 Steven M. Colvin
Steven M. Colvin Kenneth Y. Kwan
Kenneth Y. Kwan- Department of Human Genetics – The Molecular and Behavioral Neuroscience Institute, University of Michigan Medical School, Ann Arbor, MI, USA
A mechanistic understanding of the pathophysiology underpinning psychiatric disorders is essential for the development of targeted molecular therapies. For fragile X syndrome (FXS), recent mechanistic studies have been focused on the metabotropic glutamate receptor (mGluR) signaling pathway. This line of research has led to the discovery of promising candidate drugs currently undergoing various phases of clinical trial, and represents a model of how biological insights can inform therapeutic strategies in neurodevelopmental disorders. Although mGluR signaling is a key mechanism at which targeted treatments can be directed, it is likely to be one of many mechanisms contributing to FXS. A more complete understanding of the molecular and neural underpinnings of the disorder is expected to inform additional therapeutic strategies. Alterations in the assembly of neural circuits in the neocortex have been recently implicated in genetic studies of autism and schizophrenia, and may also contribute to FXS. In this review, we explore dysregulated nitric oxide signaling in the developing neocortex as a novel candidate mechanism of FXS. This possibility stems from our previous work demonstrating that neuronal nitric oxide synthase 1 (NOS1 or nNOS) is regulated by the FXS protein FMRP in the mid-fetal human neocortex. Remarkably, in the mid-late fetal and early postnatal neocortex of human FXS patients, NOS1 expression is severely diminished. Given the role of nitric oxide in diverse neural processes, including synaptic development and plasticity, the loss of NOS1 in FXS may contribute to the etiology of the disorder. Here, we outline the genetic and neurobiological data that implicate neocortical dysfunction in FXS, review the evidence supporting dysregulated nitric oxide signaling in the developing FXS neocortex and its contribution to the disorder, and discuss the implications for targeting nitric oxide signaling in the treatment of FXS and other psychiatric illnesses.
Introduction
Although a single locus genetic disorder, fragile X syndrome (FXS), the leading inherited cause of intellectual disability (ID) and monogenic syndromic form of autism spectrum disorder (ASD), remains incompletely understood at the mechanistic level. Since the identification of FMR1 as the X-linked gene affected in FXS (Warren et al., 1987; Oberlé et al., 1991; Verkerk et al., 1991; Vincent et al., 1991), significant research efforts have focused on FMRP, the protein encoded by FMR1 (Ashley et al., 1993). FMRP is an RNA-binding protein that controls the localization, stability, and translation of neuronal mRNAs (Bagni and Greenough, 2005; Zalfa et al., 2007; Bassell and Warren, 2008). It is highly expressed in neurons and nearly ubiquitously present in the central nervous system (Devys et al., 1993; Hinds et al., 1993; Bakker et al., 2000; O’Donnell and Warren, 2002). Because FMRP is expressed in the brain without preference for particular regions or neuronal subtypes, its expression pattern does not provide insights into the potential neural or neuronal substrates underlying the intellectual and behavioral deficits of the disorder. At the molecular level, FMRP interacts with and controls the translation of a large number of neuronal mRNAs (Brown et al., 2001; Darnell et al., 2011), estimated to be more than 800 transcripts. Consistent with the significant localization of FMRP to dendritic spines (Feng et al., 1997; Antar et al., 2004; Gabel et al., 2004), which are specializations at post-synaptic sites, many of these transcripts play key roles in synapse formation and function (Brown et al., 2001; Darnell et al., 2001, 2011; Miyashiro et al., 2003; Kindler and Kreienkamp, 2012). The converging localization and function of many FMRP target mRNAs to the synapses has illuminated FMRP neurobiology; however, precisely which of the myriad transcripts are mechanistically most significant for the psychopathology of FXS, or most suitable as targets of molecular therapeutics, remains to be fully explored.
One potential mechanism, coined the metabotropic glutamate receptor (mGluR) theory of FXS, was proposed following observations linking mGluR signaling, long-term depression (LTD), protein synthesis, and FMRP function (Weiler et al., 1997; Huber et al., 2000, 2001, 2002) and has been a focus of recent basic and clinical research (Bear et al., 2004; Dölen et al., 2007; Ronesi and Huber, 2008; Berry-Kravis, 2014). At the time of writing, drugs targeting several molecular players in the mGluR signaling pathway are being studied in clinical trials (Berry-Kravis, 2014; Erickson et al., 2014; Jacquemont et al., 2014; Pop et al., 2014).
Since its original proposal, several lines of converging evidence have corroborated the mGluR theory. The majority of the data supporting excessive LTD in FXS is derived from studies of the hippocampus (Huber et al., 2002; Bear et al., 2004), a critical brain region for learning and memory. The specific contribution of overactive hippocampal LTD to the pathophysiology of FXS, however, remains incompletely explored. Furthermore, the precise identities of the de novo proteins necessary for LTD have remained largely mysterious, and they are likely to represent only a subset of FMRP targets. Therefore, it is possible that additional mechanisms or brain regions are involved in the intellectual and behavioral manifestations of FXS. Elucidating these mechanisms is expected to uncover the neurobiology pertinent to FXS and provide novel targets toward molecular therapy.
In this review, we explore disrupted nitric oxide (NO) signaling as a new candidate mechanism of FXS centered in the developing neocortex. In our recent work, we showed neuronal nitric oxide synthase 1 (NOS1 or nNOS) to be translationally regulated by FMRP in the fetal human neocortex and severely reduced in the fetal and post-natal developing neocortex of FXS patients (Kwan et al., 2012a). As NO is a multifunctional signaling molecule with diverse neural functions, including synapse development, neural transmission, and synaptic plasticity, the disruption of NOS1 protein translation in FXS may underlie the pathogenesis of the disorder. Here, we (1) outline the genetic and neurobiological data that implicate neocortical dysfunction in the etiology of FXS; (2) review the evidence supporting dysregulated NO signaling in the developing FXS neocortex and its potential functional contribution to the disorder; and (3) discuss the implications for targeting NO signaling in the treatment of FXS and other psychiatric disorders, including autism and schizophrenia (SCZ).
Neocortical Dysfunction as a Neural Basis of FXS
Research into the mechanisms underlying FXS has long been focused on the biology of FMRP, a key aspect of which is centered on the identities of its mRNA targets (Brown et al., 2001; Darnell et al., 2011). Although the number of direct FMRP-interacting mRNAs is numerous and their individual contributions to the disorder remain largely unknown, some neurobiological aspects behind their function and dysfunction have emerged. FMRP regulates the translation of many transcripts encoded by genes implicated in ASD (Darnell et al., 2011), and de novo mutations in ASD simplex families preferentially affect genes encoding FMRP targets (Iossifov et al., 2012). This convergence suggests that the pathophysiologies of ASD and FXS may be mediated by common mechanisms, and is consistent with the broad behavioral overlap between FXS and autism (Rogers et al., 2001; Hagerman et al., 2010). Similarly, genes disrupted by de novo variants in SCZ are also significantly enriched for those that encode FMRP target transcripts (Fromer et al., 2014; Purcell et al., 2014). Although a potential link between FXS and SCZ has not been extensively explored, decreased FMRP levels have been associated with intellectual deficits and earlier onset in SCZ (Kovács et al., 2013). Furthermore, recent findings have shown that the genetic underpinnings of ASD and SCZ exhibit significant overlap (Carroll and Owen, 2009; McCarthy et al., 2014). This putative mechanistic intersection between FXS, ASD, and SCZ implicates a partial convergence in the molecular and neural substrates underlying these disorders.
Because FMRP is expressed in most neuronal subtypes from early fetal development to aging (Abitbol et al., 1993; Devys et al., 1993; Hinds et al., 1993; Kang et al., 2011), its spatiotemporal expression pattern does not implicate any particular neural structure or developmental stage to be especially important to the etiology of the disorder. Recent advances in the fields of ASD and SCZ genetics have, however, begun to reveal the neurobiology and neural substrates that underlie complex disorders of brain development. Historically, the genetic heterogeneity of ASD, SCZ, and other psychiatric diseases has been a major challenge in identifying the genetic loci that carry definitive genetic risk. Ironically, the large number of risk loci that has impeded progress may now provide an opportunity to better pinpoint the neural substrates and developmental stages that are central to the pathophysiology of these disorders (State and Šestan, 2012). Because of the pleiotropic nature of gene function, the majority of genes in mammalian genomes are expressed in multiple organs over multiple time points during the development, maturation, and aging of the organism. Accordingly, expression analyses of ASD or SCZ risk genes using comprehensive spatiotemporal transcriptomic data from the human brain (Johnson et al., 2009; Colantuoni et al., 2011; Kang et al., 2011), have revealed highly disparate spatial patterns and developmental trajectories. However, gene co-expression network analyses, which can be used to identify clusters of highly correlated genes (Eisen et al., 1998; Stuart et al., 2003; Geschwind and Konopka, 2009), have uncovered key points of convergence in the timing and location of risk gene expression. Expression network analyses of high-confidence ASD loci have identified the mid-fetal neocortex to be a significant point of spatiotemporal intersection (Parikshak et al., 2013; Willsey et al., 2013), suggesting a significant contribution of the developing neocortex to ASD circuit dysfunction (Kwan, 2013). In SCZ, a similar analysis has also implicated the fetal frontal neocortex (Gulsuner et al., 2013). Mid-gestation is a critical developmental period during which key neocortical axon tracts are being established prior to the onset of significant synaptogenesis (Figure 1; Kang et al., 2011). This convergence of ASD and SCZ gene expression, therefore, strongly implicates miswiring of neocortical circuits in the pathophysiology of these neurodevelopmental disorders.
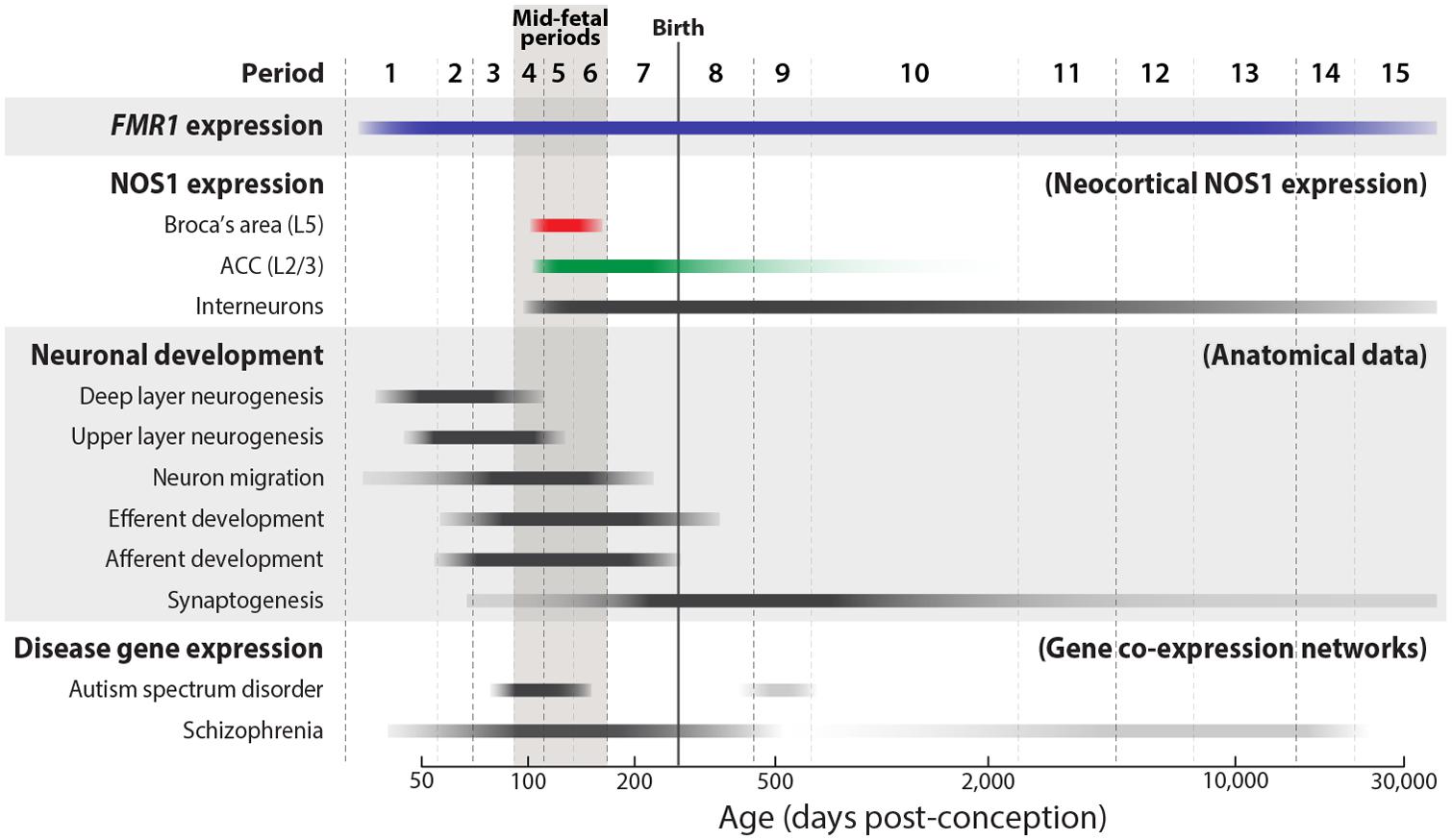
FIGURE 1. Trajectories of NOS1 and FMR1 expression and key developmental events in the mid-fetal human neocortex. The approximate timing and duration of selected neocortical developmental processes, as defined by anatomical findings, are illustrated (Bourgeois, 1997; de Graaf-Peters and Hadders-Algra, 2006; Clancy et al., 2007; Workman et al., 2013). FMR1 (blue) is widely expressed in the brain from early fetal development into adult aging. During the mid-fetal ages, periods 4–6 (Kang et al., 2011), NOS1 is expressed in the pyramidal neurons of Broca’s area L5 (red) and ACC L2/L3 (green) (Kwan et al., 2012a). Unlike its transient pyramidal expression, NOS1 is expressed in interneurons from fetal development to late adulthood. Supporting a potential role in neuropsychiatric disorders, mid-fetal neocortical development has been recently implicated in analyses of gene co-expression networks of ASD- and SCZ-associated genes (Gulsuner et al., 2013; Parikshak et al., 2013; Willsey et al., 2013).
Neocortical dysfunction may similarly contribute to FXS etiology. The neocortex underlies conscious experience, thoughts, and actions in mammals, and is the seat of higher cognitive functions including speech and language and social cognition (Geschwind and Rakic, 2013), which are often affected in FXS and other neurodevelopmental disorders. The proper functioning of the neocortex depends on the precise wiring of its neural circuits during fetal and early post-natal development. Dysregulation of neocortical circuit assembly and function has long been thought to contribute to neuropsychiatric disorders. In FXS, cognitive and language development are delayed or impaired (Abbeduto et al., 2007), phenotypes that are consistent with disrupted neocortical function. With 30% of FXS patients meeting the full diagnostic criteria for autism and an additional 30% falling elsewhere on the autism spectrum (Harris et al., 2008), deficits in social cognition, another higher function centered in the neocortex, are also a key impairment in FXS. Anatomically, this possibility is further supported by findings that revealed altered dendritic spine morphologies in the neocortical pyramidal neurons of human FXS patients and Fmr1 null mice (Comery et al., 1997; Irwin et al., 2000).
With recent advances in neuroimaging, deficits in neural connectivity are being mapped and alterations in neocortical circuitry have been reported in multiple disorders, including ASD, SCZ, and bipolar disorder (Kuperberg et al., 2008; Henin et al., 2009; Henseler et al., 2010; Hall et al., 2013; Zikopoulos and Barbas, 2013). Functional and anatomical imaging studies have also supported neocortical dysfunction in FXS. One analysis found that, during the counting Stroop interference task, normal activation in the inferior/middle frontal gyrus and inferior/superior parietal lobe was absent, and an aberrant activation in the anterior region of the prefrontal cortex (PFC) was present instead (Tamm et al., 2002). In the Go/NoGo task, FXS patients exhibited abnormal activation patterns in multiple brain regions, including significantly reduced activation in the supplementary motor area and anterior cingulate cortex (ACC; Menon et al., 2004). Furthermore, white-matter abnormalities were found in frontostriatal circuitry (Haas et al., 2009), and a recent analysis of functional connectivity and large-scale brain networks in FXS revealed widespread reductions in neocortical connectivity (Hall et al., 2013). Interestingly, maturation of the PFC was found to be aberrant in adolescent FXS patients, an alteration that is correlated with delayed cognitive maturation (Bray et al., 2011). The PFC, often considered to be the prototypical center of higher-order cognitive processing, is critical to working memory, executive function, and inhibition, and has been robustly implicated in multiple psychiatric disorders (Weinberger et al., 1986; Goldman-Rakic, 1996; Miller et al., 2002). Together, these lines of evidence converge on the possibility that alterations in neocortical circuitry underlie some aspects of FXS pathogenesis. The molecular mechanisms that may contribute to these alterations in FXS, however, have not been extensively explored.
FMRP Regulation of NOS1 Translation in the Human Fetal Neocortex
Recently, we identified neuronal NOS1 or nNOS to be a novel FMRP target mRNA in the mid-fetal human neocortex (Kwan et al., 2012a). This interaction was mediated by two G-quartet motifs within the coding region of human NOS1 that are necessary and sufficient for FMRP binding. Consistent with a direct interaction, FMRP was found to be a positive regulator of NOS1 translation, controlling NOS1 protein levels in a dose-dependent manner in vitro and in vivo. FMRP is mostly known to be a translation repressor through a ribosome stalling mechanism that was elegantly shown using an in vitro system (Darnell et al., 2011). In some cases, it has been shown to function as a positive translational regulator (e.g., SOD1, ASCL1 (hASH1), Kcnd2 (Kv4.2), and DLG4 (PSD95)) (Todd et al., 2003; Bechara et al., 2009; Fähling et al., 2009; Gross et al., 2011), and may mediate this translational control in a context- or activity-dependent manner.
Interestingly, FMRP binding and regulation of NOS1 is species-dependent, being present in primate species, which exhibit conserved G-quartet sequences in NOS1, but absent in rodent species, which lack the NOS1 G-quartets. Consistent with this species difference, NOS1 has not been previously identified in the mouse brain to be an FMRP target mRNA (Brown et al., 2001; Darnell et al., 2011). The absence of the G-quartet motifs from rodent Nos1 and the high degree of amino acid identity in FMRP among mammals suggest that the interaction between FMRP and NOS1 evolved via changes in the NOS1 sequence, as opposed to changes in FMRP function. This suggests the possibility of additional examples of evolutionary changes that can impact FMRP interaction with target mRNAs. Indeed, consistent with this possibility, a recent analysis of brain-expressed exons under purifying selection demonstrated enrichment for ASD-associated genes and ASD-relevant FMRP targets (Uddin et al., 2014).
The dose-dependence upregulation of NOS1 translation by FMRP in the normal human neocortex suggests the possibility of disrupted NOS1 protein synthesis in human FXS patients. Examination of FXS patient brains revealed, remarkably, that NOS1 is absent from the neocortex at fetal ages, and significantly decreased in the neocortex during postnatal development (Figure 2). Although a large number of FMRP target mRNAs have been identified in the mouse brain (Brown et al., 2001; Darnell et al., 2011), the majority of these transcripts have not been assessed for potential translational dysregulation in FXS patient materials. Therefore, NOS1 represents the first mRNA that is found to interact with FMRP in the fetal human brain and translationally disrupted in the developing FXS patient brain. NOS1 is an enzyme that mediates the tightly-regulated biosynthesis of NO, a multifunctional gaseous signaling molecule (Bredt and Snyder, 1994; Garthwaite, 2008). Given the key role of NO in diverse neural processes including synaptic plasticity, retrograde signaling, development, excitotoxicity, and mental health (Brenman et al., 1996; Hölscher, 1997; Contestabile, 2000; Calabrese et al., 2007; Garthwaite, 2008; Steinert et al., 2010), the loss of NOS1 in the developing FXS neocortex may contribute to the pathophysiology of the disorder. Here, we assess disrupted NO signaling as a candidate mechanism underlying FXS, focusing on the highly specific spatiotemporal expression pattern of NOS1 in the fetal human neocortex and the potential neurobiological contribution of disrupted NO signaling to FXS and other neurodevelopmental disorders.
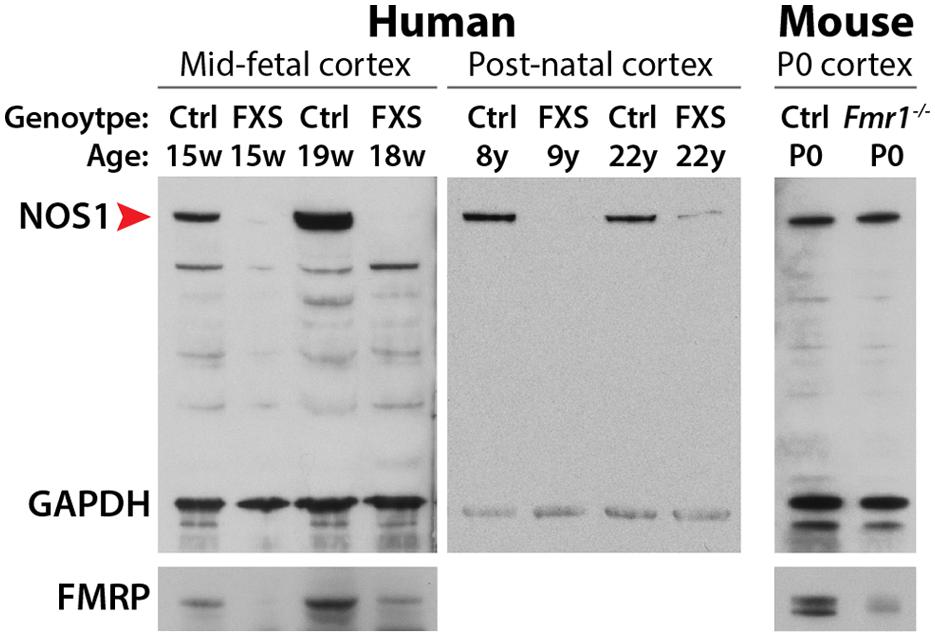
FIGURE 2. Immunoblot analysis of NOS1 expression in the developing neocortex. In the developing neocortex, the expression levels of NOS1 (red arrowhead) are severely reduced in human FXS patients. This loss is species-dependent. NOS1 levels are unaltered in the neocortex of neonatal Fmr1 null mice, an animal model of FXS. Adapted from Kwan et al. (2012a).
Spatiotemporal and Species-Dependent Expression of NOS1 in the Human Neocortex
To determine the potential contribution of NOS1 to FXS, it is necessary to consider the time and location of NOS1 function during brain development. In contrast to the widespread expression of FMRP, NOS1 expression exhibits spatial, temporal, and neuronal subtype specificity, providing insights into its potential neural function. In the neocortex of several mammalian species, NOS1 has been known to be a selective marker of inhibitory interneurons (Vruwink et al., 2001) while being absent from the principal neurons of the neocortex, the excitatory projection (pyramidal) neurons. In the human mid-fetal (13–24 weeks post-conception; Figure 1) neocortex, however, we found NOS1 to be present not only in the interneurons, but also in the pyramidal neurons of two specific neocortical areas underlying higher cognitive functions: the fetal Broca’s area and orofacial motor cortex, and the ACC (Figure 3; Kwan et al., 2012a). The pyramidal projection neurons occupy a central position in neocortical circuitry, being the source of long distance axonal projections that connect distal areas within the neocortex (via intracortical connections) and the neocortex with subcortical brain structures (via corticofugal connections). The newly identified pyramidal expression of NOS1 is particularly surprising because pyramidal neurons are derived from distinct progenitors and have greatly divergent chemical and electrophysiological properties compared to interneurons (Markram et al., 2004; Kwan et al., 2012b). Interestingly, the expression of NOS1 in the human neocortex sharply contrasts that in the mouse neocortex, wherein NOS1 is exclusively present in inhibitory interneurons (Figure 3; Kwan et al., 2012a). NOS1 expression analysis in the fetal macaque neocortex and sequence conservation analysis in mammals revealed that pyramidal NOS1 expression is likely to be primate-specific. The recent origin of this expression pattern is consistent with the possibility that pyramidal NOS1 plays a key role in the development or function of neocortical circuitry underlying higher cognitive functions.
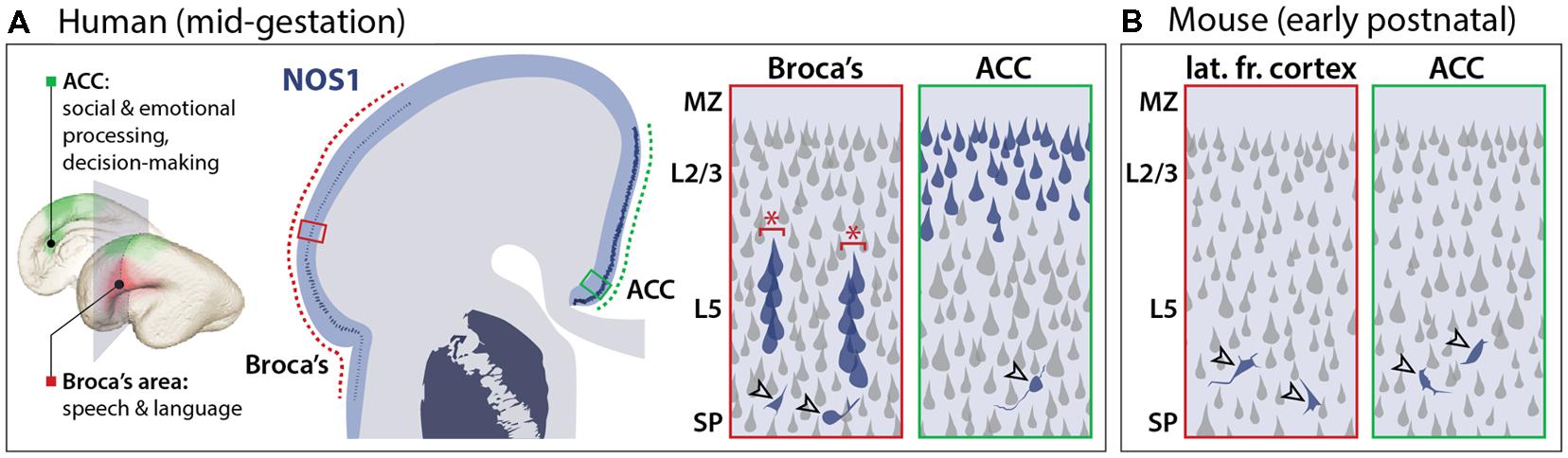
FIGURE 3. Species-dependent NOS1 expression in the developing neocortex. (A) In the mid-fetal human neocortex, NOS1 is specifically expressed in the pyramidal neurons of two neocortical regions underlying language and cognitive functions. In the Broca’s area (red boxes), NOS1 expression (blue), is selectively present in the pyramidal neurons of neocortical layer 5 (L5). NOS1-expressing neurons are assembled into minicolumns (red asterisks) separated by NOS1-negative neurons. In the anterior cingulate cortex (ACC; green boxes), many L2/L3 pyramidal neurons express high levels of NOS1. NOS1 expression in interneurons (open arrowheads) is present throughout neocortical areas and layers with no significant selectivity. (B) In the early-postnatal mouse neocortex, which is developmentally equivalent to the human mid-fetal neocortex, NOS1 is exclusively expressed in interneurons (open arrowheads) and absent from pyramidal neurons.
In addition to this notable spatial selectivity, NOS1 expression in the human neocortex further exhibits remarkable temporal specificity, occurring only transiently within a small mid-fetal window and becoming dramatically downregulated by late gestation (Figures 1 and 3). During this mid-gestation period, the genesis of neocortical pyramidal neurons is largely complete and the deep neocortical layers have been formed, whereas many upper layers neurons are migrating toward their laminar destination. Importantly, neocortical axon tracts are undergoing outgrowth to reach their target destinations, a process that largely precedes the onset of peak synaptogenesis (Hadley et al., 1983; Kang et al., 2011; Xu and Henkemeyer, 2012). Overall, the spatiotemporal specificity of pyramidal NOS1 expression is consistent with a key role for NOS1 in the formation of specific neocortical circuits important for higher cognition. The location, connectivity, and neural function of these circuits provide crucial insights into the potential consequences of NOS1 dysregulation in the FXS neocortex.
Broca’s Speech and Language Area and Orofacial Motor Cortex
In the mid-gestation frontal and frontoparietal operculum, the fetal anlage of the adult Broca’s speech and language area and orofacial motor cortex, NOS1 is specifically expressed in layer 5 (L5) corticofugal projection neurons, the output neurons of the neocortex (Figure 3; Kwan et al., 2012a). Broca’s area, located in the inferior frontal gyrus, plays a key role in the generation of speech and is also important in language comprehension (Judas and Cepanec, 2007; Keller et al., 2009). The L5 expression of NOS1 in the fetal Broca’s area is developmentally regulated, being exclusively present from approximately 16 to 24 weeks post-conception. The expression of NOS1 in these neurons during the 7–8 week fetal window when they differentiate and acquire their axonal connectivities implies a role for NOS1 in the assembly of motor circuits involved in speech and language. Several lines of evidence, including studies of gene co-expression, microscopic neuroanatomy, and functional imaging, point to a putative contribution of disrupted NOS1 expression in Broca’s area to the psychopathology of FXS and provide insights into the potential mechanistic underpinnings of this disruption.
In the mid-fetal Broca’s area, NOS1 neurons coexpress BCL11B (previously CTIP2), a transcription factor that is specifically expressed in and required for the development of corticofugal projection neurons (Arlotta et al., 2005). This coexpression is consistent with the L5 pyramidal identity of NOS1 neurons and a potential role for NOS1 in neocortical output circuits originating from this area. Abnormal activation of the inferior frontal gyrus, which encompasses the Broca’s area, in FXS patients during the counting Stroop interference task (Tamm et al., 2002) has suggested altered Broca’s area connectivity in the disorder. Furthermore, functional imaging studies of ASD patients have identified specific alterations in Broca’s area connections. For example, in a recent analysis of network connectivity in 24-month old ASD infants, alterations in network efficiencies were reported in all four cortical lobes, with a significant reduction in the Broca’s area in the frontal lobe (Lewis et al., 2014). Importantly, reduced network efficiency in Broca’s area correlated with increased ASD symptom severity. In another recent study, naturally sleeping ASD toddlers exhibited significantly weakened interhemispheric correlations in the inferior frontal gyrus (Dinstein et al., 2011). Interestingly, the strength of the interhemispheric synchrony was negatively correlated with autism severity and positively correlated with verbal ability. Furthermore, a reversal in volume asymmetry in the Broca’s area has been reported in autistic boys (De Fossé et al., 2004), implicating both structural and functional differences in this area in ASD. It remains to be shown whether NOS1 contributes to Broca’s area alterations in ASD or similar alterations in FXS. The highly specific spatiotemporal expression of NOS1, however, is consistent with this possibility.
In the mid-fetal Broca’s area, NOS1 neurons coexpress FOXP2 (Kwan et al., 2012a), a gene altered in a severe speech disorder (Lai et al., 2001; Fisher and Scharff, 2009) and implicated in ASD and cognitive impairments (Bacon and Rappold, 2012). The presence of NOS1 in FOXP2-expressing neurons adds further support to a possible role of NOS1 in ASD and cognitive neural circuits. Furthermore, NOS1-expressing neurons are organized into microcircuits known as minicolumns (Figure 3; Kwan et al., 2012a), a vertical arrangement thought to constitute a functional unit of cortical circuitry (Mountcastle, 1997; Buxhoeveden and Casanova, 2002). Interestingly, the arrangement of minicolumns is disorganized in psychiatric disorders, including ASD and SCZ, potentially contributing to circuit dysfunction by disrupting the inhibitory architecture of the neocortex (Buxhoeveden and Casanova, 2002; Casanova et al., 2002, 2003). The expression of NOS1 in minicolumns during fetal development is consistent with a role for NO in the assembly of microcircuitry, possibly related to ASD and SCZ, in this distinctly human neocortical area.
Anterior Cingulate Cortex
In addition to the Broca’s area, a second neocortical region with important cognitive functions also exhibit pyramidal NOS1 expression during mid-fetal development. Starting from approximately 16 weeks post-conception, NOS1 is expressed in a large number of upper-layer (L2–L3) intracortical projection neurons of the ACC at significantly higher levels compared to the L5 neurons of the Broca’s area (Kwan et al., 2012a). NOS1-expressing neurons in the ACC, in contrast to those in the Broca’s areas, are not marked by BCL11B but instead coexpress SATB2, a transcription factor that labels intracortical projection neurons and is required for their normal development (Alcamo et al., 2008; Britanova et al., 2008). Unlike the corticofugal projection neurons that express NOS1 in the Broca’s area, NOS1-expressing pyramidal neurons in the ACC are likely to contribute to intracortical connectivity. Furthermore, although NOS1 expression in ACC pyramidal neurons is also temporally regulated, its timing spans a lengthier period during development, being present at high levels from mid- to late- gestation and downregulated after birth (Kwan et al., 2012a). Whereas no pyramidal neurons retain NOS1 expression in the postnatal Broca’s area, a small number of pyramidal neurons in the ACC express low levels of NOS1 into adulthood (Gao et al., 2013). The significance of NOS1 expression in adult pyramidal neurons and whether its function parallels its fetal role remains to be explored. The ACC is not only highly interconnected to cognitive and emotional circuits, but also important in a number of neuropsychiatric diseases (Devinsky et al., 1995). Functional imaging, gene expression analyses, and other studies suggest the possibility that NOS1-expressing pyramidal neurons in the ACC may contribute to the psychopathology of FXS in a manner distinct from their counterparts in the Broca’s area.
Centrally positioned in the neocortex, the ACC is a key point of integration for cognitive and emotional processes, playing a crucial role in executive, social, and affective functions (Devinsky et al., 1995). The ACC exhibits extensive intracortical connectivities. The dorsal cognitive portion of the ACC is connected with motor and sensory systems of the neocortex, and is therefore well-positioned to integrate sensory stimuli with top-down commands (Paus et al., 1993; Picard and Strick, 1996; Williams et al., 2004; Crottaz-Herbette and Menon, 2006). The ventral part of the ACC, which is involved in emotion and motivation processing, is connected with the anterior insula and amygdala, as well as the nucleus accumbens and hypothalamus (Bush et al., 2002; Etkin et al., 2011). Functional imaging studies have found the ACC to be important for performance monitoring, error detection, action selection, and reinforcement of adaptive behavior (Brown and Braver, 2005; Schweimer and Hauber, 2006; Ko et al., 2009). Alterations in ACC circuitry have been proposed to contribute to maladaptive behaviors in psychiatric disorders (Drevets et al., 2008; Davey et al., 2012; Tu et al., 2012; Zikopoulos and Barbas, 2013). The expression of NOS1 in the intracortical projection neurons of the ACC implicates NOS1 in the connectivity of cognition and emotion circuits, which likely underlie some aspects of the pathogenesis in a number of neuropsychiatric disorders.
Data from several studies lend indirect support to a potential role of ACC NOS1 dysregulation in FXS. Studies of a human NOS1 hypomorphic allele revealed homozygosity to be associated with attention deficit hyperactivity disorder (ADHD), impulsivity, and aggression (Reif et al., 2009), linking reduced NOS1 activity to behavioral features often comorbid with FXS (Rogers et al., 2001). This NOS1 hypomorphism has also been linked to reduced neural activity in the ACC (Reif et al., 2009), consistent with a potential role of NOS1 in the development of ACC connectivity. Additional functional imaging studies revealed a similar reduction in ACC activation in FXS and ADHD patients during attentional processing tasks (Bush et al., 1999; Uddin and Menon, 2009) and in autistic children in response to a familiar face (Pierce and Redcay, 2008). The convergence of neural and behavioral deficits between NOS1 hypomorphism, FXS, and FXS comorbidities provide robust support to the possibility of a functional role of NOS1 in the development and function of ACC circuitry related to FXS and ASD.
In addition to FXS and ASD, ACC dysfunction has also been associated with other psychiatric disorders. In SCZ, pathological studies have found neuronal architecture to be altered in the ACC (Salgado-Pineda et al., 2014), whereas neural imaging studies have found abnormalities in the ACC that were reversed with treatment response. In major depressive disorder, neuronal density was found to be reduced in the ACC (Rajkowska et al., 1999, 2001) and psychotherapy success was found to correlate with increased ACC activity (Mayberg et al., 1997; Quidé et al., 2012). Interestingly, a recent study of depressive patient materials revealed a significant reduction in NO metabolites in the cerebrospinal fluid (Gao et al., 2013). Furthermore, NOS1 expression in the pyramidal neurons of the adult ACC was found to be significantly reduced in depressed patient brains (Gao et al., 2013). Although FXS has not been associated with major depression, de novo variations in SCZ have been shown to preferentially affect FMRP targets and FMRP levels have been correlated with SCZ severity (Kovács et al., 2013; Fromer et al., 2014; Purcell et al., 2014), suggesting a potential mechanistic overlap.
The spatiotemporal expression pattern of NOS1 during circuit wiring is consistent with a potential role in establishing global connectivity during neocortical development. By imaging resting state coordinated activity between brain areas, a measure of functional connectivity can be inferred based on synchronous activity (Power et al., 2011). Remarkably, Broca’s area and the ACC, the two neocortical areas with significant mid-fetal pyramidal NOS1 expression, exhibit coordinated activity and are known to form a cingulo-opercular network (Power et al., 2011), one of the default mode networks of the neocortex (Vaden et al., 2013; Sestieri et al., 2014). Interestingly, a disruption in the activity of the cingulo-opercular network has been identified in SCZ (Tu et al., 2012). Although the cingulo-opercular network has not been examined in individuals with the hypomorphic NOS1 allele, these data suggest a potential contribution of reduced NOS1 activity to the altered connectivity of a default mode network. Together, these findings converge on the possibility that NOS1 dysfunction in pyramidal neurons of the ACC and Broca’s area may contribute to distinct psychiatric disorders that affect higher cognitive abilities.
A Functional Convergence on Synaptic Mechanisms
Recent advances in psychiatric genetics have reaffirmed the heterogeneity of disease loci and the complexity of these disorders. However, a functional convergence has emerged from many of these studies. In ASD, specific functional subclasses of risk loci have been identified based on gene ontology and network analysis (Krumm et al., 2014; Pinto et al., 2014). These unbiased analyses have revealed chromatin biology and synaptic function to be key processes in ASD. Furthermore, empirical evidence has also indicated that the behavioral and intellectual deficits present in ASD, and often comorbid with FXS, likely reflect an underlying defect in synaptic development, plasticity, or function (Zoghbi, 2003; Garber, 2007). Interestingly, similar analyses of SCZ genes have identified a number of shared risk loci, particularly in chromatin modifiers and synaptic molecules (McCarthy et al., 2014). The emerging data have shown many of the synaptic genes associated with ASD or SCZ to encode mRNA transcripts regulated by FMRP (Darnell et al., 2011; Iossifov et al., 2012; Fromer et al., 2014; Purcell et al., 2014). Furthermore, synaptic dysregulation is now recognized as a key pathology in a number of psychiatric disorders, suggesting the synapse to be a common neural substrate underlying distinct clinical manifestations of cognitive impairment (Penzes et al., 2011).
From the earliest functional studies, it has been clear that FXS involves deficits at the synapse. Alterations in spine morphology is well-characterized in human FXS (Comery et al., 1997) and animal models of FXS (Irwin et al., 2000; Nimchinsky et al., 2001), with both presenting significantly more morphologically immature spines and fewer mature spines. Similar deficits in spine morphology and synapse function have been reported to be present in ASD (Kaufmann and Moser, 2000; Hutsler and Zhang, 2010; Penzes et al., 2011). The localization of FMRP to dendritic spines and the putative function of many of its target mRNAs at or near synaptic contacts add further support to the possibility that FXS is a disorder of synaptic development and function (Irwin et al., 2000; Huber et al., 2002; Bear et al., 2004).
Consistent with a putative contribution of dysregulated NO signaling to FXS pathophysiology, NOS1 is likely to have a synaptic function as well. Subcellularly, NOS1 is significantly localized to the dendritic spines (Burette et al., 2002; Kwan et al., 2012a). This localization is mediated by direct interaction with the PDZ domain of post-synaptic density protein PSD-95 (Brenman et al., 1996). The catalytic activity of NOS1 requires Ca2+ influx, which is under the control of near by NMDA receptors (Brenman and Bredt, 1997; Sattler et al., 1999). Interestingly, blockade of NOS1 function has been shown to disrupt synapse formation and result in spine loss (Sánchez-Islas and León-Olea, 2004; Morales-Medina et al., 2007; Nikonenko et al., 2008). Therefore, aspects of spine morphology deficits in FXS may originate from a loss of NO signaling. Overall, given the key role of synaptic proteins in ASD, NOS1 is well positioned to mediate at least some of the mechanisms disrupted in FXS (Figure 4).
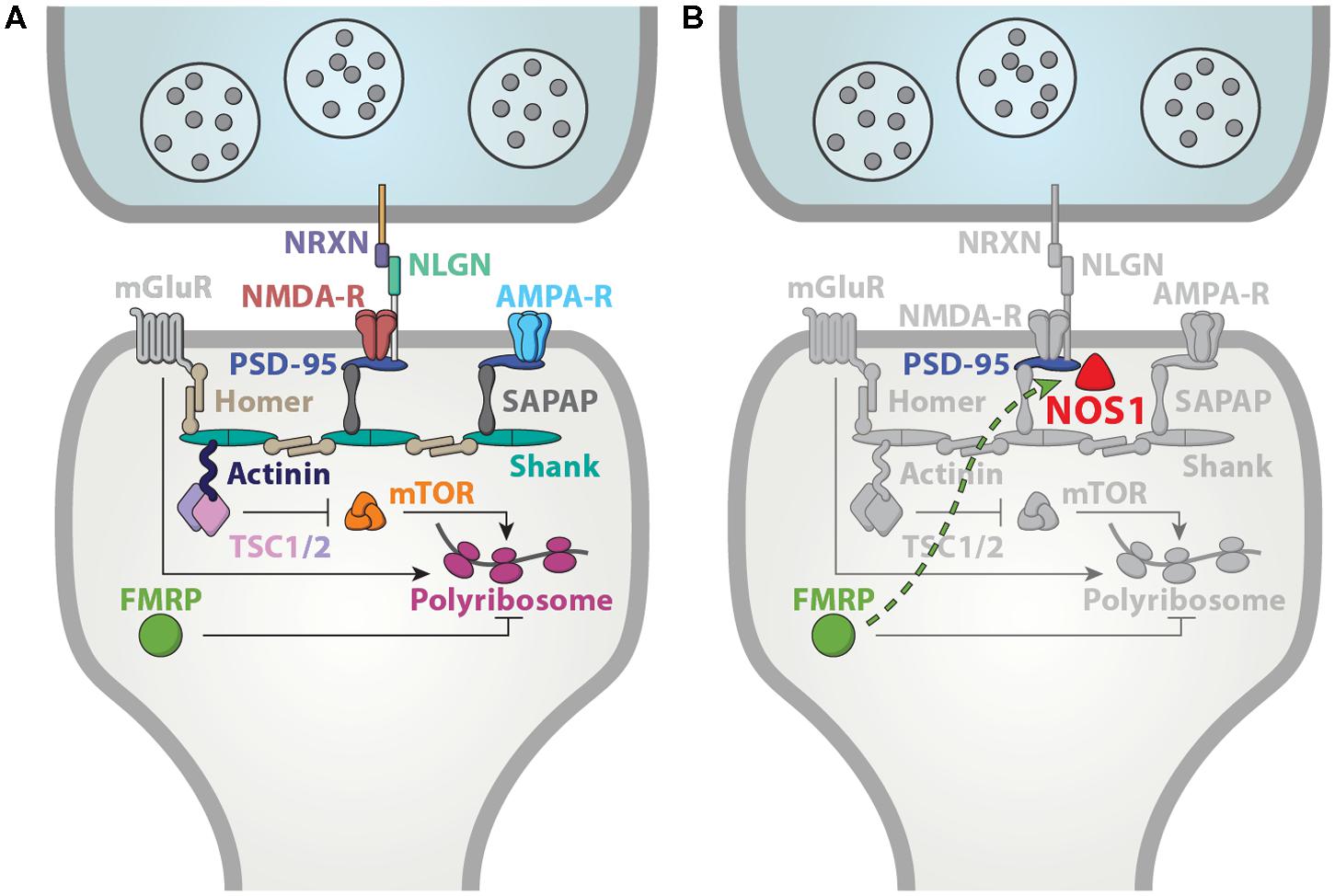
FIGURE 4. A functional convergence on synaptic mechanisms. (A) Many ASD-associated genes are localized to the synapse, robustly implicating synaptic dysfunction in ASD. Adapted from Peça et al. (2011). (B) NOS1 is anchored to the post-synaptic member via interactions with PSD-95 (Brenman et al., 1996), and therefore, well-positioned to contribute to the synaptic mechanisms underlying FXS and ASD.
Nitric Oxide Signaling as a Target for FXS Molecular Therapies
The development of molecular therapeutic interventions for neurodevelopmental disorders represents an immense challenge. In the past decades, these efforts have been impeded by the tremendous phenotypic and genotypic heterogeneity in the patient populations, the lack of specific knowledge on the molecular pathways, brain regions, or developmental timepoints most relevant to the pathophysiology of the disorders, and the identification of the appropriate primary endpoints (Haddad and Dursun, 2008; Jacquemont et al., 2014). Recent advances in psychiatric genetics have helped to pinpoint some of the molecular and neural underpinnings of these disorders, and revealed potential targets for therapeutic interventions. These strategies have focused mainly on alterations in molecular signaling pathways, which may be more targetable compared to structural deficits in brain development.
Since the proposal of the mGluR theory (Bear et al., 2004; Pop et al., 2014), a number of therapies targeting molecules up or downstream of mGluR signaling have been developed and reached various phases of clinical trial. For example, an open label trial of fenobam (NPL-2009), an mGluR5 antagonist, ameliorated anxiety, hyperarousal, and deficits in prepulse inhibition, and improved continuous performance task outcomes (Berry-Kravis et al., 2009). Arbaclofen (STX209), a GABA receptor agonist acting upstream of mGluR signaling, showed mixed promise in correcting behavioral problems, which was strengthened when adjusted for baseline severity of social withdrawal (Berry-Kravis et al., 2012; Erickson et al., 2014; Jacquemont et al., 2014). The mGluR antagonist mavoglurant (AFQ056) has shown some promise in patients with complete methylation at the FMR1 promoter and in an animal model of FXS (Jacquemont et al., 2011; Gantois et al., 2013). The commercial futures of arbaclofen and mavoglurant, however, are unclear, due to the recent discontinuation of their development.
The possibility of dysregulated NO signaling in FXS may lead to novel therapeutic strategies, including those that increase the brain levels of NO or cyclic guanosine monophosphate (cGMP), a second messenger elevated downstream of NO-mediated activation of soluble guanylate cyclase (sGC; Garthwaite and Boulton, 1995). Phosphodiesterase (PDE) inhibitors and NO donors have been used to enhance cGMP at distinct points of the NO signaling pathway. PDE type 5 inhibitors, which elevate cGMP levels by blocking their enzymatic degradation, are well known for their role in treating erectile dysfunction (Boolell et al., 1996; Sandner et al., 2008), but may have additional indications in the brain. Sildenafil has been shown to have antidepressant, anxiolytic, and cognitive effects, and to increase cGMP levels in the hippocampus (Rutten et al., 2005; Liebenberg et al., 2010, 2012; Uthayathas et al., 2013; Zhang et al., 2013). Tadalafil has similar antidepressant and anxiolytic effects, and has additionally been shown to reverse cognitive dysfunction by crossing the blood–brain barrier (Liebenberg et al., 2010, 2012; García-Barroso et al., 2013). cGMP levels may also be increased using NO donors to stimulate sGC activity. One such donor is sodium nitroprusside, a single intravenous administration of which has been shown to rapidly ameliorate the positive, negative, anxiety, and depressive symptoms in SCZ patients (Hallak et al., 2013). Interestingly, NO donors may mediate effects in addition to sGC activation. For example, the NO donor minocycline is known to inhibit matrix metalloproteinase-9 (MMP9), a protein encoded by an FMRP target mRNA (Berry-Kravis, 2014). It also promotes spine maturation and normal behavior in Fmr1 null mice (Bilousova et al., 2009) and delivers a significant improvement in anxiety and mood-related behaviors in children diagnosed with FXS (Paribello et al., 2010; Leigh et al., 2013). Additional drugs may more directly target NOS1 function or expression. For example, sapropterin, a synthetic form of the NOS cofactor tetrahydrobiopterin, was found to improve behavioral symptoms of ASD (Frye et al., 2013). A drug currently undergoing clinical trials for FXS and Rett syndrome may also affect NOS expression. NNZ-2566, a GPE peptide analog has been previously demonstrated to increase neuronal ChAT, GAD, and NOS expression (Guan et al., 1999; Guan and Gluckman, 2009; Berry-Kravis, 2014). Further studies on these and other therapies targeting NOS are expected to reveal the feasibility and efficacy of this line of treatment strategy for FXS and ASD.
No Signaling in Multiple Psychiatric Disorders
Emerging genetic studies of multiple psychiatric disorders previously thought to be mechanistically distinct have implicated the same set of genes to carry disease risk for each disorder. A recent analysis of de novo variants uncovered shared genetic underpinnings in ASD and SCZ (McCarthy et al., 2014). In a recent large genome-wide association study (GWAS), risk is correlated across five psychiatric disorders: ASD, SCZ, attention deficit-hyperactivity disorder, bipolar disorder, and major depressive disorder (Smoller et al., 2013). Remarkably, from this pooled analysis, single nucleotide polymorphisms (SNPs) within two L-type voltage-gated calcium channel (VGCC) subunits, CACNA1C and CACNB2, surpassed statistical threshold. This and previous studies associating CACNA1C with the individual disorders [Splawski et al., 2006; Ferreira et al., 2008; Walsh et al., 2008; Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium, 2011; Lu et al., 2012; Wray et al., 2012] strongly implicate VGCC signaling to be a shared neurobiological mechanism between these phenotypically divergent illnesses.
The genetic and mechanistic evidence presented in this review suggest that NOS1 may similarly play a role in multiple psychiatric disorders. NOS1 activity is dependent on calcium signaling mediated via calmodulin. This calcium signaling may partially originate from L-type VGCCs. One study found depolarization-stimulated and spontaneous cGMP formation downstream of NOS1 activity to be blocked by the L-type calcium channel blocker nifedipine (Oka et al., 1999). Furthermore, there is evidence that NOS1, in turn, regulates VGCCs including CACNA1C. The protein levels of CACNA1C and CACNA1D were found to be significantly reduced in the cerebellum of Nos1 null mice (Kim et al., 2004). Together, these data suggest that the potential involvement of NOS1 in multiple psychiatric disorders may be mediated in part via interactions with VGCCs.
The pursuit of convergent mechanisms in multiple psychiatric disorders may help identify points at which disease pathways intersect and may lead to a more mechanistic nosology in psychiatry beyond descriptive classification. These studies are expected to not only lead to conceptual advances in the neurobiology of psychiatric disorders, but also to targets of molecular therapies with widespread utility. In particular, because NO signaling has been targeted pharmacologically for other indications, understanding NOS1 dysfunction in psychiatric disorders has tremendous potential to lead to the repurposing of efficacious, tested agents to treat these disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Kwan laboratory for critical reading of the manuscript. This work was supported by the National Institutes of Health (MH096939).
References
Abbeduto, L., Brady, N., and Kover, S. T. (2007). Language development and fragile X syndrome: profiles, syndrome-specificity, and within-syndrome differences. Ment. Retard. Dev. Disabil. Res. Rev. 13, 36–46. doi: 10.1002/mrdd.20142
Abitbol, M., Menini, C., Delezoide, A. L., Rhyner, T., Vekemans, M., and Mallet, J. (1993). Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat. Genet. 4, 147–153. doi: 10.1038/ng0693-147
Alcamo, E. A., Chirivella, L., Dautzenberg, M., Dobreva, G., Fariñas, I., Grosschedl, R.,et al. (2008). Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57, 364–377. doi: 10.1016/j.neuron.2007.12.012
Antar, L. N., Afroz, R., Dictenberg, J. B., Carroll, R. C., and Bassell, G. J. (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 24, 2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004
Arlotta, P., Molyneaux, B. J., Chen, J., Inoue, J., Kominami, R., and Macklis, J. D. (2005). Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45, 207–221. doi: 10.1016/j.neuron.2004.12.036
Ashley, C. T., Wilkinson, K. D., Reines, D., and Warren, S. T. (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. doi: 10.1126/science.7692601
Bacon, C., and Rappold, G. A. (2012). The distinct and overlapping phenotypic spectra of FOXP1 and FOXP2 in cognitive disorders. Hum. Genet. 131, 1687–1698. doi: 10.1007/s00439-012-1193-z
Bagni, C., and Greenough, W. T. (2005). From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 6, 376–387. doi: 10.1038/nrn1667
Bakker, C. E., de Diego Otero, Y., Bontekoe, C., Raghoe, P., Luteijn, T., Hoogeveen, A. T.,et al. (2000). Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp. Cell Res. 258, 162–170. doi: 10.1006/excr.2000.4932
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Bechara, E. G., Didiot, M. C., Melko, M., Davidovic, L., Bensaid, M., Martin, P.,et al. (2009). A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 7:e16. doi: 10.1371/journal.pbio.1000016
Berry-Kravis, E. (2014). Mechanism-based treatments in neurodevelopmental disorders: fragile X syndrome. Pediatr. Neurol. 50, 297–302. doi: 10.1016/j.pediatrneurol.2013.12.001
Berry-Kravis, E., Hessl, D., Coffey, S., Hervey, C., Schneider, A., Yuhas, J.,et al. (2009). A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 46, 266–271. doi: 10.1136/jmg.2008.063701
Berry-Kravis, E. M., Hessl, D., Rathmell, B., Zarevics, P., Cherubini, M., Walton-Bowen, K.,et al. (2012). Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci. Transl. Med. 4, 152ra127. doi: 10.1126/scitranslmed.3004214
Bilousova, T. V., Dansie, L., Ngo, M., Aye, J., Charles, J. R., Ethell, D. W.,et al. (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 46, 94–102. doi: 10.1136/jmg.2008.061796
Boolell, M., Allen, M. J., Ballard, S. A., Gepi-Attee, S., Muirhead, G. J., Naylor, A. M.,et al. (1996). Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int. J. Impot. Res. 8, 47–52.
Bourgeois, J. P. (1997). Synaptogenesis, heterochrony and epigenesis in the mammalian neocortex. Acta Paediatr. Suppl. 422, 27–33. doi: 10.1111/j.1651-2227.1997.tb18340.x
Bray, S., Hirt, M., Jo, B., Hall, S. S., Lightbody, A. A., Walter, E.,et al. (2011). Aberrant frontal lobe maturation in adolescents with fragile X syndrome is related to delayed cognitive maturation. Biol. Psychiatry 70, 852–858. doi: 10.1016/j.biopsych.2011.05.038
Bredt, D. S., and Snyder, S. H. (1994). Nitric oxide: a physiologic messenger molecule. Annu. Rev. Biochem. 63, 175–195. doi: 10.1146/annurev.bi.63.070194.001135
Brenman, J. E., and Bredt, D. S. (1997). Synaptic signaling by nitric oxide. Curr. Opin. Neurobiol. 7, 374–378. doi: 10.1016/S0959-4388(97)80065-7
Brenman, J. E., Chao, D. S., Gee, S. H., McGee, A. W., Craven, S. E., Santillano, D. R.,et al. (1996). Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 84, 757–767. doi: 10.1016/S0092-8674(00)81053-3
Britanova, O., de Juan Romero, C., Cheung, A., Kwan, K. Y., Schwark, M., Gyorgy, A.,et al. (2008). Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 57, 378–392. doi: 10.1016/j.neuron.2007.12.028
Brown, J. W., and Braver, T. S. (2005). Learned predictions of error likelihood in the anterior cingulate cortex. Science 307, 1118–1121. doi: 10.1126/science.1105783
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A.,et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. doi: 10.1016/S0092-8674(01)00568-2
Burette, A., Zabel, U., Weinberg, R. J., Schmidt, H. H., and Valtschanoff, J. G. (2002). Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J. Neurosci. 22, 8961–8970.
Bush, G., Frazier, J. A., Rauch, S. L., Seidman, L. J., Whalen, P. J., Jenike, M. A.,et al. (1999). Anterior cingulate cortex dysfunction in attention-deficit/hyperactivity disorder revealed by fMRI and the Counting Stroop. Biol. Psychiatry 45, 1542–1552. doi: 10.1016/S0006-3223(99)00083-9
Bush, G., Vogt, B. A., Holmes, J., Dale, A. M., Greve, D., Jenike, M. A.,et al. (2002). Dorsal anterior cingulate cortex: a role in reward-based decision making. Proc. Natl. Acad. Sci. U.S.A. 99, 523–528. doi: 10.1073/pnas.012470999
Buxhoeveden, D. P., and Casanova, M. F. (2002). The minicolumn hypothesis in neuroscience. Brain 125, 935–951. doi: 10.1093/brain/awf110
Calabrese, V., Mancuso, C., Calvani, M., Rizzarelli, E., Butterfield, D. A., and Stella, A. M. (2007). Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 8, 766–775. doi: 10.1038/nrn2214
Carroll, L. S., and Owen, M. J. (2009). Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 1, 102. doi: 10.1186/gm102
Casanova, M. F., Buxhoeveden, D., and Gomez, J. (2003). Disruption in the inhibitory architecture of the cell minicolumn: implications for autism. Neuroscientist 9, 496–507. doi: 10.1177/1073858403253552
Casanova, M. F., Buxhoeveden, D. P., Switala, A. E., and Roy, E. (2002). Minicolumnar pathology in autism. Neurology 58, 428–432. doi: 10.1212/WNL.58.3.428
Clancy, B., Kersh, B., Hyde, J., Darlington, R. B., Anand, K. J., and Finlay, B. L. (2007). Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics 5, 79–94. doi: 10.1385/NI:5:1:79
Colantuoni, C., Lipska, B. K., Ye, T., Hyde, T. M., Tao, R., Leek, J. T.,et al. (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523. doi: 10.1038/nature10524
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J.,et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Contestabile, A. (2000). Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Res. Brain Res. Rev. 32, 476–509. doi: 10.1016/S0165-0173(00)00018-7
Crottaz-Herbette, S., and Menon, V. (2006). Where and when the anterior cingulate cortex modulates attentional response: combined fMRI and ERP evidence. J. Cogn. Neurosci. 18, 766–780. doi: 10.1162/jocn.2006.18.5.766
Darnell, J. C., Jensen, K. B., Jin, P., Brown, V., Warren, S. T., and Darnell, R. B. (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499. doi: 10.1016/S0092-8674(01)00566-9
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E.,et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Davey, C. G., Harrison, B. J., Yücel, M., and Allen, N. B. (2012). Regionally specific alterations in functional connectivity of the anterior cingulate cortex in major depressive disorder. Psychol. Med. 42, 2071–2081. doi: 10.1017/S0033291712000323
De Fossé, L., Hodge, S. M., Makris, N., Kennedy, D. N., Caviness, V. S., McGrath, L.,et al. (2004). Language-association cortex asymmetry in autism and specific language impairment. Ann. Neurol. 56, 757–766. doi: 10.1002/ana.20275
de Graaf-Peters, V. B., and Hadders-Algra, M. (2006). Ontogeny of the human central nervous system: what is happening when? Early Hum. Dev. 82, 257–266. doi: 10.1016/j.earlhumdev.2005.10.013
Devinsky, O., Morrell, M. J., and Vogt, B. A. (1995). Contributions of anterior cingulate cortex to behaviour. Brain 118(Pt 1), 279–306. doi: 10.1093/brain/118.1.279
Devys, D., Lutz, Y., Rouyer, N., Bellocq, J. P., and Mandel, J. L. (1993). The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 4, 335–340. doi: 10.1038/ng0893-335
Dinstein, I., Pierce, K., Eyler, L., Solso, S., Malach, R., Behrmann, M.,et al. (2011). Disrupted neural synchronization in toddlers with autism. Neuron 70, 1218–1225. doi: 10.1016/j.neuron.2011.04.018
Drevets, W. C., Savitz, J., and Trimble, M. (2008). The subgenual anterior cingulate cortex in mood disorders. CNS Spectr. 13, 663–681.
Dölen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S.,et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U.S.A. 95, 14863–14868. doi: 10.1073/pnas.95.25.14863
Erickson, C. A., Veenstra-Vanderweele, J. M., Melmed, R. D., McCracken, J. T., Ginsberg, L. D., Sikich, L.,et al. (2014). STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J. Autism Dev. Disord. 44, 958–964. doi: 10.1007/s10803-013-1963-z
Etkin, A., Egner, T., and Kalisch, R. (2011). Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn. Sci. 15, 85–93. doi: 10.1016/j.tics.2010.11.004
Feng, Y., Gutekunst, C. A., Eberhart, D. E., Yi, H., Warren, S. T., and Hersch, S. M. (1997). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 17, 1539–1547.
Ferreira, M. A., O’Donovan, M. C., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L.,et al. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058. doi: 10.1038/ng.209
Fisher, S. E., and Scharff, C. (2009). FOXP2 as a molecular window into speech and language. Trends Genet. 25, 166–177. doi: 10.1016/j.tig.2009.03.002
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P.,et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Frye, R. E., DeLatorre, R., Taylor, H. B., Slattery, J., Melnyk, S., Chowdhury, N.,et al. (2013). Metabolic effects of sapropterin treatment in autism spectrum disorder: a preliminary study. Transl. Psychiatry 3, e237. doi: 10.1038/tp.2013.14
Fähling, M., Mrowka, R., Steege, A., Kirschner, K. M., Benko, E., Förstera, B.,et al. (2009). Translational regulation of the human achaete-scute homologue-1 by fragile X mental retardation protein. J. Biol. Chem. 284, 4255–4266. doi: 10.1074/jbc.M807354200
Gabel, L. A., Won, S., Kawai, H., McKinney, M., Tartakoff, A. M., and Fallon, J. R. (2004). Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J. Neurosci. 24, 10579–10583. doi: 10.1523/JNEUROSCI.2185-04.2004
Gantois, I., Pop, A. S., de Esch, C. E., Buijsen, R. A., Pooters, T., Gomez-Mancilla, B.,et al. (2013). Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain Res. 239, 72–79. doi: 10.1016/j.bbr.2012.10.059
Gao, S. F., Qi, X. R., Zhao, J., Balesar, R., Bao, A. M., and Swaab, D. F. (2013). Decreased NOS1 expression in the anterior cingulate cortex in depression. Cereb. Cortex 23, 2956–2964. doi: 10.1093/cercor/bhs285
Garber, K. (2007). Neuroscience. Autism’s cause may reside in abnormalities at the synapse. Science 317, 190–191. doi: 10.1126/science.317.5835.190
García-Barroso, C., Ricobaraza, A., Pascual-Lucas, M., Unceta, N., Rico, A. J., Goicolea, M. A.,et al. (2013). Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 64, 114–123. doi: 10.1016/j.neuropharm.2012.06.052
Garthwaite, J. (2008). Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 27, 2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x
Garthwaite, J., and Boulton, C. L. (1995). Nitric oxide signaling in the central nervous system. Annu. Rev. Physiol. 57, 683–706. doi: 10.1146/annurev.ph.57.030195.003343
Geschwind, D., and Konopka, G. (2009). Neuroscience in the era of functional genomics and systems biology. Nature 461, 908–915. doi: 10.1038/nature08537
Geschwind, D. H., and Rakic, P. (2013). Cortical evolution: judge the brain by its cover. Neuron 80, 633–647. doi: 10.1016/j.neuron.2013.10.045
Goldman-Rakic, P. S. (1996). The prefrontal landscape: implications of functional architecture for understanding human mentation and the central executive. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 1445–1453. doi: 10.1098/rstb.1996.0129
Gross, C., Yao, X., Pong, D. L., Jeromin, A., and Bassell, G. J. (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci. 31, 5693–5698. doi: 10.1523/JNEUROSCI.6661-10.2011
Guan, J., and Gluckman, P. D. (2009). IGF-1 derived small neuropeptides and analogues: a novel strategy for the development of pharmaceuticals for neurological conditions. Br. J. Pharmacol. 157, 881–891. doi: 10.1111/j.1476-5381.2009.00256.x
Guan, J., Waldvogel, H. J., Faull, R. L., Gluckman, P. D., and Williams, C. E. (1999). The effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate in different regions following hypoxic-ischemic brain injury in adult rats. Neuroscience 89, 649–659. doi: 10.1016/S0306-4522(98)00338-8
Gulsuner, S., Walsh, T., Watts, A. C., Lee, M. K., Thornton, A. M., Casadei, S.,et al. (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529. doi: 10.1016/j.cell.2013.06.049
Haas, B. W., Barnea-Goraly, N., Lightbody, A. A., Patnaik, S. S., Hoeft, F., Hazlett, H.,et al. (2009). Early white-matter abnormalities of the ventral frontostriatal pathway in fragile X syndrome. Dev. Med. Child Neurol. 51, 593–599. doi: 10.1111/j.1469-8749.2009.03295.x
Haddad, P. M., and Dursun, S. M. (2008). Neurological complications of psychiatric drugs: clinical features and management. Hum. Psychopharmacol. 23(Suppl. 1), 15–26. doi: 10.1002/hup.918
Hadley, R. D., Kater, S. B., and Cohan, C. S. (1983). Electrical synapse formation depends on interaction of mutually growing neurites. Science 221, 466–468. doi: 10.1126/science.6867723
Hagerman, R., Hoem, G., and Hagerman, P. (2010). Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol. Autism 1, 12. doi: 10.1186/2040-2392-1-12
Hall, S. S., Jiang, H., Reiss, A. L., and Greicius, M. D. (2013). Identifying large-scale brain networks in fragile X syndrome. JAMA Psychiatry 70, 1215–1223. doi: 10.1001/jamapsychiatry.2013.247
Hallak, J. E., Maia-de-Oliveira, J. P., Abrao, J., Evora, P. R., Zuardi, A. W., Crippa, J. A.,et al. (2013). Rapid improvement of acute schizophrenia symptoms after intravenous sodium nitroprusside: a randomized, double-blind, placebo-controlled trial. JAMA Psychiatry 70, 668–676. doi: 10.1001/jamapsychiatry.2013.1292
Harris, S. W., Hessl, D., Goodlin-Jones, B., Ferranti, J., Bacalman, S., Barbato, I.,et al. (2008). Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 113, 427–438. doi: 10.1352/2008.113:427-438
Henin, A., Micco, J. A., Wozniak, J., Briesch, J. M., Narayan, A. J., and Hirshfeld-Becker, D. R. (2009). Neurocognitive functioning in bipolar disorder. Clin. Psychol. Sci. Pract. 16, 231–250. doi: 10.1111/j.1468-2850.2009.01162.x
Henseler, I., Falkai, P., and Gruber, O. (2010). Disturbed functional connectivity within brain networks subserving domain-specific subcomponents of working memory in schizophrenia: relation to performance and clinical symptoms. J. Psychiatr. Res. 44, 364–372. doi: 10.1016/j.jpsychires.2009.09.003
Hinds, H. L., Ashley, C. T., Sutcliffe, J. S., Nelson, D. L., Warren, S. T., Housman, D. E.,et al. (1993). Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat. Genet. 3, 36–43. doi: 10.1038/ng0193-36
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257. doi: 10.1126/science.288.5469.1254
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and protein synthesis – dependent long-term depression in hippocampal area CA1. J. Neurophysiol. 86, 321–325.
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Hölscher, C. (1997). Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 20, 298–303. doi: 10.1016/S0166-2236(97)01065-5
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J.,et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Irwin, S. A., Galvez, R., and Greenough, W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044. doi: 10.1093/cercor/10.10.1038
Jacquemont, S., Berry-Kravis, E., Hagerman, R., von Raison, F., Gasparini, F., Apostol, G.,et al. (2014). The challenges of clinical trials in fragile X syndrome. Psychopharmacology (Berl.) 231, 1237–1250. doi: 10.1007/s00213-013-3289-0
Jacquemont, S., Curie, A., des Portes, V., Torrioli, M. G., Berry-Kravis, E., Hagerman, R. J.,et al. (2011). Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 3, 64ra1. doi: 10.1126/scitranslmed.3001708
Johnson, M. B., Kawasawa, Y. I., Mason, C. E., Krsnik, Z., Coppola, G., Bogdanovic, D.,et al. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509. doi: 10.1016/j.neuron.2009.03.027
Judas, M., and Cepanec, M. (2007). Adult structure and development of the human fronto-opercular cerebral cortex (Broca’s region). Clin. Linguist. Phon. 21, 975–989. doi: 10.1080/02699200701617175
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M.,et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. doi: 10.1038/nature10523
Kaufmann, W. E., and Moser, H. W. (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10, 981–991. doi: 10.1093/cercor/10.10.981
Keller, S. S., Crow, T., Foundas, A., Amunts, K., and Roberts, N. (2009). Broca’s area: nomenclature, anatomy, typology and asymmetry. Brain Lang. 109, 29–48. doi: 10.1016/j.bandl.2008.11.005
Kim, M. J., Chung, Y. H., Joo, K. M., Oh, G. T., Kim, J., Lee, B.,et al. (2004). Immunohistochemical study of the distribution of neuronal voltage-gated calcium channels in the nNOS knock-out mouse cerebellum. Neurosci. Lett. 369, 39–43. doi: 10.1016/j.neulet.2004.07.047
Kindler, S., and Kreienkamp, H. J. (2012). The role of the postsynaptic density in the pathology of the fragile X syndrome. Results Probl. Cell Differ. 54, 61–80. doi: 10.1007/978-3-642-21649-7_5
Ko, J. H., Ptito, A., Monchi, O., Cho, S. S., Van Eimeren, T., Pellecchia, G.,et al. (2009). Increased dopamine release in the right anterior cingulate cortex during the performance of a sorting task: a [11C]FLB 457 PET study. Neuroimage 46, 516–521. doi: 10.1016/j.neuroimage.2009.02.031
Kovács, T., Kelemen, O., and Kéri, S. (2013). Decreased fragile X mental retardation protein (FMRP) is associated with lower IQ and earlier illness onset in patients with schizophrenia. Psychiatry Res. 210, 690–693. doi: 10.1016/j.psychres.2012.12.022
Krumm, N., O’Roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi: 10.1016/j.tins.2013.11.005
Kuperberg, G. R., West, W. C., Lakshmanan, B. M., and Goff, D. (2008). Functional magnetic resonance imaging reveals neuroanatomical dissociations during semantic integration in schizophrenia. Biol. Psychiatry 64, 407–418. doi: 10.1016/j.biopsych.2008.03.018
Kwan, K. Y. (2013). Transcriptional dysregulation of neocortical circuit assembly in ASD. Int. Rev. Neurobiol. 113, 167–205. doi: 10.1016/B978-0-12-418700-9.00006-X
Kwan, K. Y., Lam, M. M., Johnson, M. B., Dube, U., Shim, S., Rašin, M. R.,et al. (2012a). Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell 149, 899–911. doi: 10.1016/j.cell.2012.02.060
Kwan, K. Y., Sestan, N., and Anton, E. S. (2012b). Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139, 1535–1546. doi: 10.1242/dev.069963
Lai, C. S., Fisher, S. E., Hurst, J. A., Vargha-Khadem, F., and Monaco, A. P. (2001). A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413, 519–523. doi: 10.1038/35097076
Leigh, M. J., Nguyen, D. V., Mu, Y., Winarni, T. I., Schneider, A., Chechi, T.,et al. (2013). A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J. Dev. Behav. Pediatr. 34, 147–155. doi: 10.1097/DBP.0b013e318287cd17
Lewis, J. D., Evans, A. C., Pruett, J. R., Botteron, K., Zwaigenbaum, L., Estes, A.,et al. (2014). Network inefficiencies in autism spectrum disorder at 24 months. Transl. Psychiatry 4, e388. doi: 10.1038/tp.2014.24
Liebenberg, N., Harvey, B. H., Brand, L., and Brink, C. B. (2010). Antidepressant-like properties of phosphodiesterase type 5 inhibitors and cholinergic dependency in a genetic rat model of depression. Behav. Pharmacol. 21, 540–547. doi: 10.1097/FBP.0b013e32833befe5
Liebenberg, N., Harvey, B. H., Brand, L., Wegener, G., and Brink, C. B. (2012). Chronic treatment with the phosphodiesterase type 5 inhibitors sildenafil and tadalafil display anxiolytic effects in Flinders Sensitive Line rats. Metab. Brain Dis. 27, 337–340. doi: 10.1007/s11011-012-9284-z
Lu, A. T., Dai, X., Martinez-Agosto, J. A., and Cantor, R. M. (2012). Support for calcium channel gene defects in autism spectrum disorders. Mol. Autism 3, 18. doi: 10.1186/2040-2392-3-18
Markram, H., Toledo-Rodriguez, M., Wang, Y., Gupta, A., Silberberg, G., and Wu, C. (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807. doi: 10.1038/nrn1519
Mayberg, H. S., Brannan, S. K., Mahurin, R. K., Jerabek, P. A., Brickman, J. S., Tekell, J. L.,et al. (1997). Cingulate function in depression: a potential predictor of treatment response. Neuroreport 8, 1057–1061. doi: 10.1097/00001756-199703030-00048
McCarthy, S. E., Gillis, J., Kramer, M., Lihm, J., Yoon, S., Berstein, Y.,et al. (2014). De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658. doi: 10.1038/mp.2014.29
Menon, V., Leroux, J., White, C. D., and Reiss, A. L. (2004). Frontostriatal deficits in fragile X syndrome: relation to FMR1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 101, 3615–3620. doi: 10.1073/pnas.0304544101
Miller, E. K., Freedman, D. J., and Wallis, J. D. (2002). The prefrontal cortex: categories, concepts and cognition. Philos. Trans. R Soc. Lond. B Biol. Sci. 357, 1123–1136. doi: 10.1098/rstb.2002.1099
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L.,et al. (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. doi: 10.1016/S0896-6273(03)00034-5
Morales-Medina, J. C., Mejorada, A., Romero-Curiel, A., and Flores, G. (2007). Alterations in dendritic morphology of hippocampal neurons in adult rats after neonatal administration of N-omega-nitro-L-arginine. Synapse 61, 785–789. doi: 10.1002/syn.20406
Mountcastle, V. B. (1997). The columnar organization of the neocortex. Brain 120(Pt 4), 701–722. doi: 10.1093/brain/120.4.701
Nikonenko, I., Boda, B., Steen, S., Knott, G., Welker, E., and Muller, D. (2008). PSD-95 promotes synaptogenesis and multiinnervated spine formation through nitric oxide signaling. J. Cell Biol. 183, 1115–1127. doi: 10.1083/jcb.200805132
Nimchinsky, E. A., Oberlander, A. M., and Svoboda K. (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146.
O’Donnell, W. T., and Warren, S. T. (2002). A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 25, 315–338. doi: 10.1146/annurev.neuro.25.112701.142909
Oberlé, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A.,et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi: 10.1126/science.252.5009.1097
Oka, M., Itoh, Y., Ukai, Y., and Kimura, K. (1999). Blockade by NS-7, a neuroprotective compound, of both L-type and P/Q-type Ca2+ channels involving depolarization-stimulated nitric oxide synthase activity in primary neuronal culture. J. Neurochem. 72, 1315–1322. doi: 10.1046/j.1471-4159.1999.0721315.x
Paribello, C., Tao, L., Folino, A., Berry-Kravis, E., Tranfaglia, M., Ethell, I. M.,et al. (2010). Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 10:91. doi: 10.1186/1471-2377-10-91
Parikshak, N. N., Luo, R., Zhang, A., Won, H., Lowe, J. K., Chandran, V.,et al. (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. doi: 10.1016/j.cell.2013.10.031
Paus, T., Petrides, M., Evans, A. C., and Meyer, E. (1993). Role of the human anterior cingulate cortex in the control of oculomotor, manual, and speech responses: a positron emission tomography study. J. Neurophysiol. 70, 453–469.
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Peça, J., Ting, J., and Feng, G. (2011). SnapShot: autism and the synapse. Cell 147, 706–706.e1. doi: 10.1016/j.cell.2011.10.015
Picard, N., and Strick, P. L. (1996). Motor areas of the medial wall: a review of their location and functional activation. Cereb. Cortex 6, 342–353. doi: 10.1093/cercor/6.3.342
Pierce, K., and Redcay, E. (2008). Fusiform function in children with an autism spectrum disorder is a matter of “who.” Biol. Psychiatry 64, 552–560. doi: 10.1016/j.biopsych.2008.05.013
Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L.,et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694. doi: 10.1016/j.ajhg.2014.03.018
Pop, A. S., Gomez-Mancilla, B., Neri, G., Willemsen, R., and Gasparini, F. (2014). Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology (Berl.) 231, 1217–1226. doi: 10.1007/s00213-013-3330-3
Power, J. D., Cohen, A. L., Nelson, S. M., Wig, G. S., Barnes, K. A., Church, J. A.,et al. (2011). Functional network organization of the human brain. Neuron 72, 665–678. doi: 10.1016/j.neuron.2011.09.006
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P. C.,et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Quidé, Y., Witteveen, A. B., El-Hage, W., Veltman, D. J., and Olff, M. (2012). Differences between effects of psychological versus pharmacological treatments on functional and morphological brain alterations in anxiety disorders and major depressive disorder: a systematic review. Neurosci. Biobehav. Rev. 36, 626–644. doi: 10.1016/j.neubiorev.2011.09.004
Rajkowska, G., Halaris, A., and Selemon, L. D. (2001). Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol. Psychiatry 49, 741–752. doi: 10.1016/S0006-3223(01)01080-0
Rajkowska, G., Miguel-Hidalgo, J. J., Wei, J., Dilley, G., Pittman, S. D., Meltzer, H. Y.,et al. (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45, 1085–1098. doi: 10.1016/S0006-3223(99)00041-4
Reif, A., Jacob, C. P., Rujescu, D., Herterich, S., Lang, S., Gutknecht, L.,et al. (2009). Influence of functional variant of neuronal nitric oxide synthase on impulsive behaviors in humans. Arch. Gen. Psychiatry 66, 41–50. doi: 10.1001/archgenpsychiatry.2008.510
Rogers, S. J., Wehner, D. E., and Hagerman, R. (2001). The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 22, 409–417. doi: 10.1097/00004703-200112000-00008
Ronesi, J. A., and Huber, K. M. (2008). Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci. Signal. 1, pe6. doi: 10.1126/stke.15pe6
Rutten, K., Vente, J. D., Sik, A., Ittersum, M. M., Prickaerts, J., and Blokland, A. (2005). The selective PDE5 inhibitor, sildenafil, improves object memory in Swiss mice and increases cGMP levels in hippocampal slices. Behav. Brain Res. 164, 11–16. doi: 10.1016/j.bbr.2005.04.021
Salgado-Pineda, P., Landin-Romero, R., Fakra, E., Delaveau, P., Amann, B. L., and Blin, O. (2014). Structural abnormalities in schizophrenia: further evidence on the key role of the anterior cingulate cortex. Neuropsychobiology 69, 52–58. doi: 10.1159/000356972
Sánchez-Islas, E., and León-Olea, M. (2004). Nitric oxide synthase inhibition during synaptic maturation decreases synapsin I immunoreactivity in rat brain. Nitric Oxide 10, 141–149. doi: 10.1016/j.niox.2004.04.001
Sandner, P., Svenstrup, N., Tinel, H., Haning, H., and Bischoff, E. (2008). Phosphodiesterase 5 inhibitors and erectile dysfunction. Exp. Opin. Ther. Pat. 18, 21–33. doi: 10.1517/13543776.18.1.21
Sattler, R., Xiong, Z., Lu, W. Y., Hafner, M., MacDonald, J. F., and Tymianski, M. (1999). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284, 1845–1848. doi: 10.1126/science.284.5421.1845
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976. doi: 10.1038/ng.940
Schweimer, J., and Hauber, W. (2006). Dopamine D1 receptors in the anterior cingulate cortex regulate effort-based decision making. Learn. Mem. 13, 777–782. doi: 10.1101/lm.409306
Sestieri, C., Corbetta, M., Spadone, S., Romani, G. L., and Shulman, G. L. (2014). Domain-general signals in the cingulo-opercular network for visuospatial attention and episodic memory. J. Cogn. Neurosci. 26, 551–568. doi: 10.1162/jocn_a_00504
Smoller, J. W., Craddock, N., Kendler, K., Lee, P. H., Neale, B. M., Nurnberger, J. I.,et al. (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. doi: 10.1016/S0140-6736(12)62129-1
Splawski, I., Yoo, D. S., Stotz, S. C., Cherry, A., Clapham, D. E., and Keating, M. T. (2006). CACNA1H mutations in autism spectrum disorders. J. Biol. Chem. 281, 22085–22091. doi: 10.1074/jbc.M603316200
State, M. W., and Šestan, N. (2012). Neuroscience. The emerging biology of autism spectrum disorders. Science 337, 1301–1303. doi: 10.1126/science.1224989
Steinert, J. R., Chernova, T., and Forsythe, I. D. (2010). Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 16, 435–452. doi: 10.1177/1073858410366481
Stuart, J. M., Segal, E., Koller, D., and Kim, S. K. (2003). A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. doi: 10.1126/science.1087447
Tamm, L., Menon, V., Johnston, C. K., Hessl, D. R., and Reiss, A. L. (2002). fMRI study of cognitive interference processing in females with fragile X syndrome. J. Cogn. Neurosci. 14, 160–171. doi: 10.1162/089892902317236812
Todd, P. K., Mack, K. J., and Malter, J. S. (2003). The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. U.S.A. 100, 14374–14378. doi: 10.1073/pnas.2336265100
Tu, P. C., Hsieh, J. C., Li, C. T., Bai, Y. M., and Su, T. P. (2012). Cortico-striatal disconnection within the cingulo-opercular network in schizophrenia revealed by intrinsic functional connectivity analysis: a resting fMRI study. Neuroimage 59, 238–247. doi: 10.1016/j.neuroimage.2011.07.086
Uddin, L. Q., and Menon, V. (2009). The anterior insula in autism: under-connected and under-examined. Neurosci. Biobehav. Rev. 33, 1198–1203. doi: 10.1016/j.neubiorev.2009.06.002
Uddin, M., Tammimies, K., Pellecchia, G., Alipanahi, B., Hu, P., Wang, Z.,et al. (2014). Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat. Genet. 46, 742–747. doi: 10.1038/ng.2980
Uthayathas, S., Parameshwaran, K., Karuppagounder, S. S., Ahuja, M., Dhanasekaran, M., and Suppiramaniam, V. (2013). Selective inhibition of phosphodiesterase 5 enhances glutamatergic synaptic plasticity and memory in mice. Synapse 67, 741–747. doi: 10.1002/syn.21676
Vaden, K. I., Kuchinsky, S. E., Cute, S. L., Ahlstrom, J. B., Dubno, J. R., and Eckert, M. A. (2013). The cingulo-opercular network provides word-recognition benefit. J. Neurosci. 33, 18979–18986. doi: 10.1523/JNEUROSCI.1417-13.2013
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A.,et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Vincent, A., Heitz, D., Petit, C., Kretz, C., Oberlé, I., and Mandel, J. L. (1991). Abnormal pattern detected in fragile-X patients by pulsed-field gel electrophoresis. Nature 349, 624–626. doi: 10.1038/349624a0
Vruwink, M., Schmidt, H. H., Weinberg, R. J., and Burette, A. (2001). Substance P and nitric oxide signaling in cerebral cortex: anatomical evidence for reciprocal signaling between two classes of interneurons. J. Comp. Neurol. 441, 288–301. doi: 10.1002/cne.1413
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M.,et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Warren, S. T., Zhang, F., Licameli, G. R., and Peters, J. F. (1987). The fragile X site in somatic cell hybrids: an approach for molecular cloning of fragile sites. Science 237, 420–423. doi: 10.1126/science.3603029
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K.,et al. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400. doi: 10.1073/pnas.94.10.5395
Weinberger, D. R., Berman, K. F., and Zec, R. F. (1986). Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch. Gen. Psychiatry 43, 114–124. doi: 10.1001/archpsyc.1986.01800020020004
Williams, Z. M., Bush, G., Rauch, S. L., Cosgrove, G. R., and Eskandar, E. N. (2004). Human anterior cingulate neurons and the integration of monetary reward with motor responses. Nat. Neurosci. 7, 1370–1375. doi: 10.1038/nn1354
Willsey, A. J., Sanders, S. J., Li, M., Dong, S., Tebbenkamp, A. T., Muhle, R. A.,et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Workman, A. D., Charvet, C. J., Clancy, B., Darlington, R. B., and Finlay, B. L. (2013). Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 33, 7368–7383. doi: 10.1523/JNEUROSCI.5746-12.2013
Wray, N. R., Pergadia, M. L., Blackwood, D. H., Penninx, B. W., Gordon, S. D., Nyholt, D. R.,et al. (2012). Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol. Psychiatry 17, 36–48. doi: 10.1038/mp.2010.109
Xu, N. J., and Henkemeyer, M. (2012). Ephrin reverse signaling in axon guidance and synaptogenesis. Semin. Cell Dev. Biol. 23, 58–64. doi: 10.1016/j.semcdb.2011.10.024
Zalfa, F., Eleuteri, B., Dickson, K. S., Mercaldo, V., De Rubeis, S., di Penta, A.,et al. (2007). A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci. 10, 578–587. doi: 10.1038/nn1893
Zhang, J., Guo, J., Zhao, X., Chen, Z., Wang, G., Liu, A.,et al. (2013). Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav. Brain Res. 250, 230–237. doi: 10.1016/j.bbr.2013.05.017
Zikopoulos, B., and Barbas, H. (2013). Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci. 7:609. doi: 10.3389/fnhum.2013.00609
Keywords: cerebral neocortex, pyramidal neurons, neurodevelopmental disorders, nitric oxide signaling, Broca’s speech and language area, anterior cingulate cortex, neural circuit assembly, human fetal brain
Citation: Colvin SM and Kwan KY (2014) Dysregulated nitric oxide signaling as a candidate mechanism of fragile X syndrome and other neuropsychiatric disorders. Front. Genet. 5:239. doi: 10.3389/fgene.2014.00239
Received: 23 May 2014; Accepted: 03 July 2014;
Published online: 22 July 2014.
Edited by:
Peter Kennedy Todd, University of Michigan, USAReviewed by:
Maria Paola Lombardi, University of Amsterdam, NetherlandsRhiannon Meredith, VU University Amsterdam, Netherlands
Christina Gross, Cincinnati Children’s Hospital Medical Center, USA
Copyright © 2014 Colvin and Kwan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth Y. Kwan, Department of Human Genetics – The Molecular and Behavioral Neuroscience Institute, University of Michigan Medical School, 5041 BSRB, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA e-mail:a3lrd2FuQHVtaWNoLmVkdQ==