 Thomas M. Keane
Thomas M. Keane Kim Wong1
Kim Wong1 Alexandre Reymond
Alexandre Reymond Binnaz Yalcin
Binnaz Yalcin- 1Wellcome Trust Sanger Institute, Hinxton, Cambridge, UK
- 2Wellcome Trust Centre for Human Genetics, Oxford, UK
- 3Center for Integrative Genomics, University of Lausanne, Lausanne, Switzerland
- 4Institute of Genetics and Molecular and Cellular Biology, Illkirch, France
Structural variation is variation in structure of DNA regions affecting DNA sequence length and/or orientation. It generally includes deletions, insertions, copy-number gains, inversions, and transposable elements. Traditionally, the identification of structural variation in genomes has been challenging. However, with the recent advances in high-throughput DNA sequencing and paired-end mapping (PEM) methods, the ability to identify structural variation and their respective association to human diseases has improved considerably. In this review, we describe our current knowledge of structural variation in the mouse, one of the prime model systems for studying human diseases and mammalian biology. We further present the evolutionary implications of structural variation on transposable elements. We conclude with future directions on the study of structural variation in mouse genomes that will increase our understanding of molecular architecture and functional consequences of structural variation.
Introduction
Structural variation (SV) is generally considered as rearrangements of DNA regions affecting DNA sequence length and/or orientation in the genome of one species, and includes deletions, insertions, copy-number gains, inversions, and transposable elements. Structural variation has long been known to be pathogenic, resulting in rare genomic disorders such as well-known Charcot-Marie Tooth disease (Lupski et al., 1991; reviewed in Lupski, 1998, 2009), or more recently Koolen de Vries and 16p11.2 micro-deletion syndromes (Walters et al., 2010; Jacquemont et al., 2011; Koolen et al., 2012). Population-based SV has also begun to emerge as an important source of genomic variation contributing to common human diseases (Sebat et al., 2007; Hollox et al., 2008; Stefansson et al., 2008; Conrad et al., 2010; Pinto et al., 2010; Girirajan et al., 2011; Jarick et al., 2011; Malhotra et al., 2011; Elia et al., 2012; Helbig et al., 2014; Ramos-Quiroga et al., 2014), cancer development (Diskin et al., 2009; Stephens et al., 2011; Northcott et al., 2012; Rausch et al., 2012a; Malhotra et al., 2013; Ni et al., 2013), neuronal mosaicism in the human brain (McConnell et al., 2013) and genomic evolution (Perry et al., 2007; Itsara et al., 2010; Sudmant et al., 2013). However, the characterization of sequence flanking the breakpoints of structural variants (we call this breakpoint features), including for example micro-deletion and micro-insertion of 1 base pair (bp) up to several hundreds of bp, has remained challenging but is important with respect to not only their accurate identification, but also interpretation of their function and prediction of mechanisms by which structural variants arose (Yalcin et al., 2012a).
SVs have traditionally been observed by array comparative genome hybridization (aCGH), a method for analyzing copy number variations by measuring fluorescence between two differentially labeled DNA samples (DNA of a test sample compared to a reference sample). Using aCGH, the extent of genome-wide SV in the mouse was first demonstrated in 2007 with the detection of 80 high-confident copy number variants in 20 inbred strains of mice (Graubert et al., 2007), subsequently followed by other studies, summarized in Table 1 (Cutler et al., 2007; Akagi et al., 2008; Cahan et al., 2009; Henrichsen et al., 2009; Agam et al., 2010; Quinlan et al., 2010). These studies, however, have proven to be difficult to interpret due to their poor reproducibility (Agam et al., 2010) and inability to detect certain types of structural variants. For example inversions and insertions of novel sequence are blind to aCGH technology because inversions do not affect copy number, which is what is detected by aCGH technique, and novel sequence insertions have no copy in the reference genome.
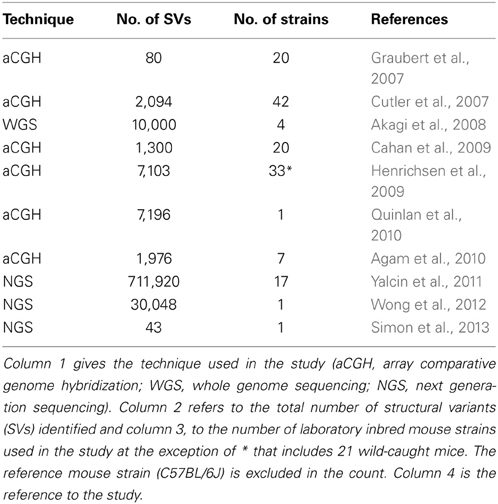
Table 1. Summary of mouse studies reporting genome-wide structural variants.
With the emergence of next-generation sequencing (NGS) (Mardis, 2011), the Mouse Genomes Project (http://www.sanger.ac.uk/resources/mouse/genomes/) was able to sequence the entire genomes of 18 classical laboratory strains and wild-derived lines of inbred strains of mice, producing detailed maps of SV and retro-transposon elements in each mouse strain, relative to the reference mouse strain C57BL/6J (Keane et al., 2011; Nellaker et al., 2012; Wong et al., 2012; Simon et al., 2013). For the first time, this resulted in the detection of an extraordinarily larger number of structural variants than previously observed using aCGH, totaling 710,000 novel structural variants affecting 1% of the mouse genome and encompassing 10 times more total nucleotides than single nucleotide polymorphisms (Yalcin et al., 2011). As a comparison, we had identified 121 deletions in a previous aCGH study of SV in DBA/2J, with SV length ranging between minimum size of 5 kilobases (Kb) and maximum of 260 Kb (median size 48 Kb) (Agam et al., 2010), whereas in a latest NGS study of SV we found far more deletions (a total of 16,318) in that same strain, of much smaller size (minimum size of 100 bp, maximum of 10 Kb, median of 400 bp) (Figure 1).
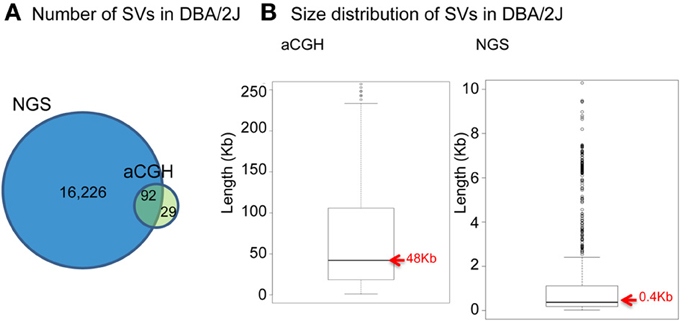
Figure 1. Comparison between NGS and aCGH in inbred mouse strain DBA/2J. (A) Venn diagram of the number of deletions detected. (B) Boxplot showing the size distribution of deletions.
Such genome-wide abundance in structural variation has led to several important questions: what is the molecular architecture of these variants, what are the mechanisms of SV formation and how do they impact gene function? In this review, we address these questions and redefine what we have learnt so far about the nature, origins, and role of structural variation from current studies in the mouse. Finally, we discuss the promises of novel methods which are likely to facilitate access to repeat-rich regions and assembly of complex genomic regions, in order to assess the origins and functional impact of structural variation in the most challenging regions of the mouse genome.
Detection of Structural Variants using Paired-End Mapping Methods
While most deep-sequencing applications focus on the identification of single-nucleotide polymorphisms (SNPs) or small insertion deletion polymorphisms, structural variation can also be identified from the same data. However, while the basic types of structural variants (deletions, insertions, inversions, and duplications) can be identified using a combination of computational methods, the detection of complex rearrangements remains challenging. We define complex rearrangements as those structural variants consisting of a combination of basic types that directly about each other or that are nested within each other (e.g., an inversion directly flanked by insertions, or a deletion nested within a tandem duplication).
Typically, genomic DNA of a test genome is sheared into fragments of 300–500 bp to generate a sequencing library. Short paired-reads (50–250 bp) from either extremity of the fragment (called paired-end reads) are sequenced and mapped to the reference genome. Structural variants are then called based on orientation, distance, and depth of the mapped paired-reads (also reviewed in Medvedev et al., 2009; Alkan et al., 2011). Depending on the size and type of structural variant, these methods exploit read pairs (Korbel et al., 2007; Chen et al., 2009), split-reads (Ye et al., 2009; Albers et al., 2011), single end clusters and read depth (Simpson et al., 2010).
The most widely used methods are read pair and read depth methods. Read pair based methods analyze distance and orientation of paired reads to infer deletion, insertion, inversion and tandem duplication events as shown in Figure 2. When the paired-end reads are mapping in the correct orientation (“+/−” is normal) but to a distance that is significantly larger than the average fragment length, this suggests a deletion, whereas if the distance is smaller than the fragment length, it suggests an insertion. When the two sequenced ends map back to the reference genome in the wrong orientation (“+/+” and “−/−”), and at a distance that is significantly larger than the size of the fragment itself, this indicates an inversion. Finally, when paired-end reads map with orientation “−/+” to a large distance, it suggest tandem duplication. In the single-end cluster analysis, one of the paired-end reads maps to the reference while its mate map to the inserted sequence (de novo sequence or repeat element insertion). Read depth methods take advantage of the high coverage of next generation sequencing to infer increase or decrease of reads at a locus. When the coverage is higher than the expected genome coverage, duplication is inferred, whereas when it is smaller or null, deletion is inferred. Once the structural variant is detected using these analyses, breakpoint refinement is typically achieved using local sequence assembly.
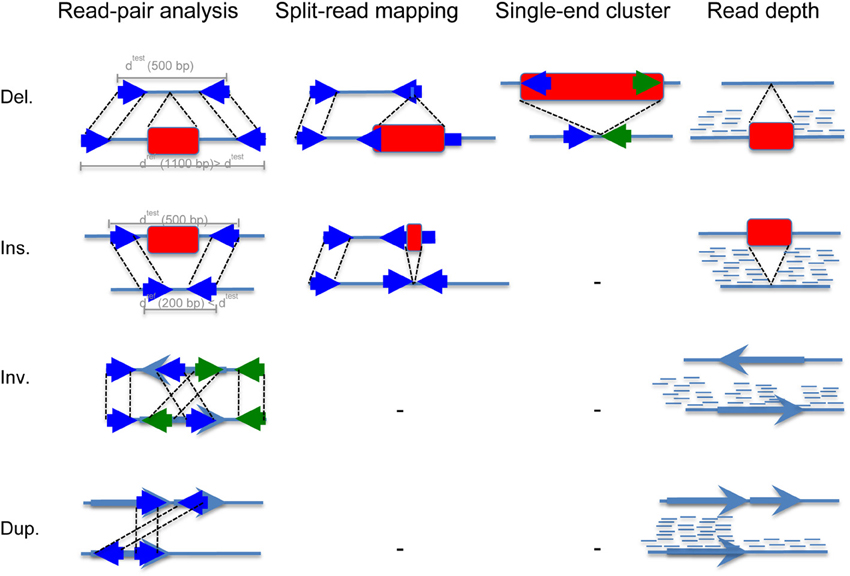
Figure 2. Read mapping patterns used by computational methods to detect basic structural variation from NGS data. This figure shows the principle of SV identification using (i) read-pair analysis, (ii) split-read mapping, (iii) single end cluster analysis, and (iv) read depth analysis. Deletions and insertions are represented using red rectangles, and inversions and duplications using light blue arrows. Reads are represented using solid dark blue arrows. The first step consists in sequencing a test genome. Typically, the genomic test DNA is fragmented into chunks of 300–500 bp. Then, reads of 50–250 bp are sequenced from either side of each fragment (we call these paired-end reads). The second step consists in mapping these paired-end reads to the mouse reference genome. A rightward facing arrow denotes a positive strand alignment, and leftward a negative strand alignment. (i) In the read-pair analysis approach, when the paired-end reads are mapping in the correct orientation (“+/−” is normal) but to a distance that is significantly larger than the average fragment length. If we suppose this distance to be 1100 bp, it suggests a deletion of 600 bp, whereas if the distance is smaller than the fragment length, for example 200 bp, it suggests an insertion of 300 bp. When the two sequenced ends of two fragments map back to the reference genome in the wrong orientation (“+/+” and “−/−”), and at a distance that is significantly larger than the size of the fragment itself, this indicates an inversion. Finally, when paired-end reads map with orientation “−/+” to a large distance, it suggest tandem duplication. (ii) In the split-read approach, one of the paired-end reads map to the reference genome while its mate contains the structural variant, typically a deletion or an insertion of small length. (iii) In the single-end cluster analysis, one of the paired-end reads maps to the reference while its mate map to the inserted sequence that can be either de novo sequence or repeat element such as LINE, SINE, or ERV. (iv) Finally, the read depth approach takes advantage of the high coverage of next generation sequencing that makes it possible to detect copy number changes. Of note, the coverage drops at insertion and inversion breakpoints, which when combined with paired-end reads analysis makes the SV call highly reliable.
Remarkably, in the past several years many algorithms have been developed to discover basic structural variation in paired-end next generation sequencing data. There are over 50 programs to date (Table 2), however none is as yet considered to reach a community standard and only a handful combine multiple methods for the detection of structural variation (Medvedev et al., 2010; Wong et al., 2010; Rausch et al., 2012b; Sindi et al., 2012; Hart et al., 2013). Accurate structural variant calling depends on many factors such as sequencing library biases, read length, uniform sequencing coverage, and proximity of SVs to repeat sequences. Some of the most frequent sequencing library biases that can detrimentally affect SV detection are high PCR duplicates, non-normal fragment size distributions, and uneven representation of the genome at varying levels of GC content. Therefore, false negative rates of most studies remain high (20–30%) compared to SNP calling (<5%). False positive rates are also high and are often caused by misalignment of the short reads and sometimes by reference genome assembly errors.
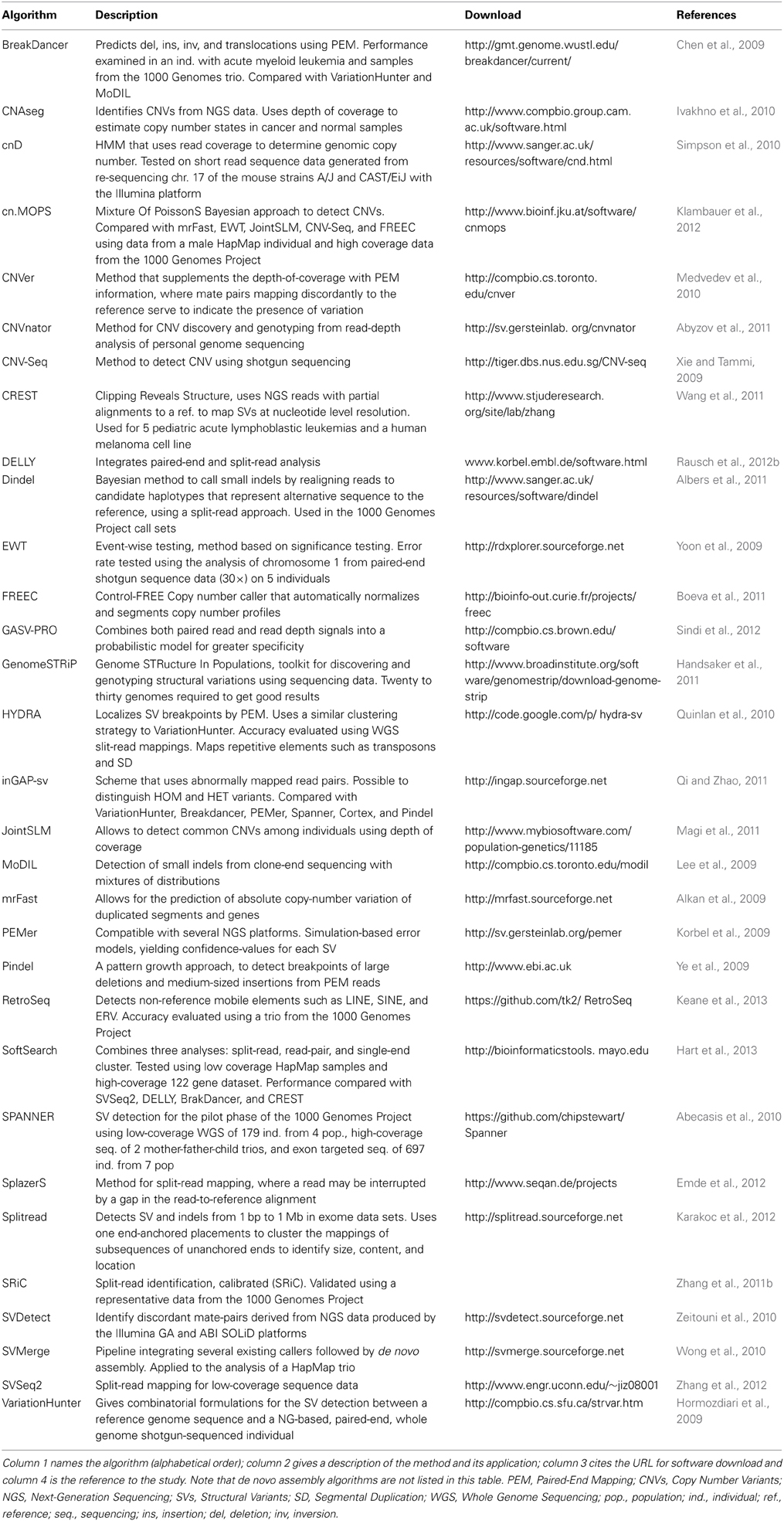
Table 2. Algorithms for the detection of structural variation.
There is a growing awareness of complex structural variants (Berger et al., 2011; Stephens et al., 2011; Quinlan and Hall, 2012; Yalcin et al., 2012a; Malhotra et al., 2013), however, their genome-wide detection is much more challenging and less intuitive as they often generate ambiguous paired-end mapping patterns. Complex structural variants are very often completely or partially missed, or incorrectly classified because a single method on its own might not be sufficient to capture the whole complexity of the structural variant (e.g., an apparent deletion and inversion may be simultaneously part of a tandem duplication region). Thus, it is important to combine multiple methods, something that the community has begun to do. Sindi and colleagues, for example, used an algorithm combining both read pairs and read depth signals into a probabilistic model implemented in a software GASV-PRO that significantly improves detection specificity (Sindi et al., 2012). Rausch and colleagues have developed DELLY that integrates short insert paired-ends, long-range mate-pairs and split-read alignments to accurately delineate genomic rearrangements at single-nucleotide resolution (Rausch et al., 2012b). In our studies, we used SVMerge (Wong et al., 2010), a pipeline that integrates structural variation calls from five existing software, and validates breakpoints using local de novo assembly.
Unbiased exploration of next-generation sequencing data is laborious, however it is essential for deciphering the true complex nature of structural variants. Toward this goal, we visualized read mappings to the whole of mouse chromosome 19 as well as a random set of regions on other chromosomes using the short-read visualization tool LookSeq (Manske and Kwiatkowski, 2009) in 17 inbred strains of laboratory mice (Yalcin et al., 2012a) as well as in C57BL/6J mice (Simon et al., 2013). We were able to recognize classical paired-end mapping (PEM) patterns, but unexpectedly we were also able to detect a number of other patterns, of greater diversity and complexity that would have been missed or miscalled by existing computational SV detection methods. When two (or more) structural variants co-localize at a locus in the genome (right next to each other), or when one or more structural variants are embedded within another one of larger size (nested), it creates confusing paired-end mapping patterns and incoherent read depth. Figure 3 highlights some complex rearrangements that cause conflicting signals during automatic detection. For example, a deletion directly flanked by a large insertion is characterized by null read depth as expected, however paired reads supporting the deletion are missing because of the insertion. However, we showed that it is possible to train genome-wide computational analysis to detect most of these atypical patterns using integration of multiple detection methods (Wong et al., 2010).
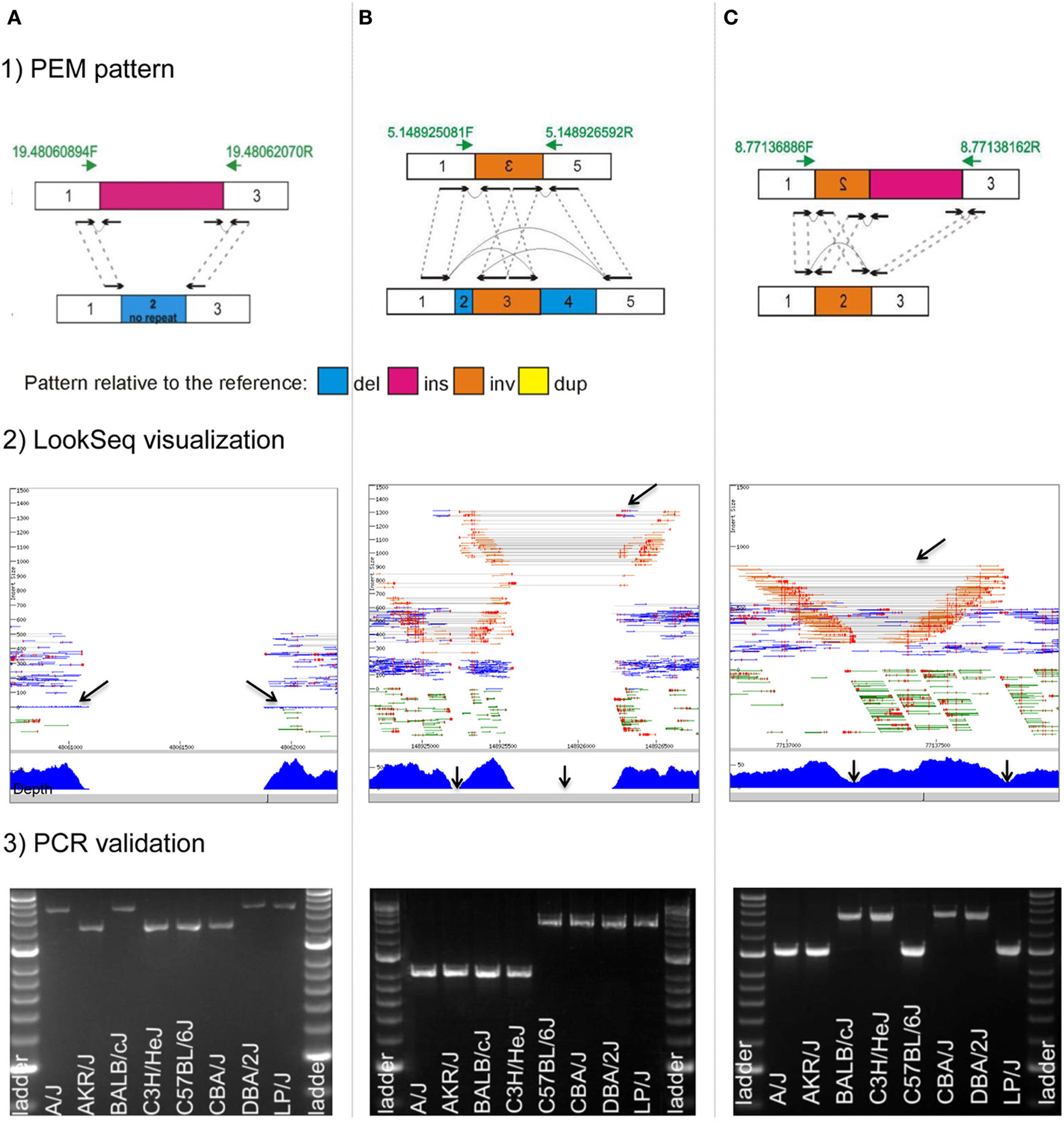
Figure 3. Complex rearrangements in mouse genomes. We highlight three examples of complex rearrangements that cause ambiguous signals during their detection (for a full list of complex rearrangements see Yalcin et al., 2012a): (A), a deletion directly flanked by an insertion; (B), an inversion directly flanked by two deletions; and (C), an inversion directly flanked by an insertion. For each complex rearrangements, we provide: (1) a drawing of the paired-end mapping (PEM) pattern, (2) an illustration using the short read visualization tool LookSeq (Manske and Kwiatkowski, 2009), and (3) PCR validation. We draw paired-end reads (black arrows) and how they map to the reference genome (dashed gray lines). Green arrows represent primer pairs used for PCR validation. PCR amplification was carried out across eight inbred strains of mice (A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, CBA/J, DBA/2J, and LP/J), which are the parental strains of the Heterogenesous Stock population (Valdar et al., 2006). Hyperladder II is the size marker. Genomic coordinates refer to the mm9 mouse assembly. (A) Deletion of 836 bp directly flanked by an insertion of 1200 bp on mouse chromosome 19 (chr19: 48,061,057–48,061,892 bp) in mouse strains A/J, BALB/cJ, DBA/2J, and LP/J. In LookSeq, the two back arrows show singleton reads suggesting an insertion (their mates are within the inserted sequence). Read depth is null but paired-end reads in support of the deletion are missing because of the insertion. PCR in four strains (A/J, BALB/cJ, DBA/2J, and LP/J) does not show directly the presence of the 836-bp deletion but instead reveals the presence of an insertion of about 400 bp that is in fact the size difference between the deletion and the insertion. (B) Inversion of 325 bp on mouse chromosome 5 (chr5: 148,925,249–148,925,573 bp), directly flanked on the left by a deletion of 71 bp (chr5: 148,925,178–148,925,248 bp) and on the right by another deletion of 645 bp (chr5: 148,925,574–148,926,218 bp). In LookSeq, the top arrow shows the PEM pattern of the deletion. Normally, the underlying read depth should be null, however, it is only null at the regions shown by the two bottom arrows. This is caused by an intervening inversion. PCR in four strains (A/J, AKR/J, BALB/cJ, and C3H/HeJ) confirms the presence of the two deletions. (C) An inversion of 548 bp on mouse chromosome 8 (chr8: 77,137,213–77,137,760 bp) directly flanked by an insertion of 400 bp in mouse strain BALB/cJ, C3H/HeJ, CBA/J, and DBA/2J. In LookSeq, the bottom arrows show a dip in the coverage; on the right, it is caused by an insertion and on the left by an inversion. The presence of the insertion results in missing reads (“−/−”), supporting the inversion. PCR shows an amplification band of about 1400 bp in BALB/cJ, C3H/HeJ, CBA/J, and DBA/2J, whereas, in the remaining strains, the band is at about 1000 bp. This confirms the insertion of 400 bp in BALB/cJ, C3H/HeJ, CBA/J, and DBA/2J.
In conclusion, to study the whole diversity and complexity of structural variants, future algorithms need to integrate multiple signals and sequence analyses features based on what we have learnt so far about the architecture of structural variants, while visual approaches will continue to increase our understanding of complex forms of structural variants such as inversions and translocations that remain to be fully resolved. It is important to gain better sensitivity and specificity in the identification of structural variants especially those that have complex architecture to study accurately their impact on diseases such as tumor heterogeneity (Russnes et al., 2011), and on the evolution of genomes.
Functional Impact of Structural Variants
The functional impact of structural variants is still controversial in the literature. On one hand, some studies showed that SNPs are more likely to contribute to individual phenotypic differences than structural variants (Conrad et al., 2010; Keane et al., 2011); on the other hand, several studies have estimated the impact of structural variation using its effect on gene expression, and these estimates ranged from 10 to 74% (Stranger et al., 2007; Cahan et al., 2009; Henrichsen et al., 2009; Yalcin et al., 2011). It has also been reported that structural variation can influence gene expression both spatially and temporally (Chaignat et al., 2011), including genes outside of SV margins (Henrichsen et al., 2009), and can do so through chromatin conformation changes (Gheldof et al., 2013). The influence of structural variation on gene expression is specifically reviewed in Harewood et al. (2012).
Interpreting the phenotypic consequences of structural variation can be done using different methods. In this review, we describe three methods with specific emphasis on genome wide association studies. Genome wide association studies (GWASs) identify genomic loci associated with individual differences (these regions are called Quantitative Trait Loci, QTLs) using large populations of outbred mice, while taking advantage of recombinants that have naturally accumulated during breeding (Valdar et al., 2006; Yalcin et al., 2010). When combined with the availability of full genome sequences, GWASs in outbred mice are providing significant advances into the understanding of the genotype-phenotype relationship (reviewed in Yalcin and Flint, 2012), especially the impact of structural variants on phenotypic differences.
To test causality of a structural variant within a QTL region, Richard Mott and colleagues have developed a statistical test (called merge) to identify genomic variants likely to be functional from those less likely to be functional (Yalcin et al., 2005). Unexpectedly, very few SVs (only 12) out of about 100,000 SVs present in classical inbred strains of mice (Yalcin et al., 2011) overlapped with a gene within QTL regions identified using an outbred population of mice known as the Heterogenous Stock mice (Talbot et al., 1999; Valdar et al., 2006; Yalcin et al., 2011). Table 3 lists these structural variants associated with quantitative traits in outbred mice. These were amongst the larger effect size QTLs. Although the number of SVs causing phenotypic differences is small, it is expected that these SVs will provide significant insights into gene function. We highlight two examples in the next paragraph.
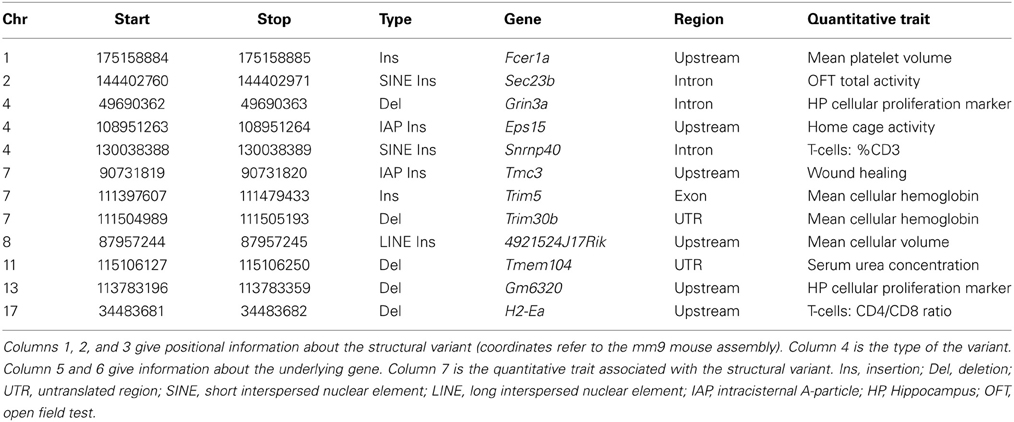
Table 3. Structural variants associated with quantitative traits in outbred mice.
Figure 4 shows a deletion of 600 bp lying within the promoter region of H2-Ea (histocompatibility 2, class II antigen E alpha) that is affecting CD4+/CD8+ ratio in T lymphocytes. This locus was fine-mapped to single-gene resolution using a population of commercial outbred mice (CFW) (Yalcin et al., 2010). Causality was confirmed using mouse transgenic data with and without the deletion. The ratio of CD4+/CD8+ was significantly increased in transgene positive mice with the deletion when compared to transgene negative mice (without the deletion), both in the spleen and in the thymus. Figure 5 illustrates a transposable element, an intracisternal A-particle (IAP) element of 6400 bp, which has inserted in the promoter region of Eps15 (Epidermal Growth Factor Receptor Pathway Substrate 15). This variant modulates home cage activity in outbred mice. There is a decrease of expression in the brain in mice with the IAP element. Data from the mouse knockout of Eps15 also show a significant decrease of home cage activity when compared to matched wildtype mice.
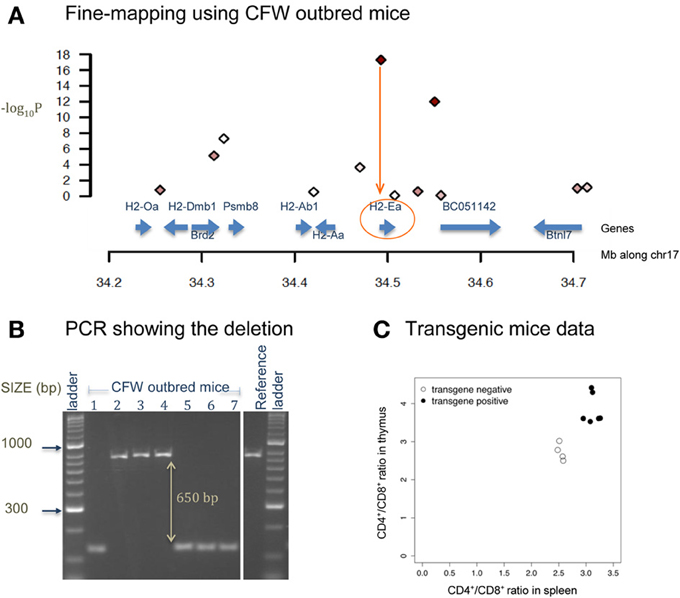
Figure 4. Deletion in H2-Ea affects CD4+/CD8+ ratio. (A) The x-axis is the position along mouse chromosome 17 (Mb). The y-axis shows the significance level of the association between CD4+/CD8+ ratio and a set of bi-allelic markers (represented using polygons) using a population of 200 commercially available outbred mice (CFW mice Yalcin et al., 2010). Markers with strong association (−log10P > 10) are colored in red. Strongest association is within the promoter region of H2-Ea. (B) PCR image of H2-Ea reveals a 600-bp deletion in 8 CFW mice. (C) Plot of mouse transgenic data with and without the deletion. The x-axis is the CD4+/CD8+ ratio in the spleen and the y-axis in the thymus. White circles are measures from transgene negative mice so with no deletion. Black circles are measures from transgene positive mice (with the deletion). Apart from the deletion, the genetic background of these mice is identical.
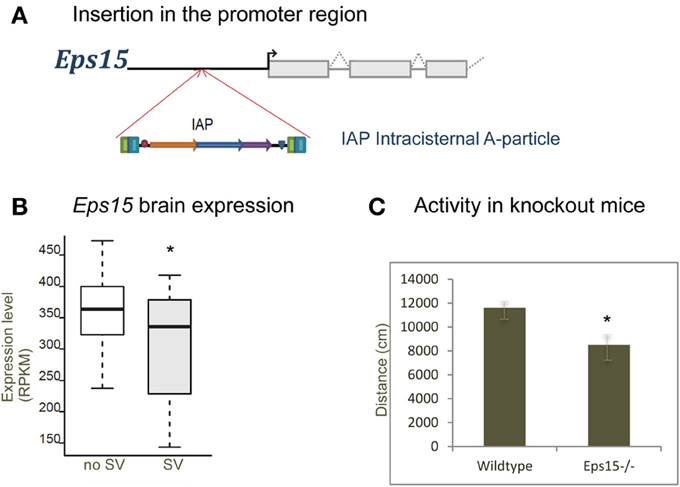
Figure 5. Insertion in Eps15 modulates activity. (A) A transposable element (Intracisternal A-particle) of 6400 bp has inserted in the promoter region of Eps15. (B) Boxplot showing expression in the brain measured using RNA-Seq in mice with and without the structural variant (RPKM, reads per kilobase per million mapped reads). There is a decrease of expression with the presence of the insertion. (C) Data from the mouse knockout of Eps15, showing a significant decrease of home cage activity compared to matched wildtype mice (*p-value < 0.05).
A second way to assess the phenotypic consequences of structural variation is to undertake a comprehensive phenotypic comparison between two closely related sub-strains of mice, and examine the relationship between structural variants and phenotypic changes between these strains. In a recent study, comparing phenotypic and genomic analysis of C57BL/6J and C57BL/6N sub-strains, 15 structural variants differentiating C57BL/6J and C57BL/6N were identified encompassing genic regions (Table 4). It includes three structural variants that have MP (Mammalian Phenotype) terms that coincide with the phenotype differentiating C57BL/6J and C57BL/6N. The first is an intronic LINE insertion found in the intron of Chl1 (Cell adhesion molecule with homology to L1CAM). C57BL/6N mice displayed abnormal spatial memory in the Morris water maze test compared to C57BL/6J mice. Interestingly, knockout mice of Chl1 also show abnormal spatial working memory. The second is an intronic ERV insertion in Rptor (Regulatory associated protein of MTOR, complex 1) in C57BL/6J mice. These mice were characterized by decreased fat mass and blood glucose. Knockout mice of Rptor interestingly also showed decreased fat mass and blood glucose amongst other metabolic phenotypes. The third is the well-known deletion at the Nnt (Nicotinamide nucleotide transhydrogenase) locus (Freeman et al., 2006) in C57BL/6J, which is associated with significantly impaired glucose tolerance.
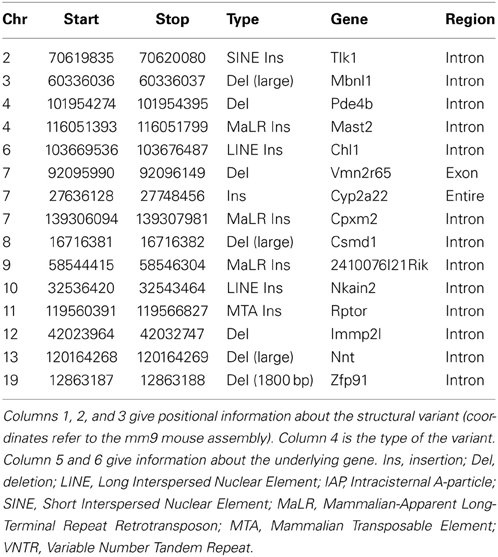
Table 4. Structural variants differentiating C57BL/6J and C57BL/6N.
A third way is to search for structural variants that affect a coding region of a gene, potentially creating a null or hypomorphic allele. We found about 50 structural variants encompassing a coding segment (Yalcin et al., 2011; reviewed in Yalcin et al., 2012b), affecting eleven already known genes (Amd2, Defb8, Fv1, Skint4, Skint3, Skint9, Soat1, Tas2r103, Tas2r120, Trim5, and Trim12a) (Best et al., 1996; Persson et al., 1999; Bauer et al., 2001; Nelson et al., 2005; Boyden et al., 2008; Tareen et al., 2009; Wu et al., 2010) and, in some cases, are giving rise to specific phenotype in mice. For example, a deletion of 1342 bp affecting the fourth coding exon of Fv1 (Friend-virus-susceptibility-1) is associated with retrovirus replication (Best et al., 1996; Yalcin et al., 2011), and a deletion of 6817 bp on the first exon of Soat1 (Sterol O-acyltransferase 1) results in hair interior defects (Wu et al., 2010; Yalcin et al., 2011).
Human GWAS have shown that common SNPs (minor allele frequency >5%) explain only some fraction of the heritability, suggesting that SVs might also be contributing to individual phenotypic variation (Manolio et al., 2009). Results presented in this review suggest that, given the abundance of structural variants in mouse genomes, SVs make less of a contribution to individual phenotypic variation than SNPs. However, when they do, structural variants have a large effect size on the phenotype, providing a unique opportunity to investigate the relationship between structural variants and phenotypic differences, at a molecular as well as mechanistic level.
Evolutionary Implications and Transposable Elements
Transposable elements (TEs) have been highly influential in shaping the structure and evolution of mammalian genomes, as exemplified by TE-derived sequence contributing between 38 and 69% of genomic sequence (Buzdin, 2004; Cordaux and Batzer, 2009; Shapiro, 2010; de Koning et al., 2011). TE insertions also can influence the transcription, translation or function of genes. Functional effects of TE insertions include their regulation of transcription by acting as alternative promoters or as enhancer elements and via the generation of antisense transcripts, or of transcriptional silencers. TEs are classified on the basis of their transposition mechanism (Goodier and Kazazian, 2008). Class I retrotransposon propagates in the host genome through an intermediate RNA step, requiring a reverse transcriptase to revert it to DNA before insertion into the genome. Class II DNA transposons do not have an RNA intermediate, and translocate with the aid of transposases and DNA polymerase. The overwhelming majority, over 96%, of TEs in the mouse genome, are of the retrotransposon type. These are further classified into three distinct classes: short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), and the endogenous retrovirus (ERV) superfamily (Stocking and Kozak, 2008). The ERV elements are ancient remnants of exogenous virus infections, consisting of internal sequence that encodes viral genes that are flanked by long terminal repeats (LTRs). Therefore, TEs provide a potential source of variants detrimental to the host by altering pre-existing gene function.
Previous studies examined two ERV families in eight mouse strains (IAP or ETn/MusD elements in C57BL/6J, A/J, DBA/2J, SPRET/EiJ, CAST/EiJ, MOLF/EiJ, WSB/EiJ, and 129X1/SvJ) (van de Lagemaat et al., 2006; Quinlan et al., 2010; Li et al., 2012), with one study in particular focusing on intronic insertions (Zhang et al., 2011a) and another exploring LINE variation in four strains (A/J, DBA/2J, 129S1/SvImJ, and 129X1/SvJ) (Akagi et al., 2008). However, the largest genome-wide survey of TE polymorphism in multiple laboratory mouse strains was carried out as part of the Mouse Genomes Project (Yalcin et al., 2011; Nellaker et al., 2012). There were two types of polymorphic TE to be cataloged; those that are present in the reference genome and not present in one or more other strain; and those that are not present in the reference genome and present in one or more other strain. In total, 103,798 TE variants (TEVs) (28,951 SINEs, 40,074 LINEs, and 34,773 ERVs) were computationally predicted among the 17 sequenced mouse strains in addition to the C57BL/6J reference strain. By placing the TE insertions within a primary phylogeny, it was possible to observe the relative expansions of all the TE families over an approximate 2 million years time period. This primary phylogeny matched the phylogeny expected from the heritage of the mouse strains (Beck et al., 2000). This analysis revealed the historic expansion of ERV families, most notably IAPs, in laboratory strains. Another interesting family are the MuLV family which arose recently and thus is found in a smaller number of copies that together show a higher fraction of variable elements.
TEV density varies by chromosome, by local nucleotide composition (G + C content) (Filipski et al., 1973; Macaya et al., 1976; Thiery et al., 1976), and by position relative to functional sequence, such as exons. LINE TEVs show a bias for being located in A + T-rich sequence, whilst SINE TEVs tend to reside in G + C-rich sequence (Korenberg and Rykowski, 1988; Boyle et al., 1990). It was also observed that ERV TEVs are more heterogeneous than SINEs or LINEs in their G + C bias, with MuLV TEVs being as enriched in high G + C sequence as SINEs. Interestingly, by contrast to monomorphic TEs, polymorphic TEVs are more unevenly distributed among the chromosomes (having accounted for G + C content) with, for example, chromosome 19 exhibiting a significant enrichment of SINEs and the X chromosome showing a strong deficit of all three TEV classes (Nellaker et al., 2012). The depletion of polymorphic LINEs on the X chromosome was previously seen in a study of four mouse strains (A/J, DBA/2J, 129S1/SvImJ, and 129X1/SvJ) (Akagi et al., 2008). TEVs from all three classes show strong and significant depletions in protein-coding gene exons, implying that such insertions are strongly deleterious (assuming that most TEVs across the noncoding genome are neutral or deleterious). The significant deficits of ERV or LINE TEVs in introns indicate that many were deleterious and thus were selectively purged over these strains' evolutionary history. These observations agree with previous findings that LINE TE insertions are less tolerated within gene-rich sequence (Kvikstad and Makova, 2010).
A strong orientation bias is evident for each of the three TE classes (32.6, 41.7, and 41.6% for ERV, LINE, and SINE TEVs, respectively) (Nellaker et al., 2012). The orientation bias for IAP TEVs was recently reported to be 25.9% for a redundant set of 3317 intronic IAPs (Li et al., 2012). The strong biases for ERVs and, to a lesser extent for LINEs, are consistent with these elements being depleted from introns. The large set of TEVs examined in the genome-wide analysis allowed the authors to infer whether the location of a TEV within a gene structure affects the strength by which it is purified from the population. Orientation bias was significantly stronger for ERV TEVs within middle or last introns, and for SINE TEVs within first introns (Nellaker et al., 2012). A recent study of 161 mouse ERV TEVs identified their strongest intronic orientation bias to be in the close vicinity of exon boundaries (Zhang et al., 2011a).
Indeed, using a stringent statistical re-sampling approach to take into account confounding influences of strain and expression divergence, TEVs were found to be twice as likely to reside in a differentially expressed gene as expected by chance (Nellaker et al., 2012). However, when TEVs are considered with other forms of potential co-segregating mutations (SNPs, indels, and other structural variations), only 34 TEVs passed a stringent genome-wide test, and these TEVs contain significantly fewer LINEs than the null expectation that all TEV classes have equal effects (Nellaker et al., 2012). While it has been extensively documented in the literature that de novo LINE insertions can cause changes in gene expression, it appears that, in Mus musculus, purifying selection has preferentially purged such variants. However, given that the proportion of expression heritability attributable to TEVs generally is no more than 10% (Yalcin et al., 2011).
To summarize, transposable elements make up almost half of the mouse genome (Gogvadze and Buzdin, 2009) and importantly their activity is the most prevalent mechanism for generating large structural variations in laboratory inbred mouse strains (Yalcin et al., 2011). However, as we demonstrated in this review, transposable elements appear to be under strong purifying selection for deleterious insertions with the majority of insertions observable in present day mouse strains having little phenotypic effects (Nellaker et al., 2012).
Data Access and Visualization
The entire set of structural variation calls across 18 mouse genomes (129P2/OlaHsd, 129S1/SvImJ, 129S5SvEvBrd, A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6NJ, CAST/EiJ, CBA/J, DBA/2J, FVB/NJ, LP/J, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, SPRET/EiJ, and WSB/EiJ) have been posted on the following ftp site ftp://ftp-mouse.sanger.ac.uk/. Data sets described in this review are also available under accession numbers “estd118” (Yalcin et al., 2011), “estd185” (Yalcin et al., 2012a), “estd200” (Wong et al., 2012), and “estd204” (Simon et al., 2013) from the Database of Genomic Variants Archive (DGVa).
The project website (http://www.sanger.ac.uk/resources/mouse/genomes/) provides tools to automatically search for structural variants by location, gene, strain, type, and functional impact. A workflow of the procedure is explained in Figure 6A. Results can be exported as TSV and CSV format. Specificity and sensitivity of automatic SV calls are described in detail in Yalcin et al. (2011). To access and query the data manually, visualization of alignments (both at base-pair and read-pair levels) can be done using LookSeq (Figure 6B) (Manske and Kwiatkowski, 2009), a Web-based tool to visualize paired end reads NGS data or using the Integrative Genomics Viewer (IGV) (Robinson et al., 2011; Thorvaldsdottir et al., 2013). Structural variants can be visually identified using our comprehensive catalog of paired end mapping (PEM) patterns described in Yalcin et al. (2012a).
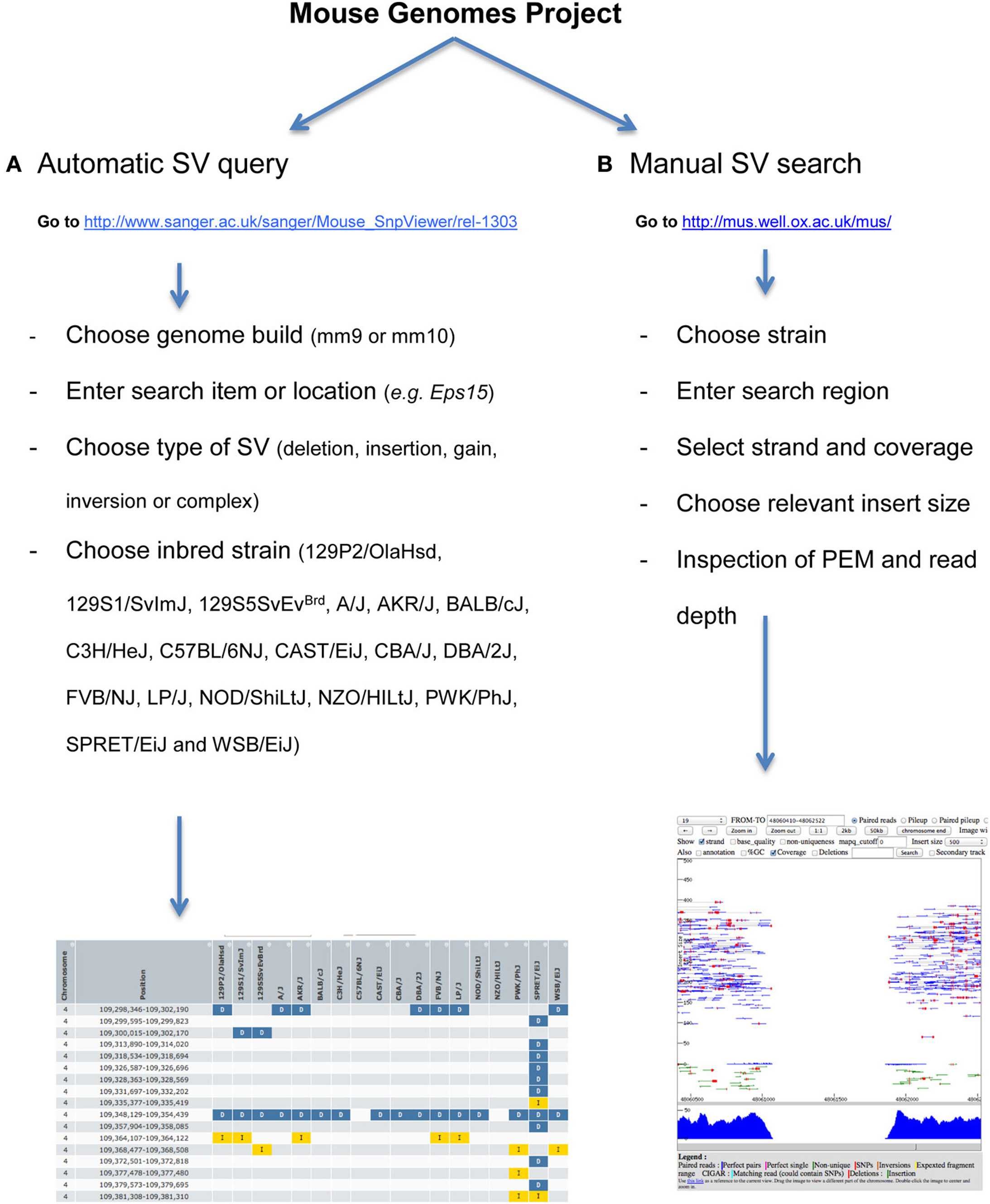
Figure 6. How to access and query the data automatically and manually. (A) Workflow of how to automatically query structural variants. Our work was published relative to mm9 Genome Build, but data can also be visualized directly onto mm10. A gene name or genomic region can be searched for simple and complex structural variants. Results can be exported as TSV and CSV format. (B) Workflow of how to manually search for structural variants. To do this, we use LookSeq (Manske and Kwiatkowski, 2009) as a Web-based tool to visualize paired end reads NGS data. The choice of the insert size depends on the size of the underlying structural variant, so that when the variant is large the insert size should also be large. Types of structural variants can be recognized using our comprehensive catalog of paired end mapping (PEM) patterns described in Yalcin et al. (2012a).
Future Work and Concluding Remarks
The current approaches for cataloging mutations are primarily based on aligning sequencing reads to the appropriate reference genome to identify SNPs, indels, and structural variations. The majority of SV discovery methods to date have been based on observing patterns of clusters of aberrant read mappings to the reference genome. However, for many groups of strains or individuals there are many haplotypes that are not present on the reference genome and therefore are excluded from the catalog of mutations. This is especially true for the wild-derived mouse strains such as SPRET/EiJ, CAST/EiJ, and PWK/PhJ. So while the current approaches can often detect the presence of a non-reference haplotype in the form of a large insertion, they are blind to sequence variation occurring on the haplotype.
One solution to this problem is to create data structures capable of representing all of the haplotypes present in a group of related samples. In a recent study, Iqbal et al. developed de Bruijn graph methods for detecting and genotyping simple and complex genetic variants in an individual or population without a reference genome and were able to discover more than 3 Mb of sequence absent from the human reference genome (Iqbal et al., 2012).
The String Graph Assembler (SGA) was the first sequence assembly pipeline for next-generation data based on sequence overlaps (Simpson and Durbin, 2012). At the heart of SGA is the use of a compressed data structure called the FM-index, which is used to model the read sequence overlap graph of all the samples. Recently, work has been carried out to investigate building these structures using reads from multiple samples to represent all of the haplotypes present in the samples (Simpson, 2012).
An alternative approach is to first create individual whole-genome de novo assemblies for each sample and then subsequently carry out whole-genome alignments of the pre-assembled sequences. Several algorithms have been proposed for creating whole-genome alignments taking into account substitutions, insertions, deletions, and larger structural rearrangements. One such implementation of this approach is the combined Progressive Cactus and Hierarchical Alignment (HAL) graph pipeline (Paten et al., 2011). HAL is a graph-based hierarchical alignment format for storing multiple genome alignments arranged phylogenetically with the corresponding ancestral sequence reconstructions as internal nodes (Hickey et al., 2013).
The Mouse Genomes Project (http://www.sanger.ac.uk/resources/mouse/genomes/) has made a substantial contribution toward our understanding of structural variation diversity in mouse genomes and in their correlation to phenotypic variation. However, as explained in this review, there are ongoing challenges in computational detection of SVs with complex molecular architecture. Improved sequencing technologies with longer read lengths, along with the completion of de novo assemblies of mouse genomes, will be crucial in the identification of the remaining structural variants. De novo assembly also avoids reference bias in ascertainment of SVs (Sousa and Hey, 2013). Using longer fragments in sequencing library construction also aids in de novo assembly and SV detection in genomic regions that are “inaccessible” to short-read mapping due to their repetitive nature.
Author Contributions
All authors read and approved the final manuscript. Thomas M. Keane and Binnaz Yalcin wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Medical Research Council and the Wellcome Trust. Binnaz Yalcin is supported by the French National Research Agency and the Swiss National Science Foundation.
References
Abecasis, G. R., Altshuler, D., Auton, A., Brooks, L. D., Durbin, R. M., Gibbs, R. A., et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. doi: 10.1038/nature09534
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. doi: 10.1101/gr.114876.110
Agam, A., Yalcin, B., Bhomra, A., Cubin, M., Webber, C., Holmes, C., et al. (2010). Elusive copy number variation in the mouse genome. PLoS ONE 5:e12839. doi: 10.1371/journal.pone.0012839
Akagi, K., Li, J., Stephens, R. M., Volfovsky, N., and Symer, D. E. (2008). Extensive variation between inbred mouse strains due to endogenous L1 retrotransposition. Genome Res. 18, 869–880. doi: 10.1101/gr.075770.107
Albers, C. A., Lunter, G., MacArthur, D. G., McVean, G., Ouwehand, W. H., and Durbin, R. (2011). Dindel: accurate indel calls from short-read data. Genome Res. 21, 961–973. doi: 10.1101/gr.112326.110
Alkan, C., Coe, B. P., and Eichler, E. E. (2011). Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376. doi: 10.1038/nrg2958
Alkan, C., Kidd, J. M., Marques-Bonet, T., Aksay, G., Antonacci, F., Hormozdiari, F., et al. (2009). Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41, 1061–1067. doi: 10.1038/ng.437
Bauer, F., Schweimer, K., Kluver, E., Conejo-Garcia, J. R., Forssmann, W. G., Rosch, P., et al. (2001). Structure determination of human and murine beta-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 10, 2470–2479. doi: 10.1110/ps.ps.24401
Beck, J. A., Lloyd, S., Hafezparast, M., Lennon-Pierce, M., Eppig, J. T., Festing, M. F., et al. (2000). Genealogies of mouse inbred strains. Nat. Genet. 24, 23–25. doi: 10.1038/71641
Berger, M. F., Lawrence, M. S., Demichelis, F., Drier, Y., Cibulskis, K., Sivachenko, A. Y., et al. (2011). The genomic complexity of primary human prostate cancer. Nature 470, 214–220. doi: 10.1038/nature09744
Best, S., Le Tissier, P., Towers, G., and Stoye, J. P. (1996). Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 382, 826–829. doi: 10.1038/382826a0
Boeva, V., Zinovyev, A., Bleakley, K., Vert, J. P., Janoueix-Lerosey, I., Delattre, O., et al. (2011). Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics 27, 268–269. doi: 10.1093/bioinformatics/btq635
Boyden, L. M., Lewis, J. M., Barbee, S. D., Bas, A., Girardi, M., Hayday, A. C., et al. (2008). Skint1, the prototype of a newly identified immunoglobulin superfamily gene cluster, positively selects epidermal gammadelta T cells. Nat. Genet. 40, 656–662. doi: 10.1038/ng.108
Boyle, A. L., Ballard, S. G., and Ward, D. C. (1990). Differential distribution of long and short interspersed element sequences in the mouse genome: chromosome karyotyping by fluorescence in situ hybridization. Proc. Natl. Acad. Sci. U.S.A. 87, 7757–7761. doi: 10.1073/pnas.87.19.7757
Buzdin, A. A. (2004). Retroelements and formation of chimeric retrogenes. Cell. Mol. Life Sci. 61, 2046–2059. doi: 10.1007/s00018-004-4041-z
Cahan, P., Li, Y., Izumi, M., and Graubert, T. A. (2009). The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells. Nat. Genet. 41, 430–437. doi: 10.1038/ng.350
Chaignat, E., Yahya-Graison, E. A., Henrichsen, C. N., Chrast, J., Schutz, F., Pradervand, S., et al. (2011). Copy number variation modifies expression time courses. Genome Res. 21, 106–113. doi: 10.1101/gr.112748.110
Chen, K., Wallis, J. W., McLellan, M. D., Larson, D. E., Kalicki, J. M., Pohl, C. S., et al. (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681. doi: 10.1038/nmeth.1363
Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y., et al. (2010). Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712. doi: 10.1038/nature08516
Cordaux, R., and Batzer, M. A. (2009). The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703. doi: 10.1038/nrg2640
Cutler, G., Marshall, L. A., Chin, N., Baribault, H., and Kassner, P. D. (2007). Significant gene content variation characterizes the genomes of inbred mouse strains. Genome Res. 17, 1743–1754. doi: 10.1101/gr.6754607
de Koning, A. P., Gu, W., Castoe, T. A., Batzer, M. A., and Pollock, D. D. (2011). Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7:e1002384. doi: 10.1371/journal.pgen.1002384
Diskin, S. J., Hou, C., Glessner, J. T., Attiyeh, E. F., Laudenslager, M., Bosse, K., et al. (2009). Copy number variation at 1q21.1 associated with neuroblastoma. Nature 459, 987–991. doi: 10.1038/nature08035
Elia, J., Glessner, J. T., Wang, K., Takahashi, N., Shtir, C. J., Hadley, D., et al. (2012). Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat. Genet. 44, 78–84. doi: 10.1038/ng.1013
Emde, A. K., Schulz, M. H., Weese, D., Sun, R., Vingron, M., Kalscheuer, V. M., et al. (2012). Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics 28, 619–627. doi: 10.1093/bioinformatics/bts019
Filipski, J., Thiery, J. P., and Bernardi, G. (1973). An analysis of the bovine genome by Cs2SO4-Ag density gradient centrifugation. J. Mol. Biol. 80, 177–197. doi: 10.1016/0022-2836(73)90240-4
Freeman, H. C., Hugill, A., Dear, N. T., Ashcroft, F. M., and Cox, R. D. (2006). Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 55, 2153–2156. doi: 10.2337/db06-0358
Gheldof, N., Witwicki, R. M., Migliavacca, E., Leleu, M., Didelot, G., Harewood, L., et al. (2013). Structural variation-associated expression changes are paralleled by chromatin architecture modifications. PLoS ONE 8:e79973. doi: 10.1371/journal.pone.0079973
Girirajan, S., Brkanac, Z., Coe, B. P., Baker, C., Vives, L., Vu, T. H., et al. (2011). Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 7:e1002334. doi: 10.1371/journal.pgen.1002334
Gogvadze, E., and Buzdin, A. (2009). Retroelements and their impact on genome evolution and functioning. Cell. Mol. Life Sci. 66, 3727–3742. doi: 10.1007/s00018-009-0107-2
Goodier, J. L., and Kazazian, H. H. Jr. (2008). Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135, 23–35. doi: 10.1016/j.cell.2008.09.022
Graubert, T. A., Cahan, P., Edwin, D., Selzer, R. R., Richmond, T. A., Eis, P. S., et al. (2007). A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS Genet. 3:e3. doi: 10.1371/journal.pgen.0030003
Handsaker, R. E., Korn, J. M., Nemesh, J., and McCarroll, S. A. (2011). Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 43, 269–276. doi: 10.1038/ng.768
Harewood, L., Chaignat, E., and Reymond, A. (2012). Structural variation and its effect on expression. Methods Mol. Biol. 838, 173–186. doi: 10.1007/978-1-61779-507-7_8
Hart, S. N., Sarangi, V., Moore, R., Baheti, S., Bhavsar, J. D., Couch, F. J., et al. (2013). SoftSearch: integration of multiple sequence features to identify breakpoints of structural variations. PLoS ONE 8:e83356. doi: 10.1371/journal.pone.0083356
Helbig, I., Swinkels, M. E., Aten, E., Caliebe, A., van't Slot, R., Boor, R., et al. (2014). Structural genomic variation in childhood epilepsies with complex phenotypes. Eur. J. Hum. Genet. 22, 896–901. doi: 10.1038/ejhg.2013.262
Henrichsen, C. N., Vinckenbosch, N., Zollner, S., Chaignat, E., Pradervand, S., Schutz, F., et al. (2009). Segmental copy number variation shapes tissue transcriptomes. Nat. Genet. 41, 424–429. doi: 10.1038/ng.345
Hickey, G., Paten, B., Earl, D., Zerbino, D., and Haussler, D. (2013). HAL: a hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics 29, 1341–1342. doi: 10.1093/bioinformatics/btt128
Hollox, E. J., Huffmeier, U., Zeeuwen, P. L., Palla, R., Lascorz, J., Rodijk-Olthuis, D., et al. (2008). Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 40, 23–25. doi: 10.1038/ng.2007.48
Hormozdiari, F., Alkan, C., Eichler, E. E., and Sahinalp, S. C. (2009). Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 19, 1270–1278. doi: 10.1101/gr.088633.108
Iqbal, Z., Caccamo, M., Turner, I., Flicek, P., and McVean, G. (2012). De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 44, 226–232. doi: 10.1038/ng.1028
Itsara, A., Wu, H., Smith, J. D., Nickerson, D. A., Romieu, I., London, S. J., et al. (2010). De novo rates and selection of large copy number variation. Genome Res. 20, 1469–1481. doi: 10.1101/gr.107680.110
Ivakhno, S., Royce, T., Cox, A. J., Evers, D. J., Cheetham, R. K., and Tavare, S. (2010). CNAseg–a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics 26, 3051–3058. doi: 10.1093/bioinformatics/btq587
Jacquemont, S., Reymond, A., Zufferey, F., Harewood, L., Walters, R. G., Kutalik, Z., et al. (2011). Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 478, 97–102. doi: 10.1038/nature10406
Jarick, I., Vogel, C. I., Scherag, S., Schafer, H., Hebebrand, J., Hinney, A., et al. (2011). Novel common copy number variation for early onset extreme obesity on chromosome 11q11 identified by a genome-wide analysis. Hum. Mol. Genet. 20, 840–852. doi: 10.1093/hmg/ddq518
Karakoc, E., Alkan, C., O'Roak, B. J., Dennis, M. Y., Vives, L., Mark, K., et al. (2012). Detection of structural variants and indels within exome data. Nat. Methods 9, 176–178. doi: 10.1038/nmeth.1810
Keane, T. M., Goodstadt, L., Danecek, P., White, M. A., Wong, K., Yalcin, B., et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. doi: 10.1038/nature10413
Keane, T. M., Wong, K., and Adams, D. J. (2013). RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics 29, 389–390. doi: 10.1093/bioinformatics/bts697
Klambauer, G., Schwarzbauer, K., Mayr, A., Clevert, D. A., Mitterecker, A., Bodenhofer, U., et al. (2012). cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40, e69. doi: 10.1093/nar/gks003
Koolen, D. A., Kramer, J. M., Neveling, K., Nillesen, W. M., Moore-Barton, H. L., Elmslie, F. V., et al. (2012). Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 44, 639–641. doi: 10.1038/ng.2262
Korbel, J. O., Abyzov, A., Mu, X. J., Carriero, N., Cayting, P., Zhang, Z., et al. (2009). PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 10:R23. doi: 10.1186/gb-2009-10-2-r23
Korbel, J. O., Urban, A. E., Affourtit, J. P., Godwin, B., Grubert, F., Simons, J. F., et al. (2007). Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426. doi: 10.1126/science.1149504
Korenberg, J. R., and Rykowski, M. C. (1988). Human genome organization: alu, lines, and the molecular structure of metaphase chromosome bands. Cell 53, 391–400. doi: 10.1016/0092-8674(88)90159-6
Kvikstad, E. M., and Makova, K. D. (2010). The (r)evolution of SINE versus LINE distributions in primate genomes: sex chromosomes are important. Genome Res. 20, 600–613. doi: 10.1101/gr.099044.109
Lee, S., Hormozdiari, F., Alkan, C., and Brudno, M. (2009). MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nat. Methods 6, 473–474. doi: 10.1038/nmeth.f.256
Li, J., Akagi, K., Hu, Y., Trivett, A. L., Hlynialuk, C. J., Swing, D. A., et al. (2012). Mouse endogenous retroviruses can trigger premature transcriptional termination at a distance. Genome Res. 22, 870–884. doi: 10.1101/gr.130740.111
Lupski, J. R. (1998). Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 14, 417–422. doi: 10.1016/S0168-9525(98)01555-8
Lupski, J. R., de Oca-Luna, R. M., Slaugenhaupt, S., Pentao, L., Guzzetta, V., Trask, B. J., et al. (1991). DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232. doi: 10.1016/0092-8674(91)90613-4
Macaya, G., Thiery, J. P., and Bernardi, G. (1976). An approach to the organization of eukaryotic genomes at a macromolecular level. J. Mol. Biol. 108, 237–254. doi: 10.1016/S0022-2836(76)80105-2
Magi, A., Benelli, M., Yoon, S., Roviello, F., and Torricelli, F. (2011). Detecting common copy number variants in high-throughput sequencing data by using JointSLM algorithm. Nucleic Acids Res. 39, e65. doi: 10.1093/nar/gkr068
Malhotra, A., Lindberg, M., Faust, G. G., Leibowitz, M. L., Clark, R. A., Layer, R. M., et al. (2013). Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res. 23, 762–776. doi: 10.1101/gr.143677.112
Malhotra, D., McCarthy, S., Michaelson, J. J., Vacic, V., Burdick, K. E., Yoon, S., et al. (2011). High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron 72, 951–963. doi: 10.1016/j.neuron.2011.11.007
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Manske, H. M., and Kwiatkowski, D. P. (2009). LookSeq: a browser-based viewer for deep sequencing data. Genome Res. 19, 2125–2132. doi: 10.1101/gr.093443.109
Mardis, E. R. (2011). A decade's perspective on DNA sequencing technology. Nature 470, 198–203. doi: 10.1038/nature09796
McConnell, M. J., Lindberg, M. R., Brennand, K. J., Piper, J. C., Voet, T., Cowing-Zitron, C., et al. (2013). Mosaic copy number variation in human neurons. Science 342, 632–637. doi: 10.1126/science.1243472
Medvedev, P., Fiume, M., Dzamba, M., Smith, T., and Brudno, M. (2010). Detecting copy number variation with mated short reads. Genome Res. 20, 1613–1622. doi: 10.1101/gr.106344.110
Medvedev, P., Stanciu, M., and Brudno, M. (2009). Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods 6, S13–S20. doi: 10.1038/nmeth.1374
Nellaker, C., Keane, T. M., Yalcin, B., Wong, K., Agam, A., Belgard, T. G., et al. (2012). The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 13:R45. doi: 10.1186/gb-2012-13-6-r45
Nelson, T. M., Munger, S. D., and Boughter, J. D. Jr. (2005). Haplotypes at the Tas2r locus on distal chromosome 6 vary with quinine taste sensitivity in inbred mice. BMC Genet. 6:32. doi: 10.1186/1471-2156-6-32
Ni, X., Zhuo, M., Su, Z., Duan, J., Gao, Y., Wang, Z., et al. (2013). Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. U.S.A. 110, 21083–21088. doi: 10.1073/pnas.1320659110
Northcott, P. A., Shih, D. J., Peacock, J., Garzia, L., Morrissy, A. S., Zichner, T., et al. (2012). Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56. doi: 10.1038/nature11327
Paten, B., Earl, D., Nguyen, N., Diekhans, M., Zerbino, D., and Haussler, D. (2011). Cactus: algorithms for genome multiple sequence alignment. Genome Res. 21, 1512–1528. doi: 10.1101/gr.123356.111
Perry, G. H., Dominy, N. J., Claw, K. G., Lee, A. S., Fiegler, H., Redon, R., et al. (2007). Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 39, 1256–1260. doi: 10.1038/ng2123
Persson, K., Heby, O., and Berger, F. G. (1999). The functional intronless S-adenosylmethionine decarboxylase gene of the mouse (Amd-2) is linked to the ornithine decarboxylase gene (Odc) on chromosome 12 and is present in distantly related species of the genus Mus. Mamm. Genome 10, 784–788. doi: 10.1007/s003359901092
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Qi, J., and Zhao, F. (2011). inGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res. 39, W567–W575. doi: 10.1093/nar/gkr506
Quinlan, A. R., Clark, R. A., Sokolova, S., Leibowitz, M. L., Zhang, Y., Hurles, M. E., et al. (2010). Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 20, 623–635. doi: 10.1101/gr.102970.109
Quinlan, A. R., and Hall, I. M. (2012). Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 28, 43–53. doi: 10.1016/j.tig.2011.10.002
Ramos-Quiroga, J. A., Sanchez-Mora, C., Casas, M., Garcia-Martinez, I., Bosch, R., Nogueira, M., et al. (2014). Genome-wide copy number variation analysis in adult attention-deficit and hyperactivity disorder. J. Psychiatr. Res. 49, 60–67. doi: 10.1016/j.jpsychires.2013.10.022
Rausch, T., Jones, D. T., Zapatka, M., Stutz, A. M., Zichner, T., Weischenfeldt, J., et al. (2012a). Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148, 59–71. doi: 10.1016/j.cell.2011.12.013
Rausch, T., Zichner, T., Schlattl, A., Stutz, A. M., Benes, V., and Korbel, J. O. (2012b). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. doi: 10.1093/bioinformatics/bts378
Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Russnes, H. G., Navin, N., Hicks, J., and Borresen-Dale, A. L. (2011). Insight into the heterogeneity of breast cancer through next-generation sequencing. J. Clin. Invest. 121, 3810–3818. doi: 10.1172/JCI57088
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. doi: 10.1126/science.1138659
Shapiro, J. A. (2010). Mobile DNA and evolution in the 21st century. Mob. DNA 1:4. doi: 10.1186/1759-8753-1-4
Simon, M. M., Greenaway, S., White, J. K., Fuchs, H., Gailus-Durner, V., Wells, S., et al. (2013). A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 14:R82. doi: 10.1186/gb-2013-14-7-r82
Simpson, J. T. (2012). Efficient Sequence Assembly and Variant Calling Using Compressed Data Structures. Ph.D thesis, University of Cambridge. Available online at: ftp://ftp.sanger.ac.uk/pub/resources/theses/js18/
Simpson, J. T., and Durbin, R. (2012). Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556. doi: 10.1101/gr.126953.111
Simpson, J. T., McIntyre, R. E., Adams, D. J., and Durbin, R. (2010). Copy number variant detection in inbred strains from short read sequence data. Bioinformatics 26, 565–567. doi: 10.1093/bioinformatics/btp693
Sindi, S. S., Onal, S., Peng, L. C., Wu, H. T., and Raphael, B. J. (2012). An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol. 13:R22. doi: 10.1186/gb-2012-13-3-r22
Sousa, V., and Hey, J. (2013). Understanding the origin of species with genome-scale data: modelling gene flow. Nat. Rev. Genet. 14, 404–414. doi: 10.1038/nrg3446
Stefansson, H., Rujescu, D., Cichon, S., Pietilainen, O. P., Ingason, A., Steinberg, S., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236. doi: 10.1038/nature07229
Stephens, P. J., Greenman, C. D., Fu, B., Yang, F., Bignell, G. R., Mudie, L. J., et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. doi: 10.1016/j.cell.2010.11.055
Stocking, C., and Kozak, C. A. (2008). Murine endogenous retroviruses. Cell. Mol. Life Sci. 65, 3383–3398. doi: 10.1007/s00018-008-8497-0
Stranger, B. E., Forrest, M. S., Dunning, M., Ingle, C. E., Beazley, C., Thorne, N., et al. (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853. doi: 10.1126/science.1136678
Sudmant, P. H., Huddleston, J., Catacchio, C. R., Malig, M., Hillier, L. W., Baker, C., et al. (2013). Evolution and diversity of copy number variation in the great ape lineage. Genome Res. 23, 1373–1382. doi: 10.1101/gr.158543.113
Talbot, C. J., Nicod, A., Cherny, S. S., Fulker, D. W., Collins, A. C., and Flint, J. (1999). High-resolution mapping of quantitative trait loci in outbred mice. Nat. Genet. 21, 305–308. doi: 10.1038/6825
Tareen, S. U., Sawyer, S. L., Malik, H. S., and Emerman, M. (2009). An expanded clade of rodent Trim5 genes. Virology 385, 473–483. doi: 10.1016/j.virol.2008.12.018
Thiery, J. P., Macaya, G., and Bernardi, G. (1976). An analysis of eukaryotic genomes by density gradient centrifugation. J. Mol. Biol. 108, 219–235. doi: 10.1016/S0022-2836(76)80104-0
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform.14, 178–192. doi: 10.1093/bib/bbs017
Valdar, W., Solberg, L. C., Gauguier, D., Burnett, S., Klenerman, P., Cookson, W. O., et al. (2006). Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 38, 879–887. doi: 10.1038/ng1840
van de Lagemaat, L. N., Medstrand, P., and Mager, D. L. (2006). Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol. 7:R86. doi: 10.1186/gb-2006-7-9-r86
Walters, R. G., Jacquemont, S., Valsesia, A., de Smith, A. J., Martinet, D., Andersson, J., et al. (2010). A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 463, 671–675. doi: 10.1038/nature08727
Wang, J., Mullighan, C. G., Easton, J., Roberts, S., Heatley, S. L., Ma, J., et al. (2011). CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 8, 652–654. doi: 10.1038/nmeth.1628
Wong, K., Bumpstead, S., Van Der Weyden, L., Reinholdt, L. G., Wilming, L. G., Adams, D. J., et al. (2012). Sequencing and characterization of the FVB/NJ mouse genome. Genome Biol. 13:R72. doi: 10.1186/gb-2012-13-8-r72
Wong, K., Keane, T. M., Stalker, J., and Adams, D. J. (2010). Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol. 11:R128. doi: 10.1186/gb-2010-11-12-r128
Wu, B., Potter, C. S., Silva, K. A., Liang, Y., Reinholdt, L. G., Alley, L. M., et al. (2010). Mutations in sterol O-acyltransferase 1 (Soat1) result in hair interior defects in AKR/J mice. J. Invest. Dermatol. 130, 2666–2668. doi: 10.1038/jid.2010.168
Xie, C., and Tammi, M. T. (2009). CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10:80. doi: 10.1186/1471-2105-10-80
Yalcin, B., Adams, D. J., Flint, J., and Keane, T. M. (2012b). Next-generation sequencing of experimental mouse strains. Mamm. Genome 23, 490–498. doi: 10.1007/s00335-012-9402-6
Yalcin, B., and Flint, J. (2012). Association studies in outbred mice in a new era of full-genome sequencing. Mamm. Genome 23, 719–726. doi: 10.1007/s00335-012-9409-z
Yalcin, B., Flint, J., and Mott, R. (2005). Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics 171, 673–681. doi: 10.1534/genetics.104.028902
Yalcin, B., Nicod, J., Bhomra, A., Davidson, S., Cleak, J., Farinelli, L., et al. (2010). Commercially available outbred mice for genome-wide association studies. PLoS Genet. 6:e1001085. doi: 10.1371/journal.pgen.1001085
Yalcin, B., Wong, K., Agam, A., Goodson, M., Keane, T. M., Gan, X., et al. (2011). Sequence-based characterization of structural variation in the mouse genome. Nature 477, 326–329. doi: 10.1038/nature10432
Yalcin, B., Wong, K., Bhomra, A., Goodson, M., Keane, T. M., Adams, D. J., et al. (2012a). The fine-scale architecture of structural variants in 17 mouse genomes. Genome Biol. 13:R18. doi: 10.1186/gb-2012-13-3-r18
Ye, K., Schulz, M. H., Long, Q., Apweiler, R., and Ning, Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. doi: 10.1093/bioinformatics/btp394
Yoon, S., Xuan, Z., Makarov, V., Ye, K., and Sebat, J. (2009). Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 19, 1586–1592. doi: 10.1101/gr.092981.109
Zeitouni, B., Boeva, V., Janoueix-Lerosey, I., Loeillet, S., Legoix-ne, P., Nicolas, A., et al. (2010). SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 26, 1895–1896. doi: 10.1093/bioinformatics/btq293
Zhang, J., Wang, J., and Wu, Y. (2012). An improved approach for accurate and efficient calling of structural variations with low-coverage sequence data. BMC Bioinformatics 13(Suppl. 6):S6. doi: 10.1186/1471-2105-13-S6-S6
Zhang, Y., Romanish, M. T., and Mager, D. L. (2011a). Distributions of transposable elements reveal hazardous zones in mammalian introns. PLoS Comput. Biol. 7:e1002046. doi: 10.1371/journal.pcbi.1002046
Keywords: array comparative genome hybridization (aCGH), next-generation sequencing (NGS), structural variation (SV), paired-end mapping (PEM), inbred strains of mice, Heterogeneous Stock (HS), Sanger Mouse Genomes Project
Citation: Keane TM, Wong K, Adams DJ, Flint J, Reymond A and Yalcin B (2014) Identification of structural variation in mouse genomes. Front. Genet. 5:192. doi: 10.3389/fgene.2014.00192
Received: 14 March 2014; Accepted: 12 June 2014;
Published online: 02 July 2014.
Edited by:
Frederic J. J. Chain, McGill University, CanadaReviewed by:
Gabriel Da Rocha Fernandes, Universidade Católica de Brasília, BrazilOmer Gokcumen, University at Buffalo, USA
Hindrik Kerstens, Radboud University Nijmegen, Netherlands
Copyright © 2014 Keane, Wong, Adams, Flint, Reymond and Yalcin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas M. Keane, Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1HH, UK e-mail:dGhvbWFzLmtlYW5lQHNhbmdlci5hYy51aw==;
Binnaz Yalcin, Center for Integrative Genomics, University of Lausanne, Lausanne, CH-1015, Switzerland e-mail:YmlubmF6LnlhbGNpbkB1bmlsLmNo