 Josée Guirouilh-Barbat1
Josée Guirouilh-Barbat1 Bernard S. Lopez
Bernard S. Lopez- 1CNRS, UMR 8200, Institut de Cancérologie Gustave Roussy, Équipe Labélisée, Université Paris-Sud, «LIGUE 2014», Villejuif, France
- 2Institut Curie, CNRS, UMR 3348, Orsay, France
- 3CEA DSV, UMR 967 CEA-INSERM-Université Paris Diderot-Université Paris Sud, Institut de Radiobiologie Cellulaire et Moléculaire, Fontenay-aux-Roses, France
Homologous recombination (HR) is an evolutionarily conserved process that plays a pivotal role in the equilibrium between genetic stability and diversity. HR is commonly considered to be error-free, but several studies have shown that HR can be error-prone. Here, we discuss the actual accuracy of HR. First, we present the product of genetic exchanges (gene conversion, GC, and crossing over, CO) and the mechanisms of HR during double strand break repair and replication restart. We discuss the intrinsic capacities of HR to generate genome rearrangements by GC or CO, either during DSB repair or replication restart. During this process, abortive HR intermediates generate genetic instability and cell toxicity. In addition to genome rearrangements, HR also primes error-prone DNA synthesis and favors mutagenesis on single stranded DNA, a key DNA intermediate during the HR process. The fact that cells have developed several mechanisms protecting against HR excess emphasize its potential risks. Consistent with this duality, several pro-oncogenic situations have been consistently associated with either decreased or increased HR levels. Nevertheless, this versatility also has advantages that we outline here. We conclude that HR is a double-edged sword, which on one hand controls the equilibrium between genome stability and diversity but, on the other hand, can jeopardize the maintenance of genomic integrity. Therefore, whether non-homologous end joining (which, in contrast with HR, is not intrinsically mutagenic) or HR is the more mutagenic process is a question that should be re-evaluated. Both processes can be “Dr. Jekyll” in maintaining genome stability/variability and “Mr. Hyde” in jeopardizing genome integrity.
Introduction
Genomes are routinely challenged with exogenous or endogenous insults of enzymatic, chemical or physical origins. These DNA alterations can generate genetic instability, leading to cell death, senescence, developmental abnormalities and tumor initiation and progression. However, while it is vital to maintain genomic stability, genetic diversity is essential to physiological processes, such as the generation of the immune repertoire or the mixing of parental alleles during meiosis. Additionally, the absence of genetic diversity would constitute an evolutionary dead end. Thus, DNA repair should maintain genomic stability and allow for genetic diversity. Therefore, the accuracy of DNA repair processes is an essential issue.
Homologous recombination (HR) is a process that is conserved in all organisms, playing an essential and pivotal role in genome stability and plasticity. Notably, HR is involved in the reactivation of replication forks that have been blocked and in the repair of DNA double strand breaks (DSBs) (reviewed in Haber, 2014).
Replication fork progression is routinely challenged by diverse exogenous or endogenous stresses, which ultimately leads to replication fork stalling, collapse or breakage, and triggers the DNA damage response (DDR) (Hyrien, 2000; Lambert and Carr, 2005, 2013; Tourriere and Pasero, 2007; Aguilera and Garcia-Muse, 2013). Failures in chromosome replication are thus a primary source of genetic instability. Consistently, in many organisms, including yeast and human cells, both slowing down and blocking fork progression are associated with chromosome breakage and genome rearrangement (reviewed in Aguilera and Gomez-Gonzalez, 2008; Branzei and Foiani, 2010). Moreover, impediments to fork progression might also challenge the completion of DNA replication, resulting in mitotic defects and multipolar mitotic cells, which then lead to uneven chromosome segregation and thus amplifying the genome instability to the whole genome, including fully replicated regions (Wilhelm et al., 2014). Consistently with the existence of endogenous replication stresses, DDR activation resulting from spontaneous endogenous replication stress has also been detected in the early stages of carcinogenesis and senescence (Bartkova et al., 2005, 2006; Gorgoulis et al., 2005; Halazonetis et al., 2008; Gorgoulis and Halazonetis, 2010).
DSBs are harmful lesions that are produced through exposure to exogenous treatments, such as ionizing radiation (IR), byproducts of endogenous cellular metabolisms and, importantly, replication forks arrest (Seigneur et al., 1998; Featherstone and Jackson, 1999; Saintigny et al., 2001; Rothkamm and Lobrich, 2003; Mahaney et al., 2009). DSBs can trigger profound genomic rearrangements or, in contrast, generate genetic diversity in essential biological processes. In the latter case, programmed DSBs are physiologically produced through controlled cellular enzymes during meiotic differentiation, mating-type switching in Saccharomyces cerevisiae or in V(D)J and class switch recombination, which ensures the diversity of the immune response (reviewed in Haber, 1992; Jung and Alt, 2004; Lieber et al., 2004; Rooney et al., 2004; Dudley et al., 2005; Buard and de Massy, 2007).
Two primary strategies are used to repair DSBs: (1) HR, which requires a sequence-homologous partner and, in fact, corresponds to different processes involving both common and distinct mechanisms (see below and Figure 1); and (2) NHEJ (non-homologous end joining), which ligates the DNA ends of a DSB without requiring extended homologies (Haber, 2014). Note that a highly mutagenic alternative end-joining pathway (A-EJ) has recently been identified (for review Grabarz et al., 2012; Rass et al., 2012; Betermier et al., 2014).
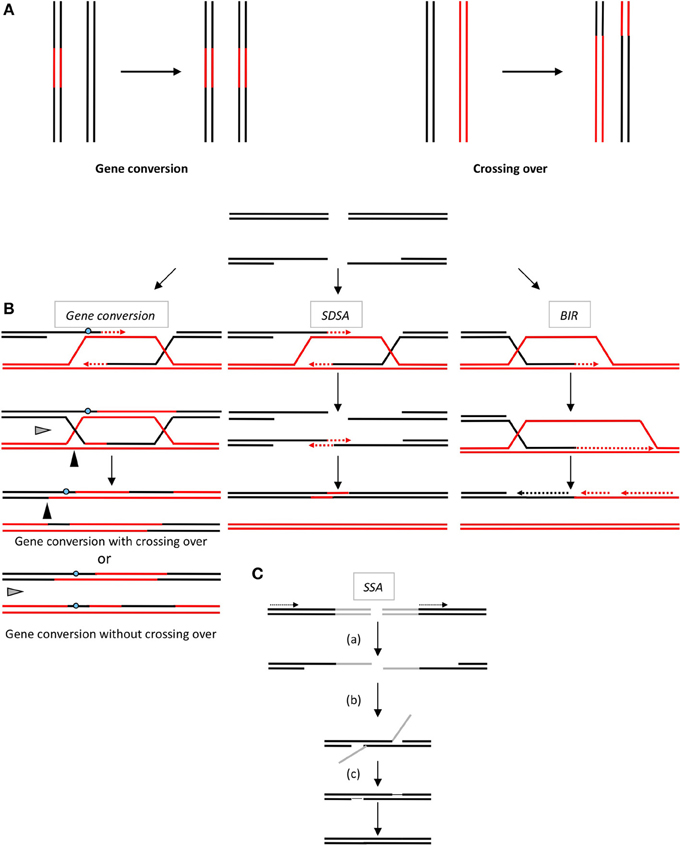
Figure 1. (A) The products of HR. Gene conversion (left panel) leading to non-reciprocal exchange of a DNA sequence (in red). Crossing over (right panel): reciprocal exchanges of adjacent sequences (black and red). Note that gene conversion can be associated with or without crossing over. (B) The double-strand break repair models through HR. Left panel: Gene conversion. After resection, the single-stranded 3′-tail invades a homologous, intact double-stranded DNA, forming a D-loop (displacement loop). This process tolerates limited imperfect sequence homologies, thus creating heteroduplex intermediates bearing mismatches (blue circle). The invading 3′-end primes DNA synthesis, which then fills in the gaps. The cruciform junctions (Holliday junctions, HJ) migrate. Resolution (or dissolution) of the HJ occurs in two different orientations (black or gray triangles), resulting in gene conversion either with or without crossing over. Middle panel: Synthesis-dependent strand annealing. Initiation is similar to that of the previous model, but the invading strand de-hybridizes and re-anneals at the other end of the injured molecule; no HJ is formed. Right panel: Break-induced replication (BIR). The initiation is similar to that of the previous models, but the synthesis continues over longer distances on the chromosome arms, even reaching the end of the chromosome. Here, there is neither resolution of the HR nor crossover. (C) Single-strand annealing (SSA). When a double-strand break is generated between two homologous sequences in tandem in the same orientation (dotted arrows), an extended single-strand resection (a) reveals two complementary DNA strands that can hybridize (b). (c) Resolution of the intermediate and gap filling complete the repair, leading to the deletion of the intergenic sequences between the initial repetitions.
In most of the literature, HR is described as an error-free process, while NHEJ is described as an error-prone DSB repair process. This statement is largely based on the fact that the mechanism of HR requires the search for a homologous partner to repair DNA, in contrast to NHEJ. Careful examination of the data from the literature might challenge these assumptions, which requires revisiting the current view. Indeed, recent data points to the intrinsic precision of canonical NHEJ (C-NHEJ; KU-Ligase 4-dependent) in contrast to A-EJ. In fact, C-NHEJ is conservative but adaptable, and the accuracy of the repair is dictated by the structure of the DNA ends rather than by the C-NHEJ machinery itself (Grabarz et al., 2012; Rass et al., 2012; Betermier et al., 2014).
Here, in a reciprocal view, we discuss the accuracy of HR and we present several situations of mutagenesis generated by HR. We conclude that HR is a double-edged sword, which on the one hand controls the equilibrium of genomic stability vs. diversity, but on the other hand can jeopardize the maintenance of genomic integrity. The importance of the versatility of HR and its impact on genomic integrity are discussed.
The Products of HR (Gene Conversion and Crossing Over) and Models
Consistently with the implication of HR in genome stability maintenance, mutant cells that are defective in HR show elevated mutagenesis and genetic instability. However, in contrast, HR can appear as a mutagenic process per se, in many situations. Such concepts can be understood when considering the products and molecular mechanisms of HR.
The products of HR are gene conversion (GC: non-reciprocal exchange of genetic material) associated or not with crossing-over (CO: reciprocal exchange of the adjacent sequences) (Figure 1A). Such products can account for genetic diversity or instability arising through HR.
Models of HR for DSB Repair
All HR processes are initiated through the 5′ to 3′-single-stranded resection of double stranded DNA ends, creating a 3′-single-stranded DNA (ssDNA), on which the pivotal RecA/Rad51 recombinase is loaded (Figure 1B). The RecA/Rad51 nucleofilament carries out the subsequent invasion of a homologous DNA duplex that primes DNA synthesis and copies the intact DNA molecule. At this point, the HR processes differ in the processing of the intermediates, leading to either gene conversion, associated or not with crossing-over, or to SDSA (synthesis-dependent strand annealing) and BIR (break-induced replication) (Figure 1B). In addition, an alternative process (SSA, single-strand annealing) is also initiated by resection; however, the following step does not require Rad51 nor strand invasion of an intact duplex DNA, but the annealing of two complementary ssDNAs (Figure 1C). SSA is a non-conservative process that systematically leads to the deletion of the intervening sequence between the two interacting DNA molecules (reviewed in Haber, 2014).
HR and Replication Forks Reactivation
HR contributes to the robustness of DNA replication by multiple mechanisms (Figure 2) and might be viewed as a pathway escorting fork progression (reviewed in Costes and Lambert, 2012) (Figure 2). HR can act either at replication forks or at replicated chromatids to ensure the completion of chromosome duplication. First, HR efficiently seals ssDNA gaps that have been left within replicated chromatids after fork passage through DNA lesions. Second, HR is involved in the recovery of arrested replication forks and has the potential to reassemble a functional replisome. While the mechanism of origin-independent loading of a replisome by HR has been extensively characterized in bacteria, its counterpart in eukaryotic cells has only recently begun to emerge.
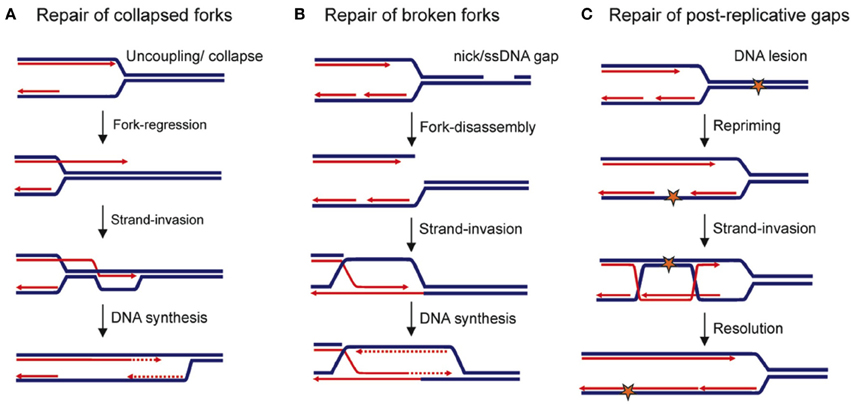
Figure 2. Replication-maintenance by homologous recombination. Blue and red lines indicate parental and neo-synthesized strands, respectively. (A) Replication-restart following collapse of the replication fork. (B) Repair of a broken replication fork. (C) Repair of ssDNA gaps that are left behind the moving fork after it has encountered a DNA lesion. Star: DNA damage.
Fork passage over a ssDNA nick or gaps in the parental DNA leads to a broken fork, with one of the sister chromatids being disconnected from the fork. Some components of the replisome are thus lost (Roseaulin et al., 2008; Hashimoto et al., 2010; Moriel-Carretero and Aguilera, 2010). HR ensures the repair of such broken forks through a mechanism that is thought to be similar to BIR (Bosco and Haber, 1998; Kraus et al., 2001; Hashimoto et al., 2010). In Xenopus, HR-mediated fork repair leads to the reassembly of a replisome (Hashimoto et al., 2012). But BIR that requires most of the components of canonic replisomes (Lydeard et al., 2007, 2010) is highly mutagenic in yeast (Deem et al., 2011). An inter-strand cross-link (ICLs) is a type of lesion that interferes with the progression of replication forks by preventing the unwinding of the parental DNA. ICLs are cleaved by specific nucleases, thus resulting in a broken fork that is then repaired by HR (Long et al., 2011).
Many chromosomal elements can behave as fork obstacles, and it remains unclear whether fork breakages occur systematically. For example, DNA-bound proteins represent more than 1400 potential sites of fork arrest in budding yeast, and HR efficiently rescues replication forks blocked by protein complexes tightly bound to DNA in fission yeast (Ivessa et al., 2003; Lambert et al., 2010; Iraqui et al., 2012). In this case, replication restart is initiated by the loading of HR factors at ssDNA exposed at blocked forks (Mizuno et al., 2009; Lambert et al., 2010). The mechanisms by which HR ensures replication restart remain to be determined. Nevertheless, the resumption of DNA synthesis at inactivated forks via the HR pathway is also mutagenic (see below).
Finally, in addition to rescuing DNA synthesis at replication forks, HR is also involved in the stability and protection of forks that are impeded in their progression. HR defects lead to the accumulation of ssDNA gaps at replication forks, perhaps due to an uncoupling between lagging and leading strand synthesis (Hashimoto et al., 2010). Additionally, resection of neo-synthesized strands has been observed in mammalian and bacterial HR-deficient cells (Courcelle and Hanawalt, 2003; Schlacher et al., 2011). While this fork-stabilizer function of HR during DNA replication appears to be evolutionarily conserved, its importance in ensuring the robustness of DNA replication remains to be established in eukaryotes.
Therefore, because HR acts through multiple pathways at the replication fork or in its vicinity, it should play an essential role in protecting cells against spontaneous replication stress and thus against the resulting genetic instability, as discussed below.
Role of HR in the Maintenance of Genome Stability
HR Defects Result in Higher Levels of Mutagenesis and Genetic Instability
In all organisms, HR-deficient cells exhibit a higher level of mutagenesis and genome rearrangements, both spontaneous and upon exposure to exogenous genotoxic agents (Quah et al., 1980; Liu et al., 1998; Takata et al., 2001; Thompson and Schild, 2001; Lambert and Lopez, 2002; Popova et al., 2012). These data suggest that HR (like NHEJ) maintains genome stability.
HR Protects Mitosis from Replication Stress
Replication stress covers many events that impact the accuracy of DNA replication and then jeopardize chromosome segregation during mitosis. Low levels of replication stress can generate mitotic defects, including anaphase bridges, supernumerary centrosomes and multipolar mitosis, which then lead to uneven chromosome segregation (Wilhelm et al., 2014). Because HR plays a pivotal role in the resumption of arrested replication forks, defects in HR should thus reveal endogenous replication stress. Consistently, HR-deficient cells are associated with spontaneous slowed replication fork progression (Daboussi et al., 2008; Wilhelm et al., 2014), anaphase bridges (Lahkim Bennani-Belhaj et al., 2010; Laulier et al., 2011b; Rodrigue et al., 2013; Wilhelm et al., 2014), common fragile sites (Ingvarsson et al., 1999; Turner et al., 2002), supernumerary centrosomes (Griffin et al., 2000; Deng, 2002; Kraakman-van der Zwet et al., 2002; Bertrand et al., 2003; Dodson et al., 2004; Daboussi et al., 2005; Katsura et al., 2009; Plo and Lopez, 2009; Rodrigue et al., 2013; Wilhelm et al., 2014), and multipolar mitosis (Wilhelm et al., 2014). Similarly, fission yeast recombination factors are necessary to ensure successful chromosome segregation following the slowdown of fork progression (Bailis et al., 2008).
These data underline the essential role played by HR in protecting genome stability at the interface between replication and mitosis, as reviewed elsewhere (Wilhelm et al., 2014).
HR: A Factor of Genetic Instability
Because of its intrinsic properties (genetic exchanges through GC and CO), HR can generate genetic instability. More surprisingly, several reports have noted a type of genome instability mediated by micro-homology in an HR-dependent manner. These types of genetic instability were initially assigned to the error-proneness of end joining. Consequently, the actual view on the accuracy of HR has been challenged in many reports.
HR Possesses the Intrinsic Capacity of Genetic Modification
HR is initiated through the invasion of a duplex DNA by a homologous single-stranded molecule, which then primes DNA synthesis (Figure 1B). The strand invasion, promoted by RecA/Rad51, is able to occur with homologous sequences containing few heterologies (although the divergences should be limited), thus generating heteroduplex DNA molecules bearing mismatches (Figure 1B). The repair of these mismatched structures can transfer sequence polymorphisms and modify the genetic information of the recipient molecule, resulting in an apparent mutagenic event. Additionally, the DNA synthesis initiated by the invading strand (Figure 3A) can duplicate a sequence that was absent in the donor molecule and thereby transfer this genetic information, resulting in modifications of the original recipient DNA sequence. Moreover, the resolution of the HR intermediate (Holliday junctions) can facilitate the exchange of adjacent sequences, leading to genetic rearrangements. Thus, both GC and CO intrinsically possess the capacity to modify genetic information. This has been used to target gene replacement and gene correction using exogenous DNA. Note that when involving identical sequences (for instance sister chromatids exchange: SCE), HR does not impact the genetic information. However, unequal SCE can lead to sequence duplication or deletion (Figure 3B). One can object that unequal SCEs should be less frequent than equal SCEs (Gonzalez-Barrera et al., 2003). Therefore, genome stability should not be strongly impacted by SCEs. In contrast, when involving repeated sequences (which are not identical) dispersed throughout the genome (non-allelic recombination, NAHR), HR can affect the genetic information (see below). Note that, if the final product of an equal SCE is error-free, this is not due to the accuracy of the HR process, but to the fact that the DNA are identical (indeed HR can efficiently processes with imperfectly homologous sequences) and because associated mechanisms orientate such kinds of events: 1-HR is restricted to the S and G2 phases (which correspond to the cell cycle phases presenting sister chromatids) and 2-the tight cohesion of the sister chromatids, through the cohesins complex, orientates the event to an equal SCE. Thus, the structure of the DNA and accessory associated mechanisms, rather than HR itself, favor such an error-free event. In addition, HR can initiate mutagenic DNA synthesis even when the interacting DNA molecules are fully identical such as sister chromatids (see discussion below). Finally, we can point out that, in yeast as well as in mammalian cells, spontaneous SCE have been described to be largely independent of the main actors of HR (Rad51, Rad52, Rad54), in contrast with induced SCE (Dronkert et al., 2000; Fasullo et al., 2001; Lambert and Lopez, 2001; Dong and Fasullo, 2003). Noteworthy, at meiosis, which aims at creating genetic diversity, equal SCEs are repressed and HR between homologous chromosomes (which are not identical) is favored. Therefore, in this situation, HR is used to generate genetic diversity.
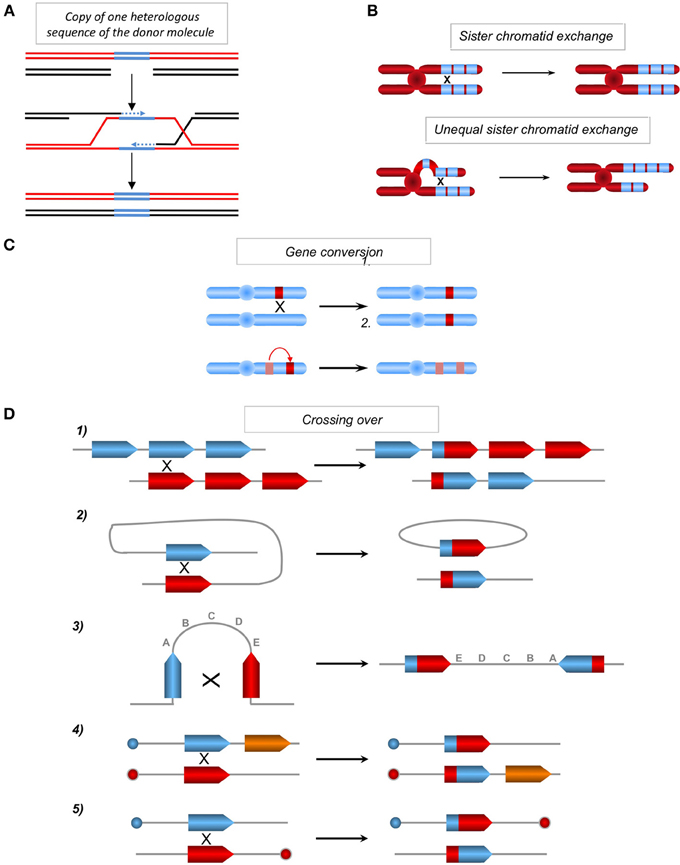
Figure 3. (A) Copy of one sequence of the donor absent on the recipient molecule. One of two homologous molecules (red and black) can contain one heterologous sequence (blue). Upon gene conversion or SDSA (see Figure 1) the heterologous (blue) sequence can be copied and transferred from the donor sequence (red) to the homologous recipient sequence (black), resulting in a genetic modification of the recipient sequence. (B) Sister chromatid exchanges. Between repeat sequences (blue boxes) without misalignment (upper panel) or with misalignment resulting in unequal sister chromatid exchanges (lower panel) and amplification and loss of genetic material. (C) Impact of gene conversion. Non-reciprocal exchange of genetic information between two heteroalleles, leading to a loss of heterozygosity (upper panel) and between a pseudogene (hatched), which often contains nonsense mutations and a gene (in red), leading to the inactivation of the latter (lower panel). (D) Chromosomal rearrangements resulting from crossing-over (CO) between repeat sequences. (1) Between homologous sequences on two chromosomes or following unequal sister chromatid exchange on the same chromosome, resulting in the amplification of one molecule and the deletion of the other. (2) Intramolecular CO between two homologous sequences in a direct orientation, resulting in the excision of the intervening sequence. (3) Intramolecular CO between two homologous sequences in an inverted orientation, resulting in the inversion of the internal fragment. (4) and (5) Inter-chromosomal CO, depending upon the orientation of the homologous sequences with respect to their centromeres (blue or red circles); this process generates a translocation (4) or a dicentric and an acentric chromosome (5).
Thus, in the cases discussed above, associated processes, rather than the HR machinery itself, in fact control the accuracy of the final outcome of HR.
Genetic Alterations through GC and/or CO
Gene conversion is able to transfer genetic information in a non-reciprocal manner between two hetero-alleles, resulting in loss of heterozygosity; gene conversion can also transfer one stop codon from a pseudogene to a related coding sequence, leading to its extinction (Figure 3C) (Amor et al., 1988; Fusco et al., 2012). Moreover, crossing over between repeated sequences that are dispersed throughout the genome (non-allelic HR) could lead to genomic rearrangements, such as translocations, deletions, amplifications and inversions (Figure 3D). These models account for genome rearrangements responsible for different human pathologies, attesting to the existence of these processes in vivo (Purandare and Patel, 1997; Chen et al., 2007; Fusco et al., 2012).
HR-Mediated Genome Rearrangements by BIR and Non-Allelic HR
In Saccharomyces cerevisiae, using an intron-based chromosomal translocation assay, it has been reported that DSB-induced translocation occurs via triparental recombination events. A short homologous sequence in the third chromosome serves as a bridge template for recombination events occurring between two non-homologous chromosomes. These events give rise mainly to reciprocal translocations that require the HR proteins Rad52 and Rad51 and the BIR-specific protein Pol32. Rad59 and Srs2 are also required, although to a lesser extent, whereas KU70 plays no role. These data suggest that BIR-mediated triparental recombination could be a major mechanism for chromosomal translocations in eukaryotic cells (Schmidt et al., 2006; Ruiz et al., 2009). Using a newly designed substrate for the analysis of DSB-induced chromosomal translocation, the group of Aguilera shows that Mus81 and Yen1 endonucleases promote BIR, thus causing non-reciprocal translocations. These endonucleases, as well as Slx4, promote replication template switching during BIR, thus participate in the generation of complex rearrangements when repeated sequences dispersed throughout the genome are involved (Pardo and Aguilera, 2012).
BIR can also induce genome instability in mammalian cells. It was recently reported that replicative stress induced by the overexpression of cyclin E in human cells led to copy number alteration (CNA). One third of these genome alterations (duplications less than 200 kb) have been attributed to BIR events or to microhomology-induced replication (MMBIR), a BIR-related mechanism (see below). The depletion of Pol D3, which encodes a subunit of pol delta, decreases the frequency of these events. The authors propose that BIR repair of damaged replication forks might explain the presence of segmental genomic duplication in human cancers. The larger amplification (>200 kb) and deletion observed after the overexpression of cyclin E may arise from other repair mechanisms, such as non-allelic HR (Costantino et al., 2014).
Replication fork arrest has also been reported to promote non-allelic HR between repeated sequences. In budding yeast, a reduced level of replicative polymerases, which can potentially alter the progression of replication forks, leads to recombination between an inverted Ty element and translocation (Lemoine et al., 2005, 2008). A more direct connection between fork arrest and HR-mediated genome rearrangements has been established in fission yeast, in which the block of a single replication fork leads to translocation and genomic deletion that results from HR between repeated sequences (Lambert et al., 2005; Iraqui et al., 2012). Such chromosomal rearrangements are a direct consequence of replication restart at unbroken forks by HR and not a consequence of failure in restarting forks and subsequent aberrant processing (Mizuno et al., 2009).
Given the potential role of HR in mediating chromosomal rearrangement, factors that prevent non-allelic HR might thus be considered as factors protecting against homology-mediated genomic instability. For example, increasing the distance between repeated sequences reduced the frequency of non-allelic HR (Lichten and Haber, 1989; Godwin et al., 1994). In fission yeast, CENP-B factors facilitate fork passage across LTR repeats that are prone to fork blockage. In the absence of CENP-B, LTR behaves as an HR hot spot prone to deletion events (Zaratiegui et al., 2011).
HR-Induced Mutagenesis
Mutagenesis associated with HR was first reported in E. coli (Cairns and Foster, 1991; Harris et al., 1994; Rosenberg et al., 1994). Repair of DSBs by HR in E. coli is non-mutagenic in unstressed cells, but under stress, switches to a mutagenic mode that is activated by stress responses (Ponder et al., 2005; Shee et al., 2011). This mutagenic repair of DNA breaks requires proteins that mend DSBs by HR, error-prone DNA polymerases, activation of SOS DDR, the controlled general and starvation stress response (RpoS), and a membrane protein stress response (RpoE), that promotes spontaneous DNA breakage in some DNA regions (Gibson et al., 2010). RpoS controls the switch that changes the normally high-fidelity process of DSBR via HR to an error-prone one. In this pathway, three steps are required: (1) DSB repair initiated by HR proteins (RecBCD, RecA); (2) the activation of SOS upregulates PolIV/DinB error-prone DNA polymerase; and (3) a second stress that activates RpoS, which allows Pol I, II, V, and/or PolI to participate in break repair instead of (or in addition to) the high fidelity DNA polIII (for review Rosenberg et al., 2012). This mechanism limits genetic instability to the stress response and to regions near a DSB, and therefore produces localized mutations rather than dispersed mutations. This could be an important evolutionary strategy, both for the minimization of deleterious mutations in cells that acquire a rare adaptive mutation and also for concerted evolution within genes and gene clusters (reviewed in Rosenberg et al., 2012).
Using HO-generated DSBs, it has been shown that mitotic recombination is mutagenic, which has been referred to as break-repair-induced mutation (BRIMs) (Strathern et al., 1995; Rattray et al., 2002; and reviewed in Abdulovic et al., 2006). Both error-prone DNA synthesis associated with DSB repair and stretches of ssDNA might account for BRIMs. During DSB repair, the DNA-end-resection machinery generates intermediates containing ssDNA that are highly sensitive to mutations due to the activity of the trans-lesion synthesis DNA polymerase Zeta (Yang et al., 2008). In addition, it has recently been shown that the DNA synthesis step during elongation of the invading strand is highly mutagenic in Saccharomyces cerevisiae, with the mutation rate increasing by up to 1400-fold, and exhibits a mutation signature (primarily microhomology-mediated inter-strand template switching). These mutations result from errors that are made by Polδ and Polε (Hicks et al., 2010). Importantly, HR can be mutagenic even when involving a long tract of DNA synthesis. Indeed, BIR, one of the HR-type processes that are thought to restart replication forks, duplicates DNA over a long distance, even to the end of the chromosome arm, by establishing a replication fork-like structure (Figure 1B). Strikingly, in Saccharomyces cerevisiae, DNA synthesis that is induced through BIR is highly inaccurate over the entire path of the replication fork. The high level of mutation results from the combinatorial effects of an increase of the nucleotide pool induced by the DDR, the uncoupling of DNA synthesis with mismatch repair, and the exposure of ssDNA (Deem et al., 2011). Recently, BIR has been proposed to proceed via a migrating D-loop mediated by the helicase Pif1. The migration of the D-loop results in the extrusion of the synthesized DNA and the exposure of a long stretch of ssDNA, which can become a hot spot for lesions leading to mutations (Saini et al., 2013; Wilson et al., 2013). In support of this hypothesis, BIR-induced mutations are largely dependent on Pif1 (Saini et al., 2013; Wilson et al., 2013).
One essential role of HR is to reactivate arrested replication forks. In Schizosaccharomyces pombe, this process is error-prone. As mentioned above, replication restart by HR mediates non-allelic HR. More surprisingly, it also leads to small deletions and duplications flanked by micro-homology. Indeed, replication forks restarted by HR are associated with error-prone DNA synthesis, liable to template switch events at micro-homologies (Iraqui et al., 2012). When progressing across small inverted repeats or palindromes, forks recovered by HR are prone to generate large chromosomal inversions (Mizuno et al., 2013).
Anti-HR Mechanisms for Protection Against Genetic Instability and Cell Toxicity
One mechanism avoiding potential genetic instability promoted by HR is to orientate it to equal SCEs, while unequal SCEs are mutagenic (see Figure 3B). Indeed, sister chromatids are identical, thus GC cannot transfer mutation and CO will not have any genetic impact. This is done by associating two processes (as discussed above): (1) restriction of HR in S and G2 phase and (2) the cohesion of the sister chromatids.
Excess HR can also lead to the accumulation of HR intermediates, which generates genomic instability and cell death (Gangloff et al., 2000). Thus, HR is a double-edged sword; on the one hand, it protects against genetic instability, but on the other hand, it can trigger cell lethality as well as profound genomic rearrangements and point mutations. Therefore, the HR process should be tightly controlled to avoid unnecessary HR events. Helicases, by destabilizing abortive HR intermediates, protect against the genomic instability generated by HR (reviewed in Barber et al., 2008; Chu and Hickson, 2009; Bernstein et al., 2010). Additionally, it has been proposed that restricting the initiation of unscheduled HR can also prevent against the accumulation of such toxic HR intermediates. In mammalian cells, this protective role against excessive HR initiation has been proposed for p53, Bcl-2, and AKT1 (Bertrand et al., 2004; Plo et al., 2008; Guirouilh-Barbat et al., 2010; Laulier et al., 2011a; Laulier and Lopez, 2012).
Of note, the fact that protective systems have evolved to counteract excess HR underlines the potential risks of this pathway.
Relationships with Chromothripsis and Kataegis
The classical theory of cancer development proposed that cells gradually and randomly accumulate mutations and rearrangements that increase their survival (reviewed in Stratton et al., 2009). However, recent studies have revealed that critical aspects of cancer development can occur on a much shorter timescale. In a process called chromothripsis (from the Greek chromos for chromosome and thripsis, shattering into pieces), tens to thousands of genomic rearrangements occur in one cellular crisis (Berger et al., 2011; Stephens et al., 2011). In kataegis, mutations accumulate in hotspots of hundreds of bases to megabases in a single cell cycle (Nik-Zainal et al., 2012; Roberts et al., 2012). Interestingly, both processes are linked to DSB repair events.
In chromothripsis, cells undergo tens to thousands of genomic rearrangements clustered into discrete subchromosomal territories, as first described in a small set of tumors (Berger et al., 2011; Stephens et al., 2011) and subsequently observed in a wide variety of tumors (Kloosterman and Kuipers, 2011; Magrangeas et al., 2011; Lapuk et al., 2012; Molenaar et al., 2012; Rausch et al., 2012). What causes such a dramatic remodeling of the genome is still unknown. However, the implicated regions are sharply circumscribed and this suggests that the original DNA damage occurs during mitosis when DNA is highly condensed. Although several mechanisms have been suggested to explain the clustered rearrangements, the most plausible cause is replicative stress on regions difficult to replicate (e.g., fragile sites). In particular, replication intermediates that do not expose long stretches of ssDNA and therefore do not activate the checkpoints allow cells to enter mitosis in their presence (Chan et al., 2009). A recent study suggested that chromosome shattering might arise from an error in mitotic chromosome segregation that leads to the production of micronuclei (Crasta et al., 2012). These micronuclei are at high risk for the integrity of the genome. First, they exhibit a defective DDR and delayed or defective DNA repair (Terradas et al., 2009, 2012; Crasta et al., 2012). Second, most micronuclei replicate more slowly than the major nucleus and therefore most micronuclei are still replicating when the major nucleus is already in the G2 phase (Crasta et al., 2012). Finally, entry in mitosis when the micronucleus is still replicating is associated with a massive induction of DSBs (Crasta et al., 2012).
The DNA repair machinery then reassembles the chromosomal pieces in a disordered fashion (see example in Figure 4A). The possible mechanisms of chromosome reassembly first implicated NHEJ and A-EJ because the junction sequences exhibited tracts of microhomology, as well as insertions or deletions of variable sizes (Rausch et al., 2012; Stephens et al., 2012). However, these mechanisms can account for the loss of genetic information but not for amplification of some genomic regions (Magrangeas et al., 2011; Rausch et al., 2012; Stephens et al., 2012). Replication-based repair pathways are more plausible, accounting for both genomic gains and losses. A hybrid of replication-independent mechanisms and replication-dependent processes has been proposed to explain the complex rearrangements found in chromothripsis, the MMBIR (microhomology-mediated break induced replication) (Figure 4B) (Hastings et al., 2009; Liu et al., 2011) associated with a specific mechanism linked to replication block, FoSTeS (for Fork Stalling and Template Switching) (Lee et al., 2007; Zhang et al., 2009). These processes begin with the conversion of a DSB (or a replication fork stall) in a ssDNA 3′ stretch. This free 3′DNA end can then anneal using a region of micro-homology on a ssDNA region exposed on an adjacent replication fork. Replication can then occur. However, such replication forks are weakly processive and can undergo several rounds of template switching, generating complex rearrangements with deletions, amplifications and non-reciprocal translocations. The use of this low fidelity repair process to manage the high level of DSBs generated during chromothripsis could be explained by the overwhelming of reliable repair processes and DDRs. It is worth mentioning that not all chromothripsis events are explainable by FoSTeS or MMBIR; some of them might be the result of chromosome shattering followed by NHEJ or A-EJ.
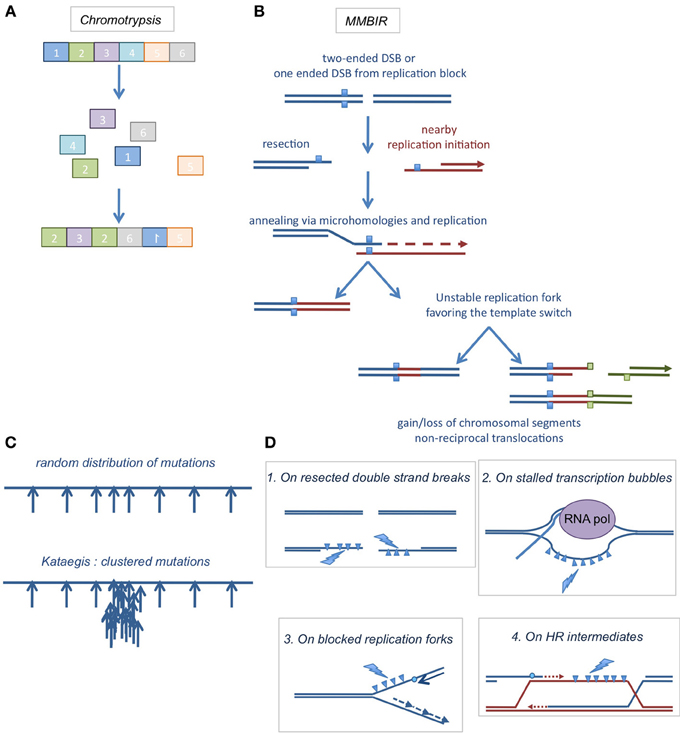
Figure 4. (A) Chromothripsis. Chromosomal shattering into pieces and abnormal re-ligation events, resulting in intra- or inter-chromosomal rearrangements. (B) A suggested model for chromothripsis occurrence, the MMBIR (microhomology mediated break induced replication). A DNA double strand end is resected to generate a 3′ overhang that will anneal with microhomologies elsewhere in the genome to initiate replication. This mechanism can lead to more complex rearrangements if it is coupled to multiple cycles of template switches. (C) Kataegis. When mutations are expected to be distributed randomly in the genome (upper cartoon), clustered mutations were found in the genomes of several cancers (lower cartoon). (D) Where kataegis occurs. These clustered mutations were at least in part correlated with the action of DNA deaminases of the APOBEC family, which deaminate cytosines on ssDNA areas found on resected DNA ends (1), stalled transcription bubbles (2), blocked replication forks (3), or HR intermediates (4).
In 2012 has been reported the occurrence of somatic localized mutation hotspots in tumor genome, called kataegis (from the Greek for thunderstorm) (Nik-Zainal et al., 2012; Roberts et al., 2012). This mechanism was then observed in a broad range of cancers (Alexandrov et al., 2013). In kataegis, mutations accumulate rapidly at somatic mutation hotspots (Figure 4C) at a critical step of tumorigenesis. Several mutation signatures were identified, particularly mutations on guanines and cytosines.
The mutation pattern matched the signatures of the RNA- and DNA-editing deaminases of the AID/APOBEC family that act on ssDNA molecules. Indeed these enzymes deaminate cytosines and generate uracils that are a substrate for Base Excision repair, generating abasic sites, causing C-to T-transitions or driving polymerase eta misincorporations. Before kataegis was described, genome sequencing studies had revealed that many cancers have somatic mutations dominated by C-to-T transitions (Sjoblom et al., 2006; Greenman et al., 2007; Jones et al., 2010; Berger et al., 2011; Kumar et al., 2011; Parsons et al., 2011; Stransky et al., 2011; Taylor et al., 2013) and that overexpression of APOBEC1 was associated with cancer development (Yamanaka et al., 1995) when overexpression of APOBEC3A induced genomic damage and mutations (Stenglein et al., 2010; Landry et al., 2011; Suspene et al., 2011). The implication of APOBEC deaminases in kataegis was validated by several groups in yeast models (Taylor et al., 2004; Chan et al., 2012; Roberts et al., 2012) and in human cells (Burns et al., 2013), where overexpression of APOBEC3B was correlated with an elevated level of mutations in breast tumors and cell lines. Knockdown experiments showed that endogenous APOBEC3B was responsible for increased mutation frequencies and C-to-T transitions when APOBEC3B overexpression induced DNA damage and C-to-T mutations in human cells.
As mentioned above, AID/APOBEC enzymes deaminate only cytosines in ssDNA. It was therefore proposed that these deamination reactions could occur on stabilized ssDNA stretches formed on stabilized transcription bubbles or after the occurrence of DSBs or replication fork blockage (Figure 4D). In the last case, the uncoupling between helicases and polymerases generates and stabilizes long patches of ssDNA.
Interestingly these strand coordinated clusters of mutated cytosines or guanines were often localized next to chromosome rearrangement breakpoints and extended up to 200 kb (Roberts et al., 2012) suggesting that they were correlated to the occurrence of DSB and DSB repair pathways generating ssDNA stretches, like HR (see Figure 1). The correlation between DSB induction and kataegis was confirmed in yeast treated with alkylating agents (Roberts et al., 2012) or even more directly, in yeast where DSB were induced by the meganuclease I-SceI (Taylor et al., 2013): In these studies, the authors observed a strand bias in the mutations observed. Cytosines were preferentially mutated on the 5′ side of a DSB and guanines on the 3′ side of the DSB. As resection only occurs in the 5′ to 3′ direction, this pattern in mirror was correlated to the generation of ssDNA stretches in Homology directed repair. It is noteworthy that HR is not the only mechanism leading to ssDNA stretches that are a template for kataegis; uncoupled replication forks that expose long stretches of ssDNA are also a template for deaminases (Roberts et al., 2012).
The association of the timescale between kataegis and chromothripsis suggests that both could occur simultaneously at certain chromosomal regions, resulting in an even more catastrophic event for the cell.
The Importance of Being Versatile
HR is versatile because it tolerates limited divergences between the interacting partners. Remarkably, this capacity to modify genetic information has been used by cells to generate beneficial genetic diversity. HR has therefore been implicated in numerous essential biological processes, from molecular evolution to DNA repair and meiotic differentiation, and is also relevant to targeted gene replacement.
At meiosis, HR ensures that allele mixing creates genetic diversity. In chickens, gene conversion of the expression allele with pseudo-genes generates the complexity of the immune repertoire (Reynaud et al., 1987).
In pathogens, antigenic variation is a widely used strategy for immune evasion. Gene conversion is a prominent system for antigenic variation through recombination between one silent copy of a gene and the expressed copy, resulting in the formation of a chimeric gene. Several pathogens, such as Trypanossoma brucei, Anaplasma marginale, Borrelia burgdorferi, Helicobacter pylori, and Neisseria gonorrhoeae, use this strategy (Palmer and Brayton, 2007; Stockdale et al., 2008; Wisniewski-Dye and Vial, 2008). For example, trypanosomes are coated with a variant surface glycoprotein (VSG). Antigenic variation involves switches in the composition of the VSG coat driven by gene conversion between the expressed allele and an archive of silent VSG genes (Marcello et al., 2007; Morrison et al., 2009). In Candida albicans, recombination generates homozygous hyperactive alleles conferring resistance to antifungals (Coste et al., 2006).
HR is a driving-force in the evolution of multi-gene families; crossovers leading to unequal exchanges between sister chromatids are responsible for variation in the repetition of duplicated sequences. During evolution, most duplicated sequences diverge; the genes of one species derived from a common ancestor are paralogs. Due to selective pressure, there are generally fewer divergences between homologous genes of two different species (orthologs) than between their respective paralogs. However, in some families of repeated genes, the divergence between the duplicated units is less significant within one species than when compared to a different species, even one that is evolutionarily close. In this case, the duplicated genes did not evolve independently but instead co-evolved; this phenomenon is called “concerted evolution” (reviewed Arnheim, 1983; Liao, 1999). Gene conversion is the driving force behind homogenization of duplicated sequences, and therefore of concerted evolution. Concerted evolution is a universal biological phenomenon that occurs in bacteria, yeast, plants and animals. Because HR should be tightly controlled, some processes exist to limit it. Indeed, sequence heterologies block gene conversion and should therefore be barriers to concerted evolution; it has been suggested that introns, which can interrupt the length of sequence homology without affecting the function of the encoded protein, can be protective barriers against HR between repeated sequences, thereby favoring the maintenance of the structural organization of the genome (Kourilsky, 1983; Kricker et al., 1992). In this context, it is tempting to speculate that introns are an evolutionary force antagonistic to concerted evolution, directing evolution toward the divergence of repeated sequences.
Up- and Down-Regulation of HR in Cancer
Genetic instability is a hallmark of cancer cells. Both inhibition and stimulation of HR have been reported in tumors or cancer-prone situations. This is consistent with the duality of HR, and this underlines that inhibition as well as stimulation of HR confer increased risks of genetic instability. More precisely, both down- and up-regulation of the recombinase RAD51 affects genomic stability.
For instance, the expression of a non-lethal dominant negative form of RAD51 in cells injected into nude mice favors tumor take and growth (Bertrand et al., 2003). The overexpression of RAD51 stimulates HR (Vispé et al., 1998; Huang et al., 1999; Lambert and Lopez, 2000) and induces a strong chromosome instability (Richardson et al., 2004), underlying the potential risks of excess HR. These data highlight the importance of tight control of the level of HR.
HR Defects Associated with Predisposition to Cancers
Most of the mutations responsible for familial breast or ovarian cancers affect genes that control HR and/or the replication/HR interface directly or indirectly (Walsh and King, 2007; Walsh et al., 2011). The two genes most often mutated, BRCA1 and BRCA2, are two major players in HR (Moynahan et al., 1999, 2001). This overrepresentation of genes involved in the response to DNA damage and the communication between replication and recombination suggests the importance of these specific metabolic pathways in the etiology of breast cancer and raises the question of characteristics common to the causation of sporadic and hereditary breast cancer. Several studies have reported the hyperactivation of the oncogenic kinase AKT1 in 40-60% of sporadic breast cancers and in 40% of sporadic ovarian cancers (Sun et al., 2001; Yang et al., 2006; Plo et al., 2008). It must be noted that PTEN, one of the genes mutated in familial breast cancer, is an antagonist of AKT1. Several studies have shown connections between AKT1 and responses to DNA damage (for a review, see Guirouilh-Barbat et al., 2010). In particular, overexpression of AKT1 induces the sequestration of BRCA1 and RAD51 in the cytoplasm, leading to the inhibition of HR (Plo et al., 2008; Plo and Lopez, 2009). Taken together, these data underline the importance of HR in protection against breast cancer and reveal the AKT1 signaling pathway as a missing link between hereditary and sporadic breast cancers.
Other examples of HR inhibition exist in situations of predisposition to cancer. For example, Bc1-2 is an inhibitor of the intrinsic pathway of apoptosis induction, and its activation confers a predisposition for lymphomas. Bc1-2 was initially found to be overexpressed in B cell lymphoma with the recurrent translocation t(14:18), but it is also overexpressed in numerous tumors. Remarkably, overexpression of Bc1-2 leads to the relocalization of BRCA1 in endomembranes (endoplasmic reticulum, mitochondria), resulting in an inhibition of HR (Laulier et al., 2011a; and reviewed in Laulier and Lopez, 2012).
Stimulation of HR in Cancer
Conversely, there are also situations associating a predisposition for tumors and hyper-recombinogenic phenotypes.
For example, in Bloom syndrome, there is a greatly elevated predisposition to spontaneous tumors in all tissues. Bloom syndrome results from the inactivation of the BLM protein, a member of the RecQ helicase family, that plays an important role in the resolution of HR intermediates, in the processing of blocked replication forks, and at the initiation of DNA double strand break repair (Bernstein et al., 2010; Grabarz et al., 2013). Cells from patients afflicted with Bloom syndrome show increased levels of exchange between sister chromatids and hyper-recombination phenotypes (reviewed in Chu and Hickson, 2009).
The tumor-suppressing p53 gene is the most frequently mutated gene in all types of cancers. It has been shown that the p53 protein represses HR; cells deficient in p53 show a hyper-recombination phenotype (for a review, see Bertrand et al., 2004).
The fusion oncogene BCR/ABL derives from the translocation of the cABL gene from chromosome 9 to the BCR gene locus on chromosome 22: Philadelphia chromosome t(9:22). This translocation is present in chronic myelogenous leukemia (CML) patients and in many acute lymphocytic leukemia patients. The BCR/ABL fusion proteins (p230, p210, or p185) exhibit constitutive tyrosine kinase activity. The resistance of BCR/ABL tumors to DNA damage induced by therapeutic drugs depends on the kinase activity of the fusion protein. The expression of BCR/ABL increases the intracellular level of RAD51 protein by different mechanisms (Slupianek et al., 2001). First, signaling from the BCR/ABL src homogy-3 (SH3) and SH2 domains stimulates RAD51 transcription via the activation of the signal transducer and activation transcription 5 (STAT5). The transcription of the paralogs RAD51B, RAD51D, and XRCC2 is also stimulated, whereas transcription of RAD51C and XRCC3 is decreased. Second, BCR/ABL inhibits caspase-3 activation and thus RAD51 protein degradation. Indeed, BCR/ABL stimulates HR between tandem repeat sequences. Additionally, BCR/ABL interacts with RAD51 and results in a high level of constitutive Tyr315 phosphorylation. This Tyr315 phosphorylation and RAD51-dependent HR seem to control resistance to cisplatin and mitomycin C (Slupianek et al., 2001). BCR/ABL expression inhibits DNA-PK activity, which is involved in non-homologous end joining, a competitor pathway to HR for DNA DSB repair (Deutsch et al., 2001). This suggests that the regulation of the balance between HR and NHEJ can be modified by BCR/ABL.
Conclusions
HR: A Double-Edged Sword
Regulation of HR should permit the maintenance of genomic stability, allowing genetic diversity but avoiding genetic instability. Depending on the structure of the interacting DNA partners, GC and CO intrinsically possess the capacity to generate genetic variability/instability. In addition to cell cycle regulation, which inhibits HR in the G1 phase and restricts it at the S-G2 phase (during which the sister chromatids are generated) and the tight cohesion of the sister chromatids that orientates exchange to equal SCE, several additional mechanisms repress HR: mismatch repair, helicases, and p53. Defects in these systems are associated with genome instability and cancer predisposition. The fact that living organisms develop strategies to repress HR underlines the potential dangers of HR excess. Indeed, excess HR does generate mutagenesis and genomic rearrangements. These capacities have been used by cell to generate beneficial genetic diversity, but conversely, many pathological rearrangements are explained by accidental HR.
Strikingly, ablation of replication origins in Archaea bacteria results in faster growth thanks to the initiation of replication by HR (Hawkins et al., 2013). This raises the question as to why organisms use replication origins to duplicate the entire genetic material, instead of HR. Considering the potential risks of HR both for the accuracy of DNA replication and for genomic architecture, the choice of replication origins should ensure a more stable and accurate duplication through generations; note that this should allow for the maintenance of the minimum common genomic structure defining a given species. In contrast, HR, especially CO, would lead to highly rearranged DNA in offspring, resulting in genetic separation between ancestors and progeny. While genome modification is a driving force for evolution giving opportunity to generate individual genetic diversity, an intergenerational maintenance of the genome should facilitate speciation.
Accuracy of HR vs. NHEJ: The World Turns Upside Down
In many scientific reports (publications, reviews, thesis dissertations, conferences), HR is claimed to be error-free, whereas NHEJ is said to be error-prone. However, the two processes share similarities:
- Both HR and NHEJ are required for genome stability maintenance.
- Both are involved in processes generating genome diversity.
- Both can generate genome rearrangements.
- In both cases, the structure of the DNA molecules determines the final product.
However, they also show differences:
- In contrast with the common view, HR contains the intrinsic capacity to modify genetic material through GC and CO (this has been used to generate genetic diversity in meiosis or V(D)J recombination in chicken) and by promoting error-prone DNA synthesis, while NHEJ is not intrinsically error-prone and can join fully complementary DNA ends mainly in a faithful manner (for review see Betermier et al., 2014).
Therefore, HR, which can generate genetic alteration, should be tightly control to limits its potential danger and to lead to accurate outcomes. However processes aiming at generating genetic diversity take advantage of these intrinsic capacities of HR.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all the people who made helpful comments. We apologize to our colleagues whose contributions are not cited due to space constraints. Bernard S. Lopez and Josée Guirouilh-Barbat are supported by funding from the Ligue Nationale Française contre le Cancer and the INCa (Institut National du Cancer). Sarah Lambert was supported by ANRJCJC10-1203 01 and la Ligue contre le cancer (comité Essonne). Pascale Bertrand was supported by the Ligue contre le Cancer, Comité Ile de France; Fondation ARC and DSV-CEA “Radiobiology grant.”
References
Abdulovic, A., Kim, N., and Jinks-Robertson, S. (2006). Mutagenesis and the three R's in yeast. DNA Repair (Amst.) 5, 409–421. doi: 10.1016/j.dnarep.2005.11.006
Aguilera, A., and Garcia-Muse, T. (2013). Causes of genome instability. Annu. Rev. Genet. 47, 1–32. doi: 10.1146/annurev-genet-111212-133232
Aguilera, A., and Gomez-Gonzalez, B. (2008). Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217. doi: 10.1038/nrg2268
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi: 10.1038/nature12477
Amor, M., Parker, K. L., Globerman, H., New, M. I., and White, P. C. (1988). Mutation in the CYP21B gene (Ile-172—Asn) causes steroid 21- hydroxylase deficiency. Proc. Natl. Acad. Sci. U.S.A. 85, 1600–1604. doi: 10.1073/pnas.85.5.1600
Arnheim, N. (1983). “Concerted evolution of multigene families,” in Evolution of Genes and Proteins, eds R. K. Koehn and M. Nei (Sunderland, MA: Sinauer Associates), 38–61.
Bailis, J. M., Luche, D. D., Hunter, T., and Forsburg, S. L. (2008). Minichromosome maintenance proteins interact with checkpoint and recombination proteins to promote s-phase genome stability. Mol. Cell. Biol. 28, 1724–1738. doi: 10.1128/MCB.01717-07
Barber, L. J., Youds, J. L., Ward, J. D., McIlwraith, M. J., O'Neil, N. J., Petalcorin, M. I., et al. (2008). RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135, 261–271. doi: 10.1016/j.cell.2008.08.016
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. doi: 10.1038/nature03482
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. doi: 10.1038/nature05268
Berger, M. F., Lawrence, M. S., Demichelis, F., Drier, Y., Cibulskis, K., Sivachenko, A. Y., et al. (2011). The genomic complexity of primary human prostate cancer. Nature 470, 214–220. doi: 10.1038/nature09744
Bernstein, K. A., Gangloff, S., and Rothstein, R. (2010). The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 44, 393–417. doi: 10.1146/annurev-genet-102209-163602
Bertrand, P., Lambert, S., Joubert, C., and Lopez, B. S. (2003). Overexpression of mammalian Rad51 does not stimulate tumorigenesis while a dominant-negative Rad51 affects centrosome fragmentation, ploidy and stimulates tumorigenesis, in p53-defective CHO cells. Oncogene 22, 7587–7592. doi: 10.1038/sj.onc.1206998
Bertrand, P., Saintigny, Y., and Lopez, B. S. (2004). p53's double life: transactivation-independent repression of homologous recombination. Trends Genet. 20, 235–243. doi: 10.1016/j.tig.2004.04.003
Betermier, M., Bertrand, P., and Lopez, B. S. (2014). Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 10:e1004086. doi: 10.1371/journal.pgen.1004086
Bosco, G., and Haber, J. E. (1998). Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150, 1037–1047.
Branzei, D., and Foiani, M. (2010). Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219. doi: 10.1038/nrm2852
Buard, J., and de Massy, B. (2007). Playing hide and seek with mammalian meiotic crossover hotspots. Trends Genet. 23, 301–309. doi: 10.1016/j.tig.2007.03.014
Burns, M. B., Lackey, L., Carpenter, M. A., Rathore, A., Land, A. M., Leonard, B., et al. (2013). APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. doi: 10.1038/nature11881
Cairns, J., and Foster, P. L. (1991) Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128, 695–701.
Chan, K., Sterling, J. F., Roberts, S. A., Bhagwat, A. S., Resnick, M. A., and Gordenin, D. A. (2012). Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 8:e1003149. doi: 10.1371/journal.pgen.1003149
Chan, K. L., Palmai-Pallag, T., Ying, S., and Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760. doi: 10.1038/ncb1882
Chen, J. M., Cooper, D. N., Chuzhanova, N., Ferec, C., and Patrinos, G. P. (2007). Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet. 8, 762–775. doi: 10.1038/nrg2193
Chu, W. K., and Hickson, I. D. (2009). RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654. doi: 10.1038/nrc2682
Costantino, L., Sotiriou, S. K., Rantala, J. K., Magin, S., Mladenov, E., Helleday, T., et al. (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. doi: 10.1126/science.1243211
Coste, A., Turner, V., Ischer, F., Morschhauser, J., Forche, A., Selmecki, A., et al. (2006). A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172, 2139–2156. doi: 10.1534/genetics.105.054767
Costes, A., and Lambert, S. A. (2012). Homologous Recombination as a Replication Fork Escort: Fork-Protection and Recovery. Biomolecules 1, 39–71. doi: 10.3390/biom3010039
Courcelle, J., and Hanawalt, P. C. (2003). RecA-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 37, 611–646. doi: 10.1146/annurev.genet.37.110801.142616
Crasta, K., Ganem, N. J., Dagher, R., Lantermann, A. B., Ivanova, E. V., Pan, Y., et al. (2012). DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58. doi: 10.1038/nature10802
Daboussi, F., Courbet, S., Benhamou, S., Kannouche, P., Zdzienicka, M. Z., Debatisse, M., et al. (2008). A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 121, 162–166. doi: 10.1242/jcs.010330
Daboussi, F., Thacker, J., and Lopez, B. S. (2005). Genetic interactions between RAD51 and its paralogues for centrosome fragmentation and ploidy control, independently of the sensitivity to genotoxic stresses. Oncogene 24, 3691–3696. doi: 10.1038/sj.onc.1208438
Deem, A., Keszthelyi, A., Blackgrove, T., Vayl, A., Coffey, B., Mathur, R., et al. (2011). Break-induced replication is highly inaccurate. PLoS Biol. 9:e1000594. doi: 10.1371/journal.pbio.1000594
Deng, C. X. (2002). Roles of BRCA1 in centrosome duplication. Oncogene 21, 6222–6227. doi: 10.1038/sj.onc.1205713
Deutsch, E., Dugray, A., AbdulKarim, B., Marangoni, E., Maggiorella, L., Vaganay, S., et al. (2001). BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 97, 2084–2090. doi: 10.1182/blood.V97.7.2084
Dodson, H., Bourke, E., Jeffers, L. J., Vagnarelli, P., Sonoda, E., Takeda, S., et al. (2004). Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 23, 3864–3873. doi: 10.1038/sj.emboj.7600393
Dong, Z., and Fasullo, M. (2003). Multiple recombination pathways for sister chromatid exchange in Saccharomyces cerevisiae: role of RAD1 and the RAD52 epistasis group genes. Nucleic Acids Res. 31, 2576–2585. doi: 10.1093/nar/gkg352
Dronkert, M. L., Beverloo, H. B., Johnson, R. D., Hoeijmakers, J. H., Jasin, M., and Kanaar, R. (2000). Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol. Cell. Biol. 20, 3147–3156. doi: 10.1128/MCB.20.9.3147-3156.2000
Dudley, D. D., Chaudhuri, J., Bassing, C. H., and Alt, F. W. (2005). Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv. Immunol. 86, 43–112. doi: 10.1016/S0065-2776(04)86002-4
Fasullo, M., Giallanza, P., Dong, Z., Cera, C., and Bennett, T. (2001). Saccharomyces cerevisiae rad51 mutants are defective in DNA damage-associated sister chromatid exchanges but exhibit increased rates of homology-directed translocations. Genetics 158, 959–972.
Featherstone, C., and Jackson, S. P. (1999). DNA double-strand break repair. Curr. Biol. 9, R759–R761. doi: 10.1016/S0960-9822(00)80005-6
Fusco, F., Paciolla, M., Napolitano, F., Pescatore, A., D'Addario, I., Bal, E., et al. (2012). Genomic architecture at the Incontinentia Pigmenti locus favours de novo pathological alleles through different mechanisms. Hum. Mol. Genet. 21, 1260–1271. doi: 10.1093/hmg/ddr556
Gangloff, S., Soustelle, C., and Fabre, F. (2000). Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 25, 192–194. doi: 10.1038/76055
Gibson, J. L., Lombardo, M. J., Thornton, P. C., Hu, K. H., Galhardo, R. S., Beadle, B., et al. (2010). The sigma(E) stress response is required for stress-induced mutation and amplification in Escherichia coli. Mol. Microbiol. 77, 415–430. doi: 10.1111/j.1365-2958.2010.07213.x
Godwin, A. R., Bollag, R. J., Christie, D. M., and Liskay, R. M. (1994). Spontaneous and restriction enzyme-induced chromosomal recombination in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 91, 12554–12558. doi: 10.1073/pnas.91.26.12554
Gonzalez-Barrera, S., Cortes-Ledesma, F., Wellinger, R. E., and Aguilera, A. (2003). Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol. Cell 11, 1661–1671. doi: 10.1016/S1097-2765(03)00183-7
Gorgoulis, V. G., and Halazonetis, T. D. (2010). Oncogene-induced senescence: the bright and dark side of the response. Curr. Opin. Cell Biol. 22, 816–827. doi: 10.1016/j.ceb.2010.07.013
Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913. doi: 10.1038/nature03485
Grabarz, A., Barascu, A., Guirouilh-Barbat, J., and Lopez, B. S. (2012). Initiation of DNA double strand break repair: signaling and single-stranded resection dictate the choice between homologous recombination, non-homologous end-joining and alternative end-joining. Am. J. Cancer Res. 2, 249–268.
Grabarz, A., Guirouilh-Barbat, J., Barascu, A., Pennarun, G., Genet, D., Rass, E., et al. (2013). A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 5, 21–28. doi: 10.1016/j.celrep.2013.08.034
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. doi: 10.1038/nature05610
Griffin, C. S., Simpson, P. J., Wilson, C. R., and Thacker, J. (2000). Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nat. Cell Biol. 2, 757–761. doi: 10.1038/35036399
Guirouilh-Barbat, J., Wilhelm, T., and Lopez, B. S. (2010). AKT1/BRCA1 in the control of homologous recombination and genetic stability: the missing link between hereditary and sporadic breast cancers. Oncotarget 1, 691–699.
Haber, J. E. (1992). Mating-type gene switching in Saccharomyces cerevisiae. Trends Genet. 8, 446–452. doi: 10.1016/0168-9525(92)90329-3
Haber, J. E. (2014). Genome Stability: DNA Repair and Recombinaition. New York, NY: Garland Science.
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. doi: 10.1126/science.1140735
Harris, R. S., Longerich, S., and Rosenberg, S. M. (1994). Recombination in adaptive mutation. Science 264, 258–260. doi: 10.1126/science.8146657
Hashimoto, Y., Chaudhuri, A. R., Lopes, M., and Costanzo, V. (2010). Rad51 protects nascent DNA from MRE11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 7, 1305–1311. doi: 10.1038/nsmb.1927
Hashimoto, Y., Puddu, F., and Costanzo, V. (2012). RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 19, 17–24. doi: 10.1038/nsmb.2177
Hastings, P. J., Ira, G., and Lupski, J. R. (2009). A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5:e1000327. doi: 10.1371/journal.pgen.1000327
Hawkins, M., Malla, S., Blythe, M. J., Nieduszynski, C. A., and Allers, T. (2013). Accelerated growth in the absence of DNA replication origins. Nature 503, 544–547. doi: 10.1038/nature12650
Hicks, W. M., Kim, M., and Haber, J. E. (2010). Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329, 82–85. doi: 10.1126/science.1191125
Huang, Y., Nakada, S., Ishiko, T., Utsugisawa, T., Datta, R., Kharbanda, S., et al. (1999). Role for caspase-mediated cleavage of Rad51 in induction of apoptosis by DNA damage. Mol. Cell. Biol. 19, 2986–2997.
Hyrien, O. (2000). Mechanisms and consequences of replication fork arrest. Biochimie 82, 5–17. doi: 10.1016/S0300-9084(00)00344-8
Ingvarsson, S., Agnarsson, B. A., Sigbjornsdottir, B. I., Kononen, J., Kallioniemi, O. P., Barkardottir, R. B., et al. (1999). Reduced Fhit expression in sporadic and BRCA2-linked breast carcinomas. Cancer Res. 59, 2682–2689.
Iraqui, I., Chekkal, Y., Jmari, N., Pietrobon, V., Freon, K., Costes, A., et al. (2012). Recovery of arrested replication forks by homologous recombination is error-prone. PLoS Genet. 8:e1002976. doi: 10.1371/journal.pgen.1002976
Ivessa, A. S., Lenzmeier, B. A., Bessler, J. B., Goudsouzian, L. K., Schnakenberg, S. L., and Zakian, V. A. (2003). The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell. 12, 1525–1536. doi: 10.1016/S1097-2765(03)00456-8
Jones, S., Wang, T. L., Shih, I., Mao, T. L., Nakayama, K., Roden, R., et al. (2010). Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231. doi: 10.1126/science.1196333
Jung, D., and Alt, F. W. (2004). Unraveling V(D)J recombination; insights into gene regulation. Cell 116, 299–311. doi: 10.1016/S0092-8674(04)00039-X
Katsura, M., Tsuruga, T., Date, O., Yoshihara, T., Ishida, M., Tomoda, Y., et al. (2009). The ATR-Chk1 pathway plays a role in the generation of centrosome aberrations induced by Rad51C dysfunction. Nucleic Acids Res. 37, 3959–3968. doi: 10.1093/nar/gkp262
Kloosterman, T. G., and Kuipers, O. P. (2011). Regulation of arginine acquisition and virulence gene expression in the human pathogen Streptococcus pneumoniae by transcription regulators ArgR1 and AhrC. J. Biol. Chem. 286, 44594–44605. doi: 10.1074/jbc.M111.295832
Kourilsky, P. (1983). Genetic exchanges between partially homologous nucleotide sequences: possible implications for multigene families. Biochimie 65, 85–93. doi: 10.1016/S0300-9084(83)80178-3
Kraakman-van der Zwet, M., Overkamp, W. J., van Lange, R. E., Essers, J., van Duijn-Goedhart, A., Wiggers, I., et al. (2002). Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol. Cell. Biol. 22, 669–679. doi: 10.1128/MCB.22.2.669-679.2002
Kraus, E., Leung, W. Y., and Haber, J. E. (2001). Break-induced replication: a review and an example in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 98, 8255–8262. doi: 10.1073/pnas.151008198
Kricker, M. C., Drake, J. W., and Radman, M. (1992). Duplication-targeted DNA methylation and mutagenesis in the evolution of eukaryotic chromosomes. Proc. Natl. Acad. Sci. U.S.A. 89, 1075–1079. doi: 10.1073/pnas.89.3.1075
Kumar, A., White, T. A., MacKenzie, A. P., Clegg, N., Lee, C., Dumpit, R. F., et al. (2011). Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. U.S.A. 108, 17087–17092. doi: 10.1073/pnas.1108745108
Lahkim Bennani-Belhaj, K., Rouzeau, S., Buhagiar-Labarchede, G., Chabosseau, P., Onclercq-Delic, R., Bayart, E., et al. (2010). The Bloom syndrome protein limits the lethality associated with RAD51 deficiency. Mol. Cancer Res. 8, 385–394. doi: 10.1158/1541-7786.MCR-09-0534
Lambert, S., and Carr, A. M. (2005). Checkpoint responses to replication fork barriers. Biochimie. 87, 591–602. doi: 10.1016/j.biochi.2004.10.020
Lambert, S., and Carr, A. M. (2013). Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma 122, 33–45. doi: 10.1007/s00412-013-0398-9
Lambert, S., and Lopez, B. S. (2000). Characterization of mammalian RAD51 double strand break repair using non lethal dominant negative forms. EMBO J. 19, 3090–3099. doi: 10.1093/emboj/19.12.3090
Lambert, S., and Lopez, B. S. (2001). Role of RAD51 in sister-chromatid exchanges in mammalian cells. Oncogene 20, 6627–6631. doi: 10.1038/sj.onc.1204813
Lambert, S., and Lopez, B. S. (2002). Inactivation of the RAD51 recombination pathway stimulates UV-induced mutagenesis in mammalian cells. Oncogene 21, 4065–4069. doi: 10.1038/sj.onc.1205535
Lambert, S., Mizuno, K., Blaisonneau, J., Martineau, S., Chanet, R., Freon, K., et al. (2010). Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell. 39, 346–359. doi: 10.1016/j.molcel.2010.07.015
Lambert, S., Watson, A., Sheedy, D. M., Martin, B., and Carr, A. M. (2005). Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121, 689–702. doi: 10.1016/j.cell.2005.03.022
Landry, S., Narvaiza, I., Linfesty, D. C., and Weitzman, M. D. (2011). APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 12, 444–450. doi: 10.1038/embor.2011.46
Lapuk, A. V., Wu, C., Wyatt, A. W., McPherson, A., McConeghy, B. J., Brahmbhatt, S., et al. (2012). From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J. Pathol. 227, 286–297. doi: 10.1002/path.4047
Laulier, C., Barascu, A., Guirouilh-Barbat, J., Pennarun, G., Le Chalony, C., Chevalier, F., et al. (2011a). Bcl-2 inhibits nuclear homologous recombination by localizing BRCA1 to the endomembranes. Cancer Res. 71, 3590–3602. doi: 10.1158/0008-5472.CAN-10-3119
Laulier, C., Cheng, A., and Stark, J. M. (2011b). The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res. 39, 5935–5944. doi: 10.1093/nar/gkr187
Laulier, C., and Lopez, B. S. (2012). The secret life of Bcl-2: Apoptosis-independent inhibition of DNA repair by Bcl-2 family members. Mutat. Res. 751, 247–257. doi: 10.1016/j.mrrev.2012.05.002
Lee, J. A., Carvalho, C. M., and Lupski, J. R. (2007). A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247. doi: 10.1016/j.cell.2007.11.037
Lemoine, F. J., Degtyareva, N. P., Kokoska, R. J., and Petes, T. D. (2008). Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol. Cell. Biol. 28, 5359–5368. doi: 10.1128/MCB.02084-07
Lemoine, F. J., Degtyareva, N. P., Lobachev, K., and Petes, T. D. (2005). Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120, 587–598. doi: 10.1016/j.cell.2004.12.039
Liao, D. (1999). Concerted evolution: molecular mechanism and biological implications. Am. J. Hum. Genet. 64, 24–30. doi: 10.1086/302221
Lichten, M., and Haber, J. E. (1989). Position effects in ectopic and allelic mitotic recombination in Saccharomyces cerevisiae. Genetics 123, 261–268.
Lieber, M. R., Ma, Y., Pannicke, U., and Schwarz, K. (2004). The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst.) 3, 817–826. doi: 10.1016/j.dnarep.2004.03.015
Liu, N., Lamerdin, J. E., Tebbs, R. S., Schild, D., Tucker, J. D., Shen, M. R., et al. (1998). XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol. Cell. 1, 783–793. doi: 10.1016/S1097-2765(00)80078-7
Liu, P., Erez, A., Nagamani, S. C., Dhar, S. U., Kolodziejska, K. E., Dharmadhikari, A. V., et al. (2011). Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146, 889–903. doi: 10.1016/j.cell.2011.07.042
Long, D. T., Raschle, M., Joukov, V., and Walter, J. C. (2011). Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333, 84–87. doi: 10.1126/science.1204258
Lydeard, J. R., Jain, S., Yamaguchi, M., and Haber, J. E. (2007). Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448, 820–823. doi: 10.1038/nature06047
Lydeard, J. R., Lipkin-Moore, Z., Sheu, Y. J., Stillman, B., Burgers, P. M., and Haber, J. E. (2010). Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 24, 1133–1144. doi: 10.1101/gad.1922610
Magrangeas, F., Avet-Loiseau, H., Munshi, N. C., and Minvielle, S. (2011). Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 118, 675–678. doi: 10.1182/blood-2011-03-344069
Mahaney, B. L., Meek, K., and Lees-Miller, S. P. (2009). Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 417, 639–650. doi: 10.1042/BJ20080413
Marcello, L., Menon, S., Ward, P., Wilkes, J. M., Jones, N. G., Carrington, M., et al. (2007). VSGdb: a database for trypanosome variant surface glycoproteins, a large and diverse family of coiled coil proteins. BMC Bioinformatics. 8:143. doi: 10.1186/1471-2105-8-143
Mizuno, K., Lambert, S., Baldacci, G., Murray, J. M., and Carr, A. M. (2009). Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 23, 2876–2886. doi: 10.1101/gad.1863009
Mizuno, K., Miyabe, I., Schalbetter, S. A., Carr, A. M., and Murray, J. M. (2013). Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 493, 246–249. doi: 10.1038/nature11676
Molenaar, J. J., Koster, J., Zwijnenburg, D. A., van Sluis, P., Valentijn, L. J., van der Ploeg, I., et al. (2012). Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593. doi: 10.1038/nature10910
Moriel-Carretero, M., and Aguilera, A. (2010). A postincision-deficient TFIIH causes replication fork breakage and uncovers alternative Rad51- or Pol32-mediated restart mechanisms. Mol. Cell. 37, 690–701. doi: 10.1016/j.molcel.2010.02.008
Morrison, L. J., Marcello, L., and McCulloch, R. (2009). Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell. Microbiol. 11, 1724–1734. doi: 10.1111/j.1462-5822.2009.01383.x
Moynahan, M. E., Chiu, J. W., Koller, B. H., and Jasin, M. (1999). Brca1 controls homology-directed DNA repair. Mol. Cell. 4, 511–518. doi: 10.1016/S1097-2765(00)80202-6
Moynahan, M. E., Pierce, A. J., and Jasin, M. (2001). BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 7, 263–272. doi: 10.1016/S1097-2765(01)00174-5
Nik-Zainal, S., Alexandrov, L. B., Wedge, D. C., Van Loo, P., Greenman, C. D., Raine, K., et al. (2012). Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993. doi: 10.1016/j.cell.2012.04.024
Palmer, G. H., and Brayton, K. A. (2007). Gene conversion is a convergent strategy for pathogen antigenic variation. Trends Parasitol. 23, 408–413. doi: 10.1016/j.pt.2007.07.008
Pardo, B., and Aguilera, A. (2012). Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet. 8:e1002979. doi: 10.1371/journal.pgen.1002979
Parsons, D. W., Li, M., Zhang, X., Jones, S., Leary, R. J., Lin, J. C., et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439. doi: 10.1126/science.1198056
Plo, I., Laulier, C., Gauthier, L., Lebrun, F., Calvo, F., and Lopez, B. S. (2008). AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 68, 9404–9412. doi: 10.1158/0008-5472.CAN-08-0861
Plo, I., and Lopez, B. (2009). AKT1 represses gene conversion induced by different genotoxic stresses and induces supernumerary centrosomes and aneuploidy in hamster ovary cells. Oncogene 28, 2231–2237. doi: 10.1038/onc.2009.85
Ponder, R. G., Fonville, N. C., and Rosenberg, S. M. (2005). A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell. 19, 791–804. doi: 10.1016/j.molcel.2005.07.025
Popova, T., Manie, E., Rieunier, G., Caux-Moncoutier, V., Tirapo, C., Dubois, T., et al. (2012). Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462. doi: 10.1158/0008-5472.CAN-12-1470
Purandare, S. M., and Patel, P. I. (1997). Recombination hot spots and human disease. Genome Res. 7, 773–786.
Quah, S. K., von Borstel, R. C., and Hastings, P. J. (1980). The origin of spontaneous mutation in Saccharomyces cerevisiae. Genetics 96, 819–839.
Rass, E., Grabarz, A., Bertrand, P., and Lopez, B. S. (2012). [Double Strand Break Repair, one mechanism can hide another: alternative non-homologous end joining.]. Cancer Radiother. 16, 1–10. doi: 10.1016/j.canrad.2011.05.004
Rattray, A. J., Shafer, B. K., McGill, C. B., and Strathern, J. N. (2002). The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics 162, 1063–1077.
Rausch, T., Jones, D. T., Zapatka, M., Stutz, A. M., Zichner, T., Weischenfeldt, J., et al. (2012). Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148, 59–71. doi: 10.1016/j.cell.2011.12.013
Reynaud, C. A., Anquez, V., Grimal, H., and Weill, J. C. (1987). A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell 48, 379–388. doi: 10.1016/0092-8674(87)90189-9
Richardson, C., Stark, J. M., Ommundsen, M., and Jasin, M. (2004). Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 23, 546–553. doi: 10.1038/sj.onc.1207098
Roberts, S. A., Sterling, J., Thompson, C., Harris, S., Mav, D., Shah, R., et al. (2012). Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell. 46, 424–435. doi: 10.1016/j.molcel.2012.03.030
Rodrigue, A., Coulombe, Y., Jacquet, K., Gagne, J. P., Roques, C., Gobeil, S., et al. (2013). The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. J. Cell Sci. 126, 348–359. doi: 10.1242/jcs.114595
Rooney, S., Chaudhuri, J., and Alt, F. W. (2004). The role of the non-homologous end-joining pathway in lymphocyte development. Immunol. Rev. 200, 115–131. doi: 10.1111/j.0105-2896.2004.00165.x
Roseaulin, L., Yamada, Y., Tsutsui, Y., Russell, P., Iwasaki, H., and Arcangioli, B. (2008). Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 27, 1378–1387. doi: 10.1038/emboj.2008.65
Rosenberg, S. M., Longerich, S., Gee, P., and Harris, R. S. (1994). Adaptive mutation by deletions in small mononucleotide repeats. Science 265, 405–407. doi: 10.1126/science.8023163
Rosenberg, S. M., Shee, C., Frisch, R. L., and Hastings, P. J. (2012). Stress-induced mutation via DNA breaks in Escherichia coli: a molecular mechanism with implications for evolution and medicine. Bioessays 34, 885–892. doi: 10.1002/bies.201200050
Rothkamm, K., and Lobrich, M. (2003). Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. U.S.A. 100, 5057–5062. doi: 10.1073/pnas.0830918100
Ruiz, J. F., Gomez-Gonzalez, B., and Aguilera, A. (2009). Chromosomal translocations caused by either pol32-dependent or pol32-independent triparental break-induced replication. Mol. Cell. Biol. 29, 5441–5454. doi: 10.1128/MCB.00256-09
Saini, N., Ramakrishnan, S., Elango, R., Ayyar, S., Zhang, Y., Deem, A., et al. (2013). Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392. doi: 10.1038/nature12584
Saintigny, Y., Delacote, F., Vares, G., Petitot, F., Lambert, S., Averbeck, D., et al. (2001). Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 20, 3861–3870. doi: 10.1093/emboj/20.14.3861
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., and Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. doi: 10.1016/j.cell.2011.03.041
Schmidt, K. H., Wu, J., and Kolodner, R. D. (2006). Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol. Cell. Biol. 26, 5406–5420. doi: 10.1128/MCB.00161-06
Seigneur, M., Bidnenko, V., Ehrlich, S. D., and Michel, B. (1998). RuvAB acts at arrested replication forks. Cell 95, 419–430. doi: 10.1016/S0092-8674(00)81772-9
Shee, C., Gibson, J. L., Darrow, M. C., Gonzalez, C., and Rosenberg, S. M. (2011). Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 13659–13664. doi: 10.1073/pnas.1104681108
Sjoblom, T., Jones, S., Wood, L. D., Parsons, D. W., Lin, J., Barber, T. D., et al. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274. doi: 10.1126/science.1133427
Slupianek, A., Schmutte, C., Tombline, G., Nieborowska-Skorska, M., Hoser, G., Nowicki, M. O., et al. (2001). BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell. 8, 795–806. doi: 10.1016/S1097-2765(01)00357-4
Stenglein, M. D., Burns, M. B., Li, M., Lengyel, J., and Harris, R. S. (2010). APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 17, 222–229. doi: 10.1038/nsmb.1744
Stephens, P. J., Greenman, C. D., Fu, B., Yang, F., Bignell, G. R., Mudie, L. J., et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. doi: 10.1016/j.cell.2010.11.055
Stephens, P. J., Tarpey, P. S., Davies, H., Van Loo, P., Greenman, C., Wedge, D. C., et al. (2012). The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404. doi: 10.1038/nature11017
Stockdale, C., Swiderski, M. R., Barry, J. D., and McCulloch, R. (2008). Antigenic variation in Trypanosoma brucei: joining the DOTs. PLoS Biol. 6:e185. doi: 10.1371/journal.pbio.0060185
Stransky, N., Egloff, A. M., Tward, A. D., Kostic, A. D., Cibulskis, K., Sivachenko, A., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160. doi: 10.1126/science.1208130
Strathern, J. N., Shafer, B. K., and McGill, C. B. (1995). DNA synthesis errors associated with double-strand-break repair. Genetics 140, 965–972.
Stratton, M. R., Campbell, P. J., and Futreal, P. A. (2009). The cancer genome. Nature 458, 719–724. doi: 10.1038/nature07943
Sun, M., Wang, G., Paciga, J. E., Feldman, R. I., Yuan, Z. Q., Ma, X. L., et al. (2001). AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am. J. Pathol. 159, 431–437. doi: 10.1016/S0002-9440(10)61714-2
Suspene, R., Aynaud, M. M., Guetard, D., Henry, M., Eckhoff, G., Marchio, A., et al. (2011). Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl. Acad. Sci. U.S.A. 108, 4858–4863. doi: 10.1073/pnas.1009687108
Takata, M., Sasaki, M. S., Tachiiri, S., Fukushima, T., Sonoda, E., Schild, D., et al. (2001). Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol. 21, 2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001
Taylor, A. M., Groom, A., and Byrd, P. J. (2004). Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst.) 3, 1219–1225. doi: 10.1016/j.dnarep.2004.04.009
Taylor, B. J., Nik-Zainal, S., Wu, Y. L., Stebbings, L. A., Raine, K., Campbell, P. J., et al. (2013). DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2:e00534. doi: 10.7554/eLife.00534
Terradas, M., Martin, M., Hernandez, L., Tusell, L., and Genesca, A. (2012). Nuclear envelope defects impede a proper response to micronuclear DNA lesions. Mutat. Res. 729, 35–40. doi: 10.1016/j.mrfmmm.2011.09.003
Terradas, M., Martin, M., Tusell, L., and Genesca, A. (2009). DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair (Amst.) 8, 1225–1234. doi: 10.1016/j.dnarep.2009.07.004
Thompson, L. H., and Schild, D. (2001). Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 477, 131–153. doi: 10.1016/S0027-5107(01)00115-4
Tourriere, H., and Pasero, P. (2007). Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst.) 6, 900–913. doi: 10.1016/j.dnarep.2007.02.004
Turner, B. C., Ottey, M., Zimonjic, D. B., Potoczek, M., Hauck, W. W., Pequignot, E., et al. (2002). The fragile histidine triad/common chromosome fragile site 3B locus and repair-deficient cancers. Cancer Res. 62, 4054–4060.
Vispé, S., Cazaux, C., Lesca, C., and Defais, M. (1998). Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 26, 2859–2864. doi: 10.1093/nar/26.12.2859
Walsh, T., Casadei, S., Lee, M. K., Pennil, C. C., Nord, A. S., Thornton, A. M., et al. (2011). Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. U.S.A. 108, 18032–18037. doi: 10.1073/pnas.1115052108
Walsh, T., and King, M. C. (2007). Ten genes for inherited breast cancer. Cancer Cell. 11, 103–105. doi: 10.1016/j.ccr.2007.01.010
Wilhelm, T., Magdalou, I., Barascu, A., Techer, H., Debatisse, M., and Lopez, B. S. (2014). Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 111, 763–768. doi: 10.1073/pnas.1311520111
Wilson, M. A., Kwon, Y., Xu, Y., Chung, W. H., Chi, P., Niu, H., et al. (2013). Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502, 393–396. doi: 10.1038/nature12585
Wisniewski-Dye, F., and Vial, L. (2008). Phase and antigenic variation mediated by genome modifications. Antonie Van Leeuwenhoek. 94, 493–515. doi: 10.1007/s10482-008-9267-6
Yamanaka, S., Balestra, M. E., Ferrell, L. D., Fan, J., Arnold, K. S., Taylor, S., et al. (1995). Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc. Natl. Acad. Sci. U.S.A. 92, 8483–8487. doi: 10.1073/pnas.92.18.8483
Yang, H., Wen, Y. Y., Zhao, R., Lin, Y. L., Fournier, K., Yang, H. Y., et al. (2006). DNA damage-induced protein 14-3-3 sigma inhibits protein kinase B/Akt activation and suppresses Akt-activated cancer. Cancer Res. 66, 3096–3105. doi: 10.1158/0008-5472.CAN-05-3620
Yang, Y., Sterling, J., Storici, F., Resnick, M. A., and Gordenin, D. A. (2008). Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet. 4:e1000264. doi: 10.1371/journal.pgen.1000264
Zaratiegui, M., Vaughn, M. W., Irvine, D. V., Goto, D., Watt, S., Bahler, J., et al. (2011). CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature 469, 112–115. doi: 10.1038/nature09608
Keywords: Homologous recombination, mutagenesis, DNA double strand break repair, replication stress, genetic variability, genetic instability
Citation: Guirouilh-Barbat J, Lambert S, Bertrand P and Lopez BS (2014) Is homologous recombination really an error-free process? Front. Genet. 5:175. doi: 10.3389/fgene.2014.00175
Received: 21 February 2014; Accepted: 23 May 2014;
Published online: 11 June 2014.
Edited by:
Antonio Porro, University of Zurich, SwitzerlandReviewed by:
Michael Chang, European Research Institute for the Biology of Ageing, NetherlandsAnna Malkova, University of Iowa, USA
Copyright © 2014 Guirouilh-Barbat, Lambert, Bertrand and Lopez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernard S. Lopez, PR2, UMR CNRS 8200, Institut Gustave Roussy, Université Paris-Sud, 114 Rue Edouard Vaillant, 94805 Villejuif, France e-mail:YmVybmFyZC5sb3BlekBndXN0YXZlcm91c3N5LmZy