 Fabio Coppedè
Fabio Coppedè- Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
Available treatments for neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and Huntington's disease, do not arrest disease progression but mainly help keeping patients from getting worse for a limited period of time. Increasing evidence suggests that epigenetic mechanisms such as DNA methylation and histone tail modifications are dynamically regulated in neurons and play a fundamental role in learning and memory processes. In addition, both global and gene-specific epigenetic changes and deregulated expression of the writer and eraser proteins of epigenetic marks are believed to contribute to the onset and progression of neurodegeneration. Studies in animal models of neurodegenerative diseases have highlighted the potential role of epigenetic drugs, including inhibitors of histone deacetylases and methyl donor compounds, in ameliorating the cognitive symptoms and preventing or delaying the motor symptoms of the disease, thereby opening the way for a potential application in human pathology.
Introduction
Epigenetic processes, including DNA methylation and post-translational modifications of the histone tails, influence gene expression levels without involving changes of the primary DNA sequence, and play a fundamental role in embryonic development, cell differentiation, and maintenance of the cellular identity (Martín-Subero, 2011). They are also dynamically regulated in neurons in response to experiential stimuli to regulate the expression of memory-related genes, and play a role in the formation, consolidation and storage of memory (Guo et al., 2011a; Puckett and Lubin, 2011; Sultan and Day, 2011; Lattal and Wood, 2013). Therefore, it is not surprising that changes of epigenetic marks might be at least partially involved in the onset and progression of neurodegenerative diseases (Coppedè, 2014), leading researchers to hypothesize that targeting the epigenome with compounds exerting epigenetic properties could represent a promising preventative and therapeutic approach for neurodegeneration (Karagiannis and Ververis, 2012; Harrison and Dexter, 2013).
DNA Methylation
DNA methylation typically occurs in CpG islands (regions unusually enriched in the CpG dinucleotide, found in the promoter region of almost a half of human genes and relatively absent from most other places in the genome) and consists of the addition of a methyl group (CH3) to the 5′ position of the cytosine pyrimidine ring, forming 5-methylcytosine (5-mC). The reaction is mediated by DNA methyltransferases (DNMTs) using S-adenosylmethionine (SAM) as the methyl donor compound. Methylation of CpG islands in the promoter region of a gene can be specifically recognized by a set of proteins called methyl-CpG binding proteins (MBPs), that contain a transcription repression domain to interact with other proteins and enhance DNA methylation-mediated transcriptional repression (Fournier et al., 2012; Jones, 2012). Enzymes of the TET (ten-eleven translocation) family promote DNA demethylation converting 5-methylcytosine to 5-hydroxymethylcytosine (5-hmc), a modified form of cytosine hydroxymethylated at the 5′ position showing lower affinity for MBPs and present at high levels in the brain (Guo et al., 2011b).
Histone Tail Modifications
The nucleosome is the structural and functional unit of chromatin and consists of approximately 147 DNA base pairs wrapped twice around a histone octamer consisting of two copies each of the core histones H2A, H2B, H3, and H4. Nucleosomes are connected by stretches of “linker DNA” and linker histones, such as histone H1, and are involved in chromatin compaction (Luger et al., 1997). Histone tail modifications include acetylation, methylation, phosphorylation, ubiquitylation, sumoylation and other post-translational modifications (Berger, 2007). The combination of those modifications, and their crosstalk with DNA methylation and chromatin remodeling proteins, regulate the chromatin structure in a dynamic fashion. Indeed, chromatin can exist in a condensate inactive state (heterochromatin) or in a non-condensate and transcriptionally active state (euchromatin) (Berger, 2007).
Acetylation and methylation of the histone tails represent the two most studied mechanisms of histone modifications. Acetylation of lysine residues on histone tails neutralizes their positive charge and decreases their affinity for the DNA, leading to an increased accessibility of transcriptional regulatory proteins to chromatin templates. Histone acetyltransferases (HATs) and deacetylases (HDACs) catalyze lysine acetylation and deacetylation, respectively (Berger, 2007). Eighteen HDAC enzymes are known in mammals which are divided into four classes (I-IV). A summary of their function and localization is provided in Table 1, more detailed information can be found in recent research and review articles (Broide et al., 2007; Zakhary et al., 2010; Joshi et al., 2013; Lazo-Gómez et al., 2013). Methylation on either lysine or arginine residues of the histone tails can be associated with either condensation or relaxation of the chromatin structure, since several sites for methylation are present on each tail thus allowing different combinations (Martin and Zhang, 2005). Crosstalk between acetylation and methylation at specific residues further influences transcriptional activation, for example di- and tri-methylation of H3K4 (H3K4me2 or H3K4me3), and acetylation at H3/H4 (H3K9Ac and H4K9Ac) are generally linked to an open chromatin structure that allows gene transcription, while histone hypoacetylation, and tri-methylation at H3K9 (H3K9me3) and H3K27 (H3K27me3) are considered repressive marks (Berger, 2007). Histone methyltransferases (HMTs) transfer the methyl group from SAM to histone tail residues, and histone demethylases (HDMTs) are the “erasers” of that mark (Martin and Zhang, 2005).
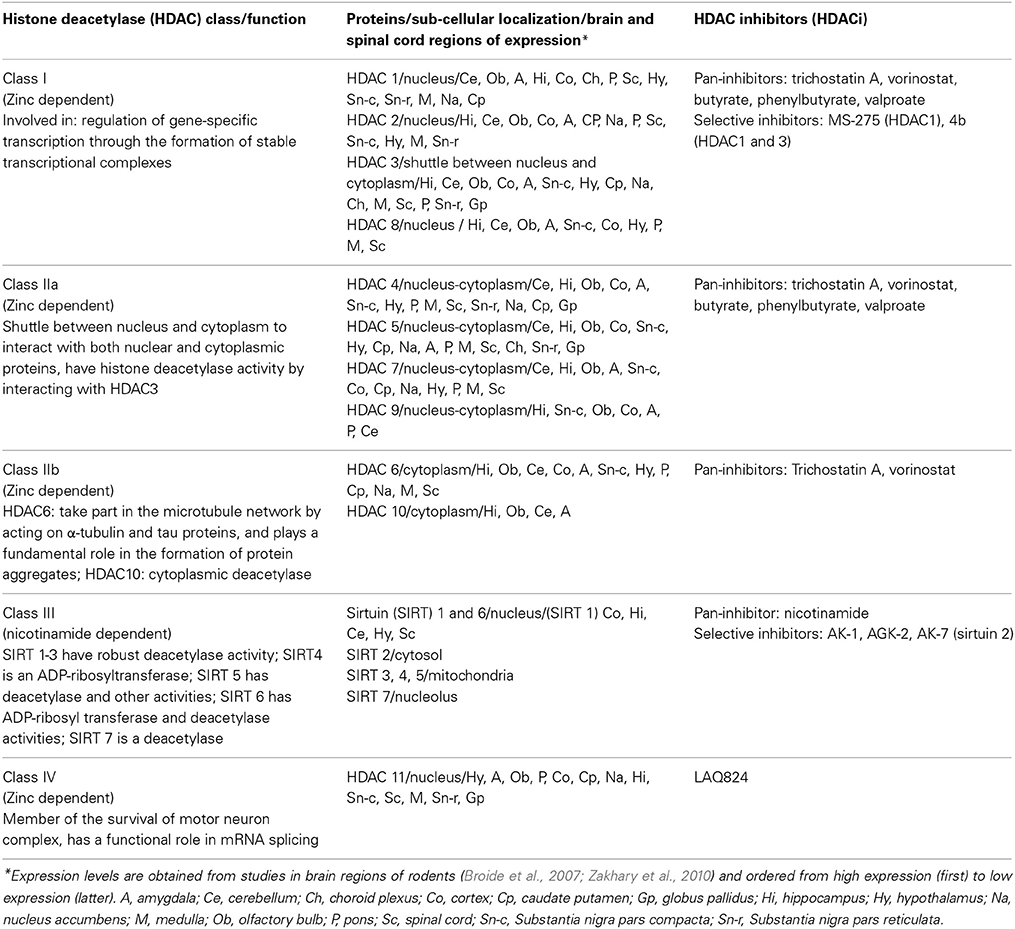
Table 1. Mammalian histone deacetylase (HDAC) proteins and some of their inhibitors (HDACi).
Alzheimer's Disease
Alzheimer's disease (AD) is the most common neurodegenerative disorder and the primary form of dementia in the elderly. The disease symptoms get worse over time and available treatments may only help keeping patients from getting worse for a short period. Affected brain regions show extracellular amyloid deposits called senile plaques (SP) and intra-neuronal aggregates of hyperphosphorylated tau protein, called neurofibrillary tangles (NFT). The primary component of SP is the amyloid β (Aβ) peptide, resulting from the proteolytic cleavage of its precursor protein (APP), mediated by the proteins β-secretase (BACE1) and γ-secretase, a protein complex composed of presenilins and other proteins (Overk and Masliah, 2014). Mutations in APP and presenilin (PSEN1 and PSEN2) genes cause early-onset (<65 years) AD, overall accounting for almost 1% of the cases. However, most of AD (95–98%) are late-onset (>65 years) sporadic forms, resulting from complex interactions among genetic, environmental and stochastic factors superimposed on the physiological decline of cognitive functions with age (Coppedè, 2014). Epigenetic mechanisms are at the core of gene-environment interactions and believed to play a role in AD (Migliore and Coppedè, 2009).
Particularly, DNA methylation changes in AD are supported by studies in cell cultures and animal models (reviewed in Coppedè, 2014), by a number of studies that have shown reduced global levels of 5-mC and 5-hmC in human post-mortem AD brains (Mastroeni et al., 2010; Chouliaras et al., 2013), and by recent epigenome-wide approaches searching for candidate genes (Bakulski et al., 2012; Sanchez-Mut et al., 2014). Also changes in histone tail modifications have been observed in post-mortem AD brains, including decreased levels of H3 acetylation in the temporal lobe (Zhang et al., 2012), and increased levels of the histone deacetylases HDAC6 and HDAC2, the first leading to increased tau phosphorylation and the latter repressing genes required for learning and memory (Ding et al., 2008; Gräff et al., 2012). However, studies in humans are scarce, limited by the small sample size of case and control brains, and often conflicting (Coppedè, 2014; Coppieters et al., 2014). Those performed with rodents and cell cultures revealed the contribution of histone modifications to learning and memory processes, and the manipulation of histone acetylation with HDAC inhibitors (HDACi) in AD animal models often resulted in both prevention of cognitive deficits and memory recovery (Karagiannis and Ververis, 2012).
Epigenetic Interventions in Alzheimer's Disease
Folate, other B group vitamins (B2, B6, and B12), and homocysteine (hcy), participate in one-carbon metabolism, the metabolic pathway required for SAM production, and there is indication of impaired one-carbon metabolism and reduced DNA methylation potential in AD individuals (Coppedè, 2010). Human neuroblastoma cells and transgenic AD mice maintained under conditions of B vitamin deficiency showed PSEN1 promoter demethylation, with subsequent increased production of presenilin1, BACE1 and APP proteins, and Aβ deposition in the animal brains. By contrast, SAM supplementation induced an opposite tendency, restored PSEN1 methylation levels, and reduced the progression of the AD-like features induced by B vitamin deficiency in mice (Fuso et al., 2005, 2008, 2012). There is some indication from elderly human subjects suggesting that B vitamin supplementation can slow the atrophy of specific brain regions that are associated with cognitive decline, but additional trials are warranted to see if the progression to dementia can be prevented (Douaud et al., 2013). In addition, dietary SAM supplementation reduced oxidative stress (Tchantchou et al., 2008) and delayed Aβ and tau pathology in transgenic AD mice (Lee et al., 2012), suggesting a possible role of SAM as a neuroprotective dietary supplement in AD.
Another line of active research involves the manipulation of histone acetylation with HDACi in AD animal models (Karagiannis and Ververis, 2012). Several inhibitors of those enzymes have been developed, including valproic acid, trichostatin A, sodium phenylbutyrate, and vorinostat that interact with zinc-dependent HDAC proteins (class I, class II, and class IV), nicotinamide that inhibits class III HDACs, and more recent compounds that selectively inhibit certain HDACs (Table 1). Early studies in the field revealed that sodium butyrate administration for 4 weeks was able to reinstate learning and memory in transgenic mice that already suffered from severe AD pathology (Fischer et al., 2007). Subsequent studies revealed that sodium butyrate injection in a transgenic mouse model of AD is correlated with decreased tau phosphorylation and restoration of dendritic spine density in hippocampal neurons (Ricobaraza et al., 2010), and prolonged treatment in a transgenic mouse model for amyloid deposition (APP/PS1 mice) improved associative memory by increasing both hippocampal histone acetylation and the expression of genes implicated in associative learning (Govindarajan et al., 2011). Others showed that after fear conditioning training, the levels of hippocampal acetylated histone 4 (H4) in APP/PS1 mice were about 50% lower than in wild-type littermates. However, an acute treatment with trichostatin A prior to training rescued both acetylated H4 levels and contextual freezing performances to wild-type values (Francis et al., 2009). Several similar examples are available in the literature concerning HDACi and memory function in AD animal models, for example 2–3 weeks treatment with either sodium valproate, sodium butyrate, or vorinostat reversed contextual memory deficits in APP/PS1 mice (Kilgore et al., 2010), a 10 days treatment with entinostat, a selective inhibitor of HDAC1, reduced neuroinflammation and amyloid plaque deposition and improved behavioral impairment in APPPS1-21 mice (Zhang and Schluesener, 2013), and a 4 week treatment with a class II inhibitor in transgenic AD mice over-expressing mutant APP, presenilin1 and tau proteins (3 × AD mice), improved memory functions and decreased Aβ and phosphorylated tau levels (Sung et al., 2013). Taken overall, those studies suggest that targeting histone modifications with HDACi can improve cognition and reduce AD-like features in AD models (Table 2).
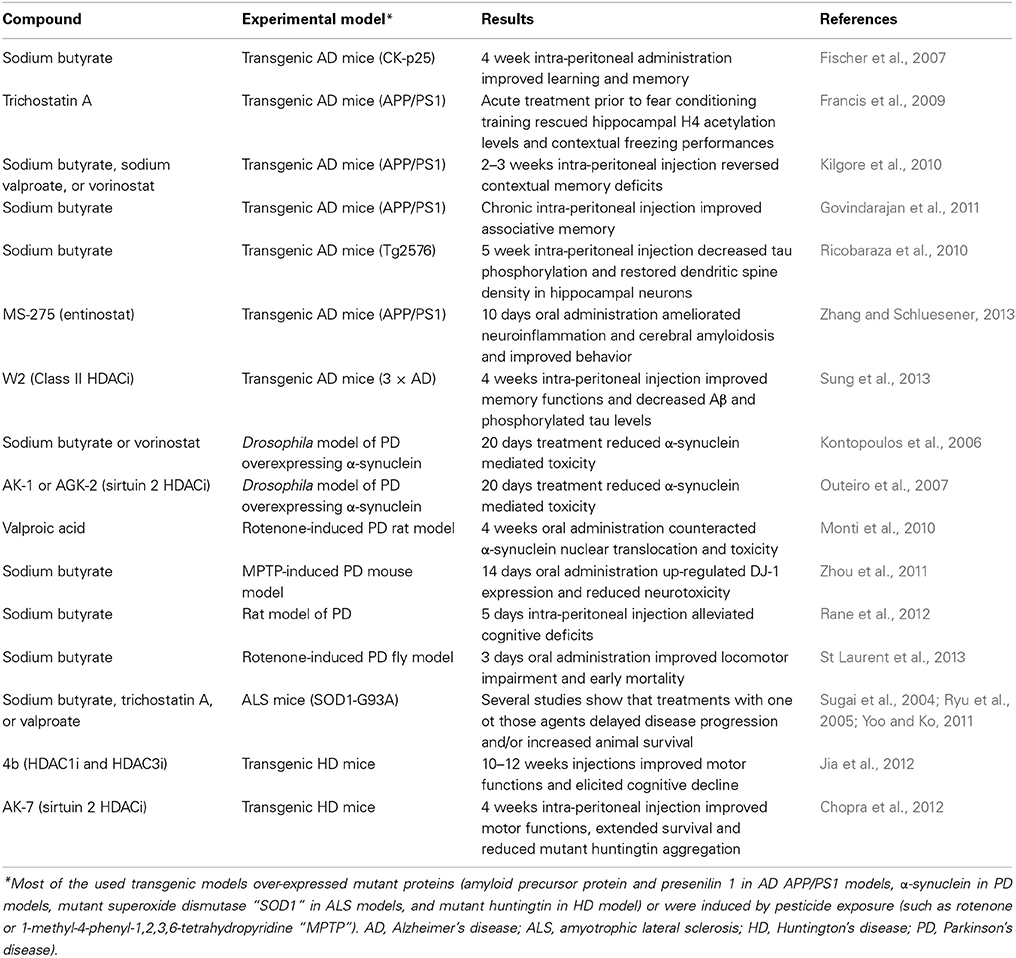
Table 2. Some examples of the effects of histone deacetylase inhibitors (HDACi) in animal models of neurodegenerative diseases.
Parkinson's Disease
Parkinson's disease (PD), the second most common neurodegenerative disorder after AD, is clinically characterized by resting tremor, rigidity, bradykinesia, and postural instability as well as non-motor symptoms such as autonomic insufficiency, cognitive impairment, and sleep disorders. Pathologically, PD is characterized by progressive and profound loss of neuromelanin containing dopaminergic neurons in the substantia nigra with the presence of eosinophilic, intracytoplasmic inclusions called Lewy bodies (LBs; containing aggregates of α-synuclein and other substances), and Lewy neurites in surviving neurons. Although some improvements of the symptoms can be achieved with levodopa and dopaminergic therapy, there is no available treatment to halt disease progression (Thomas and Beal, 2011). The majority of PD cases are sporadic, likely arising from a combination of polygenic inheritance, environmental exposures, and complex gene-environment interactions superimposed on slow and sustained neuronal dysfunction due to aging (Migliore and Coppedè, 2009). In a minority of the cases PD is inherited as a Mendelian trait, and studies in PD families allowed the identification of at least 15 PD loci (PARK1-15) and several causative genes (Nuytemans et al., 2010). Most of the studies evaluating the role of epigenetics in disease pathogenesis have focused on the analysis of promoter methylation of causative PD genes in post-mortem brains and peripheral blood cells of affected individuals (Coppedè, 2012). Particularly, reduced methylation levels of the gene coding for α-synuclein (SNCA gene), the first identified causative PD gene, were observed in several brain regions of sporadic PD patients including the substantia nigra (Jowaed et al., 2010; Matsumoto et al., 2010). In addition, it was shown that α-synuclein sequesters DNMT1 from the nucleus into the cytoplasm, and reduced DNMT1 levels were observed in post-mortem PD brains (Desplats et al., 2011).
Our knowledge of histone modifications in PD derives mainly from studies in cell cultures and animal models of the disease, such as those induced by the mitochondrial toxins 1-methyl-4-phenylpyridinium (MPP+), paraquat and rotenone, or those overexpressing human α-synuclein (Harrison and Dexter, 2013). Overall, those studies revealed that α-synuclein interacts with histones and inhibits histone acetylation, and that several HDACi are neuroprotective against α-synuclein-mediated toxicity (Table 2).
Epigenetic Interventions in Parkinson's Disease
The analysis of nigral neurons of mice exposed to the herbicide paraquat revealed that α-synuclein translocates into the nucleus and binds to histones (Goers et al., 2003). Studies in Drosophila showed that α-synuclein mediates neurotoxicity in the nucleus, binds directly to histone H3, and inhibits histone acetylation. The toxicity of α-synuclein was rescued by the administration of sodium butyrate or vorinostat (Kontopoulos et al., 2006). Selective inhibition of the histone deacetylase sirtuin 2 rescued α-synuclein-mediated toxicity in several models of PD (Outeiro et al., 2007). In addition, valproic acid resulted neuroprotective in a rotenone-induced rat model of PD counteracting α-synuclein translocation into the nuclei (Monti et al., 2010). Neurotoxic pesticides and paraquat were shown to increase histone acetylation in mice brains or cell culture models (Song et al., 2010, 2011), and an increasing number of studies reveal protective effects of HDACi on neurons following a neurotoxic-induced insult (Harrison and Dexter, 2013). For example, it was recently shown that trichostatin A selectively rescues mitochondrial fragmentation and cell death induced by MPP+ in human neuroblastoma cells (Zhu et al., 2014), and that sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of PD (St Laurent et al., 2013), alleviates cognitive deficits in a rat model of PD in the pre-motor deficit stage (Rane et al., 2012), and up-regulates DJ-1 protein expression and protects neurons in cell cultures and mouse models against MPP+ toxicity (Zhou et al., 2011). DJ-1 is involved in the protection of oxidative stress, and mutation of DJ-1 gene cause early-onset PD (Bonifati et al., 2003).
All the above studies suggest that histone modifications occur in α-synuclein-mediated as well as environmentally-induced dopaminergic neuronal cell death and suggest a protective role for HDACi in those models (Table 2).
Other Neurodegenerative Diseases
Several similar examples of the potential neuro-protective effects obtained by targeting the epigenome are available for other neurodegenerative diseases than AD or PD. For example, SAM supplementation in a transgenic mouse model (SOD1-G93A) of amyotrophic lateral sclerosis (ALS) delayed the onset of motor neuron pathology (Suchy et al., 2010). Also HDACi ameliorated disease progression in ALS animal models. For example, sodium phenylbutyrate significantly extended survival in G93A transgenic ALS mice (Ryu et al., 2005). The combination of phenylbutyrate and riluzole ameliorated gross lumbar and ventral horn atrophy, attenuated lumbar ventral horn neuronal cell death, and decreased reactive astrogliosis in ALS G93A mice (Del Signore et al., 2009). Similarly, combined lithium and valproate treatment delayed disease onset, reduced neurological deficits and prolonged survival (Feng et al., 2008), and treatment with trichostatin A (Yoo and Ko, 2011), or valproate (Sugai et al., 2004), delayed disease progression and/or increased survival in the SOD1-G93A mice. A phase II study in ALS individuals revealed that sodium phenylbutyrate was safe and tolerable, and histone acetylation was significantly increased after sodium phenylbutyrate administration (Cudkowicz et al., 2009). Conversely, a trial using valproic acid did not show a beneficial effect on survival or disease progression in patients with ALS (Piepers et al., 2009).
Research in Huntington's disease (HD), a neurodegenerative diseases caused by trinucleotide repeat expansion in the gene (HTT) coding for the huntingtin protein, revealed that mutant huntingtin directly interacts with HAT proteins, leading to altered histone acetylation (Steffan et al., 2000; Jiang et al., 2006). Numerous studies revealed that treatment with HDACi arrested the ongoing progressive neuronal degeneration in both fly and mouse models of HD (reviewed by Gray, 2010). Some recent examples using selective HDACi are listed in Table 2 (Chopra et al., 2012; Jia et al., 2012), and other compounds are under investigation (Bürli et al., 2013; Duan, 2013).
Concluding Remarks
The studies described in this manuscript provided evidence that targeting the epigenome, mainly with small drugs such as HDACi that are able to cross the blood brain barrier, delayed the onset and progression of the symptoms in animal models of neurodegenerative disease (Table 2). Several other examples are available in the literature, but cannot be described in details due to limited space allowance.
Despite the encouraging results obtained so far in animal models, highlighting the potential of HDACi for the treatment of neurodegenerative diseases, additional research is required prior to their application to humans. Indeed, as suggested by many authors, reasons of concern include the multi-targeted and multi-cellular effects exerted by most of those drugs, the possibility of unexpected side effects, and the anatomical and metabolic differences between humans and rodents, suggesting that further investigation is warranted to clarify the most appropriate therapeutical approaches, including the use of pan-HDACi or selective inhibitors, the timing, the dosage regimen, a better understanding of the interplay among histone tail modifications and other mechanisms that regulate gene expression, and the evaluation of potential side effects (Harrison and Dexter, 2013; Fischer, 2014). In addition, the mode of delivery varied among studies, and not all HDACs are similarly expressed in the brain regions affected by AD, PD, or other neurodegenerative diseases (Table 2). Indeed, albeit HDAC2 and HDAC6 could represent promising drug targets in AD, it remains unknown which drug or dosing regime present the most efficacy, and similar conclusions can be drawn for other neurodegenerative diseases (Harrison and Dexter, 2013; Fischer, 2014). Moreover, not all drugs always work. For example, valproic acid treatment in the SOD1 transgenic ALS mice only prevented motor neuron death, but failed to improve survival or motor performance (Rouaux et al., 2007), and similar results were observed in the clinical trial in humans (Piepers et al., 2009). Another limitation to our knowledge is that drugs have been tested in animal models bearing mutations in human genes, such as SOD1, that only account for only about 1–2% of the total human cases (Table 2). Moreover, other writer or eraser proteins of the histone code than HDACs could represent potential targets for drug development, but studies are scarce (Fischer, 2014), and natural compounds found in the diet, including folate, vitamins, polyphenols, and flavonoids, alter the availability of methyl groups and influence the activity of DNMTs, thereby representing potential “epigenetic” preventive factors for neurodegeneration (Coppedè, 2014).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author was supported by the Italian Ministry of Health, GR-2009-1606229 “Folate Metabolism, Epigenetics, and Alzheimer's Disease.”
References
Bakulski, K. M., Dolinoy, D. C., Sartor, M. A., Paulson, H. L., Konen, J. R., Lieberman, A. P., et al. (2012). Genome-wide DNA methylation differences between late-onset Alzheimer's disease and cognitively normal controls in human frontal cortex. J. Alzheimers Dis. 29, 571–588. doi: 10.3233/JAD-2012-111223
Berger, S. L. (2007). The complex language of chromatin regulation during transcription. Nature 447, 407–412. doi: 10.1038/nature05915
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Broide, R. S., Redwine, J. M., Aftahi, N., Young, W., Bloom, F. E., and Winrow, C. J. (2007). Distribution of histone deacetylases 1–11 in the ra tbrain. J. Mol. Neurosci. 31, 47–58. doi: 10.1007/bf02686117
Bürli, R. W., Luckhurst, C. A., Aziz, O., Matthews, K. L., Yates, D., Lyons, K. A., et al. (2013). Design, synthesis, and biological evaluation of potent and selective class IIa histone deacetylase (HDAC) inhibitors as a potential therapy for Huntington's disease. J. Med. Chem. 56, 9934–9954. doi: 10.1021/jm4011884
Chopra, V., Quinti, L., Kim, J., Vollor, L., Narayanan, K. L., Edgerly, C., et al. (2012). The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington's disease mouse models. Cell Rep. 2, 1492–1497. doi: 10.1016/j.celrep.2012.11.001
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R., et al. (2013). Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol. Aging 34, 2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021
Coppedè, F. (2010). One-carbon metabolism and Alzheimer's disease: focus on epigenetics. Curr. Genomics 11, 246–260. doi: 10.2174/138920210791233090
Coppedè, F. (2012). Genetics and epigenetics of Parkinson's disease. ScientificWorldJournal 2012:489830. doi: 10.1100/2012/489830
Coppedè, F. (2014). Advances in the genetics and epigenetics of neurodegenerative diseases. Epigenetics Neurodegener. Dis. 1, 3–31. doi: 10.2478/end-2012-0002
Coppieters, N., Dieriks, B. V., Lill, C., Faull, R. L., Curtis, M. A., and Dragunow, M. (2014). Global changes in DNA methylation and hydroxymethylation in Alzheimer's disease human brain. Neurobiol. Aging 35, 1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031
Cudkowicz, M. E., Andres, P. L., Macdonald, S. A., Bedlack, R. S., Choudry, R., Brown, R. H. Jr., et al. (2009). Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph. Lateral Scler. 10, 99–106. doi: 10.1080/17482960802320487
Del Signore, S. J., Amante, D. J., Kim, J., Stack, E. C., Goodrich, S., Cormier, K., et al. (2009). Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph. Lateral Scler. 10, 85–94. doi: 10.1080/17482960802226148
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 286, 9031–9037. doi: 10.1074/jbc.C110.212589
Ding, H., Dolan, P. J., and Johnson, G. V. (2008). Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119–2130. doi: 10.1111/j.1471-4159.2008.05564.x
Douaud, G., Refsum, H., de Jager, C. A., Jacoby, R., Nichols, T. E., Smith, S. M., et al. (2013). Preventing Alzheimer's disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. U.S.A. 110, 9523–9528. doi: 10.1073/pnas.1301816110
Duan, W. (2013). Targeting sirtuin-1 in Huntington's disease: rationale and current status. CNS Drugs 27, 345–352. doi: 10.1007/s40263-013-0055-0
Feng, H. L., Leng, Y., Ma, C. H., Zhang, J., Ren, M., and Chuang, D. M. (2008). Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 155, 567–572. doi: 10.1016/j.neuroscience.2008.06.040
Fischer, A. (2014). Targeting histone-modifications in Alzheimer's disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 80C, 95–102. doi: 10.1016/j.neuropharm.2014.01.038
Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M., and Tsai, L. H. (2007). Recovery of learning and memory after neuronal loss is associated with chromatin remodeling. Nature 447, 178–182. doi: 10.1038/nature05772
Fournier, A., Sasai, N., Nakao, M., and Defossez, P. A. (2012). The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief Funct. Genomics 11, 251–264. doi: 10.1093/bfgp/elr040
Francis, Y. I., Fà, M., Ashraf, H., Zhang, H., Staniszewski, A., Latchman, D. S., et al. (2009). Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J. Alzheimers Dis. 18, 131–139. doi: 10.3233/JAD-2009-1134
Fuso, A., Nicolia, V., Cavallaro, R. A., Ricceri, L., D'Anselmi, F., Coluccia, P., et al. (2008). B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol. Cell Neurosci. 37, 731–746. doi: 10.1016/j.mcn.2007.12.018
Fuso, A., Nicolia, V., Ricceri, L., Cavallaro, R. A., Isopi, E., Mangia, F., et al. (2012). S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol. Aging 33, 1482.e1–1482.e16. doi: 10.1016/j.neurobiolaging.2011.12.013
Fuso, A., Seminara, L., Cavallaro, R. A., D'Anselmi, F., and Scarpa, S. (2005). S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell Neurosci. 28, 195–204. doi: 10.1016/j.mcn.2004.09.007
Goers, J., Manning-Bog, A. B., McCormack, A. L., Millett, I. S., Doniach, S., Di Monte, D. A., et al. (2003). Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 42, 8465–8471. doi: 10.1021/bi0341152
Govindarajan, N., Agis-Balboa, R. C., Walter, J., Sananbenesi, F., and Fischer, A. (2011). Sodium butyrate improves memory function in an Alzheimer's disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis. 26, 187–197. doi: 10.3233/JAD-2011-110080
Gräff, J., Rei, D., Guan, J. S., Wang, W. Y., Seo, J., Hennig, K. M., et al. (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. doi: 10.1038/nature10849
Gray, S. G. (2010). Targeting histone deacetylases for the treatment of Huntington's disease. CNS Neurosci. Ther. 16, 348–361. doi: 10.1111/j.1755-5949.2010.00184.x
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., et al. (2011a). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14, 1345–1351. doi: 10.1038/nn.2900
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011b). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Harrison, I. F., and Dexter, D. T. (2013). Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson's disease? Pharmacol. Ther. 140, 34–52. doi: 10.1016/j.pharmthera.2013.05.010
Jia, H., Kast, R. J., Steffan, J. S., and Thomas, E. A. (2012). Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington's disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum. Mol. Genet. 21, 5280–5293. doi: 10.1093/hmg/dds379
Jiang, H., Poirier, M. A., Liang, Y., Pei, Z., Weiskittel, C. E., Smith, W. W., et al. (2006). Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 23, 543–551. doi: 10.1016/j.nbd.2006.04.011
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi: 10.1038/nrg3230
Joshi, P., Greco, T. M., Guise, A. J., Luo, Y., Yu, F., Nesvizhskii, A. I., et al. (2013). The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. doi: 10.1038/msb.2013.26
Jowaed, A., Schmitt, I., Kaut, O., and Wüllner, U. (2010). Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains. J. Neurosci. 30, 6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010
Karagiannis, T. C., and Ververis, K. (2012). Potential of chromatin modifying compounds for the treatment of Alzheimer's disease. Pathobiol. Aging Age Relat. Dis. 2:14980. doi: 10.3402/pba.v2i0.14980
Kilgore, M., Miller, C. A., Fass, D. M., Hennig, K. M., Haggarty, S. J., Sweatt, J. D., et al. (2010). Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology 35, 870–880. doi: 10.1038/npp.2009.197
Kontopoulos, E., Parvin, J. D., and Feany, M. B. (2006). Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. doi: 10.1093/hmg/ddl243
Lattal, K. W., and Wood, M. A. (2013). Epigenetics and persistent memory: implications for reconsolidation and silent extinction beyond the zero. Nat. Neurosci. 16, 124–129. doi: 10.1038/nn.3302
Lazo-Gómez, R., Ramírez-Jarquín, U. N., Tovar-y-Romo, L. B., and Tapia, R. (2013). Histone deacetylases and their role in motor neuron degeneration. Front. Cell. Neurosci. 7:243. doi: 10.3389/fncel.2013.00243
Lee, S., Lemere, C. A., Frost, J. L., and Shea, T. B. (2012). Dietary supplementation with S-adenosyl methionine delayed amyloid-β and tau pathology in 3xTg-AD mice. J. Alzheimers Dis. 28, 423–431. doi: 10.3233/JAD-2011-111025
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F., and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260.
Martin, C., and Zhang, Y. (2005). The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849. doi: 10.1038/nrm1761
Martín-Subero, J. I. (2011). How epigenomics brings phenotype into being. Pediatr. Endocrinol. Rev. S1, 506–510.
Mastroeni, D., Grover, A., Delvaux, E., Whiteside, C., Coleman, P. D., and Rogers, J. (2010). Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol. Aging 31, 2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005
Matsumoto, L., Takuma, H., Tamaoka, A., Kurisaki, H., Date, H., Tsuji, S., et al. (2010). CpG demethylation enhances alphasynuclein expression and affects the pathogenesis of Parkinson's disease. PLoS ONE 5:e15522. doi: 10.1371/journal.pone.0015522
Migliore, L., and Coppedè, F. (2009). Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 667, 82–97. doi: 10.1016/j.mrfmmm.2008.10.011
Monti, B., Gatta, V., Piretti, F., Raffaelli, S. S., Virgili, M., and Contestabile, A. (2010). Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of α-synuclein. Neurotox. Res. 17, 130–141. doi: 10.1007/s12640-009-9090-5
Nuytemans, K., Theuns, J., Cruts, M., and Van Broeckhoven, C. (2010). Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum. Mutat. 31, 763–780. doi: 10.1002/humu.21277
Outeiro, T. F., Kontopoulos, E., Altmann, S. M., Kufareva, I., Strathearn, K. E., Amore, A. M., et al. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science 317, 516–519. doi: 10.1126/science.1143780
Overk, C. R., and Masliah, E. (2014). Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem. Pharmacol. 88, 508–516. doi: 10.1016/j.bcp.2014.01.015
Piepers, S., Veldink, J. H., de Jong, S. W., van der Tweel, I., van der Pol, W. L., Uijtendaal, E. V., et al. (2009). Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann. Neurol. 66, 227–234. doi: 10.1002/ana.21620
Puckett, R. E., and Lubin, F. D. (2011). Epigenetic mechanisms in experience-driven memory formation and behaviour. Epigenomics 3, 649–664. doi: 10.2217/epi.11.86
Rane, P., Shields, J., Heffernan, M., Guo, Y., Akbarian, S., and King, J. A. (2012). The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 62, 2409–2412. doi: 10.1016/j.neuropharm.2012.01.026
Ricobaraza, A., Cuadrado-Tejedor, M., Marco, S., Pérez-Otaño, I., and García-Osta, A. (2010). Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22, 1040–1050. doi: 10.1002/hipo.20883
Rouaux, C., Panteleeva, I., René, F., Gonzalez de Aguilar, J. L., Echaniz-Laguna, A., Dupuis, L., et al. (2007). Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 27, 5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007
Ryu, H., Smith, K., Camelo, S. I., Carreras, I., Lee, J., Iglesias, A. H., et al. (2005). Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93, 1087–1098.
Sanchez-Mut, J. V., Aso, E., Panayotis, N., Lott, I., Dierssen, M., Rabano, A., et al. (2014). DNA methylation map of mouse and human brain identifies target genes in Alzheimer's disease. Brain 136, 3018–3027. doi: 10.1093/brain/awt237
Song, C., Kanthasamy, A., Anantharam, V., Sun, F., and Kanthasamy, A. G. (2010). Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 77, 621–632. doi: 10.1124/mol.109.062174
Song, C., Kanthasamy, A., Jin, H., Anantharam, V., and Kanthasamy, A. G. (2011). Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 32, 586–595. doi: 10.1016/j.neuro.2011.05.018
Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., et al. (2000). The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U.S.A. 97, 6763–6768. doi: 10.1073/pnas.100110097
St Laurent, R., O'Brien, L. M., and Ahmad, S. T. (2013). Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson's disease. Neuroscience 246, 382–390. doi: 10.1016/j.neuroscience.2013.04.037
Suchy, J., Lee, S., Ahmed, A., and Shea, T. B. (2010). Dietary supplementation with S-adenosyl methionine delays the onset of motor neuron pathology in a murine model of amyotrophic lateral sclerosis. Neuromolecular Med. 12, 86–97. doi: 10.1007/s12017-009-8089-7
Sugai, F., Yamamoto, Y., Miyaguchi, K., Zhou, Z., Sumi, H., Hamasaki, T., et al. (2004). Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur. J. Neurosci. 20, 3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x
Sultan, F. A., and Day, J. J. (2011). Epigenetic mechanisms in memory and synaptic function. Epigenomics 3, 157–181. doi: 10.2217/epi.11.6
Sung, Y. M., Lee, T., Yoon, H., DiBattista, A. M., Song, J., Sohn, Y., et al. (2013). Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer's disease. Exp. Neurol. 239, 192–201. doi: 10.1016/j.expneurol.2012.10.005
Tchantchou, F., Graves, M., Falcone, D., and Shea, T. B. (2008). S-adenosylmethionine mediates glutathione efficacy by increasing glutathione S-transferase activity: implications for S-adenosyl methionine as a neuroprotective dietary supplement. J. Alzheimers Dis. 14, 323–328.
Thomas, B., and Beal, M. F. (2011). Molecular insights into Parkinson's disease. F1000 Med. Rep. 3, 7. doi: 10.3410/M3-7
Yoo, Y. E., and Ko, C. P. (2011). Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 231, 147–159. doi: 10.1016/j.expneurol.2011.06.003
Zakhary, S. M., Ayubcha, D., Dileo, J. N., Jose, R., Leheste, J. R., Horowitz, J. M., et al. (2010). Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. (Hoboken) 293, 1024–1032. doi: 10.1002/ar.21116
Zhang, K., Schrag, M., Crofton, A., Trivedi, R., Vinters, H., and Kirsch, W. (2012). Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics 12, 1261–1268. doi: 10.1002/pmic.201200010
Zhang, Z., and Schluesener, Y. H. J. (2013). Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J. Neuropathol. Exp. Neurol. 72, 178–185. doi: 10.1097/NEN.0b013e318283114a
Zhou, W., Bercury, K., Cummiskey, J., Luong, N., Lebin, J., and Freed, C. R. (2011). Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 286, 14941–14951. doi: 10.1074/jbc.M110.211029
Keywords: Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease, DNA methylation, histone tail modifications, histone deacetylase inhibitors (HDACi), therapeutic approaches
Citation: Coppedè F (2014) The potential of epigenetic therapies in neurodegenerative diseases. Front. Genet. 5:220. doi: 10.3389/fgene.2014.00220
Received: 29 April 2014; Paper pending published: 23 May 2014;
Accepted: 25 June 2014; Published online: 14 July 2014.
Edited by:
Anne-Marie Baird, Queensland University of Technology, AustraliaReviewed by:
Chris Anthony Murgatroyd, Manchester Metropolitan University, UKYves Renaudineau, Brest University Medical School, France
Copyright © 2014 Coppedè. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Coppedè, Department of Translational Research and New Technologies in Medicine and Surgery, Medical School, University of Pisa, Via Roma 55, 56126 Pisa, Italy e-mail:ZmFiaW8uY29wcGVkZUBtZWQudW5pcGkuaXQ=