 Madhumathi Manickam
Madhumathi Manickam Palaniyandi Ravanan
Palaniyandi Ravanan Priti Talwar
Priti Talwar- Apoptosis and Cell Death Research Laboratory, Centre for Biomedical Research, School of Biosciences and Technology, Vellore Institute of Technology University, Vellore, India
Gaucher's disease (GD) is an autosomal recessive disorder caused by the deficiency of glucocerebrosidase, a lysosomal enzyme that catalyses the hydrolysis of the glycolipid glucocerebroside to ceramide and glucose. Polymorphisms in GBA gene have been associated with the development of Gaucher disease. We hypothesize that prediction of SNPs using multiple state of the art software tools will help in increasing the confidence in identification of SNPs involved in GD. Enzyme replacement therapy is the only option for GD. Our goal is to use several state of art SNP algorithms to predict/address harmful SNPs using comparative studies. In this study seven different algorithms (SIFT, MutPred, nsSNP Analyzer, PANTHER, PMUT, PROVEAN, and SNPs&GO) were used to predict the harmful polymorphisms. Among the seven programs, SIFT found 47 nsSNPs as deleterious, MutPred found 46 nsSNPs as harmful. nsSNP Analyzer program found 43 out of 47 nsSNPs are disease causing SNPs whereas PANTHER found 32 out of 47 as highly deleterious, 22 out of 47 are classified as pathological mutations by PMUT, 44 out of 47 were predicted to be deleterious by PROVEAN server, all 47 shows the disease related mutations by SNPs&GO. Twenty two nsSNPs were commonly predicted by all the seven different algorithms. The common 22 targeted mutations are F251L, C342G, W312C, P415R, R463C, D127V, A309V, G46E, G202E, P391L, Y363C, Y205C, W378C, I402T, S366R, F397S, Y418C, P401L, G195E, W184R, R48W, and T43R.
Introduction
Gaucher's disease (GD) is a rare genetic disease in which fatty substances accumulate in cells and certain organs (James et al., 2006). It is a common lysosomal storage disorder and results from an inborn deficiency of the enzyme glucocerebrosidase (also known as acid β-glucosidase). This enzyme is responsible for glucocerebroside (glucosylceramide) degradation. The accumulation of undegraded substrate generally happens because of enzyme deficiency, mainly within cells of the macrophage lineage or monocyte, and it is responsible for the clinical manifestations of the disease (Beutler and Grabowski, 2001). This glucosylceramide degrading enzyme is encoded by a gene named GBA, which is 7.6 kb in length and located in 1q21 locus. Recessive mutation in GBA gene affects both males and females (Horowitz et al., 1989; Zimran et al., 1991; Winfield et al., 1997). GBA protein is 497 amino acids long with the molecular weight of 55.6 KD. GBA enzyme catalyses the breakdown of glucosylceramide, a cell membrane constituent of white blood cells and red blood cells. The macrophages fail to eliminate the waste product and results in accumulation of lipids in fibrils and this turn into Gaucher cells (Aharon et al., 2004). GD can be classified into three classes namely types 1, 2, and 3. In type 1, Glycosylceramide accumulate in visceral organs whereas in type 2 and 3, the accumulation is in the central nervous system (Grabowski, 2008).
The international disease frequency of GD is 200,000 except for areas of the world with large Ashkenazi Jewish populations where 60% of the patients are estimated to be homozygous, which accounts for 75% of disease alleles (Pilar et al., 2012). Almost 300 unique mutations have been reported in the GBA gene, with distribution that spans the entire gene. These include 203 missense mutations, 18 nonsense mutations, 36 small insertions or deletions that lead to frameshift or in-frame alterations, 14 splice junction mutations and 13 complex alleles carrying two or more mutations (Hruska et al., 2008). The single nucleotide variations in the genome that occur at a frequency of more than 1% are referred as single nucleotide polymorphisms (SNPs) and in the human genome, SNPs occur in just about every 3000 base pairs (Cargill et al., 1999).
Nearly 200 mutations in the GBA gene have been described in patients with GD types 1, 2, and 3 (Jmoudiak and Futerman, 2005). L444P mutation was identified in GBA gene in patients with GD types 1, 2, and 3. The L444P substitution is one of the major SNP associated with the GBA gene. D409H, A456P, and V460V mutations were also identified in patients with GD (Tsuji et al., 1987; Latham et al., 1990). Previous findings have shown that, in 60 patients with types 1 and 3, the most common Gaucher mutations identified were N370S, L444P, and R463C. (Sidransky et al., 1994). The other mutation E326K had been identified in patients with all three types of GD, but in each instance it was found on the same allele with another GBA mutation. Also, Park et al. identified the E326K allele in 1.3% of patients with GD and in 0.9% of controls, indicating that it is a polymorphism (Park et al., 2002).
The harmful SNPs for the GBA gene have not been predicted to date in silico. Therefore we designed a strategy for analyzing the entire GBA coding region. Different algorithms such as SIFT (Ng and Henikoff, 2001), MutPred (Li et al., 2009), nsSNP Analyzer (Bao et al., 2005), PANTHER (Mi et al., 2012), PMUT (Costa et al., 2002), PROVEAN (Choi et al., 2012), and SNPs&GO (Calabrese et al., 2009) were utilized to predict high-risk nonsynonymous single nucleotide polymorphisms (nsSNPs) in coding regions that are likely to have an effect on the function and structure of the protein.
Materials and Methods
Data Set
SNPs associated with GBA gene were retrieved from the single nucleotide polymorphism database (dbSNP) (http://www.ncbi.nlm.nih.gov/snp/), and are commonly referred by their reference sequence IDs (rsID) (Wheeler et al., 2005).
Validation of Tolerated and Deleterious SNPs
The type of genetic mutation that causes a single amino acid substitution (AAS) in a protein sequence is called nsSNP. An nsSNP could potentially influence the function of the protein, subsequently altering the phenotype of carrier. This protocol describes the use of the Sorting Intolerant From Tolerant (SIFT) algorithm (http://sift.jcvi.org) for predicting whether an AAS affects protein function. To assess the effect of a substitution, SIFT assumes that important positions in a protein sequence have been conserved throughout evolution and therefore at these positions substitutions may affect protein function. Thus, by using sequence homology, SIFT predicts the effects of all possible substitutions at each position in the protein sequence. The protocol typically takes 5–20 min, depending on the input (Kumar et al., 2009).
Prediction of Harmful Mutations
MutPred (http://mutdb.org/mutpred) models structural features and functional sites changes between mutant sequences and wild-type sequence. These changes are expressed as probabilities of gain or loss of structure and function. The MutPred output contains a general score (g), i.e., the probability that the AAS is deleterious/disease-associated and top five property scores (p), where p is the P-value that certain structural and functional properties are impacted. Certain combinations of high values of general scores and low values of property scores are referred to as hypotheses (Li et al., 2009).
Identifying Disease-Associated nsSNPs
nsSNP Analyzer (http://snpanalyzer.uthsc.edu) is a tool to predict whether a nsSNP has a phenotypic effect (disease-associated vs. neutral) using a machine learning method called Random Forest, and extracting structural and evolutionary information from a query nsSNP (Bao et al., 2005).
Prediction of Deleterious nsSNPs
PANTHER (http://pantherdb.org/tools/csnpScoreForm.jsp) estimates the likelihood of a particular nsSNP to cause a functional impact on a protein (Thomas et al., 2003). It calculates the substitution position-specific evolutionary conservation (subPSEC) score based on the alignment of evolutionarily related proteins. The subPSEC score is the negative logarithm of the probability ratio of the wild-type and the mutant amino acids at a particular position. The subPSEC scores are values from 0 (neutral) to about −10 (most likely to be deleterious).
Prediction of Pathological Mutations on Proteins
PMUT (http://mmb2.pcb.ub.es:8080/PMut) uses a robust methodology to predict disease-associated mutations. PMUT method is based on the use of neural networks (NNs) trained with a large database of neutral mutations (NEMUs) and pathological mutations of mutational hot spots, which are obtained by alanine scanning, massive mutation, and genetically accessible mutations. The final output is displayed as a pathogenicity index ranging from 0 to 1 (indexes > 0.5 single pathological mutations) and a confidence index ranging from 0 (low) to 9 (high) (Costa et al., 2005).
Predicting the Functional Effect of Amino Acid Substitutions
PROVEAN (Protein Variation Effect Analyzer) (http://provean.jcvi.org) is a sequence based predictor that estimates the effect of protein sequence variation on protein function (Choi et al., 2012). It is based on a clustering method where BLAST hits with more than 75% global sequence identity are clustered together and top 30 such clusters from a supporting sequence are averaged within and across clusters to generate the final PROVEAN score. A protein variant is predicted to be “deleterious” if the final score is below a certain threshold (default is −2.5), and is predicted to be “neutral” if the score is above the threshold.
Prediction of Disease Related Mutations
The SNPs&GO algorithms (http://snps-and-go.biocomp.unibo.it/snps-and-go/) predict the impact of protein variations using functional information encoded by Gene Ontology (GO) terms of the three main roots: Molecular function, Biological process, and Cellular component (Calabrese et al., 2009). SNPs&GO is a support vector machine (SVM) based web server to predict disease related mutations from the protein sequence, scoring with accuracy of 82% and Matthews correlation coefficient equal to 0.63. SNPs&GO collects, in a unique framework, information derived from protein sequence, protein sequence profile and protein functions.
Results
nsSNPs Found by SIFT Program
Protein sequence with mutational position and amino acid residue variants associated with 97 missense nsSNPs were submitted as input to the SIFT server, and the results are shown in Table 1. The lower the tolerance index, the higher the functional impact a particular amino acid residue substitution is likely to have and vice versa. Among the 97 nsSNPs analyzed, 47 nsSNPs were identified to be deleterious with a tolerance index score ≤0.05 (Kumar et al., 2009). Among 47 deleterious nsSNPs, 25 nsSNPs were found to be highly deleterious.
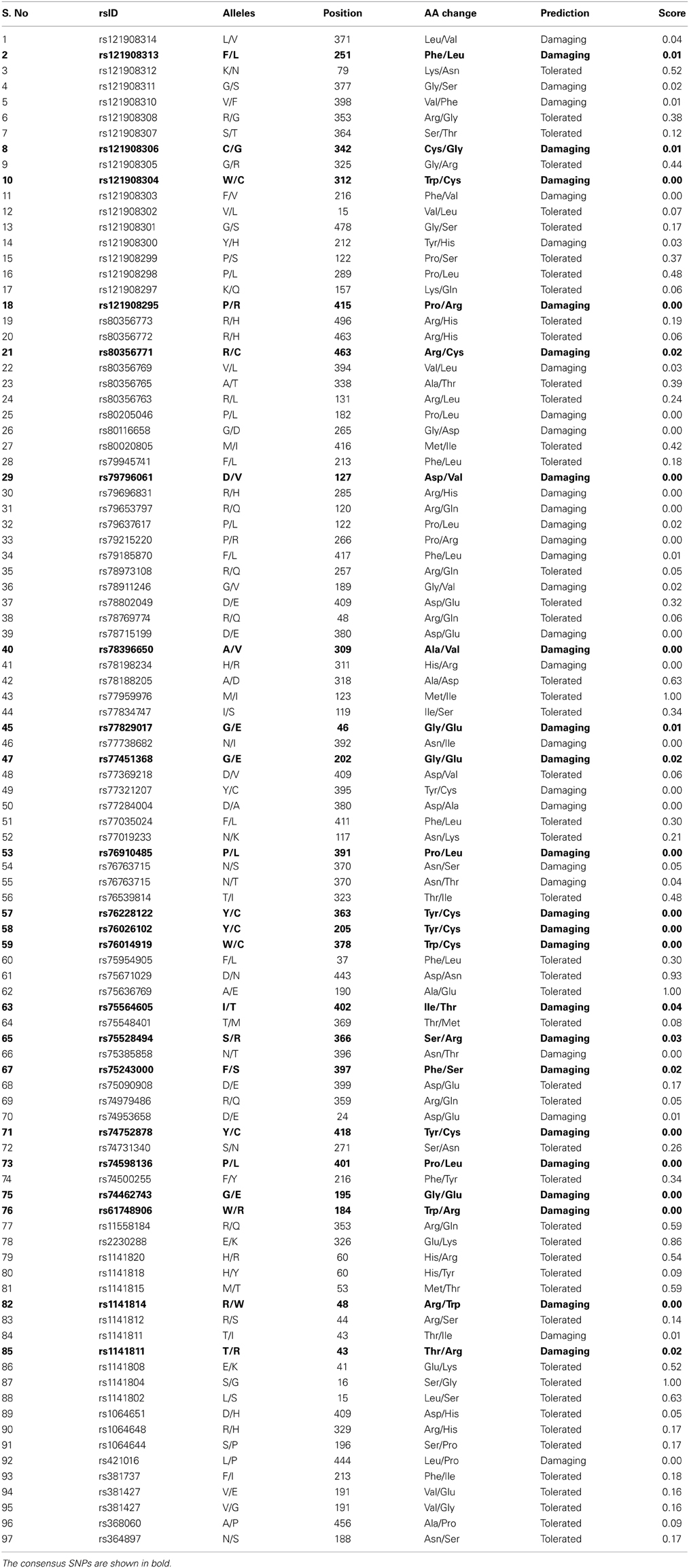
Table 1. Tolerated and deleterious nsSNPs using SIFT.
Validation of Harmful Mutations
The MutPred score is the probability that an AAS is deleterious/disease-associated. A missense mutation with a MutPred score >0.5 could be considered as “harmful,” while a MutPred score >0.75 should be considered a high confidence “harmful” prediction (Li et al., 2009). Among the 47 deleterious nsSNPs, 8 were found to be harmful mutations with a score of >0.5 and <0.75 and 38 were found to be high confidence (highly harmful) mutations and 1 nsSNP found to be normal with the score of 0.193 (Table 2).
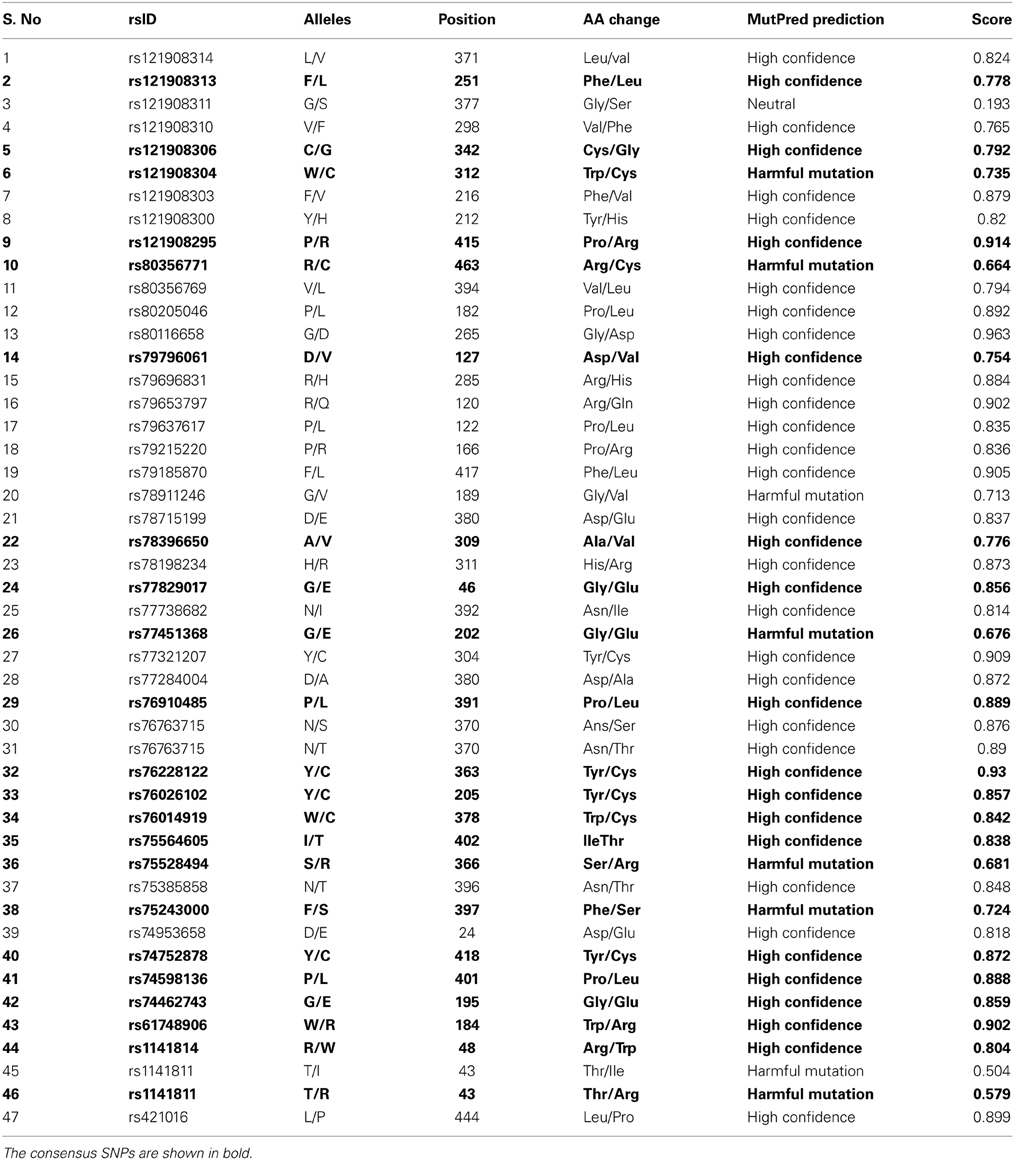
Table 2. Prediction of functional effects of nsSNPs using MutPred.
Disease-Associated nsSNPs
Out of 47 deleterious nsSNPs, 43 were found to be a disease causing nsSNPs and 4 were found to be neutral nsSNPs (Table 3).
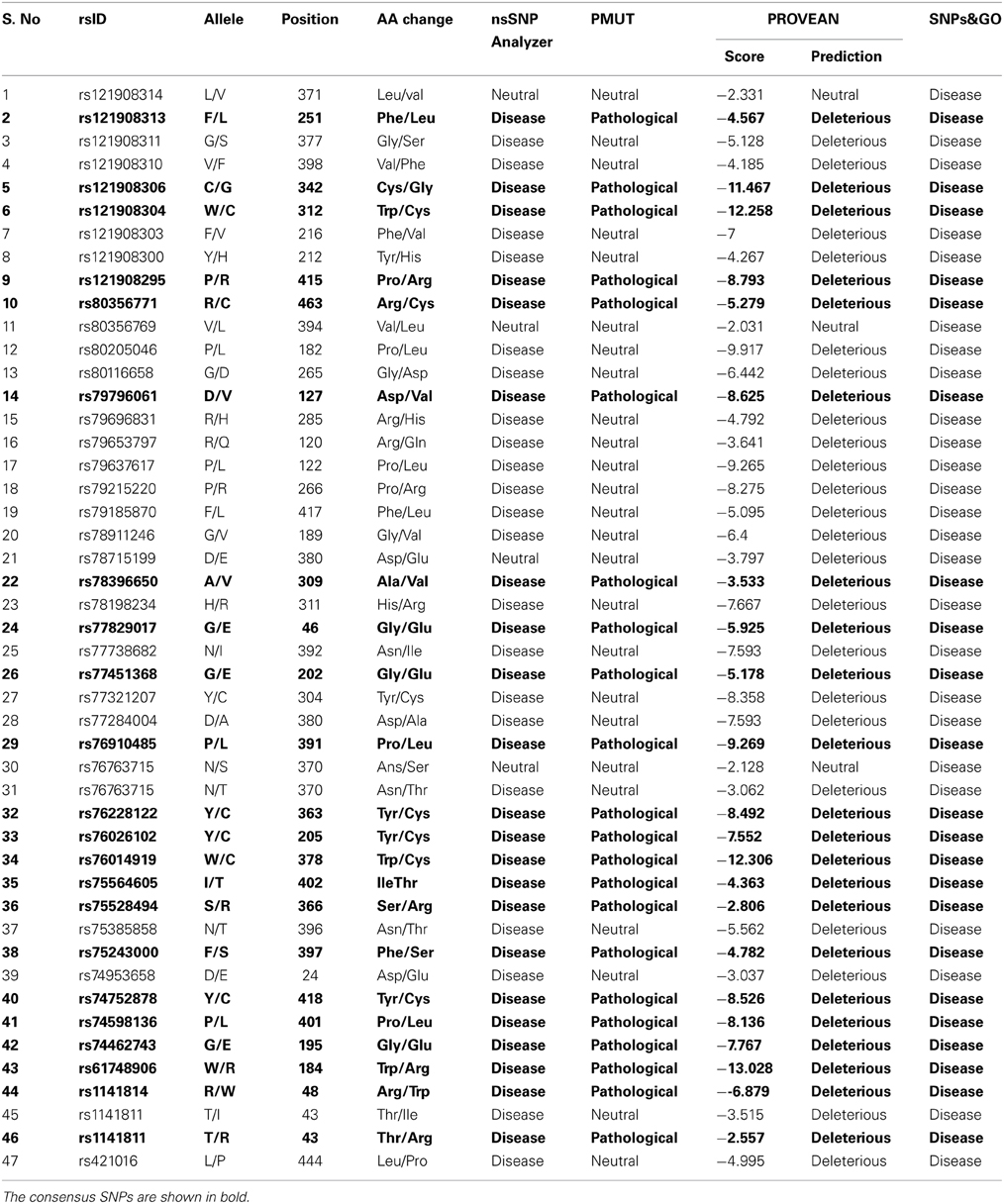
Table 3. The results from nsSNP Analyzer, PMUT, PROVEAN, and SNPs&GO.
Validation by Panther
The protein sequence was given as input and analyzed for the deleterious effect on protein function. The subPSEC scores are values from 0 (neutral) to about −10 (deleterious) (Thomas et al., 2003). Out of 47 deleterious nsSNPs, 8 were found to be more than −6 (highly deleterious) and rest were found to be less deleterious. The mutant with a greater Pdeleterious tends to have more severe destructions in function. It was found that 32 out of 47 deleterious nsSNPs scored greater than 3 and rests were below the damage threshold (Table 4).
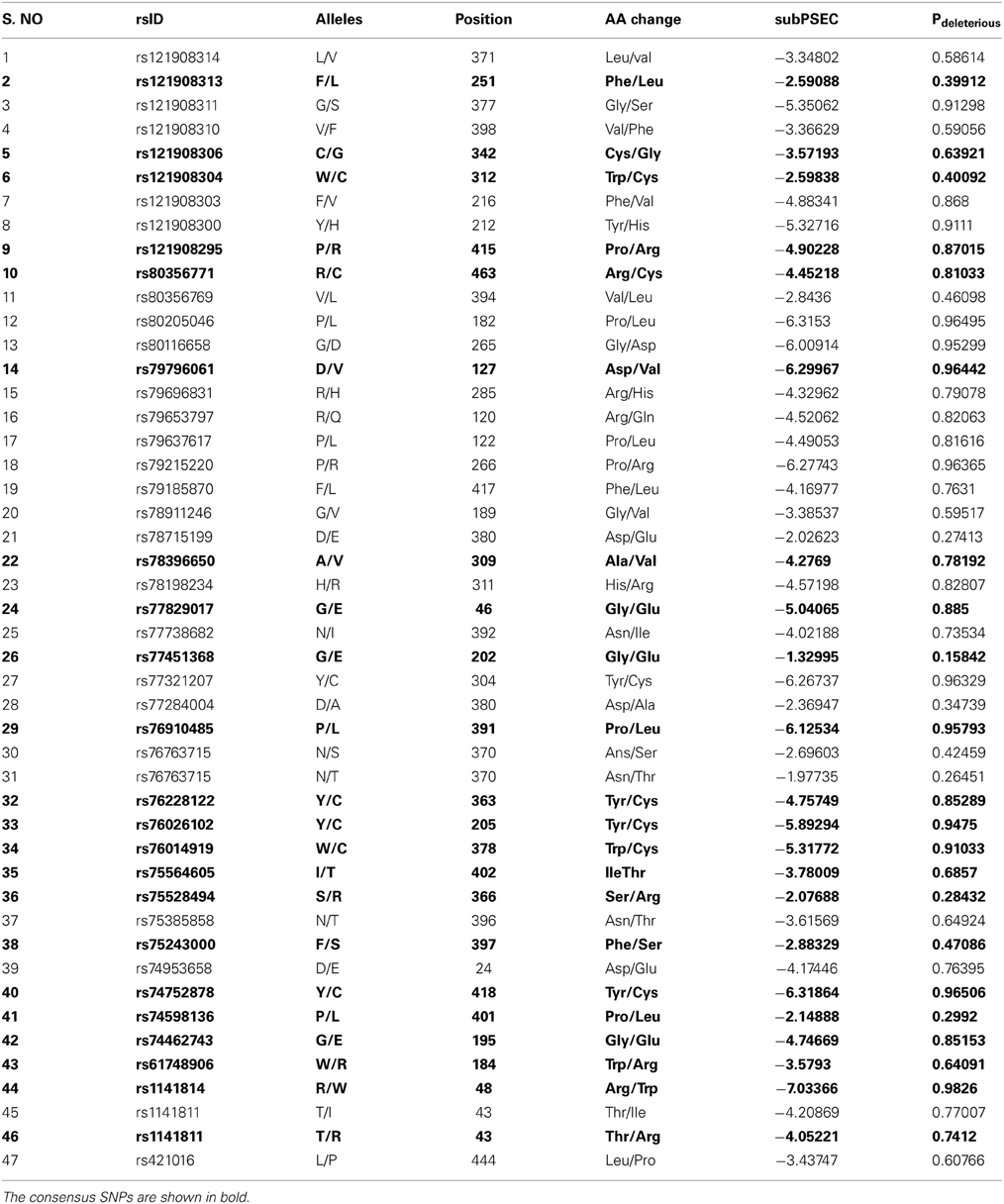
Table 4. Mutant scores from PANTHER.
Functional Impact of Mutations on Proteins
The functional impact of 47 deleterious nsSNPs in protein of GBA was analyzed using PMUT server. Of the 47 nsSNPs, 22 are classified as pathological, and the remaining were neutral (Table 3).
Protein Variation Effect Analysis
PROVEAN predicts the effect of the variant on the biological function of the protein based on sequence homology. PROVEAN scores are classified as “deleterious” if below a certain threshold (here −2.5) and “neutral” if above it (Choi et al., 2012). Out of 47 nsSNPs, 44 were predicted to be “deleterious” and 3 were found to be “neutral” (Table 3).
Prediction of Disease Related Mutations by SNPs&GO
SNPs&GO is trained and tested with cross-validation procedures in which similar proteins are placed together as a dataset to calculate the LGO score derived from the GO data base. All 47 deleterious nsSNPs showed the disease related mutations (Table 3).
Discussion
In the recent years, SNPs have emerged as the new generation molecular markers. The harmful SNPs for the GBA gene were never been predicted to date in silico. This study was designed to understand the genetic variations associated with GBA gene. We have predicted the harmful nsSNPs using SIFT, MutPred, nsSNP Analyzer, PANTHER, PMUT, PROVEAN, and SNPs&GO state of the art computational tools. Among 97 nsSNPs, 47 were found to be deleterious with a tolerance index score of ≤0.05 found by SIFT program. Among the 47 deleterious nsSNPs, 46 were found to be harmful nsSNPs found by MutPred, 43 were found to be disease causing nsSNPs by nsSNP Analyzer tool, 32 are highly deleterious found by PANTHER program, 22 are classified as pathological mutations by PMUT, 44 were predicted to be deleterious by PROVEAN server while all 47 deleterious nsSNPs showed the disease related mutations by SNPs&GO. Also, we found that SNPs&GO was most successful of all state of the art SNP prediction programs that were used for this comparative study. In this work, we found 22 nsSNPs that are common in all (SIFT, MutPred, nsSNP Analyzer, PANTHER, PMUT, PROVEAN, and SNPs&GO) prediction (Figure 1). These sets of 22 nsSNPs (F251L, C342G, W312C, P415R, R463C, D127V, A309V, G46E, G202E, P391L, Y363C, Y205C, W378C, I402T, S366R, F397S, Y418C, P401L, G195E, W184R, R48W, and T43R) are possibly the main targeted mutation for the GD (Tables 1–4). The previous work has shown that, in 60 patients with types 1 and 3, the most common Gaucher mutations identified were L444P, N370S, and R463C. L444P was the most common mutation in GD types 1, 2, and 3 (Latham et al., 1990; Sidransky et al., 1994). In our analysis, out of 7 methods, 6 methods (Sift, MutPred, PROVEAN, PANTHER, nsSNP Analyzer, and SNPs&GO) shows L444P mutation as damaging, 3 methods shows N370S mutation as damaging and all the 7 methods shows R463C mutation as damaging. D409H, A456P, E326K, and V460V mutations were also identified in patients with GD (Tsuji et al., 1987; Park et al., 2002). In our analysis SIFT result shows D409H, A456P, and E326K mutation is the tolerated mutation. Further studies using these mutations will shed light on the genetic understanding of this major lysosomal storage disease.

Figure 1. Sets of various mutations identified using various software tools. The respective locations of 44 amino acids responsible for all 47 mutations are shown in the sequence (center, colored in bold) and 22 common mutations are highlighted as consensus.
Author Contributions
Madhumathi Manickam, Priti Talwar, and Palaniyandi Ravanan wrote the main manuscript and analyzed original datasets. Pratibha Singh prepared tables and figure. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors greatly acknowledge the financial support and laboratory facilities given by VIT University, Vellore, India to carry out this research work. Additionally, authors greatly acknowledge the critical work of all reviewers on this paper.
References
Aharon, P. J., Rosenbaum, H., and Gershoni, B. R. (2004). Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977. doi: 10.1056/NEJMoa033277
Bao, L., Zhou, M., and Cui, Y. (2005). nsSNP Analyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 33, 480–482. doi: 10.1093/nar/gki372
Beutler, E., and Grabowski, G. A. (2001). “Gaucher disease,” in Metabolic and Molecular Bases of Inherited Disease, II, eds C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York, NY: McGraw-Hill), 3635–3668.0
Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., and Casadio, R. (2009). Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 8, 1237–1244. doi: 10.1002/humu.21047
Cargill, M., Altshuler, D., Ireland, J., Sklar, P., Ardlie, K., Patil, N., et al. (1999). Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 22, 231–238. doi: 10.1038/10290
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7:e46688. doi: 10.1371/journal.pone.0046688
Costa, F. C., Gelpi, J. L., Zamakola, L., Parraga, I., Cruz, D. L. X., and Orozco, M. (2005). PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 14, 3176–3178. doi: 10.1093/bioinformatics/bti486
Costa, F. C., Orozco, M., and Cruz, D. L. X. (2002). Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J. Mol. Biol. 315, 771–786. doi: 10.1006/jmbi.2001.5255
Grabowski, G. A. (2008). Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 372, 1263–1271. doi: 10.1016/S0140-6736(08)61522-6
Horowitz, M., Wilder, S., Horowitz, Z., Reiner, O., Gelbart, T., and Beutler, E. (1989). The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4, 87–96. doi: 10.1016/0888-7543(89)90319-4
Hruska, K. S., LaMarca, M. E., Scott, C. R., and Sidransky, E. (2008). Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583. doi: 10.1002/humu.20676
James, W. D., Berger, T. M., and Elston, D. M. (2006). Andrew's Diseases of the Skin: Clinical Dermatology, 10th Edn. Philadelphia, PA: Saunders; Elsevier.
Jmoudiak, M., and Futerman, A. H. (2005). Gaucher disease: pathological mechanisms and modern management. Br. J. Haematol. 129, 178–188. doi: 10.1111/j.1365-2141.2004.05351.x
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. doi: 10.1038/nprot.2009.86
Latham, T., Grabowski, G. A., Theophilus, B. D. M., and Smith, F. I. (1990). Complex alleles of the acid beta-glucosidase gene in Gaucher disease. Am. J. Hum. Genet. 47, 79–86.
Li, B., Krishnan, V. G., Mort, M. E., Xin, F., Kamati, K. K., Cooper, D. N., et al. (2009). Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750. doi: 10.1093/bioinformatics/btp528
Mi, H., Muruganujan, A., and Thomas, P. D. (2012). PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, 377–386. doi: 10.1093/nar/gks1118
Ng, P. C., and Henikoff, S. (2001). Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. doi: 10.1101/gr.176601
Park, J. K., Tayebi, N., Stubblefield, B. K., LaMarca, M. E., MacKenzie, J. J., Stone, D. L., et al. (2002). The E326K mutation and Gaucher disease: mutation or polymorphism? Clin. Genet. 61, 32–34. doi: 10.1034/j.1399-0004.2002.610106.x
Pilar, G., Pilar, A., Pilar, I., Laura, G., Amparo, C., Lluisa, V., et al. (2012). Mapping the genetic and clinical characteristics of Gaucher disease in the Iberian Peninsula. Orphanet J. Rare Dis. 7:17. doi: 10.1186/1750-1172-7-17
Sidransky, E., Bottler, A., Stubblefield, B., and Ginns, E. I. (1994). DNA mutational analysis of type 1 and type 3 Gaucher patients: how well do mutations predict phenotype? Hum. Mutat. 3, 25–28. doi: 10.1002/humu.1380030105
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., et al. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. doi: 10.1101/gr.772403
Tsuji, S., Choudary, P. V., Martin, B. M., Stubblefield, B. K., Mayor, J. A., Barranger, J. A., et al. (1987). A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher's disease. N. Engl. J. Med. 316, 570–575. doi: 10.1056/NEJM198703053161002
Wheeler, D. L., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., et al. (2005). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 34, 173–180. doi: 10.1093/nar/gkj158
Winfield, S. L., Tayebi, N., Martin, B. M., Ginns, E. I., and Sidransky, E. (1997). Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: implications for Gaucher disease. Genome Res. 7, 1020–1026.
Keywords: glucocerebrosidase, SIFT, MutPred, PANTHER, PMUT, PROVEAN, SNPs&GO
Citation: Manickam M, Ravanan P, Singh P and Talwar P (2014) In silico identification of genetic variants in glucocerebrosidase (GBA) gene involved in Gaucher's disease using multiple software tools. Front. Genet. 5:148. doi: 10.3389/fgene.2014.00148
Received: 14 March 2014; Accepted: 06 May 2014;
Published online: 27 May 2014.
Edited by:
Babajan B., King Abdulaziz University, Saudi ArabiaReviewed by:
Dhananjai M. Rao, Miami University, USAIndira Shrivastava, University of Pittsburgh, USA
Copyright © 2014 Manickam, Ravanan, Singh and Talwar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dr. Priti Talwar, Apoptosis and Cell Death Research Laboratory, Centre for Biomedical Research, School of Biosciences and Technology, Vellore Institute of Technology University, Vellore, Tamil Nadu-632 014, India e-mail:cHJpdGkudEB2aXQuYWMuaW4=