse model of Alzheimer's disease expressing mutated APP, PS1, and Mapt (3xTg-AD). Behav. Brain Res. 234, 334–342. doi: 10.1016/j.bbr.2012.07.004
Flicker, C., Bartus, R. T., Crook, T. H., and Ferris, S. H. (1984). Effects of aging and dementia upon recent visuospatial memory. Neurobiol. Aging 5, 275–283. doi: 10.1016/0197-4580(84)90003-4
Flint, J., and Shifman, S. (2008). Animal models of psychiatric disease. Curr. Opin. Genet. Dev. 18, 235–240. doi: 10.1016/j.gde.2008.07.002
Flood, D. G., Reaume, A. G., Dorfman, K. S., Lin, Y. G., Lang, D. M., Trusko, S. P., et al. (2002). FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol. Aging 23, 335–348. doi: 10.1016/S0197-4580(01)00330-X
Folstein, M. F., Folstein, S. E., and Mchugh, P. R. (1975). “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198. doi: 10.1016/0022-3956(75)90026-6
Fuld, P. (1978). Psychological Testing in the Differential Diagnosis of the Dementias. New York, NY: Raven Press.
Galvan, V., Gorostiza, O. F., Banwait, S., Ataie, M., Logvinova, A. V., Sitaraman, S., et al. (2006). Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. U.S.A. 103, 7130–7135. doi: 10.1073/pnas.0509695103
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. doi: 10.1038/373523a0
Giannakopoulos, P., Kovari, E., Gold, G., Von Gunten, A., Hof, P. R., and Bouras, C. (2009). Pathological substrates of cognitive decline in Alzheimer's disease. Front. Neurol. Neurosci. 24, 20–29. doi: 10.1159/000197881
Gibson, A. P., Bettinger, T. L., Patel, N. C., and Crismon, M. L. (2006). Atomoxetine versus stimulants for treatment of attention deficit/hyperactivity disorder. Ann. Pharmacother. 40, 1134–1142. doi: 10.1345/aph.1G582
Gil-Bea, F. J., Aisa, B., Schliebs, R., and Ramirez, M. J. (2007). Increase of locomotor activity underlying the behavioral disinhibition in tg2576 mice. Behav. Neurosci. 121, 340–344. doi: 10.1037/0735-7044.121.2.340
Gimenez-Llort, L., Blazquez, G., Canete, T., Johansson, B., Oddo, S., Tobena, A., et al. (2007). Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci. Biobehav. Rev. 31, 125–147. doi: 10.1016/j.neubiorev.2006.07.007
Glickman, S. E., and Jensen, G. D. (1961). The effects of hunger and thirst on Y-maze exploration. J. Comp. Physiol. Psychol. 54, 83–85. doi: 10.1037/h0049301
Gold, G., Bouras, C., Kovari, E., Canuto, A., Glaria, B. G., Malky, A., et al. (2000). Clinical validity of Braak neuropathological staging in the oldest-old. Acta Neuropathol. 99, 579–582. discussion: 583–574. doi: 10.1007/s004010051163
Gordon, M. N., King, D. L., Diamond, D. M., Jantzen, P. T., Boyett, K. V., Hope, C. E., et al. (2001). Correlation between cognitive deficits and Abeta deposits in transgenic APP+PS1 mice. Neurobiol. Aging 22, 377–385. doi: 10.1016/S0197-4580(00)00249-9
Gortz, N., Lewejohann, L., Tomm, M., Ambree, O., Keyvani, K., Paulus, W., et al. (2008). Effects of environmental enrichment on exploration, anxiety, and memory in female TgCRND8 Alzheimer mice. Behav. Brain Res. 191, 43–48. doi: 10.1016/j.bbr.2008.03.006
Graves, L., Pack, A., and Abel, T. (2001). Sleep and memory: a molecular perspective. Trends Neurosci. 24, 237–243. doi: 10.1016/S0166-2236(00)01744-6
Griffiths, D. P., and Clayton, N. S. (2001). Testing episodic memory in animals: a new approach. Physiol. Behav. 73, 755–762. doi: 10.1016/S0031-9384(01)00532-7
Grober, E., Dickson, D., Sliwinski, M. J., Buschke, H., Katz, M., Crystal, H., et al. (1999). Memory and mental status correlates of modified Braak staging. Neurobiol. Aging 20, 573–579. doi: 10.1016/S0197-4580(99)00063-9
Guillozet, A. L., Weintraub, S., Mash, D. C., and Mesulam, M. M. (2003). Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 60, 729–736. doi: 10.1001/archneur.60.5.729
Halagappa, V. K., Guo, Z., Pearson, M., Matsuoka, Y., Cutler, R. G., Laferla, F. M., et al. (2007). Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 26, 212–220. doi: 10.1016/j.nbd.2006.12.019
Hall, A. M., and Roberson, E. D. (2012). Mouse models of Alzheimer's disease. Brain Res. Bull. 88, 3–12. doi: 10.1016/j.brainresbull.2011.11.017
Hanna, A., Horne, P., Yager, D., Eckman, C., Eckman, E., and Janus, C. (2009). Amyloid beta and impairment in multiple memory systems in older transgenic APP TgCRND8 mice. Genes Brain Behav. 8, 676–684. doi: 10.1111/j.1601-183X.2009.00510.x
Hanna, A., Iremonger, K., Das, P., Dickson, D., Golde, T., and Janus, C. (2012). Age-related increase in amyloid plaque burden is associated with impairment in conditioned fear memory in CRND8 mouse model of amyloidosis. Alzheimers Res. Ther. 4, 21. doi: 10.1186/alzrt124
Harper, D. G., Stopa, E. G., Mckee, A. C., Satlin, A., Fish, D., and Volicer, L. (2004). Dementia severity and Lewy bodies affect circadian rhythms in Alzheimer disease. Neurobiol. Aging 25, 771–781. doi: 10.1016/j.neurobiolaging.2003.04.009
Harris, J. A., Devidze, N., Halabisky, B., Lo, I., Thwin, M. T., Yu, G. Q., et al. (2010). Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer's disease are independent of caspase cleavage of the amyloid precursor protein. J. Neurosci. 30, 372–381. doi: 10.1523/JNEUROSCI.5341-09.2010
Hartman, R. E., Izumi, Y., Bales, K. R., Paul, S. M., Wozniak, D. F., and Holtzman, D. M. (2005). Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer's disease. J. Neurosci. 25, 6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005
Heneka, M. T., Ramanathan, M., Jacobs, A. H., Dumitrescu-Ozimek, L., Bilkei-Gorzo, A., Debeir, T., et al. (2006). Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 26, 1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006
Hochgrafe, K., Sydow, A., and Mandelkow, E. M. (2013). Regulatable transgenic mouse models of Alzheimer disease: onset, reversibility and spreading of Tau pathology. FEBS J. 280, 4371–4381. doi: 10.1111/febs.12250
Hodges, J. R., and Patterson, K. (1995). Is semantic memory consistently impaired early in the course of Alzheimer's disease? Neuroanatomical and diagnostic implications. Neuropsychologia 33, 441–459. doi: 10.1016/0028-3932(94)00127-B
Hodges, J. R., Salmon, D. P., and Butters, N. (1992). Semantic memory impairment in Alzheimer's disease: failure of access or degraded knowledge? Neuropsychologia 30, 301–314. doi: 10.1016/0028-3932(92)90104-T
Hof, P. R., Vogt, B. A., Bouras, C., and Morrison, J. H. (1997). Atypical form of Alzheimer's disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res. 37, 3609–3625. doi: 10.1016/S0042-6989(96)00240-4
Holcomb, L., Gordon, M. N., Mcgowan, E., Yu, X., Benkovic, S., Jantzen, P., et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100. doi: 10.1038/nm0198-097
Holcomb, L. A., Gordon, M. N., Jantzen, P., Hsiao, K., Duff, K., and Morgan, D. (1999). Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav. Genet. 29, 177–185. doi: 10.1023/A:1021691918517
Holtzer, R., Irizarry, M. C., Sanders, J., Hyman, B. T., Wegesin, D. J., Riba, A., et al. (2006). Relation of quantitative indexes of concurrent alpha-synuclein abnormalities to clinical outcome in autopsy-proven Alzheimer disease. Arch. Neurol. 63, 226–230. doi: 10.1001/archneur.63.2.226
Hope, T., Keene, J., Gedling, K., Fairburn, C. G., and Jacoby, R. (1998). Predictors of institutionalization for people with dementia living at home with a carer. Int. J. Geriatr. Psychiatry 13, 682–690.
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. doi: 10.1126/science.274.5284.99
Huang, S. M., Mouri, A., Kokubo, H., Nakajima, R., Suemoto, T., Higuchi, M., et al. (2006). Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 281, 17941–17951. doi: 10.1074/jbc.M601372200
Huff, F. J., Becker, J. T., Belle, S. H., Nebes, R. D., Holland, A. L., and Boller, F. (1987). Cognitive deficits and clinical diagnosis of Alzheimer's disease. Neurology 37, 1119–1124. doi: 10.1212/WNL.37.7.1119
Huitron-Resendiz, S., Sanchez-Alavez, M., and Criado, J. R. (2005). Sleep-wake states in transgenic mouse models overexpressing the human beta-amyloid precursor protein. Am. J. Alzheimers Dis. Other Demen. 20, 87–90. doi: 10.1177/153331750502000204
Huitron-Resendiz, S., Sanchez-Alavez, M., Gallegos, R., Berg, G., Crawford, E., Giacchino, J. L., et al. (2002). Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Res. 928, 126–137. doi: 10.1016/S0006-8993(01)03373-X
Humby, T., Laird, F. M., Davies, W., and Wilkinson, L. S. (1999). Visuospatial attentional functioning in mice: interactions between cholinergic manipulations and genotype. Eur. J. Neurosci. 11, 2813–2823. doi: 10.1046/j.1460-9568.1999.00701.x
Hyde, L. A., Kazdoba, T. M., Grilli, M., Lozza, G., Brusa, R., Zhang, Q., et al. (2005). Age-progressing cognitive impairments and neuropathology in transgenic CRND8 mice. Behav. Brain Res. 160, 344–355. doi: 10.1016/j.bbr.2004.12.017
Hyde, L. A., Sherman, G. F., and Denenberg, V. H. (2000). Non-spatial water radial-arm maze learning in mice. Brain Res. 863, 151–159. doi: 10.1016/S0006-8993(00)02113-2
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Hyman, B. T., and Trojanowski, J. Q. (1997). Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 1095–1097. doi: 10.1097/00005072-199710000-00002
Izquierdo, A., and Jentsch, J. D. (2012). Reversal learning as a measure of impulsive and compulsive behavior in addictions. Psychopharmacology (Berl.) 219, 607–620. doi: 10.1007/s00213-011-2579-7
Jacobson, M. W., Delis, D. C., Bondi, M. W., and Salmon, D. P. (2002). Do neuropsychological tests detect preclinical Alzheimer's disease: individual-test versus cognitive-discrepancy score analyses. Neuropsychology 16, 132–139. doi: 10.1037/0894-4105.16.2.132
Jacqmin-Gadda, H., Fabrigoule, C., Commenges, D., and Dartigues, J. F. (1997). A 5-year longitudinal study of the Mini-Mental State Examination in normal aging. Am. J. Epidemiol. 145, 498–506. doi: 10.1093/oxfordjournals.aje.a009137
Jankowsky, J. L., Slunt, H. H., Ratovitski, T., Jenkins, N. A., Copeland, N. G., and Borchelt, D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165. doi: 10.1016/S1389-0344(01)00067-3
Janus, C. (2004). Search strategies used by APP transgenic mice during navigation in the Morris water maze. Learn. Mem. 11, 337–346. doi: 10.1101/lm.70104
Janus, C., Pearson, J., Mclaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., et al. (2000). A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408, 979–982. doi: 10.1038/35050110
Janus, C., and Westaway, D. (2001). Transgenic mouse models of Alzheimer's disease. Physiol. Behav. 73, 873–886. doi: 10.1016/S0031-9384(01)00524-8
Jicha, G. A., Abner, E. L., Schmitt, F. A., Kryscio, R. J., Riley, K. P., Cooper, G. E., et al. (2012). Preclinical AD Workgroup staging: pathological correlates and potential challenges. Neurobiol. Aging 33, 622.e621–622.e616. doi: 10.1016/j.neurobiolaging.2011.02.018
Jolly, D. C., Richards, J. B., and Seiden, L. S. (1999). Serotonergic mediation of DRL 72s behavior: receptor subtype involvement in a behavioral screen for antidepressant drugs. Biol. Psychiatry 45, 1151–1162. doi: 10.1016/S0006-3223(98)00014-6
Kaplan, E. F., Goodglass, H., and Weintraub, S. (1978). The Boston Naming Test. Philadelphia, PA: Lea & Febiger.
Karl, T., Bhatia, S., Cheng, D., Kim, W. S., and Garner, B. (2012). Cognitive phenotyping of amyloid precursor protein transgenic J20 mice. Behav. Brain Res. 228, 392–397. doi: 10.1016/j.bbr.2011.12.021
Karran, E., Mercken, M., and De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Katzman, R., Brown, T., Fuld, P., Peck, A., Schechter, R., and Schimmel, H. (1983). Validation of a short Orientation-Memory-Concentration Test of cognitive impairment. Am. J. Psychiatry 140, 734–739.
Kelly, P. H., Bondolfi, L., Hunziker, D., Schlecht, H. P., Carver, K., Maguire, E., et al. (2003). Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol. Aging 24, 365–378. doi: 10.1016/S0197-4580(02)00098-2
King, D. L., and Arendash, G. W. (2002). Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol. Behav. 75, 627–642. doi: 10.1016/S0031-9384(02)00639-X
King, D. L., Arendash, G. W., Crawford, F., Sterk, T., Menendez, J., and Mullan, M. J. (1999). Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer's disease. Behav. Brain Res. 103, 145–162. doi: 10.1016/S0166-4328(99)00037-6
Klingner, M., Apelt, J., Kumar, A., Sorger, D., Sabri, O., Steinbach, J., et al. (2003). Alterations in cholinergic and non-cholinergic neurotransmitter receptor densities in transgenic Tg2576 mouse brain with beta-amyloid plaque pathology. Int. J. Dev. Neurosci. 21, 357–369. doi: 10.1016/j.ijdevneu.2003.08.001
Knafo, S., Venero, C., Merino-Serrais, P., Fernaud-Espinosa, I., Gonzalez-Soriano, J., Ferrer, I., et al. (2009). Morphological alterations to neurons of the amygdala and impaired fear conditioning in a transgenic mouse model of Alzheimer's disease. J. Pathol. 219, 41–51. doi: 10.1002/path.2565
Knopman, D. S., and Ryberg, S. (1989). A verbal memory test with high predictive accuracy for dementia of the Alzheimer type. Arch. Neurol. 46, 141–145. doi: 10.1001/archneur.1989.00520380041011
Kobayashi, D. T., and Chen, K. S. (2005). Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav. 4, 173–196. doi: 10.1111/j.1601-183X.2005.00124.x
Koepsell, T. D., Kurland, B. F., Harel, O., Johnson, E. A., Zhou, X. H., and Kukull, W. A. (2008). Education, cognitive function, and severity of neuropathology in Alzheimer disease. Neurology 70, 1732–1739. doi: 10.1212/01.wnl.0000284603.85621.aa
Kraybill, M. L., Larson, E. B., Tsuang, D. W., Teri, L., Mccormick, W. C., Bowen, J. D., et al. (2005). Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64, 2069–2073. doi: 10.1212/01.WNL.0000165987.89198.65
Kryscio, R. J., Schmitt, F. A., Salazar, J. C., Mendiondo, M. S., and Markesbery, W. R. (2006). Risk factors for transitions from normal to mild cognitive impairment and dementia. Neurology 66, 828–832. doi: 10.1212/01.wnl.0000203264.71880.45
Lalonde, R., Dumont, M., Staufenbiel, M., Sturchler-Pierrat, C., and Strazielle, C. (2002). Spatial learning, exploration, anxiety, and motor coordination in female APP23 transgenic mice with the Swedish mutation. Brain Res. 956, 36–44. doi: 10.1016/S0006-8993(02)03476-5
Lalonde, R., Fukuchi, K., and Strazielle, C. (2012). Neurologic and motor dysfunctions in APP transgenic mice. Rev. Neurosci. 23, 363–379. doi: 10.1515/revneuro-2012-0041
Lalonde, R., Kim, H. D., and Fukuchi, K. (2004). Exploratory activity, anxiety, and motor coordination in bigenic APPswe + PS1/DeltaE9 mice. Neurosci. Lett. 369, 156–161. doi: 10.1016/j.neulet.2004.07.069
Lalonde, R., Kim, H. D., Maxwell, J. A., and Fukuchi, K. (2005). Exploratory activity and spatial learning in 12-month-old APP(695) SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci. Lett. 390, 87–92. doi: 10.1016/j.neulet.2005.08.028
Lalonde, R., Lewis, T. L., Strazielle, C., Kim, H., and Fukuchi, K. (2003). Transgenic mice expressing the betaAPP695SWE mutation: effects on exploratory activity, anxiety, and motor coordination. Brain Res. 977, 38–45. doi: 10.1016/S0006-8993(03)02694-5
Lambon Ralph, M. A., Patterson, K., Graham, N., Dawson, K., and Hodges, J. R. (2003). Homogeneity and heterogeneity in mild cognitive impairment and Alzheimer's disease: a cross-sectional and longitudinal study of 55 cases. Brain 126, 2350–2362. doi: 10.1093/brain/awg236
Lambourne, S. L., Humby, T., Isles, A. R., Emson, P. C., Spillantini, M. G., and Wilkinson, L. S. (2007). Impairments in impulse control in mice transgenic for the human FTDP-17 tauV337M mutation are exacerbated by age. Hum. Mol. Genet. 16, 1708–1719. doi: 10.1093/hmg/ddm119
Lange, K. L., Bondi, M. W., Salmon, D. P., Galasko, D., Delis, D. C., Thomas, R. G., et al. (2002). Decline in verbal memory during preclinical Alzheimer's disease: examination of the effect of APOE genotype. J. Int. Neuropsychol. Soc. 8, 943–955. doi: 10.1017/S1355617702870096
Larrieu, S., Letenneur, L., Orgogozo, J. M., Fabrigoule, C., Amieva, H., Le Carret, N., et al. (2002). Incidence and outcome of mild cognitive impairment in a population-based prospective cohort. Neurology 59, 1594–1599. doi: 10.1212/01.WNL.0000034176.07159.F8
Lassalle, J. M., Halley, H., Daumas, S., Verret, L., and Frances, B. (2008). Effects of the genetic background on cognitive performances of TG2576 mice. Behav. Brain Res. 191, 104–110. doi: 10.1016/j.bbr.2008.03.017
Lenoir, H., Dufouil, C., Auriacombe, S., Lacombe, J. M., Dartigues, J. F., Ritchie, K., et al. (2011). Depression history, depressive symptoms, and incident dementia: the 3C Study. J. Alzheimers Dis. 26, 27–38. doi: 10.3233/JAD-2011-101614
Locascio, J. J., Growdon, J. H., and Corkin, S. (1995). Cognitive test performance in detecting, staging, and tracking Alzheimer's disease. Arch. Neurol. 52, 1087–1099. doi: 10.1001/archneur.1995.00540350081020
Lovasic, L., Bauschke, H., and Janus, C. (2005). Working memory impairment in a transgenic amyloid precursor protein TgCRND8 mouse model of Alzheimer's disease. Genes Brain Behav. 4, 197–208. doi: 10.1111/j.1601-183X.2004.00104.x
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304, 448–452. doi: 10.1126/science.1091230
Luth, H. J., Apelt, J., Ihunwo, A. O., Arendt, T., and Schliebs, R. (2003). Degeneration of beta-amyloid-associated cholinergic structures in transgenic APP SW mice. Brain Res. 977, 16–22. doi: 10.1016/S0006-8993(03)02658-1
Ma, T., Du, X., Pick, J. E., Sui, G., Brownlee, M., and Klann, E. (2012). Glucagon-like peptide-1 cleavage product GLP-1(9-36) amide rescues synaptic plasticity and memory deficits in Alzheimer's disease model mice. J. Neurosci. 32, 13701–13708. doi: 10.1523/JNEUROSCI.2107-12.2012
Marchese, M., Cowan, D., Head, E., Ma, D., Karimi, K., Ashthorpe, V., et al. (2013). Autoimmune Manifestations in the 3xTg-AD Model of Alzheimer's Disease. J. Alzheimers Dis. 39, 191–210. doi: 10.3233/JAD-131490
Markesbery, W. R., Schmitt, F. A., Kryscio, R. J., Davis, D. G., Smith, C. D., and Wekstein, D. R. (2006). Neuropathologic substrate of mild cognitive impairment. Arch. Neurol. 63, 38–46. doi: 10.1001/archneur.63.1.38
Mckee, A. C., Kosik, K. S., and Kowall, N. W. (1991). Neuritic pathology and dementia in Alzheimer's disease. Ann. Neurol. 30, 156–165. doi: 10.1002/ana.410300206
Mckhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Mega, M. S., Cummings, J. L., Fiorello, T., and Gornbein, J. (1996). The spectrum of behavioral changes in Alzheimer's disease. Neurology 46, 130–135. doi: 10.1212/WNL.46.1.130
Meilandt, W. J., Yu, G. Q., Chin, J., Roberson, E. D., Palop, J. J., Wu, T., et al. (2008). Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 28, 5007–5017. doi: 10.1523/JNEUROSCI.0590-08.2008
Meltzer, C. C., Smith, G., Dekosky, S. T., Pollock, B. G., Mathis, C. A., Moore, R. Y., et al. (1998). Serotonin in aging, late-life depression, and Alzheimer's disease: the emerging role of functional imaging. Neuropsychopharmacology 18, 407–430. doi: 10.1016/S0893-133X(97)00194-2
Mendez, M. F., Martin, R. J., Smyth, K. A., and Whitehouse, P. J. (1990). Psychiatric symptoms associated with Alzheimer's disease. J. Neuropsychiatry Clin. Neurosci. 2, 28–33.
Mickes, L., Wixted, J. T., Fennema-Notestine, C., Galasko, D., Bondi, M. W., Thal, L. J., et al. (2007). Progressive impairment on neuropsychological tasks in a longitudinal study of preclinical Alzheimer's disease. Neuropsychology 21, 696–705. doi: 10.1037/0894-4105.21.6.696
Migliorelli, R., Petracca, G., Teson, A., Sabe, L., Leiguarda, R., and Starkstein, S. E. (1995a). Neuropsychiatric and neuropsychological correlates of delusions in Alzheimer's disease. Psychol. Med. 25, 505–513. doi: 10.1017/S0033291700033420
Migliorelli, R., Teson, A., Sabe, L., Petracchi, M., Leiguarda, R., and Starkstein, S. E. (1995b). Prevalence and correlates of dysthymia and major depression among patients with Alzheimer's disease. Am. J. Psychiatry 152, 37–44.
Moechars, D., Dewachter, I., Lorent, K., Reverse, D., Baekelandt, V., Naidu, A., et al. (1999). Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 274, 6483–6492. doi: 10.1074/jbc.274.10.6483
Moechars, D., Gilis, M., Kuiperi, C., Laenen, I., and Van Leuven, F. (1998). Aggressive behaviour in transgenic mice expressing APP is alleviated by serotonergic drugs. Neuroreport 9, 3561–3564. doi: 10.1097/00001756-199811160-00004
Moechars, D., Lorent, K., De Strooper, B., Dewachter, I., and Van Leuven, F. (1996). Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 15, 1265–1274.
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Morgan, D. (2003). Learning and memory deficits in APP transgenic mouse models of amyloid deposition. Neurochem. Res. 28, 1029–1034. doi: 10.1023/A:1023255106106
Morgan, D., Diamond, D. M., Gottschall, P. E., Ugen, K. E., Dickey, C., Hardy, J., et al. (2000). A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408, 982–985. doi: 10.1038/35050116
Mori, T., Koyama, N., Guillot-Sestier, M. V., Tan, J., and Town, T. (2013). Ferulic acid is a nutraceutical beta-secretase modulator that improves behavioral impairment and alzheimer-like pathology in transgenic mice. PLoS ONE 8:e55774. doi: 10.1371/journal.pone.0055774
Morris, J. C., Heyman, A., Mohs, R. C., Hughes, J. P., Van Belle, G., Fillenbaum, G., et al. (1989). The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology 39, 1159–1165. doi: 10.1212/WNL.39.9.1159
Morris, R. G. (2001). Episodic-like memory in animals: psychological criteria, neural mechanisms and the value of episodic-like tasks to investigate animal models of neurodegenerative disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 1453–1465. doi: 10.1098/rstb.2001.0945
Morris, R. G., Garrud, P., Rawlins, J. N., and O'keefe, J. (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683. doi: 10.1038/297681a0
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. Available online at: http://www.jneurosci.org/content/20/11/4050.long
Muir, J. L. (1996). Attention and stimulus processing in the rat. Brain Res. Cogn. Brain Res. 3, 215–225. doi: 10.1016/0926-6410(96)00008-0
Mungas, D., Reed, B. R., Ellis, W. G., and Jagust, W. J. (2001). The effects of age on rate of progression of Alzheimer disease and dementia with associated cerebrovascular disease. Arch. Neurol. 58, 1243–1247. doi: 10.1001/archneur.58.8.1243
Murakami, K., Yokoyama, S., Murata, N., Ozawa, Y., Irie, K., Shirasawa, T., et al. (2011). Insulin receptor mutation results in insulin resistance and hyperinsulinemia but does not exacerbate Alzheimer's-like phenotypes in mice. Biochem. Biophys. Res. Commun. 409, 34–39. doi: 10.1016/j.bbrc.2011.04.101
Murman, D. L., Chen, Q., Colucci, P. M., Colenda, C. C., Gelb, D. J., and Liang, J. (2002a). Comparison of healthcare utilization and direct costs in three degenerative dementias. Am. J. Geriatr. Psychiatry 10, 328–336. doi: 10.1097/00019442-200205000-00013
Murman, D. L., Chen, Q., Powell, M. C., Kuo, S. B., Bradley, C. J., and Colenda, C. C. (2002b). The incremental direct costs associated with behavioral symptoms in AD. Neurology 59, 1721–1729. doi: 10.1212/01.WNL.0000036904.73393.E4
Murray, M. E., Graff-Radford, N. R., Ross, O. A., Petersen, R. C., Duara, R., and Dickson, D. W. (2011). Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10, 785–796. doi: 10.1016/S1474-4422(11)70156-9
Musilli, M., Nicolia, V., Borrelli, S., Scarpa, S., and Diana, G. (2013). Behavioral effects of Rho GTPase modulation in a model of Alzheimer's disease. Behav. Brain Res. 237, 223–229. doi: 10.1016/j.bbr.2012.09.043
Nagy, Z., Esiri, M. M., Jobst, K. A., Morris, J. H., King, E. M., Mcdonald, B., et al. (1995). Relative roles of plaques and tangles in the dementia of Alzheimer's disease: correlations using three sets of neuropathological criteria. Dementia 6, 21–31.
Nagy, Z., Hindley, N. J., Braak, H., Braak, E., Yilmazer-Hanke, D. M., Schultz, C., et al. (1999). The progression of Alzheimer's disease from limbic regions to the neocortex: clinical, radiological and pathological relationships. Dement. Geriatr. Cogn. Disord. 10, 115–120. doi: 10.1159/000017111
Nasreddine, Z. S., Phillips, N. A., Bédirian, V., Charbonneau, S., Whitehead, V., Collin, I., et al. (2005). The Montreal Cognitive Assessment (MoCA): a brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 53, 695–699. doi: 10.1111/j.1532-5415.2005.53221.x
Nelson, P. T., Abner, E. L., Scheff, S. W., Schmitt, F. A., Kryscio, R. J., Jicha, G. A., et al. (2009a). Alzheimer's-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci. Lett. 450, 336–339. doi: 10.1016/j.neulet.2008.11.006
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Nelson, P. T., Braak, H., and Markesbery, W. R. (2009b). Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J. Neuropathol. Exp. Neurol. 68, 1–14. doi: 10.1097/NEN.0b013e3181919a48
Nelson, P. T., Jicha, G. A., Schmitt, F. A., Liu, H., Davis, D. G., Mendiondo, M. S., et al. (2007a). Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J. Neuropathol. Exp. Neurol. 66, 1136–1146. doi: 10.1097/nen.0b013e31815c5efb
Nelson, R. L., Guo, Z., Halagappa, V. M., Pearson, M., Gray, A. J., Matsuoka, Y., et al. (2007b). Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp. Neurol. 205, 166–176. doi: 10.1016/j.expneurol.2007.01.037
Nestler, E. J., and Hyman, S. E. (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169. doi: 10.1038/nn.2647
Nilsson, L. N., Arendash, G. W., Leighty, R. E., Costa, D. A., Low, M. A., Garcia, M. F., et al. (2004). Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol. Aging 25, 1153–1167. doi: 10.1016/j.neurobiolaging.2003.12.011
O'leary, T. P., and Brown, R. E. (2009). Visuo-spatial learning and memory deficits on the Barnes maze in the 16-month-old APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav. Brain Res. 201, 120–127. doi: 10.1016/j.bbr.2009.01.039
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/S0896-6273(03)00434-3
Ognibene, E., Middei, S., Daniele, S., Adriani, W., Ghirardi, O., Caprioli, A., et al. (2005). Aspects of spatial memory and behavioral disinhibition in Tg2576 transgenic mice as a model of Alzheimer's disease. Behav. Brain Res. 156, 225–232. doi: 10.1016/j.bbr.2004.05.028
Ohno, M., Chang, L., Tseng, W., Oakley, H., Citron, M., Klein, W. L., et al. (2006). Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur. J. Neurosci. 23, 251–260. doi: 10.1111/j.1460-9568.2005.04551.x
Ohno, M., Sametsky, E. A., Younkin, L. H., Oakley, H., Younkin, S. G., Citron, M., et al. (2004). BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41, 27–33. doi: 10.1016/S0896-6273(03)00810-9
Okawa, M., Mishima, K., Hishikawa, Y., Hozumi, S., Hori, H., and Takahashi, K. (1991). Circadian rhythm disorders in sleep-waking and body temperature in elderly patients with dementia and their treatment. Sleep 14, 478–485.
Olton, D. S., Becker, J. T., and Handelmann, G. E. (1979). Hippocampus, Space, and Memory. Behav. Brain Sci. 2, 313–322. doi: 10.1017/S0140525X00062713
Olton, D. S., and Feustle, W. A. (1981). Hippocampal function required for nonspatial working memory. Exp. Brain Res. 41, 380–389.
Olton, D. S., and Samuelson, R. J. (1976). Remembrance of Places Passed - Spatial Memory in Rats. J. Exp. Psychol. Anim. Behav. Process. 2, 97–116. doi: 10.1037/0097-7403.2.2.97
Oules, B., Del Prete, D., Greco, B., Zhang, X., Lauritzen, I., Sevalle, J., et al. (2012). Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 32, 11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012
Ownby, R. L., Crocco, E., Acevedo, A., John, V., and Loewenstein, D. (2006). Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 63, 530–538. doi: 10.1001/archpsyc.63.5.530
Palop, J. J., Jones, B., Kekonius, L., Chin, J., Yu, G. Q., Raber, J., et al. (2003). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 100, 9572–9577. doi: 10.1073/pnas.1133381100
Papadopoulos, P., Rosa-Neto, P., Rochford, J., and Hamel, E. (2013). Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer's disease with combined amyloid-beta and cerebrovascular pathology. PLoS ONE 8:e68612. doi: 10.1371/journal.pone.0068612
Parasuraman, R., and Nestor, P. (1993). Attention and driving. Assessment in elderly individuals with dementia. Clin. Geriatr. Med. 9, 377–387.
Park, J. H., Widi, G. A., Gimbel, D. A., Harel, N. Y., Lee, D. H., and Strittmatter, S. M. (2006). Subcutaneous Nogo receptor removes brain amyloid-beta and improves spatial memory in Alzheimer's transgenic mice. J. Neurosci. 26, 13279–13286. doi: 10.1523/JNEUROSCI.4504-06.2006
Pattij, T., Janssen, M. C., Loos, M., Smit, A. B., Schoffelmeer, A. N., and Van Gaalen, M. M. (2007). Strain specificity and cholinergic modulation of visuospatial attention in three inbred mouse strains. Genes Brain Behav. 6, 579–587. doi: 10.1111/j.1601-183X.2006.00284.x
Pearlson, G. D., Ross, C. A., Lohr, W. D., Rovner, B. W., Chase, G. A., and Folstein, M. F. (1990). Association between family history of affective-disorder and the depressive syndrome of Alzheimers-disease. Am. J. Psychiatry 147, 452–456.
Perry, R. J., and Hodges, J. R. (1999). Attention and executive deficits in Alzheimer's disease. A critical review. Brain 122(Pt 3), 383–404. doi: 10.1093/brain/122.3.383
Perry, R. J., Watson, P., and Hodges, J. R. (2000). The nature and staging of attention dysfunction in early (minimal and mild) Alzheimer's disease: relationship to episodic and semantic memory impairment. Neuropsychologia 38, 252–271. doi: 10.1016/S0028-3932(99)00079-2
Pietropaolo, S., Feldon, J., and Yee, B. K. (2008). Age-dependent phenotypic characteristics of a triple transgenic mouse model of Alzheimer disease. Behav. Neurosci. 122, 733–747. doi: 10.1037/a0012520
Platt, T. L., Reeves, V. L., and Murphy, M. P. (2013). Transgenic models of Alzheimer's disease: better utilization of existing models through viral transgenesis. Biochim. Biophys. Acta 1832, 1437–1448. doi: 10.1016/j.bbadis.2013.04.017
Prut, L., Abramowski, D., Krucker, T., Levy, C. L., Roberts, A. J., Staufenbiel, M., et al. (2007). Aged APP23 mice show a delay in switching to the use of a strategy in the Barnes maze. Behav. Brain Res. 179, 107–110. doi: 10.1016/j.bbr.2007.01.017
Pugh, P. L., Richardson, J. C., Bate, S. T., Upton, N., and Sunter, D. (2007). Non-cognitive behaviours in an APP/PS1 transgenic model of Alzheimer's disease. Behav. Brain Res. 178, 18-28. doi: 10.1016/j.bbr.2006.11.044
Reiserer, R. S., Harrison, F. E., Syverud, D. C., and Mcdonald, M. P. (2007). Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 6, 54–65. doi: 10.1111/j.1601-183X.2006.00221.x
Richter, H., Ambree, O., Lewejohann, L., Herring, A., Keyvani, K., Paulus, W., et al. (2008). Wheel-running in a transgenic mouse model of Alzheimer's disease: protection or symptom? Behav. Brain Res. 190, 74–84. doi: 10.1016/j.bbr.2008.02.005
Riley, K. P., Jicha, G. A., Davis, D., Abner, E. L., Cooper, G. E., Stiles, N., et al. (2011). Prediction of preclinical Alzheimer's disease: longitudinal rates of change in cognition. J. Alzheimers Dis. 25, 707–717. doi: 10.3233/JAD-2011-102133
Riley, K. P., Snowdon, D. A., and Markesbery, W. R. (2002). Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann. Neurol. 51, 567–577. doi: 10.1002/ana.10161
Robbins, T. W. (1996). Dissociating executive functions of the prefrontal cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 1463–1470. discussion: 1470–1461. doi: 10.1098/rstb.1996.0131
Robbins, T. W. (2002). The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology (Berl.) 163, 362–380. doi: 10.1007/s00213-002-1154-7
Robinson, A. L., Heaton, R. K., Lehman, R. A., and Stilson, D. W. (1980). The utility of the Wisconsin Card Sorting Test in detecting and localizing frontal lobe lesions. J. Consult. Clin. Psychol. 48, 605–614. doi: 10.1037/0022-006X.48.5.605
Robinson, J. K., and Crawley, J. N. (1993). Intraventricular galanin impairs delayed nonmatching-to-sample performance in rats. Behav. Neurosci. 107, 458–467. doi: 10.1037/0735-7044.107.3.458
Robinson, J. L., Geser, F., Corrada, M. M., Berlau, D. J., Arnold, S. E., Lee, V. M., et al. (2011). Neocortical and hippocampal amyloid-beta and tau measures associate with dementia in the oldest-old. Brain 134, 3708–3715. doi: 10.1093/brain/awr308
Romberg, C., Bussey, T. J., and Saksida, L. M. (2013a). Paying more attention to attention: towards more comprehensive cognitive translation using mouse models of Alzheimer's disease. Brain Res. Bull. 92, 49–55. doi: 10.1016/j.brainresbull.2012.02.007
Romberg, C., Horner, A. E., Bussey, T. J., and Saksida, L. M. (2013b). A touch screen-automated cognitive test battery reveals impaired attention, memory abnormalities, and increased response inhibition in the TgCRND8 mouse model of Alzheimer's disease. Neurobiol. Aging 34, 731–744. doi: 10.1016/j.neurobiolaging.2012.08.006
Romberg, C., Mattson, M. P., Mughal, M. R., Bussey, T. J., and Saksida, L. M. (2011). Impaired attention in the 3xTgAD mouse model of Alzheimer's disease: rescue by donepezil (Aricept). J. Neurosci. 31, 3500–3507. doi: 10.1523/JNEUROSCI.5242-10.2011
Rosen, C., Hansson, O., Blennow, K., and Zetterberg, H. (2013). Fluid biomarkers in Alzheimer's disease - current concepts. Mol. Neurodegener. 8:20. doi: 10.1186/1750-1326-8-20
Rosen, C., and Zetterberg, H. (2013). Cerebrospinal fluid biomarkers for pathological processes in Alzheimer's disease. Curr. Opin. Psychiatry 26, 276–282. doi: 10.1097/YCO.0b013e32835f6747
Rosen, W. G., Mohs, R. C., and Davis, K. L. (1984). A new rating scale for Alzheimer's disease. Am. J. Psychiatry 141, 1356–1364.
Sabbagh, M. N., Cooper, K., Delange, J., Stoehr, J. D., Thind, K., Lahti, T., et al. (2010). Functional, global and cognitive decline correlates to accumulation of Alzheimer's pathology in MCI and AD. Curr. Alzheimer Res. 7, 280–286. doi: 10.2174/156720510791162340
Sabbagh, M. N., Corey-Bloom, J., Tiraboschi, P., Thomas, R., Masliah, E., and Thal, L. J. (1999). Neurochemical markers do not correlate with cognitive decline in the Lewy body variant of Alzheimer disease. Arch. Neurol. 56, 1458–1461. doi: 10.1001/archneur.56.12.1458
Sadowski, M., Pankiewicz, J., Scholtzova, H., Ji, Y., Quartermain, D., Jensen, C. H., et al. (2004). Amyloid-beta deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 63, 418–428. Available online at: http://journals.lww.com/jneuropath/Pages/default.aspx
Salmon, D. P., and Bondi, M. W. (2009). Neuropsychological assessment of dementia. Annu. Rev. Psychol. 60, 257–282. doi: 10.1146/annurev.psych.57.102904.190024
Salmon, D. P., Heindel, W. C., and Lange, K. L. (1999). Differential decline in word generation from phonemic and semantic categories during the course of Alzheimer's disease: implications for the integrity of semantic memory. J. Int. Neuropsychol. Soc. 5, 692–703. doi: 10.1017/S1355617799577126
Salmon, D. P., Thomas, R. G., Pay, M. M., Booth, A., Hofstetter, C. R., Thal, L. J., et al. (2002). Alzheimer's disease can be accurately diagnosed in very mildly impaired individuals. Neurology 59, 1022–1028. doi: 10.1212/WNL.59.7.1022
Sanchez-Mejia, R. O., Newman, J. W., Toh, S., Yu, G. Q., Zhou, Y., Halabisky, B., et al. (2008). Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat. Neurosci. 11, 1311–1318. doi: 10.1038/nn.2213
Santacruz, K. S., Sonnen, J. A., Pezhouh, M. K., Desrosiers, M. F., Nelson, P. T., and Tyas, S. L. (2011). Alzheimer disease pathology in subjects without dementia in 2 studies of aging: the Nun Study and the Adult Changes in Thought Study. J. Neuropathol. Exp. Neurol. 70, 832–840. doi: 10.1097/NEN.0b013e31822e8ae9
Satlin, A., Volicer, L., Stopa, E. G., and Harper, D. (1995). Circadian locomotor activity and core-body temperature rhythms in Alzheimer's disease. Neurobiol. Aging 16, 765–771. doi: 10.1016/0197-4580(95)00059-N
Saura, C. A., Chen, G., Malkani, S., Choi, S. Y., Takahashi, R. H., Zhang, D., et al. (2005). Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J. Neurosci. 25, 6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. doi: 10.1038/22124
Schmitt, F. A., Davis, D. G., Wekstein, D. R., Smith, C. D., Ashford, J. W., and Markesbery, W. R. (2000). “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology 55, 370–376. doi: 10.1212/WNL.55.3.370
Schmitt, F. A., and Wichems, C. H. (2006). A systematic review of assessment and treatment of moderate to severe Alzheimer's disease. Prim. Care Companion J. Clin. Psychiatry 8, 158–159. doi: 10.4088/PCC.v08n0306
Seligman, M. E., Rosellini, R. A., and Kozak, M. J. (1975). Learned helplessness in the rat: time course, immunization, and reversibility. J. Comp. Physiol. Psychol. 88, 542–547. doi: 10.1037/h0076431
Senanarong, V., Cummings, J. L., Fairbanks, L., Mega, M., Masterman, D. M., O'connor, S. M., et al. (2004). Agitation in Alzheimer's disease is a manifestation of frontal lobe dysfunction. Dement. Geriatr. Cogn. Disord. 17, 14–20. doi: 10.1159/000074080
Shin, I. S., Carter, M., Masterman, D., Fairbanks, L., and Cummings, J. L. (2005). Neuropsychiatric symptoms and quality of life in Alzheimer disease. Am. J. Geriatr. Psychiatry 13, 469–474. doi: 10.1097/00019442-200506000-00005
Shukla, V., Zheng, Y. L., Mishra, S. K., Amin, N. D., Steiner, J., Grant, P., et al. (2013). A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer's disease phenotypes in model mice. FASEB J. 27, 174–186. doi: 10.1096/fj.12-217497
Silver, M. H., Newell, K., Brady, C., Hedley-White, E. T., and Perls, T. T. (2002). Distinguishing between neurodegenerative disease and disease-free aging: correlating neuropsychological evaluations and neuropathological studies in centenarians. Psychosom. Med. 64, 493–501.
Simard, M., and Van Reekum, R. (1999). Memory assessment in studies of cognition-enhancing drugs for Alzheimer's disease. Drugs Aging 14, 197–230. doi: 10.2165/00002512-199914030-00004
Simon, A. M., Schiapparelli, L., Salazar-Colocho, P., Cuadrado-Tejedor, M., Escribano, L., Lopez De Maturana, R., et al. (2009). Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol. Dis. 33, 369–378. doi: 10.1016/j.nbd.2008.11.005
Smith, C. (1985). Sleep states and learning: a review of the animal literature. Neurosci. Biobehav. Rev. 9, 157–168. doi: 10.1016/0149-7634(85)90042-9
Smith, G. E., Pankratz, V. S., Negash, S., Machulda, M. M., Petersen, R. C., Boeve, B. F., et al. (2007). A plateau in pre-Alzheimer memory decline: evidence for compensatory mechanisms? Neurology 69, 133–139. doi: 10.1212/01.wnl.0000265594.23511.16
Snyderman, D., and Rovner, B. (2009). Mental status exam in primary care: a review. Am. Fam. Physician 80, 809–814. Available online at: http://www.aafp.org/afp/2009/1015/p809
Song, C., and Leonard, B. E. (2005). The olfactory bulbectomised rat as a model of depression. Neurosci. Biobehav. Rev. 29, 627–647. doi: 10.1016/j.neubiorev.2005.03.010
Sood, A., Warren Beach, J., Webster, S. J., Terry, A. V., and Buccafusco, J. J. (2007). The effects of JWB1-84-1 on memory-related task performance by amyloid Abeta transgenic mice and by young and aged monkeys. Neuropharmacology 53, 588–600. doi: 10.1016/j.neuropharm.2007.06.028
Spreen, O., and Strauss, E. (1998). A compendium of neuropsychological tests, 2nd Edn. New York, NY: Oxford University Press.
Sterniczuk, R., Dyck, R. H., Laferla, F. M., and Antle, M. C. (2010). Characterization of the 3xTg-AD mouse model of Alzheimer's disease: Part 1. Circadian changes. Brain Res. 1348, 139–148. doi: 10.1016/j.brainres.2010.05.013
Stewart, S., Cacucci, F., and Lever, C. (2011). Which memory task for my mouse? A systematic review of spatial memory performance in the Tg2576 Alzheimer's mouse model. J. Alzheimers Dis. 26, 105–126. doi: 10.3233/JAD-2011-101827
Stopford, C. L., Thompson, J. C., Neary, D., Richardson, A. M., and Snowden, J. S. (2012). Working memory, attention, and executive function in Alzheimer's disease and frontotemporal dementia. Cortex 48, 429–446. doi: 10.1016/j.cortex.2010.12.002
Storandt, M. (1991). Neuropsychological assessment in Alzheimer's disease. Exp. Aging Res. 17, 100–101.
Storandt, M., Grant, E. A., Miller, J. P., and Morris, J. C. (2006). Longitudinal course and neuropathologic outcomes in original vs revised MCI and in pre-MCI. Neurology 67, 467–473. doi: 10.1212/01.wnl.0000228231.26111.6e
Storandt, M., and Hill, R. D. (1989). Very mild senile dementia of the Alzheimer type. II. Psychometric test performance. Arch. Neurol. 46, 383–386. doi: 10.1001/archneur.1989.00520400037017
Storandt, M., Kaskie, B., and Von Dras, D. D. (1998). Temporal memory for remote events in healthy aging and dementia. Psychol. Aging 13, 4–7. doi: 10.1037/0882-7974.13.1.4
Strauss, M. E., and Ogrocki, P. K. (1996). Confirmation of an association between family history of affective disorder and the depressive syndrome in Alzheimer's disease. Am. J. Psychiatry 153, 1340–1342.
Sturchler-Pierrat, C., Abramowski, D., Duke, M., Wiederhold, K. H., Mistl, C., Rothacher, S., et al. (1997). Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292. doi: 10.1073/pnas.94.24.13287
Tan, L. L., Wong, H. B., and Allen, H. (2005). The impact of neuropsychiatric symptoms of dementia on distress in family and professional caregivers in Singapore. Int. Psychogeriatr. 17, 253–263. doi: 10.1017/S1041610205001523
Thal, D. R., Rub, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Thibault, O., Pancani, T., Landfield, P. W., and Norris, C. M. (2012). Reduction in neuronal L-type calcium channel activity in a double knock-in mouse model of Alzheimer's disease. Biochim. Biophys. Acta 1822, 546–549. doi: 10.1016/j.bbadis.2012.01.004
Tiraboschi, P., Hansen, L. A., Alford, M., Merdes, A., Masliah, E., Thal, L. J., et al. (2002). Early and widespread cholinergic losses differentiate dementia with Lewy bodies from Alzheimer disease. Arch. Gen. Psychiatry 59, 946–951. doi: 10.1001/archpsyc.59.10.946
Tiraboschi, P., Hansen, L. A., Thal, L. J., and Corey-Bloom, J. (2004). The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62, 1984–1989. doi: 10.1212/01.WNL.0000129697.01779.0A
Tohda, C., Urano, T., Umezaki, M., Nemere, I., and Kuboyama, T. (2012). Diosgenin is an exogenous activator of 1,25D(3)-MARRS/Pdia3/ERp57 and improves Alzheimer's disease pathologies in 5XFAD mice. Sci. Rep. 2:535. doi: 10.1038/srep00535
Tomlinson, B. E., Blessed, G., and Roth, M. (1970). Observations on the brains of demented old people. J. Neurol. Sci. 11, 205–242. doi: 10.1016/0022-510X(70)90063-8
Trinchese, F., Liu, S., Battaglia, F., Walter, S., Mathews, P. M., and Arancio, O. (2004). Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814. doi: 10.1002/ana.20101
Tulving, E., and Markowitsch, H. J. (1998). Episodic and declarative memory: role of the hippocampus. Hippocampus 8, 198–204.
Tuokko, H., Morris, C., and Ebert, P. (2005). Mild cognitive impairment and everyday functioning in older adults. Neurocase 11, 40–47. doi: 10.1080/13554790490896802
Twamley, E. W., Ropacki, S. A. L., and Bondi, M. W. (2006). Neuropsychological and neuroimaging changes in preclinical Alzheimer's disease. J. Int. Neuropsychol. Soc. 12, 707–735. doi: 10.1017/S1355617706060863
Urano, T., and Tohda, C. (2010). Icariin improves memory impairment in Alzheimer's disease model mice (5xFAD) and attenuates amyloid beta-induced neurite atrophy. Phytother. Res. 24, 1658–1663. doi: 10.1002/ptr.3183
Van Dam, D., D'hooge, R., Staufenbiel, M., Van Ginneken, C., Van Meir, F., and De Deyn, P. P. (2003). Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 17, 388–396. doi: 10.1046/j.1460-9568.2003.02444.x
Van Der Poel, A. M. (1967). Ethological study of the behaviour of the albino rat in a passive-avoidance test. Acta Physiol. Pharmacol. Neerl. 14, 503–505.
Van Dorpe, J., Smeijers, L., Dewachter, I., Nuyens, D., Spittaels, K., Van Den Haute, C., et al. (2000). Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am. J. Pathol. 157, 1283–1298. doi: 10.1016/S0002-9440(10)64644-5
Van Someren, E. J., Hagebeuk, E. E., Lijzenga, C., Scheltens, P., De Rooij, S. E., Jonker, C., et al. (1996). Circadian rest-activity rhythm disturbances in Alzheimer's disease. Biol. Psychiatry 40, 259–270. doi: 10.1016/0006-3223(95)00370-3
Vanderwolf, C. H. (1964). Effect of combined medial thalamic and septal lesions on active-avoidance behavior. J. Comp. Physiol. Psychol. 58, 31–37. doi: 10.1037/h0042400
Vitiello, M. V., Bliwise, D. L., and Prinz, P. N. (1992). Sleep in Alzheimer's disease and the sundown syndrome. Neurology 42, 83–93. discussion: 93–84.
Vloeberghs, E., Van Dam, D., Coen, K., Staufenbiel, M., and De Deyn, P. P. (2006). Aggressive male APP23 mice modeling behavioral alterations in dementia. Behav. Neurosci. 120, 1380–1383. doi: 10.1037/0735-7044.120.6.1380
Vloeberghs, E., Van Dam, D., Engelborghs, S., Nagels, G., Staufenbiel, M., and De Deyn, P. P. (2004). Altered circadian locomotor activity in APP23 mice: a model for BPSD disturbances. Eur. J. Neurosci. 20, 2757–2766. doi: 10.1111/j.1460-9568.2004.03755.x
Volianskis, A., Kostner, R., Molgaard, M., Hass, S., and Jensen, M. S. (2010). Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1deltaE9-deleted transgenic mice model of ss-amyloidosis. Neurobiol. Aging 31, 1173–1187. doi: 10.1016/j.neurobiolaging.2008.08.005
Webster, S. J., Bachstetter, A. D., and Van Eldik, L. J. (2013). Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer's disease. Alzheimers. Res. Ther. 5, 28. doi: 10.1186/alzrt182
Wechsler, D. (1997). Wechsler Memory Scale. 3rd Edition Manual. San Antonio, TX: The Psychological Corperation.
Weinberger, D. R., Berman, K. F., and Zec, R. F. (1986). Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch. Gen. Psychiatry 43, 114–124. doi: 10.1001/archpsyc.1986.01800020020004
Weintraub, S., Wicklund, A. H., and Salmon, D. P. (2012). The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006171. doi: 10.1101/cshperspect.a006171
Welsh, K., Butters, N., Hughes, J., Mohs, R., and Heyman, A. (1991). Detection of abnormal memory decline in mild cases of Alzheimer's disease using CERAD neuropsychological measures. Arch. Neurol. 48, 278–281. doi: 10.1001/archneur.1991.00530150046016
Welsh, K. A., Butters, N., Hughes, J. P., Mohs, R. C., and Heyman, A. (1992). Detection and staging of dementia in Alzheimer's disease. Use of the neuropsychological measures developed for the consortium to establish a registry for Alzheimer's Disease. Arch. Neurol. 49, 448–452. doi: 10.1001/archneur.1992.00530290030008
Westerman, M. A., Cooper-Blacketer, D., Mariash, A., Kotilinek, L., Kawarabayashi, T., Younkin, L. H., et al. (2002). The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 22, 1858–1867. Available online at: http://www.jneurosci.org/content/22/5/1858.long
Whitwell, J. L., Josephs, K. A., Murray, M. E., Kantarci, K., Przybelski, S. A., Weigand, S. D., et al. (2008). MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology 71, 743–749. doi: 10.1212/01.wnl.0000324924.91351.7d
Wilcock, D. M., Rojiani, A., Rosenthal, A., Subbarao, S., Freeman, M. J., Gordon, M. N., et al. (2004). Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflammation 1:24. doi: 10.1186/1742-2094-1-24
Wilkinson, R. T. (1963). Interaction of noise with knowledge of results and sleep deprivation. J. Exp. Psychol. 66, 332–337. doi: 10.1037/h0044161
Wisor, J. P., Edgar, D. M., Yesavage, J., Ryan, H. S., Mccormick, C. M., Lapustea, N., et al. (2005). Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer's disease: a role for cholinergic transmission. Neuroscience 131, 375–385. doi: 10.1016/j.neuroscience.2004.11.018
Yassine, N., Lazaris, A., Dorner-Ciossek, C., Despres, O., Meyer, L., Maitre, M., et al. (2013). Detecting spatial memory deficits beyond blindness in tg2576 Alzheimer mice. Neurobiol. Aging 34, 716–730. doi: 10.1016/j.neurobiolaging.2012.06.016
Young, J. W., Light, G. A., Marston, H. M., Sharp, R., and Geyer, M. A. (2009). The 5-choice continuous performance test: evidence for a translational test of vigilance for mice. PLoS ONE 4:e4227. doi: 10.1371/journal.pone.0004227
Zhang, W., Hao, J., Liu, R., Zhang, Z., Lei, G., Su, C., et al. (2011). Soluble Abeta levels correlate with cognitive deficits in the 12-month-old APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav. Brain Res. 222, 342–350. doi: 10.1016/j.bbr.2011.03.072
Zhuo, J. M., Prakasam, A., Murray, M. E., Zhang, H. Y., Baxter, M. G., Sambamurti, K., et al. (2008). An increase in Abeta42 in the prefrontal cortex is associated with a reversal-learning impairment in Alzheimer's disease model Tg2576 APPsw mice. Curr. Alzheimer Res. 5, 385–391. doi: 10.2174/156720508785132280
Zhuo, J. M., Prescott, S. L., Murray, M. E., Zhang, H. Y., Baxter, M. G., and Nicolle, M. M. (2007). Early discrimination reversal learning impairment and preserved spatial learning in a longitudinal study of Tg2576 APPsw mice. Neurobiol. Aging 28, 1248–1257. doi: 10.1016/j.neurobiolaging.2006.05.034
Keywords: Alzheimer's disease, mouse models, neuropsychological assessment, behavior, cognition, APP mice, APP/PS1 mice, 3×TG-AD mice
Citation: Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA and Van Eldik LJ (2014) Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 5:88. doi: 10.3389/fgene.2014.00088
Received: 21 February 2014; Paper pending published: 16 March 2014;
Accepted: 01 April 2014; Published online: 23 April 2014.
Edited by:
Shin Murakami, Touro University-California, USAReviewed by:
James C. Vickers, University of Tasmania, AustraliaDanna Zimmer, University of Maryland Medical School, USA
Copyright © 2014 Webster, Bachstetter, Nelson, Schmitt and Van Eldik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda J. Van Eldik, Sanders-Brown Center on Aging, 800 S. Limestone, Lexington, KY 40536-0230, USA e-mail:bGluZGEudmFuZWxkaWtAdWt5LmVkdQ==