 Shaohua Fan
Shaohua Fan Axel Meyer*
Axel Meyer*- Lehrstuhl für Zoologie und Evolutionsbiologie, Department of Biology, University of Konstanz, Konstanz, Germany
African cichlid fishes are an ideal system for studying explosive rates of speciation and the origin of diversity in adaptive radiation. Within the last few million years, more than 2000 species have evolved in the Great Lakes of East Africa, the largest adaptive radiation in vertebrates. These young species show spectacular diversity in their coloration, morphology and behavior. However, little is known about the genomic basis of this astonishing diversity. Recently, five African cichlid genomes were sequenced, including that of the Nile Tilapia (Oreochromis niloticus), a basal and only relatively moderately diversified lineage, and the genomes of four representative endemic species of the adaptive radiations, Neolamprologus brichardi, Astatotilapia burtoni, Metriaclima zebra, and Pundamila nyererei. Using the Tilapia genome as a reference genome, we generated a high-resolution genomic variation map, consisting of single nucleotide polymorphisms (SNPs), short insertions and deletions (indels), inversions and deletions. In total, around 18.8, 17.7, 17.0, and 17.0 million SNPs, 2.3, 2.2, 1.4, and 1.9 million indels, 262, 306, 162, and 154 inversions, and 3509, 2705, 2710, and 2634 deletions were inferred to have evolved in N. brichardi, A. burtoni, P. nyererei, and M. zebra, respectively. Many of these variations affected the annotated gene regions in the genome. Different patterns of genetic variation were detected during the adaptive radiation of African cichlid fishes. For SNPs, the highest rate of evolution was detected in the common ancestor of N. brichardi, A. burtoni, P. nyererei, and M. zebra. However, for the evolution of inversions and deletions, we found that the rates at the terminal taxa are substantially higher than the rates at the ancestral lineages. The high-resolution map provides an ideal opportunity to understand the genomic bases of the adaptive radiation of African cichlid fishes.
Introduction
Cichlid fishes provide one of the most extreme examples for adaptive radiations in vertebrates (Kocher, 2004; Salzburger and Meyer, 2004; Kuraku and Meyer, 2008; Henning and Meyer, in press). More than 2000 species have evolved during the last few million years in the Great Lakes of East Africa (Meyer, 1993; Salzburger et al., 2005; Elmer et al., 2009). With >250 endemic species in Lake Tanganyika, >800 species in Lake Malawi and >500 species in Lake Victoria, these are the largest adaptive radiations in vertebrates (Meyer et al., 1990; Stiassny and Meyer, 1999; Kocher, 2004; Henning and Meyer, in press). Lake Tanganyika is maximally 7–8 million years old, and is the oldest of these lakes (Sturmbauer et al., 1994). Lake Malawi is younger at about 2–4 Million years and the current form of Lake Victoria is probably less than 100,000 years old. These young species, particularly of Lakes Malawi and Victoria are both extremely young, yet spectacularly diverse in morphology, coloration and behavior. Since most species of Lakes Malawi and Victoria can be hybridized in the laboratory for forward genetic studies, we referred to them to be “natural mutants” that can be used for studying genomic diversification by natural and sexual selection (Meyer, 1993; Meyer et al., 1993, 1995; Kuraku and Meyer, 2008; Henning and Meyer, in press). A large body of work, including transcriptome sequencing (Salzburger et al., 2008; Lee et al., 2010; Baldo et al., 2011; Gunter et al., 2013), BAC library construction (Lang et al., 2006), candidate gene sequencing (Terai et al., 2002, 2006; Hofmann et al., 2009; Fan et al., 2011), and microarrays (Gunter et al., 2011; Loh et al., 2013), has been conducted in an effort to study the molecular basis of the adaptive radiation of African cichlid fishes (reviewed by Fan et al., 2012).
Recently, the cichlid genome consortium sequenced five African cichlid genomes, including the Nile Tilapia (Oreochromis niloticus), Neolamprologus brichardi (endemic to Lake Tanganyika), Astatotilapia burtoni (lives in and around Lake Tanganyika), Metriaclima zebra (endemic to Lake Malawi), and Pundamila nyererei (endemic to Lake Victoria) (Brawand et al., submitted). The analysis of these five African cichlid genomes shows that their rapid evolution is associated not with one, but with multiple mechanisms, including an excess of gene duplications, transposable element expansions, fast evolution of conserved non-coding elements, and the evolution of novel micro RNAs (Brawand et al., submitted). However, genomic work on cichlid diversification has lacked so far a comprehensive comparative analysis of large-scale genomic variation. Ordered by size, genomic variation can take on the form of single nucleotide polymorphisms (SNPs), short insertions and deletions (indels), and larger structural variation (SVs, normally > 50 bp). Furthermore, SVs can be classified as insertions, inversions, deletions, duplications and translocations (Alkan et al., 2011a). The advent of next generation sequencing (NGS) technologies has revolutionized the study of genomic variation (Alkan et al., 2011a), as high-density maps were generated for various model systems. Such maps, reveal the presence of a wide spectrum of variation and are of important for gaining a deeper understanding of phenotypic diversification and speciation from a genomic perspective (Quinlan et al., 2010; Elmer and Meyer, 2011; Mills et al., 2011b; Zhan et al., 2011; Jones et al., 2012b; Feulner et al., 2013; Zichner et al., 2013). For example, recent studies have shown that adaptive evolution in three spine stickleback is associated with the reuse of standing variations, but also with SVs such as chromosomal inversion and deletions (Chan et al., 2010; Jones et al., 2012a,b; Feulner et al., 2013).
In this study, using the recently sequenced five African cichlid genomes, we investigated patterns of genomic variation that accompany the adaptive radiation of African cichlids. The aims of the present study were 3-fold: first, we characterized the prevalence and locations of the genomic variation in these five African cichlid genomes. Second, we analyzed the variation between these genomes in a phylogenetic context, which enabled us to gain a deeper understanding of questions such as: when did this variation originate? How do processes such as natural selection operate on different types of variation? Third, we inferred the potential functional impact of this variation using gene annotation information. These analyses of the genomic variation will not only enable us to assess the impact of the genomic differentiation on the functional portions of the genome, but also elucidate not only correlative changes, but also possibly causal structural changes as potential mechanisms for the rapid evolution of cichlid fishes.
Materials and Methods
Data Collection
The Broad Institute determined the five African cichlid genomes using Illumina technology. Briefly, one individual per species was sequenced using paired-end and mate-paired libraries. The raw reads of each species were assembled using ALLPATHS-LG pipeline (Gnerre et al., 2011). The Tilapia genome was further anchored into 22 linkage groups using linkage map information (Brawand et al., submitted). The information of the five African cichlid genomes is listed in the Supplementary Table 1. In this study, the raw sequencing data were downloaded from the NCBI SRA database (for accession numbers of the libraries see Supplementary Table 2).
Data Processing
The SRA files were converted to Fastq format using the fastq-dump (version 2.3.2) with the NCBI SRA toolkit. Sickle (https://github.com/najoshi/sickle) was used to remove the sequencing adaptors, to mask bases with quality score lower than 20, and to exclude reads less than 50 bp in length with parameters -q 20, -l 50.
We processed the reads from the paired-end and mate-paired libraries separately. The overlapping paired-end reads (insertion size: 180 bp, sequencing length: 100 bp) were first trimmed and we only kept the first and the last 50 bp in the first and second read using fastx_trimmer of the Fastx toolkit (version 0.0.13) (http://hannonlab.cshl.edu/fastx_toolkit/). For the mate-paired libraries (insertion size 3000 bp, sequencing length: 36 bp), we first mapped the raw reads to their corresponding genomes using BWA (version 0.7.3a-r367) (Li and Durbin, 2009) and excluded the read pairs that are facing each other, as these reads could be potential contaminations of paired-end reads in the mate-paired libraries. The remaining mate-paired reads were reverse-complemented, therefore the orientation of the mate pair reads are as same as the paired-end reads, to fit the requirements of the software in the downstream analyses.
The filtered paired-end and mate-paired reads from Tilapia, N. bricharid, A. burtoni, P. nyererei, and M. zebra genomes were mapped to the anchored Tilapia genome using Burrows-Wheeler Aligner (BWA) with the default parameters (version 0.7.3a-r367) (Li and Durbin, 2009). Although mapping short reads against a relative distantly related (around 4% sequence divergence in coding regions) outgroup can be a challenge for BWA, the reference genome is equidistant to N. bricharid, A. burtoni, P. nyererei, and M. zebra, thus would not bias the placement of the reads and not affect the downstream analyses. The raw mapping results were converted to BAM format and ambiguously mapped reads removed by requiring a mapping quality ≥ 20 using Samtools (version 0.1.19-44428cd) (Li et al., 2009). Duplicated read pairs were removed using the MarkDuplicates in the Picard toolkit (version 1.92) (http://picard.sourceforge.net/). The filtered bam files from the former steps were utilized for variation detection.
SNPs and Indels
SNPs and indels were genotyped using GATK (version 2.6.5) (McKenna et al., 2010; Depristo et al., 2011). Specifically, reads in the indel regions were realigned locally to minimize the number of the mismatching bases in each read; the raw SNP and indel callings were filtered with the parameters of phred-scaled quality score >30, depth of coverage between 6 and 5000, and the strand bias based on the phred-scaled p-value using Fisher's exact test <200. We intersected the coordinates of the SNPs and indels with the transposable element regions in the Tilapia genome using the intersectBed in the Bedtools version v2.17.0 (Quinlan and Hall, 2010). The SNPs and indels located in the transposable element regions were excluded in the further analyses. The effects of the SNPs and indels were estimated by the SNPEff version 3.3f based on the gene annotation information of the Tilapia genome. We classified SNPs and indels as intergenic, intronic, upstream (within 5 kb upstream of a gene), downstream (within 5 kb downstream of a gene) and exonic ones. The highest effect of a SNP or indel was selected using VariantAnnotator in the GATK toolkit (McKenna et al., 2010; Depristo et al., 2011). The presence and absence the SNPs and indels sites across different species was checked using the Seqmule pipeline (http://seqmule.usc.edu).
Structural Variation
SV was detected with Pindel (version 0.2.5a1) with default parameters. As quality control, we first detected SVs using the mapping result of Tilapia reads against the Tilapia genome. SVs then found in the Tilapia genome were considered to be potential assembly errors. Therefore, the SVs detected in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes were filtered out if they overlapped with the SVs in the Tilapia genome.
For the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes, we first estimated the SVs based on the mapping results of the paired-end and mate-paired libraries separately and merged the results with the same species using mergeBed in the Bedtools version v2.17.0 (Quinlan and Hall, 2010).
The raw results from Pindel were converted to the VCF format using pindel2vcf in the Pindel package (Ye et al., 2009). All the SVs that overlapped with the transposable element regions in the Tilapia genome were excluded from further analyses. Genes overlapping with the SV regions were identified using intersectBed in the Bedtools version v2.17.0 (Quinlan and Hall, 2010). The enrichment of the gene ontology (GO) in the SV regions was examined using the Fisher's exact test (with FDR correction <0.05) in Blast2GO (Conesa et al., 2005; Gotz et al., 2008). We used Multovl version 1.2.98 with default parameters to check whether SVs overlaps across different species (Aszodi, 2012).
Origin and Rate of Genetic Variation
The origin of the genetic variation (e.g., SNPs, indels, inversions, and deletions) was analyzed in a phylogenetic context. We mapped the variation to the phylogeny from the African cichlid genome project, which was estimated using around 2.7 million 4-fold degenerate sites from the alignments of nine teleost genomes (zebrafish, fugu, tetraodon, stickleback, medaka, and the five African cichlid genomes). Based the maximum parsimony assumption, if a variation was shared by sister taxa, we assumed the structural variant had evolved in the common ancestor of these two species or sister lineages rather than having evolved independently in two species. The origin of variation shared by non-sister taxa is hard to determine using this method, as these variants could evolve independently in different species or due to the low sample size (one individual per species) used in this study. Therefore, we excluded these variants from the further rate analyses. To compare the rates of variation at different lineages, we first estimated the divergence time of the species used in this study. By assuming the cichlids in Lake Victoria (P. nyererei) and Lake Malawi (M. zebra) diverged from their most recent common ancestor on average 2.3 million years ago (Friedman et al., 2013), the divergence time for other lineages was estimated using a non-parametric approach (Sanderson, 1997) in the TreeEdit version v.1.0a10 (Rambaut and Charleston, 2002). Then, the rate of variation was calculated by dividing the number of lineage specific variation by the divergence time.
Results
In total, 992 Gb raw sequence data were analyzed in this study (Supplementary Table 2). We achieved at least 20x coverage for all the species after filtering with the criteria mentioned before. In the following sections, we will focus on the genomic variation in 22 linkage groups, which make up about 71% of the whole Tilapia genome assembly.
Around 17 million SNPs were detected in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes using the Tilapia genome as the reference genome (Table 1, Figure 1). Whereas most of the SNPs are located in the intergenic and intronic regions of the genomes (>70%), many SNPs (around 4%) are located the exonic regions of all four species (Table 1) and the ratio between non-synonymous and synonymous SNP is around 0.7 (Table 1). Of all 20 162 genes on the 22 linkage groups, we found around 77% of the genes with at least one non-synonymous SNP (data not shown). Except for the genes in the N. brichardi genome, the top 1% of the genes with highest proportion of the non-synonymous SNPs (normalized by the exon length) is significant enriched in the pathways related to immune response in the A. burtoni, P. nyererei, and M. zebra genomes.
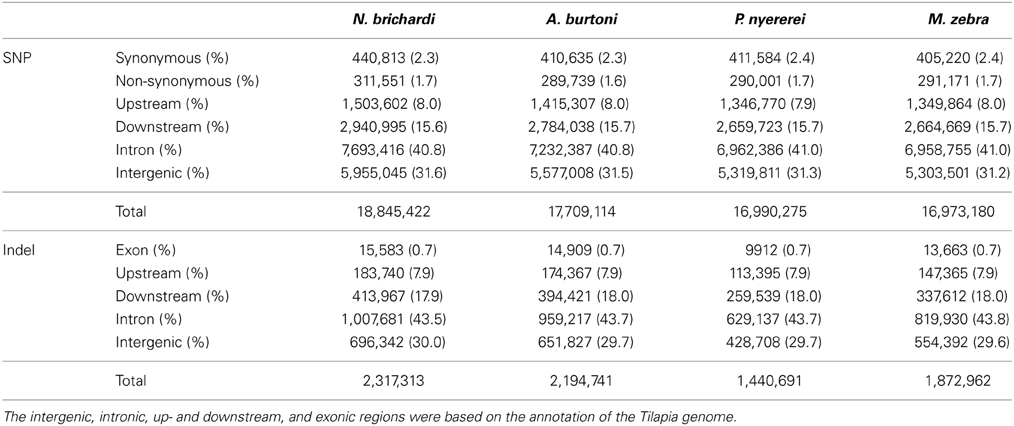
Table 1. Summary of the SNPs and indels in the African cichlid genomes.
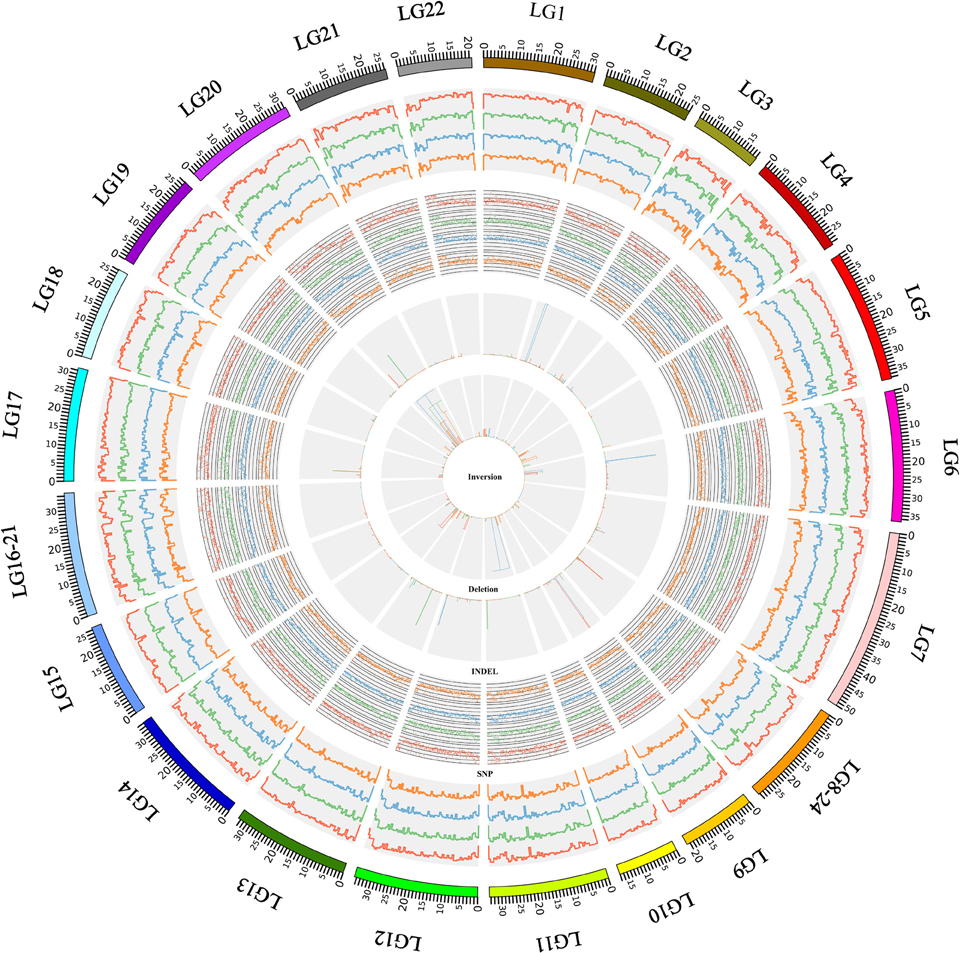
Figure 1. The distribution of genomic variation in the African cichlid genomes. This figure shows an overview of the SNPs, indels, inversions, and deletions that were identified in this study. The most outer ring showed the chromosomal ideogram in a clockwise orientation. The panels from outside to inside summarized the SNP density, indel density, and the location of the inversions and deletions, which were identified in the N. brichardi (in red), A. burtoni (in green), P. nyererei (in blue), and M. zebra (in orange) genomes. The SNP and indel densities were scanned using a 50 kb sliding window. The histograms in the inversion and deletion panels represent the position of the inversion and deletion and the widths of the histograms show the size of the variations. The results were visualized using Circos (Krzywinski et al., 2009).
The number of indels in these data sets ranges from 1,440,691 to 2,317,313 in these four species (Table 1). Proportionally, indels occur as frequently as SNPs in the intronic, intergenic, upstream and downstream regions, but are less common in the exonic regions (<1% vs. >4%, Table 1). Based on the gene annotation of the Tilapia genome, some of the exonic indels may have deleterious effects, such as causing frameshifts or the loss of start and stop codons. In comparison to the genome-wide indels, the exonic indels are enriched for sizes divisible by three, which indicates that those indel events may affect a whole codon (Figure 3).
Using paired-end and mate-paired data, Pindel identified 621, 683, 371, and 389 inversions in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes, respectively, (Table 2). Normalized by the length of linkage groups, we did not detect any linkage group that has a significantly high number of inversions than any other in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes (generalized ESD test, p-value > 0.05) (Rosner, 1983). The average length of the inverted region ranges from 98,025 to 214,075 bp in the species compared (Table 2). The longest inversion, 24,423,751 bp, was located on LG20 of the P. nyererei genome and involves 853 genes. In total, we found that 262, 306, 162, and 154 genes overlapped with the inverted regions in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes (Table 2). However, no GO terms are significantly enriched in the inverted regions.
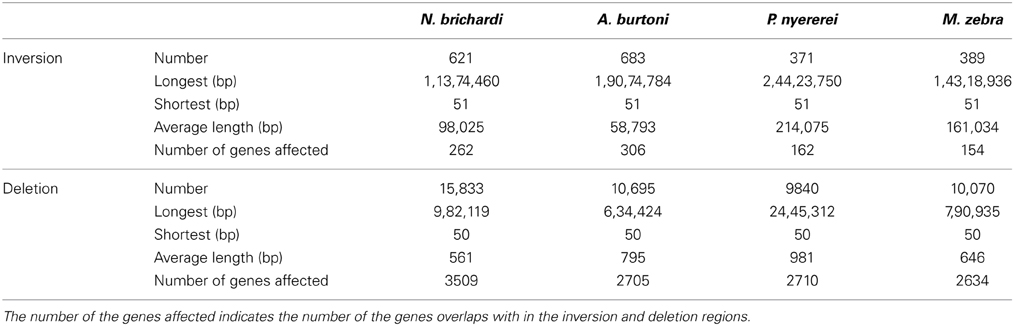
Table 2. Summary of the inversions and deletions in the African cichlid genomes.
In addition to SNPs, indels and inversions, we also assessed long deletions (>50 bp) in the African cichlid genomes. 15,833, 10,695, 9,840, and 10,070 long deletions were identified in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes, respectively, (Table 1). However, no linkage group harbors a significantly higher number of long deletions after normalizing by the length of linkage group (generalized ESD test, p-value > 0.05) (Rosner, 1983). The N. brichardi genome has the greatest number of deletions; however, their average length in the P. nyererei genome is larger than in the other species (Table 2). Investigating this further with the annotated gene regions, we found that the genes affected by the deleted regions were significantly associated with the GO terms “binding,” “cell adhesion,” and “biological adhesion” (FDR corrected p-value < 1E-4).
Different patterns of structural genetic variation were detected during the divergence of the African cichlids (Figure 2). Large numbers of SNPs (9.8 million) can be traced back to the common ancestor of N. brichardi, A. burtoni, P. nyererei, and M. zebra and indicate in a substantially higher rate of SNP evolution (from 1.7 TO 3.3-fold changes) in comparison to other lineages (Figure 2A) at the basis of the radiation of African cichlids. However, we did not detect similar patterns in the evolution of inversions and deletions. The rates of inversions and deletions in the common ancestor of these four species are the slowest rate in comparison to other lineages, especially in comparison to the haplochromine lineages (Figures 2C,D). The rate of indels evolved uniformly in all lineages (Figure 2B).
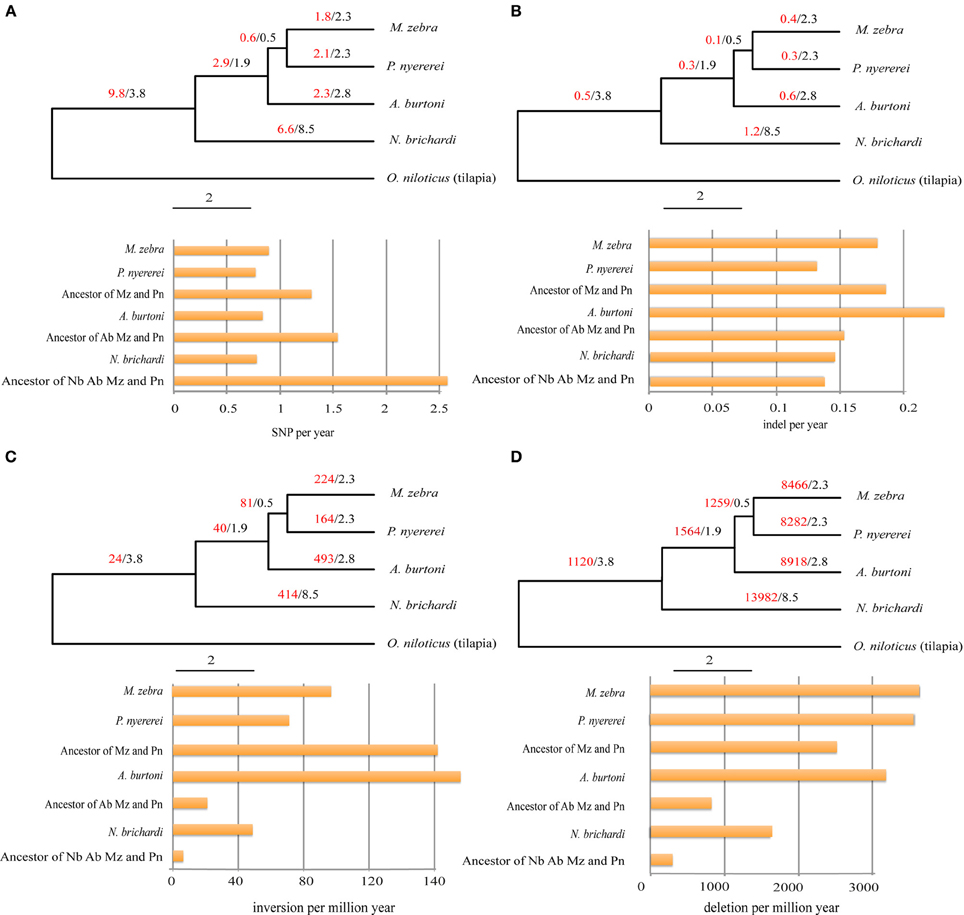
Figure 2. The evolutionary patterns of SNPs, indels, inversions, and deletion during the adaptive radiation of African cichlids. The phylogeny was adopted from the cichlid genome paper (Brawand et al., submitted). The evolution of SNP, indel, inversion, and deletion were shown in (A–D). The numbers before and after slash are the number of the lineage specific variation and the divergence time (million years). The numbers of the SNPs and indels on the phylogeny were scaled by millions. The numbers of the inversions and deletion were not scaled by millions. The histogram shows the number of the variations normalized by the corresponding divergence time. Nb, Ab, Pn, and Mz indicate Neolamprologus brichardi, Astatotilapia burtoni, Pundamila nyererei, and Metriaclima zebra.
Discussion
The genome sequence data collected by the cichlid genome consortium provides an opportunity for the study of genome evolution of African cichlid fishes. Using these whole genome sequencing data, we generated a high-resolution map of genomic variation in African cichlid fishes (Figure 1). The resolution of most of this genetic variation down to the nucleotide level enabled us to conduct this initial investigation of their potential impacts to the adaptive radiation of African cichlid fishes.
SNPs are the most common form of genetic variation in the cichlid genomes. On average, we found around 17 million SNPs in the genomes of N. brichardi, A. burtoni, P. nyererei, and M. zebra using the Tilapia genome as the reference genome. Although the relative low number in comparison to the SNPs in the other regions, many non-synonymous SNPs evolved during the adaptive radiation of African cichlids (Table 1). We found that the genes involved in immune pathways, for example, Interleukin-15, major histocompatibility complex II (MHC II), and C-C motif chemokine 19 have an excessive number of non-synonymous SNPs. Immune genes are commonly characterized by signature of positive selection during speciation process (Jansa et al., 2003; Nielsen et al., 2005; Jiggins and Kim, 2007; Jones et al., 2012a). The selection can be strongly intensified during the colonization of new habitats as new pathogens may induce primary challenges to host immune system (Matthews et al., 2010; Jones et al., 2012a).
The extensive amount of SNP sites that is shared across the genomes of N. brichardi, A. burtoni, P. nyererei, and M. zebra could be the result of two scenarios. First, these may be deeply shared polymorphisms, which originated in the common ancestor of N. brichardi, A. burtoni, P. nyererei, and M. zebra and are still present in these species. Previous study has shown that genetic polymorphisms in the endemic cichlids of Lake Victoria can be traced back to the ancestors of the modern haplochromine lineages (Elmer et al., 2009). Furthermore, genotyping a set of 280 SNPs in ~160 African cichlid species, Loh and coauthors found that around 50% of these loci are polymorphic in the cichlid lineages across East Africa (Loh et al., 2013). Second, it may be that ongoing but occasional genetic interchange among lacustrine cichlids from different great lakes is facilitated by riverine cichlid species as “transporters” (Verheyen et al., 2003; Loh et al., 2013). Such ongoing gene flow among cichlid species could transfer beneficial alleles among species, thus promoting the rapid evolution of African cichlids because these pre-existing genetic variations may have already been tested by natural selection in similar environments (e.g., in adaptation to lacustrine environments).
Comparing the rate of SNP evolution during the evolution of the adaptive radiations of African cichlids, a higher rate of SNP evolution was observed in the lineage of the common ancestor of all four representatives of the three East African cichlid radiations: N. brichardi, A. burtoni, P. nyererei, and M. zebra, compared to the terminal branches (Figure 2A). This could be the result of relaxed purifying selection or positive selection for adaptation to the novel niches in the newly colonized lakes, compared to an ancestral riverine environment that would have been inhabited by the ancestral lineages to the endemic radiations. Environmental factors often lead to chanced selection pressures that either eliminates or weakens the purified selection that might have dominated in previous habitats (Lahti et al., 2009) or increases the frequency of the beneficial alleles by positive natural selection (Nielsen et al., 2007).
In comparison to SNPs, we found much fewer indels in exons (Table 1) and if there, the length is highly likely to be divisible by three, obviously to keep the reading frame intact (Figure 3). This was also reported for the genomes of cattle (Zhan et al., 2011), human (Mills et al., 2011a), and sticklebacks (Feulner et al., 2013). The rare presence of the indels in the exonic regions reflects the strong purify selection against this form of variation, which potentially disrupts protein coding. Based on the gene annotation of the Tilapia genome, some of those exonic indels may have deleterious effects, which needs further validation in the future. However, we cannot rule out that some predicted effects of the exonic indels resulted from an incorrect gene model in the Nextgen genomes (Alkan et al., 2011b) or from sequencing errors.
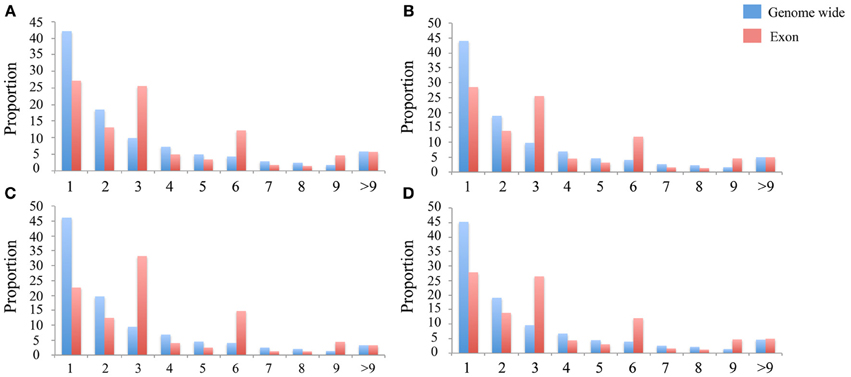
Figure 3. Indel length distribution. The Y-axis is the proportion of the indels in the exon region (Red) and genome wide (Blue). The X-axis is binned by the length of the indel. (A–D) present in the indel length distribution in the N. brichardi, A. burtoni, P. nyererei, and M. zebra genomes.
In contrast to the SNP evolution, the rates of SV in the haplochromine lineages are higher than in the ancestral lineages (Figures 2C,D). A large body of research has examined the role of genomic structure variation (including inversions and deletions) in local adaptation and speciation (Noor et al., 2001; Kirkpatrick and Barton, 2006; Feder and Nosil, 2009; Chan et al., 2010; Jones et al., 2012b; Feder et al., 2013). For example, multiple lines of evidence show that loci involved in local adaptation (Coluzzi et al., 2002; Anderson et al., 2005; Kirkpatrick and Barton, 2006; Lowry and Willis, 2010) and pre- or postzygotic isolations were mapped to inverted regions (Noor et al., 2001; Rieseberg, 2001; Ayala et al., 2013). The evolutionary importance of inversions lies in their ability to suppress recombination when they are heterozygous between population or species (Sturtevant, 1917). Therefore, co-adapted alleles may be embedded in the inverted regions and will not be eroded by introgression and recombination (Dobzhansky, 1947, 1970). Besides, structural variation can also affect the gene expression if the breakpoints of SVs overlap with regulatory regions (Wesley and Eanes, 1994; Chan et al., 2010; Harewood et al., 2012). For example, repeated deletions of the enhancer of the Pitx1 gene changed the gene expression patterns and is responsible for the repeated and independent loss of the pelvic fin in the freshwater stickleback populations (Chan et al., 2010). Our analyses of structural variation of five East Africa cichlid genomes provides a starting point for further investigations of the impact of structural variation during the adaptive radiation of African cichlid fishes. Especially, given the ongoing gene flow between cichlids from different lakes (Ruber et al., 2001; Verheyen et al., 2003; Elmer et al., 2009; Joyce et al., 2011; Loh et al., 2013), the lineage specific SV (Figures 2C,D) would be highly informative in understanding the processes and genomic consequences of speciation-with-gene-flow.
Our analyses provide a first glance at the genomic variation of genomes of cichlid fish of adaptive radiation of East Africa. Although only one individual per species was sampled so far, we detected a large amount of shared structural variation across the cichlid genomes of these closely related adaptive radiations, indicating that also this type of genomic variation is a form of shared ancestral variation that is maintained across species and lineages. Much of the structural variation of these genomes is located in functionally important regions of genes (e.g., exonic or regulatory regions). One might be tempted to speculate that also this type of variation might have contributed to both the local adaptation and speciation of cichlids—but this need further investigation through functional essays of candidate genes and structural variants, such as CRISPR-Cas (Kratochwil and Meyer, unpublished data). One next step is to evaluate the functional impacts of these regions during different waves of repeated adaptive radiations by systematically investigating the variability and selection pressures of these regions through population genomic analyses in representatives of the parallel-evolved species flocks of African cichlids.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Iker Irisarri for his help with the phylogenetic analyses and Frederico Henning for his comments on an earlier version the manuscript. This work was partially funded by a scholarship from the University of Konstanz to Shaohua Fan and the European Research Council (ERC advanced grant “GenAdap” 293700) to Axel Meyer.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2014.00163/abstract
References
Alkan, C., Coe, B. P., and Eichler, E. E. (2011a). Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376. doi: 10.1038/nrg2958
Alkan, C., Sajjadian, S., and Eichler, E. E. (2011b). Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65. doi: 10.1038/nmeth.1527
Anderson, A. R., Hoffmann, A. A., McKechnie, S. W., Umina, P. A., and Weeks, A. R. (2005). The latitudinal cline in the In(3R)Payne inversion polymorphism has shifted in the last 20 years in Australian Drosophila melanogaster populations. Mol. Ecol. 14, 851–858. doi: 10.1111/j.1365-294X.2005.02445.x
Aszodi, A. (2012). MULTOVL: fast multiple overlaps of genomic regions. Bioinformatics 28, 3318–3319. doi: 10.1093/bioinformatics/bts607
Ayala, D., Guerrero, R. F., and Kirkpatrick, M. (2013). Reproductive isolation and local adaptation quantified for a chromosome inversion in a malaria mosquito. Evolution 67, 946–958. doi: 10.1111/j.1558-5646.2012.01836.x
Baldo, L., Santos, M. E., and Salzburger, W. (2011). Comparative transcriptomics of Eastern African cichlid fishes shows signs of positive selection and a large contribution of untranslated regions to genetic diversity. Genome Biol. Evol. 3, 443–455. doi: 10.1093/gbe/evr047
Chan, Y. F., Marks, M. E., Jones, F. C., Villarreal, G., Shapiro, M. D., Brady, S. D., et al. (2010). Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305. doi: 10.1126/science.1182213
Coluzzi, M., Sabatini, A., Della Torre, A., Di Deco, M. A., and Petrarca, V. (2002). A polytene chromosome analysis of the Anopheles gambiae species complex. Science 298, 1415–1418. doi: 10.1126/science.1077769
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Depristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491. doi: 10.1038/ng.806
Dobzhansky, T. (1947). Genetics of natural populations; a response of certain gene arrangements in the third chromosome of Drosophila pseudoobscura to natural selection. Genetics 32, 142–160.
Dobzhansky, T. (1970). Genetics of the Evolutionary Process. New York, NY: Columbia University Press.
Elmer, K. R., and Meyer, A. (2011). Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends Ecol. Evol. 26, 298–306. doi: 10.1016/j.tree.2011.02.008
Elmer, K. R., Reggio, C., Wirth, T., Verheyen, E., Salzburger, W., and Meyer, A. (2009). Pleistocene desiccation in East Africa bottlenecked but did not extirpate the adaptive radiation of Lake Victoria haplochromine cichlid fishes. Proc. Natl. Acad. Sci. U.S.A. 106, 13404–13409. doi: 10.1073/Pnas.0902299106
Fan, S. H., Elmer, K. R., and Meyer, A. (2011). Positive Darwinian selection drives the evolution of the morphology-related gene, EPCAM, in particularly species-rich lineages of African cichlid fishes. J. Mol. Evol. 73, 1–9. doi: 10.1007/s00239-011-9452-5
Fan, S. H., Elmer, K. R., and Meyer, A. (2012). Genomics of adaptation and speciation in cichlid fishes: recent advances and analyses in African and Neotropical lineages. Philos. Trans. R. Soc. B-Biol. Sci. 367, 385–394. doi: 10.1098/rstb.2011.0247
Feder, J. L., Flaxman, S. M., Egan, S., Comeault, A. A., and Nosil, P. (2013). Geographic mode of speciation and genomic divergence. Annu. Rev. Ecol. Evol. Syst. 44, 73–97. doi: 10.1146/annurev-ecolsys-110512-135825
Feder, J. L., and Nosil, P. (2009). Chromosomal inversions and species differences: when are genes affecting adaptive divergence and reproductive isolation expected to reside within inversions? Evolution 63, 3061–3075. doi: 10.1111/j.1558-5646.2009.00786.x
Feulner, P. G., Chain, F. J., Panchal, M., Eizaguirre, C., Kalbe, M., Lenz, T. L., et al. (2013). Genome-wide patterns of standing genetic variation in a marine population of three-spined sticklebacks. Mol. Ecol. 22, 635–649. doi: 10.1111/j.1365-294X.2012.05680.x
Friedman, M., Keck, B. P., Dornburg, A., Eytan, R. I., Martin, C. H., Hulsey, C. D., et al. (2013). Molecular and fossil evidence place the origin of cichlid fishes long after Gondwanan rifting. Proc. Biol. Sci. 280:20131733. doi: 10.1098/rspb.2013.1733
Gnerre, S., Maccallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Gotz, S., Garcia-Gomez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Gunter, H. M., Clabaut, C., Salzburger, W., and Meyer, A. (2011). Identification and characterization of gene expression involved in the coloration of cichlid fish using microarray and qRT-PCR approaches. J. Mol. Evol. 72, 127–137. doi: 10.1007/s00239-011-9431-x
Gunter, H. M., Fan, S., Xiong, F., Franchini, P., Fruciano, C., and Meyer, A. (2013). Shaping development through mechanical strain: the transcriptional basis of diet-induced phenotypic plasticity in a cichlid fish. Mol. Ecol. 22, 4516–4531. doi: 10.1111/mec.12417
Harewood, L., Chaignat, E., and Reymond, A. (2012). Structural variation and its effect on expression. Methods Mol. Biol. 838, 173–186. doi: 10.1007/978-1-61779-507-7_8
Henning, F., and Meyer, A. (in press). Evolutionary genomics of cichlid cishes: explosive speciation and adaptation in the postgenomic era. Annu. Rev. Genomics Hum. Genet. 15. doi: 10.1146/annurev-genom-090413-025412
Hofmann, C. M., O'quin, K. E., Marshall, N. J., Cronin, T. W., Seehausen, O., and Carleton, K. L. (2009). The eyes have it: regulatory and structural changes both underlie cichlid visual pigment diversity. PLoS Biol. 7:e1000266. doi: 10.1371/journal.pbio.1000266
Jansa, S. A., Lundrigan, B. L., and Tucker, P. K. (2003). Tests for positive selection on immune and reproductive genes in closely related species of the murine genus mus. J. Mol. Evol. 56, 294–307. doi: 10.1007/s00239-002-2401-6
Jiggins, F. M., and Kim, K. W. (2007). A screen for immunity genes evolving under positive selection in Drosophila. J. Evol. Biol. 20, 965–970. doi: 10.1111/j.1420-9101.2007.01305.x
Jones, F. C., Chan, Y. F., Schmutz, J., Grimwood, J., Brady, S. D., Southwick, A. M., et al. (2012a). A genome-wide SNP genotyping array reveals patterns of global and repeated species-pair divergence in sticklebacks. Curr. Biol. 22, 83–90. doi: 10.1016/j.cub.2011.11.045
Jones, F. C., Grabherr, M. G., Chan, Y. F., Russell, P., Mauceli, E., Johnson, J., et al. (2012b). The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61. doi: 10.1038/nature10944
Joyce, D. A., Lunt, D. H., Genner, M. J., Turner, G. F., Bills, R., and Seehausen, O. (2011). Repeated colonization and hybridization characterize the Lake Malawi cichlids. Curr. Biol. 21, R108–R109. doi: 10.1016/j.cub.2010.11.029
Kirkpatrick, M., and Barton, N. (2006). Chromosome inversions, local adaptation and speciation. Genetics 173, 419–434. doi: 10.1534/genetics.105.047985
Kocher, T. D. (2004). Adaptive evolution and explosive speciation: the cichlid fish model. Nat. Rev. Genet. 5, 288–298. doi: 10.1038/nrg1316
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuraku, S., and Meyer, A. (2008). Genomic analysis of cichlid fish ‘natural mutants’. Curr. Opin. Genet. Dev. 18, 551–558. doi: 10.1016/j.gde.2008.11.002
Lahti, D. C., Johnson, N. A., Ajie, B. C., Otto, S. P., Hendry, A. P., Blumstein, D. T., et al. (2009). Relaxed selection in the wild. Trends Ecol. Evol. 24, 487–496. doi: 10.1016/j.tree.2009.03.010
Lang, M., Miyake, T., Braasch, I., Tinnemore, D., Siegel, N., Salzburger, W., et al. (2006). A BAC library of the East African haplochromin cichlid fish Astatotilapia burtoni. J. Exp. Zool. B Mol. Dev. Evol. 306B, 35–44. doi: 10.1002/jez.b.21068
Lee, B. Y., Howe, A. E., Conte, M. A., D'cotta, H., Pepey, E., Baroiller, J. F., et al. (2010). An EST resource for Tilapia based on 17 normalized libraries and assembly of 116,899 sequence tags. BMC Genomics 11:278. doi: 10.1186/1471-2164-11-278
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Loh, Y. H., Bezault, E., Muenzel, F. M., Roberts, R. B., Swofford, R., Barluenga, M., et al. (2013). Origins of shared genetic variation in African cichlids. Mol. Biol. Evol. 30, 906–917. doi: 10.1093/molbev/mss326
Lowry, D. B., and Willis, J. H. (2010). A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 8:e1000500. doi: 10.1371/journal.pbio.1000500
Matthews, B., Harmon, L. J., M'Gonigle, L., Marchinko, K. B., and Schaschl, H. (2010). Sympatric and allopatric divergence of MHC genes in threespine stickleback. PLoS ONE 5:e10948. doi: 10.1371/journal.pone.0010948
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meyer, A. (1993). Phylogenetic relationships and evolutionary processes in East African cichlid fishes. Trends Ecol. Evol. 8, 279–284. doi: 10.1016/0169-5347(93)90255-N
Meyer, A., Biermann, C. H., and Orti, G. (1993). The phylogenetic position of the zebrafish (Danio rerio), a model system in developmental biology: an invitation to the comparative method. Proc. Biol. Sci. 252, 231–236. doi: 10.1098/rspb.1993.0070
Meyer, A., Kocher, T. D., Basasibwaki, P., and Wilson, A. C. (1990). Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNA sequences. Nature 347, 550–553. doi: 10.1038/347550a0
Meyer, A., Ritchie, P. A., and Witte, K.-E. (1995). Predicting developmental processes from evolutionary patterns: a molecular phylogeny of the zebrafish (Danio rerio) and its relatives. Philos. Trans. R. Soc. Lond. B Biol. Sci. 349, 103–111. doi: 10.1098/rstb.1995.0096
Mills, R. E., Pittard, W. S., Mullaney, J. M., Farooq, U., Creasy, T. H., Mahurkar, A. A., et al. (2011a). Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 21, 830–839. doi: 10.1101/gr.115907.110
Mills, R. E., Walter, K., Stewart, C., Handsaker, R. E., Chen, K., Alkan, C., et al. (2011b). Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65. doi: 10.1038/nature09708
Nielsen, R., Bustamante, C., Clark, A. G., Glanowski, S., Sackton, T. B., Hubisz, M. J., et al. (2005). A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3:e170. doi: 10.1371/journal.pbio.0030170
Nielsen, R., Hellmann, I., Hubisz, M., Bustamante, C., and Clark, A. G. (2007). Recent and ongoing selection in the human genome. Nat. Rev. Genet. 8, 857–868. doi: 10.1038/nrg2187
Noor, M. A., Grams, K. L., Bertucci, L. A., and Reiland, J. (2001). Chromosomal inversions and the reproductive isolation of species. Proc. Natl. Acad. Sci. U.S.A. 98, 12084–12088. doi: 10.1073/pnas.221274498
Quinlan, A. R., Clark, R. A., Sokolova, S., Leibowitz, M. L., Zhang, Y., Hurles, M. E., et al. (2010). Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 20, 623–635. doi: 10.1101/gr.102970.109
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rambaut, A., and Charleston, M. (2002). TreeEdit. Phylogenetic Tree Editor v1.0 Alpha 10. Oxford: Oxford University.
Rieseberg, L. H. (2001). Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358. doi: 10.1016/S0169-5347(01)02187-5
Rosner, B. (1983). Percentage points for a generalized ESD many-outlier procedure. Technometrics 25, 165–172. doi: 10.1080/00401706.1983.10487848
Ruber, L., Meyer, A., Sturmbauer, C., and Verheyen, E. (2001). Population structure in two sympatric species of the Lake Tanganyika cichlid tribe Eretmodini: evidence for introgression. Mol. Ecol. 10, 1207–1225. doi: 10.1046/j.1365-294X.2001.01259.x
Salzburger, W., Mack, T., Verheyen, E., and Meyer, A. (2005). Out of Tanganyika: genesis, explosive speciation, key-innovations and phylogeography of the haplochromine cichlid fishes. BMC Evol. Biol. 5:17. doi: 10.1186/1471-2148-5-17
Salzburger, W., and Meyer, A. (2004). The species flocks of East African cichlid fishes: recent advances in molecular phylogenetics and population genetics. Naturwissenschaften 91, 277–290. doi: 10.1007/s00114-004-0528-6
Salzburger, W., Renn, S. C., Steinke, D., Braasch, I., Hofmann, H. A., and Meyer, A. (2008). Annotation of expressed sequence tags for the East African cichlid fish Astatotilapia burtoni and evolutionary analyses of cichlid ORFs. BMC Genomics 9:96. doi: 10.1186/1471-2164-9-96
Sanderson, M. J. (1997). A nonparametric approach to estimating divergence times in the absence of rate constancy. Mol. Biol. Evol. 14, 1218–1231. doi: 10.1093/oxfordjournals.molbev.a025731
Stiassny, M. L. J., and Meyer, A. (1999). Cichlids of the Rift lakes. Sci. Am. 280, 64–69. doi: 10.1038/scientificamerican0299-64
Sturmbauer, C., Verheyen, E., and Meyer, A. (1994). Mitochondrial phylogeny of the Lamprologini, the major substrate spawning lineage of cichlid fishes from Lake-Tanganyika in eastern Africa. Mol. Biol. Evol. 11, 691–703.
Sturtevant, A. H. (1917). Genetic factors affecting the strength of linkage in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 3, 555–558. doi: 10.1073/pnas.3.9.555
Terai, Y., Mayer, W. E., Klein, J., Tichy, H., and Okada, N. (2002). The effect of selection on a long wavelength-sensitive (LWS) opsin gene of Lake Victoria cichlid fishes. Proc. Natl. Acad. Sci. U.S.A. 99, 15501–15506. doi: 10.1073/pnas.232561099
Terai, Y., Seehausen, O., Sasaki, T., Takahashi, K., Mizoiri, S., Sugawara, T., et al. (2006). Divergent selection on opsins drives incipient speciation in Lake Victoria cichlids. PLoS Biol. 4:e433. doi: 10.1371/journal.pbio.0040433
Verheyen, E., Salzburger, W., Snoeks, J., and Meyer, A. (2003). Origin of the superflock of cichlid fishes from Lake Victoria, East Africa. Science 300, 325–329. doi: 10.1126/science.1080699
Wesley, C. S., and Eanes, W. F. (1994). Isolation and analysis of the breakpoint sequences of chromosome inversion ln(3L)Payne in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 91, 3132–3136. doi: 10.1073/pnas.91.8.3132
Ye, K., Schulz, M. H., Long, Q., Apweiler, R., and Ning, Z. M. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. doi: 10.1093/bioinformatics/btp394
Zhan, B., Fadista, J., Thomsen, B., Hedegaard, J., Panitz, F., and Bendixen, C. (2011). Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genomics 12:557. doi: 10.1186/1471-2164-12-557
Keywords: SNPs, insertions, deletions, inversions, adaptive radiation, structural variation
Citation: Fan S and Meyer A (2014) Evolution of genomic structural variation and genomic architecture in the adaptive radiations of African cichlid fishes. Front. Genet. 5:163. doi: 10.3389/fgene.2014.00163
Received: 07 February 2014; Accepted: 15 May 2014;
Published online: 03 June 2014.
Edited by:
Philine G. D. Feulner, Max Planck Institute for Evolutionary Biology, GermanyReviewed by:
Harald Schneider, Natural History Museum, UKFeng Zhang, Fudan University, China
Arne W. Nolte, Max Planck Society, Germany
Copyright © 2014 Fan and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Axel Meyer, Lehrstuhl für Zoologie und Evolutionsbiologie, Department of Biology, University of Konstanz, Universitätstraße 10, Konstanz 78464, Germany e-mail:YXhlbC5tZXllckB1bmkta29uc3RhbnouZGU=