 Sara H. Sinclair
Sara H. Sinclair Kristen E. Rennoll-Bankert
Kristen E. Rennoll-Bankert J. S. Dumler
J. S. Dumler- 1Graduate Program in Cellular and Molecular Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2Department of Microbiology and Immunology, School of Medicine, University of Maryland Baltimore, Baltimore, MD, USA
- 3Department of Pathology, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 4Department of Pathology, School of Medicine, University of Maryland Baltimore, Baltimore, MD, USA
Obligate intracellular pathogenic bacteria evolved to manipulate their host cells with a limited range of proteins constrained by their compact genomes. The harsh environment of a phagocytic defense cell is one that challenges the majority of commensal and pathogenic bacteria; yet, these are the obligatory vertebrate homes for important pathogenic species in the Anaplasmataceae family. Survival requires that the parasite fundamentally alter the native functions of the cell to allow its entry, intracellular replication, and transmission to a hematophagous arthropod. The small genomic repertoires encode several eukaryotic-like proteins, including ankyrin A (AnkA) of Anaplasma phagocytophilum and Ank200 and tandem-repeat containing proteins of Ehrlichia chaffeensis that localize to the host cell nucleus and directly bind DNA. As a model, A. phagocytophilum AnkA appears to directly alter host cell gene expression by recruiting chromatin modifying enzymes such as histone deacetylases and methyltransferases or by acting directly on transcription in cis. While cis binding could feasibly alter limited ranges of genes and cellular functions, the complex and dramatic alterations in transcription observed with infection are difficult to explain on the basis of individually targeted genes. We hypothesize that nucleomodulins can act broadly, even genome-wide, to affect entire chromosomal neighborhoods and topologically associating chromatin domains by recruiting chromatin remodeling complexes or by altering the folding patterns of chromatin that bring distant regulatory regions together to coordinate control of transcriptional reprogramming. This review focuses on the A. phagocytophilum nucleomodulin AnkA, how it impacts host cell transcriptional responses, and current investigations that seek to determine how these multifunctional eukaryotic-like proteins facilitate epigenetic alterations and cellular reprogramming at the chromosomal level.
Introduction
In order to infect mammalian hosts, bacterial pathogens evolved an array of mechanisms that serve to create an environment conducive for survival, replication, and spread. While many bacterial species survive in an extracellular environment, intracellular pathogens must be capable of both entering their host cells undetected and altering the cellular milieu in order to replicate. Traditionally, the ability of bacterial-derived proteins to induce disruptions of cell signaling or major cellular processes such as NF-κB, MAPK, and JAK/STAT pathways, are the predominant focus of host–pathogen interaction studies (Brodsky and Medzhitov, 2009). Recently, there has been an increasing interest in the ability of these intracellular pathogens to direct alterations in host cell gene expression that promote survival and replication (Paschos and Allday, 2010; Bierne et al., 2012; Silmon de Monerri and Kim, 2014). It is now well recognized that bacterial pathogens can reprogram host gene expression either directly or indirectly, by altering the accessibility of gene promoters via epigenetic modifications.
Eukaryotic DNA is highly organized and gene expression tightly regulated by an orchestrated network of proteins, RNAs, and other modulators. Histone octamers, or nucleosomes, organize DNA by acting as a bobbin on which the DNA winds. The charge of the histone proteins can be covalently modified in order to more tightly or loosely associate with DNA. Histone acetylation, which imparts a negative charge, is predominantly associated with an open configuration where promoters are easily accessed by RNA polymerases. Histone methylation or phosphorylation which impart a positive charge, cause DNA to more tightly associate with histone proteins, reducing promoter accessibility to transcription activating machinery. The process of modulating open and closed chromatin is further complicated by (i) the residue(s) of which histone protein(s) is/are modified, (ii) methylation of cytosine residues in DNA, and (iii) non-coding RNAs, and by the manner in which these mechanisms are intertwined. It therefore is no surprise that bacterial-derived proteins have evolved to interfere with host gene expression that improves bacterial fitness.
Over the last decade, examples of secreted bacterial effector proteins, ranging from those produced by Listeria monocytogenes, Chlamydia trachomatis, Shigella flexneri, and others, were found to target the host cell nucleus (Paschos and Allday, 2010; Bierne et al., 2012; Silmon de Monerri and Kim, 2014). These discoveries demonstrate the relationship of altered transcription and function of infected host cells and microbial survival, and in some cases, pathogenicity. S. flexneri, a bacterial pathogen that can cause dysentery, prevents NF-κB from binding its target gene promoters by altering the phosphorylation state of histone H3 at serine 10. The bacterium does so by secreting outer membrane protein F (OspF) that de-phosphorylates MAPKs in the nucleus resulting in a lack of histone H3 phosphorylation at serine 10 at a number of NF-κB-dependent genes (Arbibe et al., 2007). By altering NF-κB target gene transcription, S. flexneri suppresses the host cell inflammatory response promoting microbial survival and transmission (Arbibe et al., 2007). L. monocytogenes nuclear targeted protein A (LntA), blocks binding of heterochromatin inducing protein BAHD1 at interferon-stimulated genes resulting in upregulated expression (Rohde, 2011). Nuclear effector E (NUE) of C. trachomatis and RomA of L. pneumophila have methyltransferase activity and induce methylation of eukaryotic histones and altered host cell gene expression (Pennini et al., 2010; Rolando et al., 2013). Plant pathogens in the genus Xanthomonas, as well as Ralstonia solanacearum, Burkholderia rhizoxinica, and an endosymbiont of the plant pathogenic fungus Rhizopus microsporus all possess genes encoding transcription activator-like (TAL) protein effectors that when expressed, bind plant DNA and alter transcription and disease susceptibility (Sugio et al., 2007; Bogdanove, 2014). These examples are part of an expanding array of bacterial-derived proteins termed nucleomodulins that target host cell chromatin or chromatin-linked pathways to alter transcription, typically at one or a few host genes. To date, the only prokaryotic nucleomodulins shown to directly bind mammalian DNA and influence surrounding chromatin are from the Anaplasmataceae family. Ankyrin A (AnkA) of Anaplasma phagocytophilum, as well as Ank200 and several tandem-repeat containing proteins (TRPs) from Ehrlichia chaffeensis, have been shown to enter the nucleus and bind DNA, and interact with host epigenetic machinery or alter nearby histone octamers (Garcia-Garcia et al., 2009a; Luo et al., 2011; Zhu et al., 2011; Luo and McBride, 2012; Dunphy et al., 2013).
Targeting of individual genes or binding of an effector at a single chromatin region or small numbers of promoter loci could, in theory, lead to cis regulation of those loci and in part explain transcriptional alterations induced by infection. However, the degree of transcriptional alterations and the coordination of these events that dramatically affect cellular function programs are unlikely to be explained by individual targets given the complexity and repertoire of the human genome, at approximately 3251 Mb vs. genome sizes of S. flexneri (≤4.83 Mb; Jin et al., 2002), C. trachomatis (≤1.04 Mb; Stephens et al., 1998), L. monocytogenes (≤3.00 Mb; Evans et al., 2004), L. pneumophila (≤3.40 Mb; Chien et al., 2004), and A. phagocytophilum (≤1.47 Mb; Lin et al., 2011). We hypothesize that nucleomodulins can act broadly, even genome-wide, to affect entire chromosomal neighborhoods and topologically associating chromatin domains by recruiting chromatin remodeling complexes or by altering the folding patterns of chromatin that bring distant regulatory regions together to coordinate control of transcriptional reprogramming. This review will discuss the current knowledge of A. phagocytophilum subversion of host cells by nucleomodulins and how AnkA could play an even larger role than its documented effects on transcription.
Anaplasma phagocytophilum
Anaplasma phagocytophilum, transmitted by Ixodes spp. ticks, was discovered as the causative agent of human granulocytic anaplasmosis (HGA) in 1990 (Chen et al., 1994). While the infection is usually subclinical, manifestations in humans range from mild fever to severe infection requiring intensive care or even death (Chen et al., 1994). A. phagocytophilum is a Gram negative, obligate intracellular bacterium that is a parasite of neutrophils in mammalian hosts. Owing to their roles in protective inflammatory and immune responses toward microbial infections and their innate ability to recognize and kill pathogens, neutrophils are unlikely host cells for any microorganism. Yet, A. phagocytophilum colonizes these cells and thwarts their normal functions to create a hospitable environment for intracellular replication and subsequent transmission via tick bite. With a limited genome of approximately 1.5 Mb (Lin et al., 2011) the potential genetic reservoir for controlling an infected host cell, whether in the tick or mammal, is only a fraction of the tick or human genome. This simple observation suggests that to circumvent such extremes, its control mechanisms must be highly efficient, multifunctional, or target master regulators or similar checkpoints in eukaryotic cells.
One remarkable feature of A. phagocytophilum-infected cells is the marked change in transcriptional profiles that belies aberrant regulation of many key host pathways (Borjesson et al., 2005; de la Fuente et al., 2005; Lee et al., 2008). A. phagocytophilum-infected neutrophils are characterized by an “activated-deactivated” phenotype with major functional aberrations including decreased respiratory burst, delayed apoptosis, reduced transmigration across endothelial cell barriers, and decreased antimicrobial activities such as phagocytosis and microbial killing, while simultaneous increases in degranulation of vesicle contents including proteases and increased production of chemokines, are observed (Carlyon et al., 2002; Carlyon and Fikrig, 2003; Choi et al., 2005; Dumler et al., 2005; Garyu et al., 2005; Ge et al., 2005; Carlyon and Fikrig, 2006; Garcia-Garcia et al., 2009b). The net result of these changes is an increase in (i) inflammatory responses that recruit new host cells, (ii) prolonged survival of infected cells, (iii) an inability to kill internalized microbes, and (iv) net sequestration of infected cells within the intravascular compartment that is more readily accessed by the next tick bite (Rennoll-Bankert and Dumler, 2012). In fact, interference with any of these processes using in vitro and in vivo models leads to reduced fitness of the microbe underscoring how important these functional changes are in mammalian hosts.
The biological basis for such dramatic changes in neutrophil function is increasingly studied by methods that range from examination of individual pathways to genome-wide systems biology approaches. Transcriptional profiling of infected neutrophils and HL-60 cells, the latter a commonly used cell model for A. phagocytophilum infection, reveals altered transcription genome-wide, confirming that changes in transcription are not restricted to a few genes and limited cellular functions, but likely play a role in most of the known functional changes induced by infection (Borjesson et al., 2005; de la Fuente et al., 2005; Lee et al., 2008). Transcriptional downregulation of two components of the NADPH oxidase, CYBB encoding NOX2 or gp91phox and the GTPase RAC2, that assemble in the membranes of activated cells to generate superoxide production and promote microbial killing plays a role in prolonged inhibition of respiratory burst (Banerjee et al., 2000; Carlyon et al., 2002). In addition, delayed apoptosis is achieved by maintained transcription of BCL2 family members (Choi et al., 2005; Ge et al., 2005) and increased proinflammatory responses are due to upregulated transcription of cytokine and chemokine genes such as IL8 (Klein et al., 2000; Akkoyunlu et al., 2001; Scorpio et al., 2004).
While many studies document these changes at functional and transcriptional levels, the precise mechanisms that organize and coordinate alterations to support improved A. phagocytophilum fitness are not well addressed. The biggest advance in understanding these processes occurred with the discovery of secreted prokaryotic effector proteins and their secretion systems. The genome of A. phagocytophilum encodes a type IV secretion system (T4SS) that allows protein effectors to translocate into infected host cells where they likely act by mechanisms similar to those of other bacterial secreted effectors that target cytosolic pathways such as signal transduction, cytoskeletal rearrangements, intracellular trafficking, etc. (Ohashi et al., 2002; IJdo et al., 2007; Gillespie et al., 2009). These observations also hold true for A. phagocytophilum effectors. However, not all effector proteins remain localized within the host cytosol, and those that enter the nucleus have direct access to genes as well as to a distinct and diverse array of proteins that could impact cellular function on a global scale.
AnkA of Anaplasma phagocytophilum
Anaplasma phagocytophilum expresses a number of major immunoreactive proteins, including major surface protein 2/p44 (Msp2/p44) and AnkA, the latter of which was found by Caturegli et al. (2000) using immunoelectron microscopy to be transported into the host cell nucleus and bound within heterochromatin (Park et al., 2004). AnkA contains many EPIYA motifs that become tyrosine phosphorylated and recruit SHP-1 and ABL1 when introduced into mammalian cells which in turn regulates endosomal entry and intracellular infection (IJdo et al., 2007; Lin et al., 2011). Aside from this, AnkA contains multiple eukaryotic motifs including 8–15 or more ankyrin repeats, a putative bipartite nuclear localization signal (NLS), and a putative high mobility group N-chromatin unfolding domain (HMGN-CHUD; Caturegli et al., 2000). The ankyrin repeat domains are organized tandemly, creating highly stable spring-like structures that allow protein–protein or protein–DNA interactions and are commonly found in transcription factors and their regulatory proteins (Bork, 1993; Jernigan and Bordenstein, 2014). HMGN-CHUD domains facilitate binding to nucleosomes to alter chromatin structure and transcription of surrounding genes (Bustin, 1999). The presence of these motifs lends credence to the idea that AnkA plays a role in altering neutrophil transcription.
Park et al. (2004) confirmed that AnkA directly binds both DNA and nuclear proteins, and provided limited evidence of its capacity to bind broadly throughout the human genome. AnkA binds to regions at the promoter of CYBB, encoding the gp91phox component of phagocyte oxidase (Garcia-Garcia et al., 2009b). CYBB is known to be transcriptionally repressed with A. phagocytophilum infection, further suggesting that AnkA acts in cis to alter gene transcription (Thomas et al., 2005; Garcia-Garcia et al., 2009b). Moreover, transcription of CYBB is decreased in a dose dependent manner as nuclear AnkA concentrations increase (Garcia-Garcia et al., 2009b). When AnkA-expressing plasmids are transfected into HL-60 cells, CYBB expression is dampened, strengthening the link to transcriptional regulation (Garcia-Garcia et al., 2009b). In electrophoretic mobility shift assays, AnkA binds to the CYBB promoter at the same locations as other known transcriptional regulators such as CAATT displacement protein (CDP) and special-AT rich binding protein-1 (SATB1; Thomas et al., 2005; Garcia-Garcia et al., 2009b; Rennoll-Bankert and Dumler, 2012). Surprisingly, DNA binding by AnkA does not target a conserved DNA sequence motif or signature; rather it binds to regions rich in AT nucleotides that have specific structural qualities, including the ability to uncoil under superhelical stress (Park et al., 2004; Garcia-Garcia et al., 2009b). The latter feature is often observed in eukaryotic proteins that tether DNA strands together into the nuclear matrix at matrix attachment regions (MARs) to regulate transcription from distantly located but functionally related chromosomal regions; proteins that bind these regions are called MAR-binding proteins (MARBPs).
Interestingly, Garcia-Garcia et al. (2009a) showed that multiple downregulated defense genes in infected granulocytes were clustered on chromosomes. The spatial clustering of similarly regulated genes suggests that, in addition to cis-regulation as with CYBB, long ranges of chromosomes are affected – a process often attributed to epigenetic mechanisms such as DNA methylation and histone chromatin modifications. Using chromatin immunoprecipitation (ChIP) to investigate histone marks at defense gene promoters, acetylation of histone H3 was dramatically reduced, a finding often associated with silenced gene transcription (Garcia-Garcia et al., 2009a). To explain this, binding of histone deacetylase-1 (HDAC1) was found increased across multiple defense gene promoters (Garcia-Garcia et al., 2009a). Moreover, A. phagocytophilum-infected granulocytes have increased HDAC activity most likely explained by the increased quantity of both HDAC1 and HDAC2. Inhibition of HDAC1, but not HDAC2 expression by siRNA or pharmacologic inhibition of HDAC activity impairs A. phagocytophilum propagation, whereas overexpression leads to increased intracellular propagation (Garcia-Garcia et al., 2009a). We modeled this process by using the wild type CYBB promoter, to which AnkA binds, and mutated forms unable to bind AnkA in order to demonstrate that AnkA binding leads to HDAC1 recruitment and silencing of expression at CYBB (Garcia-Garcia et al., 2009b). These results clearly link AnkA with HDAC1 toward facilitating widespread downregulation of antimicrobial responses.
Currently, most data examining the ability of HDAC1 and AnkA to modulate transcription focuses on a small number of loci, including CYBB and up to 17 other gene promoters. While AnkA and HDAC1 are important contributors to transcriptional downregulation of some genes in A. phagocytophilum-infected cells, neither has been determined to provide a broad mechanism that coordinates the transcriptional or functional alterations observed. Importantly, transcriptional profiling of A. phagocytophilum-infected granulocytes, including both primary neutrophils and cell lines, demonstrates that the majority of differentially expressed genes are upregulated (Borjesson et al., 2005). Thus, while HDAC1 recruitment by AnkA is important for down-regulating some genes, the majority of DEGs are likely regulated by alternative or additional mechanisms.
Of considerable interest is the interplay between chromatin structure and DNA methylation. Histone modifications, DNA methylation at CpG islands, and binding of methyl CpG DNA-binding proteins (MeCPs) via their methyl-DNA binding domains (MBDs) occur in concert during biological responses including neoplasia and cellular differentiation (Baylin and Herman, 2000; Baylin et al., 2001; Stirzaker et al., 2004; Srivastava et al., 2010; Reddington et al., 2013). Cytosine residues of CpG dinucleotides can by methylated by DNA methyltransferases (DNMTs) leading to sustained DNA methylation over multiple generations of cell divisions. It is believed in part that the methylation protects genes and transcriptional programs from inappropriate ectopic expression as cells enter various stages of tissue differentiation (Srivastava et al., 2010). DNMT1 is highly conserved and is thought to be primarily responsible for maintaining existing methyl CpGs (Baylin et al., 2001). In contrast, DNMT3a and 3b are believed to undergo transient de novo expression that induces CpG methylation in response to cellular stimuli (Okano et al., 1999). MBDs can recognize these newly methylated regions of DNA and are often associated with HDACs in MeCP1 and MeCP2 complexes, illustrating the potential for a direct link between DNA methylation and alterations in chromatin structure (Jones et al., 1998; Nan et al., 1998; Ng et al., 1999). Generally, hypermethylation of gene promoters occurs synchronously with histone deacetylation and decreased gene expression of many tumor suppressor genes (Baylin and Herman, 2000; Baylin et al., 2001; Stirzaker et al., 2004). This complex interplay has been best studied in cancer genomes, where it is currently unclear whether DNA methylation induces alterations in chromatin structure or if chromatin structure induces changes in DNA methylation. However, given the interplay between HDAC activity and DNA methylation, it is not unreasonable to hypothesize that A. phagocytophilum infection induces hypermethylation of the host genome during the process of global transcriptome reprogramming (Figure 1).
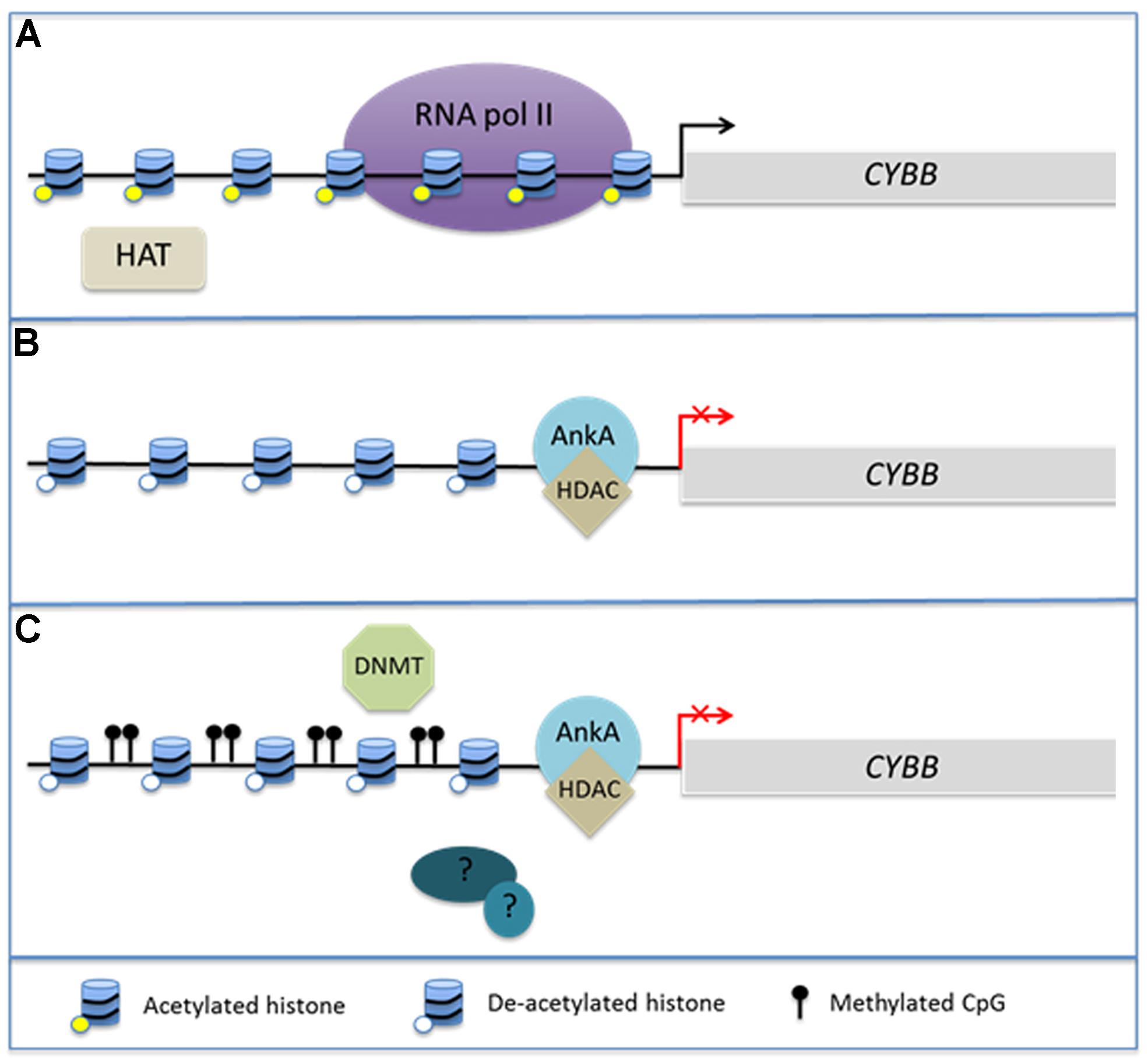
FIGURE 1. AnkA alters chromatin structure at the CYBB promoter. During Anaplasma phagocytophilum infection, AnkA accumulates in the host cell nucleus where it can directly bind DNA and influence the transcription of CYBB, which encodes the gp91phox component of the NADPH oxidase. (A) In the absence of infection, CYBB is activated by inflammatory signals and is easily transcribed. (B) During infection, AnkA binds the proximal promoter and recruits HDAC which deacetylates nearby histones in order to inhibit transcription. (C) We propose that additional host or bacterial-derived chromatin modifying enzymes, such as DNMT methylation of host CpGs, may also be involved in altering the host epigenome to the bacterium’s advantage.
In addition to chromatin alterations induced by HDACs, MARs are responsible for dictating the three-dimensional architecture of chromatin loops and serve as tethering points for DNA to the nuclear matrix (Spector, 2003; Kumar et al., 2007; Kohwi-Shigematsu et al., 2012). Arranging chromatin into loops allows transcription factors attached to the matrix access to promoters and brings distal genomic loci and regulatory regions into a position of proximity for coordinated regulation (Arope et al., 2013). Interestingly, the AT-rich DNA docking sites of AnkA are similar to those of MARs. In fact, several transcription factors including SATB1, bind MARs to coordinately modulate transcription from large genomic regions (Kumar et al., 2007). Like AnkA, SATB1 occupies the CYBB promoter during myeloid differentiation where it represses the transcription of CYBB, is involved in maintaining expression of BCL2 (Gong et al., 2010) which is transcriptionally sustained during A. phagocytophilum infection (Ge et al., 2005) and is implicated in activation of cytokine expression in T cells (Hawkins et al., 2001; Beyer et al., 2011). SATB1 interactions with chromatin remodeling complexes include HDACs which are likely involved in some of these processes (Yasui et al., 2002). The complex looping of chromatin facilitated by MARs and MAR binding proteins allows for entire chromatin domains and territories to be remodeled despite relatively few binding sites (Cai et al., 2006). For example, SATB1 binds to only nine regions across the 200 kb TH2 locus, yet it is a major regulator of TH2 lymphocyte differentiation and function (Cai et al., 2006). Complex protein interactions involving chromatin remodeling and histone modification mediated by anchoring proteins like SATB1 make it plausible that DNA-binding bacterial nucleomodulins such as AnkA could target broad transcriptional programs that belie functions of host cells. While only limited data currently exist, AnkA binds to at least 23 distinct sites on 12 separate chromosomes with A. phagocytophilum infection of the human HL-60 promyelocytic cell line (Park et al., 2004) detailed mapping of AnkA genomic binding sites is now in progress. We therefore think it is plausible that AnkA binds throughout the genome and exerts its effects, like SATB1, to both repress and activate transcription by tethering chromatin to the nuclear matrix and exposing promoters to chromatin remodeling complexes.
Anaplasma phagocytophilum Infection is Associated with Transcriptional Changes Over Megabases in the Human Genome
As a relatively new field, studies that examine the epigenome of cells infected by or as a consequence of bacterial infection, whether parasitic or symbiotic, are few and far between. Given the proven interaction of A. phagocytophilum AnkA with gene promoters, epigenetic alterations of nearby chromatin, and its MAR-binding attributes, we reanalyzed publicly available transcription microarray data generated from A. phagocytophilum-infected human peripheral blood neutrophils (Borjesson et al., 2005). We used updated bioinformatics tools, including an analysis of the SD estimates for differential transcription at each locus, and wherever possible, improved gene and gene feature localization using the NCBI GRCh38 human genome assembly (release date December 24, 2013). The differential expression of all 18,400 interrogated transcripts and variants, including 14,500 well-characterized human genes, was mapped by chromosomal position so that the relationship between the linear chromosome landscape and transcriptional activity could be visualized and investigated. To display physical locations on chromosomes with long regions of altered transcription, the fold change of expression for each locus was averaged with the nearest neighboring genes to create sliding windows for each chromosome. Overall, the median sliding window included 10.0 genes/gene features (IQR 2.0) spanning a median interval of 2.04 Mb (IQR 2.16) corresponding approximately to the lower end dimensions of chromosomal or gene expression dysregulation domains (Letourneau et al., 2014). The sliding window average along with individual gene transcriptional fold changes were plotted for each chromosome. To assess significance of fold changes in the sliding window, a similar sliding window was created to simultaneously show the average estimated SDs over each window.
All chromosomes showed linear regions of marked differential transcription covering megabases in scale on both p and q arms (Figure 2). In keeping with the original observations of Borjesson et al., most clusters were upregulated; however, the analysis also demonstrated a similar pattern of downregulated genes over long linear regions. Of interest was chromosome 17 with a 9.4 Mb cluster containing myeloid peroxidase (MPO), eosinophil peroxidase (EPX), and lactoperoxidase (LPO) which showed marked downregulation, and a 3.2 Mb cluster containing multiple upregulated chemokine genes (Figure 3). In addition to reduced transcript levels, MPO and EPX promoters were previously shown to be deacetylated upon A. phagocytophilum infection (Garcia-Garcia et al., 2009a). Furthermore, a 3.6 Mb cluster on chromosome 6 containing FLOT1, TNF, and the major histocompatibility complex (MHC) was upregulated (Figure 4). The chromosomal landscape of the MHC locus is well documented with respect to chromatin looping via MARS and histone marks (Ottaviani et al., 2008). It is difficult to determine with certainty whether these changes are the result of manipulation by microbial effectors like AnkA, or whether these represent host cellular responses to infection, or an amalgam of both. The limited genome-wide transcriptional alterations originally documented with exposure to heat-killed A. phagocytophilum argue for the former (Borjesson et al., 2005). Regardless, these observations clearly suggest that A. phagocytophilum infection targets large chromosomal territories to induce transcriptional alterations, in addition to specific genes. Given that HL-60 cells transfected to express AnkA have nearly identical differential gene expression patterns (Garcia-Garcia et al., 2009b) and reductions in induced respiratory burst compared to A. phagocytophilum-infected cells, a hypothesis that prokaryotic nucleomodulins evolved to modulate cell function by epigenetic alterations is compelling.
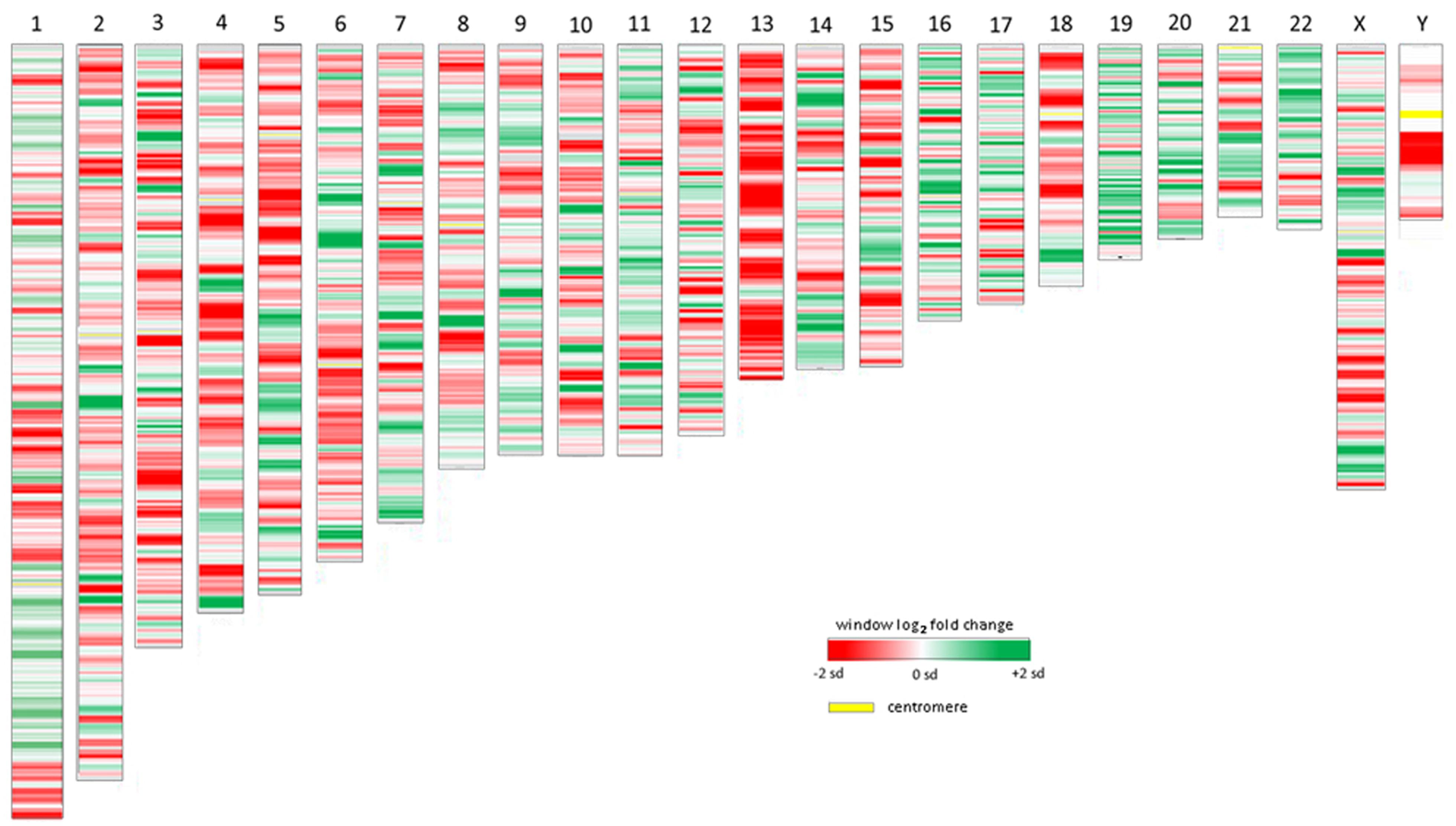
FIGURE 2. Heat map demonstrations of long linear chromosomal differential transcription of A. phagocytophilum-infected human neutrophils vs. uninfected neutrophils. The human chromosome number is shown at the top. The colors represent estimates of the SD of differential transcription of the window at each position compared to the overall differential transcription> for that chromosome; dark red = -2 SDs; dark green = +2 SDs.
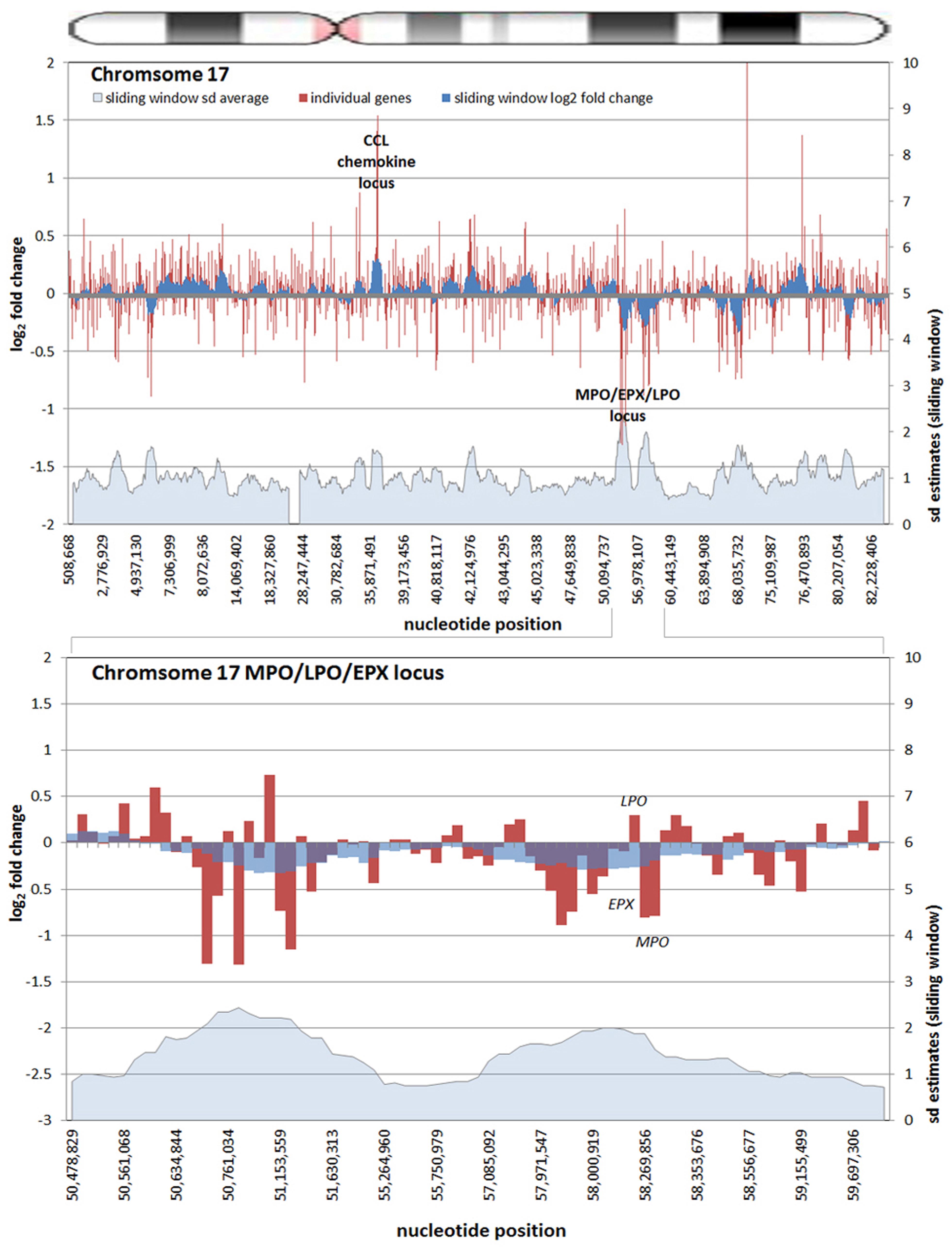
FIGURE 3. Differential gene expression patterns cluster over large linear genomic regions in A. phagocytophilum-infected human peripheral blood neutrophils. The top panel shows human chromosome 17; the bottom panel shows the MPO/EPX/LPO locus on human chromosome 17 and the large genomic region that is upregulated with infection. The chromosome ideogram is shown at the top. Red bars (left axes) represent differential transcription of individual genes, including some replicates; the dark blue zones (left axes) show the sliding window average log2-fold differential transcription over the contiguous 9–11 genes (or 0.38–1.25 Mb). The light blue zone at the bottom (right axis) shows the sliding window average over the same region for estimated SDs of log2-fold differential transcription at each gene or gene feature. Data re-analyzed from Borjesson et al. (2005) www.ncbi.nlm.nih.gov/geo, accession no. GSE2405.
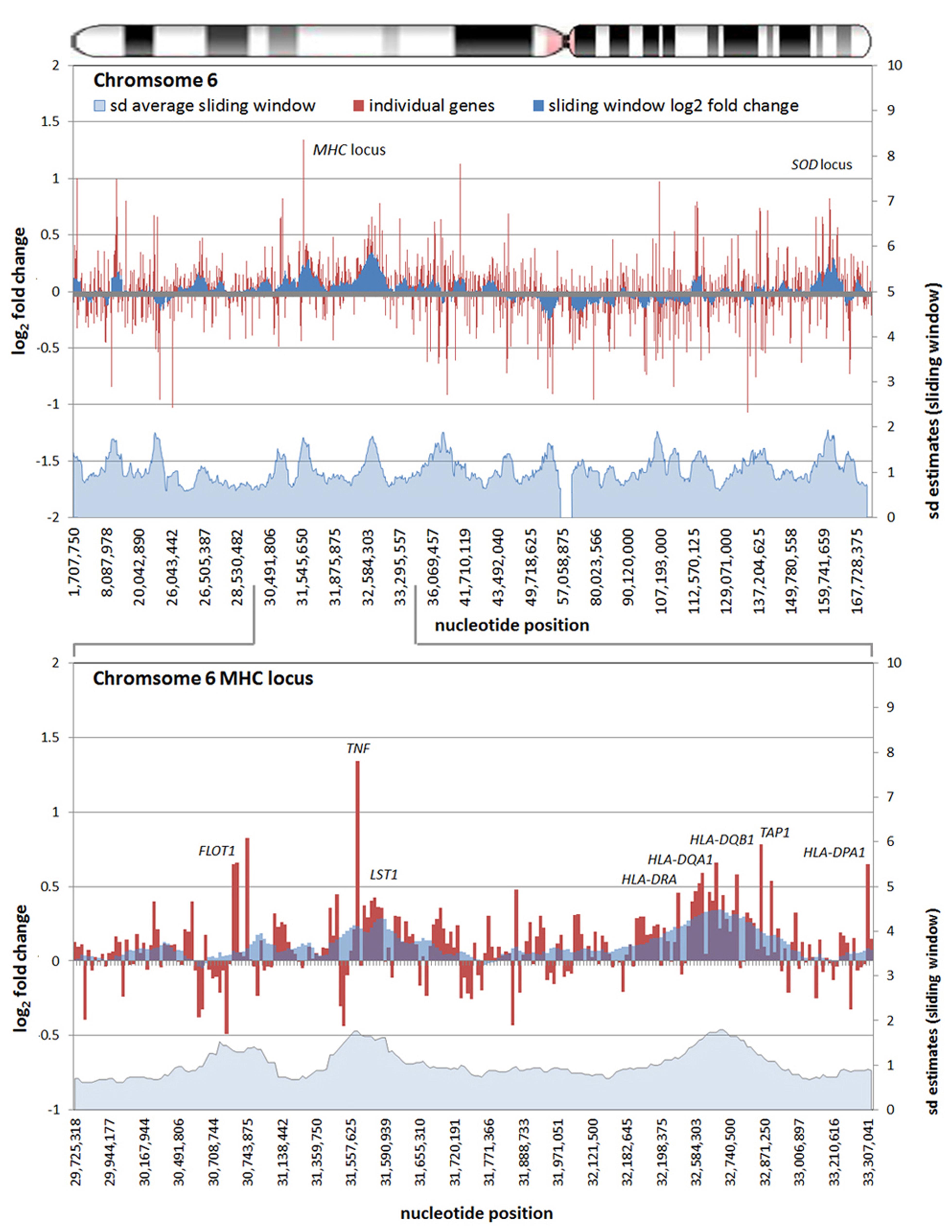
FIGURE 4. Differential gene expression patterns comparing A. phagocytophilum-infected vs. non-infected human peripheral blood neutrophils are clustered over large linear genomic regions. The top panel shows human chromosome 6; the bottom panel shows the MHC locus on human chromosome 6 including a large genomic region spanning the TNF and HLAD loci that are upregulated with infection. The chromosome ideogram is shown at the top. Red bars (left axes) represent differential transcription of individual genes, including some replicates, the dark blue zones (left axes) show the sliding window average log2-fold differential transcription over the contiguous 9–12 genes (or 0.42–3.24 Mb). The light blue zone at the bottom (right axis) shows the sliding window average over the same region for estimated SDs of log2-fold differential transcription at each gene or gene feature. Data re-analyzed from Borjesson et al. (2005) www.ncbi.nlm.nih.gov/geo, accession no. GSE2405.
Conclusion and Future Directions
Given the limited nature of bacterial genome repertoire, and the expansive genome repertoire, organization and complexity of transcriptional regulation in eukaryotes, the ability of prokaryotes to alter eukaryotic host cell functions at the epigenetic level is truly a remarkable phenomenon. The processes by which bacteria-derived proteins alter host cell signaling, endocytic or vesicular trafficking, and cis transcription of genes are important. However, these pathways become challenging as paradigms that can account for the marked and diverse changes in host cell transcription and functions during the course of infection. We believe that successful intracellular prokaryotes, whether parasitic and pathogenic, mutualistic or symbiotic, evolved multifunctional and complex proteins with broad effects that target key master regulators or checkpoints that regulate major cellular reprogramming events. The A. phagocytophilum genome is a fraction of the size of its human host, yet it manages to disrupt core functions of the cell by transforming the transcriptome and reprogramming the cell for improved microbial fitness, survival, and transmission. It is increasingly apparent that A. phagocytophilum and perhaps other intracellular prokaryotes manipulate their hosts with a high degree of efficacy, but by altering the “on” or “off” state of genes in a cis only fashion or targeting individual or small numbers of signaling pathways is unlikely to account for this alone, in essence, an “effector bottleneck”. An analysis of available infection transcriptome data demonstrates that large chromosomal territories appear to be coordinately regulated – up or down. This observation, along with HDAC recruitment by AnkA, lends strong evidence to the idea that A. phagocytophilum induces global changes in host gene expression via a broad mechanism that involves epigenetic regulation of transcriptional programs which belie cellular functions.
It is currently unclear as to whether HDAC recruitment is the predominant mechanism by which AnkA exerts its chromatin modulating effects, including whether there are other host factors [e.g., polycomb repressive or hematopoietic associated factor-1 (HAF1) complexes] or additional bacterial-derived nucleomodulins that further contribute to the dramatic changes observed. To examine this, we designed a genome-level bioinformatics approach to predict nucleomodulins based on the combination of secretory and NLSs and studied the genomes of 12 phylogenetically diverse intracellular prokaryotic pathogens, including Chlamydia pneumoniae, Mycobacterium tuberculosis, and Yersinia pestis among others, and identified between 7 and 35 candidates for each (Borroto et al., 2009). Among these and by using iTRAQ proteomics approaches, we identified other proteins encoded in the A. phagocytophilum genome that localize to the host cell nucleus and are examining whether these too contribute to chromosomal structure alterations or work in synergy to enhance AnkA function (Gilmore et al., 2012). Elucidating the exact locations where AnkA or other nucleomodulins bind throughout the genome, and determining the architecture of the surrounding chromatin will be crucial to understanding this process. The interplay between HDACs, DNMTs, and methyl binding proteins suggests that A. phagocytophilum infection could also induce widespread DNA methylation perhaps as a mechanism for obtaining broad epigenetic changes and functional reprogramming.
Understanding the role of prokaryotic control over complex eukaryotic transcription machinery in lieu of the bottleneck that would occur with single effector targets at signaling pathways will allow for better understanding of the essential components of whole cell transcriptional re-programming. However, this has implications not only for host–pathogen or host–symbiont interactions, but across biology. The ability of proteins like AnkA of A. phagocytophilum and OspF of S. flexneri to alter inflammatory responses could provide opportunities for engineering of therapeutic agents which interfere with cellular responses in inflammatory diseases or other conditions where epigenetic factors control or contribute to disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by grant R01AI044102 from the US National Institutes of Allergy and Infectious Diseases, National Institutes of Health and by Mid-Atlantic Regional Center for Excellence in Biodefense and Emerging Infectious Diseases (MARCE) Career Development award 0002374. The authors thank J. C. Garcia-Garcia for creating the algorithm to identify potential nucleofactors in prokaryotic genomes and for work on iTRAQ studies.
References
Akkoyunlu, M., Malawista, S. E., Anguita, J., and Fikrig, E. (2001). Exploitation of interleukin-8-induced neutrophil chemotaxis by the agent of human granulocytic ehrlichiosis. Infect. Immun. 69, 5577–5588. doi: 10.1128/IAI.69.9.5577-5588.2001
Arbibe, L., Kim, D. W., Batsche, E., Pedron, T., Mateescu, B., Muchardt, C.,et al. (2007). An injected bacterial effector targets chromatin access for transcription factor NF-κB to alter transcription of host genes involved in immune responses. Nat. Immunol. 8, 47–56. doi: 10.1038/ni1423
Arope, S., Harraghy, N., Pjanic, M., and Mermod, N. (2013). Molecular characterization of a human matrix attachment region epigenetic regulator. PLoS ONE 8:e79262. doi: 10.1371/journal.pone.0079262
Banerjee, R., Anguita, J., Roos, D., and Fikrig, E. (2000). Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J. Immunol. 164, 3946–3949. doi: 10.4049/jimmunol.164.8.3946
Baylin, S. B., Esteller, M., Rountree, M. R., Bachman, K. E., Schuebel, K., and Herman, J. G. (2001). Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 10, 687–692. doi: 10.1093/hmg/10.7.687
Baylin, S. B., and Herman, J. G. (2000). DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 16, 168–174. doi: 10.1016/S0168-9525(99)01971-X
Beyer, M., Thabet, Y., Muller, R. U., Sadlon, T., Classen, S., Lahl, K.,et al. (2011). Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat. Immunol. 12, 898–907. doi: 10.1038/ni.2084
Bierne, H., Hamon, M., and Cossart, P. (2012). Epigenetics and bacterial infections. Cold Spring Harb. Perspect. Med. 2:a010272. doi: 10.1101/cshperspect.a010272
Bogdanove, A. J. (2014). Principles and applications of TAL effectors for plant physiology and metabolism. Curr. Opin. Plant Biol. 19C, 99–104. doi: 10.1016/j.pbi.2014.05.007
Borjesson, D. L., Kobayashi, S. D., Whitney, A. R., Voyich, J. M., Argue, C. M., and Deleo, F. R. (2005). Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J. Immunol. 174, 6364–6372. doi: 10.4049/jimmunol.174.10.6364
Bork, P. (1993). Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17, 363–374. doi: 10.1002/prot.340170405
Borroto, C. J., Cole R., Dumler, J. S., and Garcia-Garcia, J. C. (2009). Identification of candidate nuclear effectors among intracellular bacteria. Paper Presented at the Mid-Atlantic Regional Center for Excellence for Biodefense and Emerging Infectious Diseases [MARCE-2] Investigators’ Meeting, Airlie Center, Warrenton, VA.
Brodsky, I. E., and Medzhitov, R. (2009). Targeting of immune signalling networks by bacterial pathogens. Nat. Cell Biol. 11, 521–526. doi: 10.1038/ncb0509-521
Bustin, M. (1999). Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19, 5237–5246.
Cai, S., Lee, C. C., and Kohwi-Shigematsu, T. (2006). SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 38, 1278–1288. doi: 10.1038/ng1913
Carlyon, J. A., Chan, W. T., Galan, J., Roos, D., and Fikrig, E. (2002). Repression of Rac2 mRNA expression by Anaplasma phagocytophila is essential to the inhibition of superoxide production and bacterial proliferation. J. Immunol. 169, 7009–7018. doi: 10.4049/jimmunol.169.12.7009
Carlyon, J. A., and Fikrig, E. (2003). Invasion and survival strategies of Anaplasma phagocytophilum. Cell Microbiol. 5, 743–754. doi: 10.1046/j.1462-5822.2003.00323.x
Carlyon, J. A., and Fikrig, E. (2006). Mechanisms of evasion of neutrophil killing by Anaplasma phagocytophilum. Curr. Opin. Hematol. 13, 28–33. doi: 10.1097/01.moh.0000190109.00532.56
Caturegli, P., Asanovich, K. M., Walls, J. J., Bakken, J. S., Madigan, J. E., Popov, V. L.,et al. (2000). ankA: an Ehrlichia phagocytophila group gene encoding a cytoplasmic protein antigen with ankyrin repeats. Infect. Immun. 68, 5277–5283. doi: 10.1128/IAI.68.9.5277-5283.2000
Chen, S. M., Dumler, J. S., Bakken, J. S., and Walker, D. H. (1994). Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J. Clin. Microbiol. 32, 589–595.
Chien, M., Morozova, I., Shi, S., Sheng, H., Chen, J., Gomez, S. M.,et al. (2004). The genomic sequence of the accidental pathogen Legionella pneumophila. Science 305, 1966–1968. doi: 10.1126/science.1099776
Choi, K. S., Park, J. T., and Dumler, J. S. (2005). Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect. Immun. 73, 8209–8218. doi: 10.1128/IAI.73.12.8209-8218.2005
de la Fuente, J., Ayoubi, P., Blouin, E. F., Almazan, C., Naranjo, V., and Kocan, K. M. (2005). Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell Microbiol. 7, 549–559. doi: 10.1111/j.1462-5822.2004.00485.x
Dumler, J. S., Choi, K. S., Garcia-Garcia, J. C., Barat, N. S., Scorpio, D. G., Garyu, J. W.,et al. (2005). Human granulocytic anaplasmosis and Anaplasma phagocytophilum. Emerg. Infect. Dis. 11, 1828–1834. doi: 10.3201/eid1112.050898
Dunphy, P. S., Luo, T., and McBride, J. W. (2013). Ehrlichia moonlighting effectors and interkingdom interactions with the mononuclear phagocyte. Microbes Infect. 15, 1005–1016. doi: 10.1016/j.micinf.2013.09.011
Evans, M. R., Swaminathan, B., Graves, L. M., Altermann, E., Klaenhammer, T. R., Fink, R. C.,et al. (2004). Genetic markers unique to Listeria monocytogenes serotype 4b differentiate epidemic clone II (hot dog outbreak strains) from other lineages. Appl. Environ. Microbiol. 70, 2383–2390. doi: 10.1128/AEM.70.4.2383-2390.2004
Garcia-Garcia, J. C., Barat, N. C., Trembley, S. J., and Dumler, J. S. (2009a). Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLoS Pathog. 5:e1000488. doi: 10.1371/journal.ppat.1000488
Garcia-Garcia, J. C., Rennoll-Bankert, K. E., Pelly, S., Milstone, A. M., and Dumler, J. S. (2009b). Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect. Immun. 77, 2385–2391. doi: 10.1128/IAI.00023-09
Garyu, J. W., Choi, K. S., Grab, D. J., and Dumler, J. S. (2005). Defective phagocytosis in Anaplasma phagocytophilum-infected neutrophils. Infect. Immun. 73, 1187–1190. doi: 10.1128/IAI.73.2.1187-1190.2005
Ge, Y., Yoshiie, K., Kuribayashi, F., Lin, M., and Rikihisa, Y. (2005). Anaplasma phagocytophilum inhibits human neutrophil apoptosis via upregulation of Bfl-1, maintenance of mitochondrial membrane potential and prevention of caspase 3 activation. Cell Microbiol. 7, 29–38. doi: 10.1111/j.1462-5822.2004.00427.x
Gillespie, J. J., Ammerman, N. C., Dreher-Lesnick, S. M., Rahman, M. S., Worley, M. J., Setubal, J. C.,et al. (2009). An anomalous type IV secretion system in rickettsia is evolutionarily conserved. PloS ONE 4:e4833. doi: 10.1371/journal.pone.0004833
Gilmore, S. H., Garcia-Garcia, J. C., and Dumler, J. S. (2012). Identification of 6 novel nuclear translocated proteins in A. phagocytophilum. Paper Presented at the 25th Meeting of the American Society of Rickettsiology, Park City, UT, Abstract 130.
Gong, F., Sun, L., and Sun, Y. (2010). A novel SATB1 binding site in the BCL2 promoter region possesses transcriptional regulatory function. J. Biomed. Res. 24, 452–459. doi: 10.1016/S1674-8301(10)60060-7
Hawkins, S. M., Kohwi-Shigematsu, T., and Skalnik, D. G. (2001). The matrix attachment region-binding protein SATB1 interacts with multiple elements within the gp91phox promoter and is down-regulated during myeloid differentiation. J. Biol. Chem. 276, 44472–44480. doi: 10.1074/jbc.M104193200
IJdo, J. W., Carlson, A. C., and Kennedy, E. L. (2007). Anaplasma phagocytophilum AnkA is tyrosine-phosphorylated at EPIYA motifs and recruits SHP-1 during early infection. Cell Microbiol. 9, 1284–1296. doi: 10.1111/j.1462-5822.2006.00871.x
Jernigan, K. K., and Bordenstein, S. R. (2014). Ankyrin domains across the tree of life. Peer J. 2:e264. doi: 10.7717/peerj.264
Jin, Q., Yuan, Z., Xu, J., Wang, Y., Shen, Y., Lu, W.,et al. (2002). Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 30, 4432–4441. doi: 10.1093/nar/gkf566
Jones, P. L., Veenstra, G. J., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N.,et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191. doi: 10.1038/561
Klein, M. B., Hu, S., Chao, C. C., and Goodman, J. L. (2000). The agent of human granulocytic ehrlichiosis induces the production of myelosuppressing chemokines without induction of proinflammatory cytokines. J. Infect. Dis. 182, 200–205. doi: 10.1086/315641
Kohwi-Shigematsu, T., Kohwi, Y., Takahashi, K., Richards, H. W., Ayers, S. D., Han, H. J.,et al. (2012). SATB1-mediated functional packaging of chromatin into loops. Methods 58, 243–254. doi: 10.1016/j.ymeth.2012.06.019
Kumar, P. P., Bischof, O., Purbey, P. K., Notani, D., Urlaub, H., Dejean, A.,et al. (2007). Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC Class I locus. Nat. Cell Biol. 9, 45–56. doi: 10.1038/ncb1516
Lee, H. C., Kioi, M., Han, J., Puri, R. K., and Goodman, J. L. (2008). Anaplasma phagocytophilum-induced gene expression in both human neutrophils and HL-60 cells. Genomics 92, 144–151. doi: 10.1016/j.ygeno.2008.05.005
Letourneau, A., Santoni, F. A., Bonilla, X., Sailani, M. R., Gonzalez, D., Kind, J.,et al. (2014). Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–350. doi: 10.1038/nature13200
Lin, M., Kikuchi, T., Brewer, H. M., Norbeck, A. D., and Rikihisa, Y. (2011). Global proteomic analysis of two tick-borne emerging zoonotic agents: Anaplasma phagocytophilum and Ehrlichia chaffeensis. Front. Microbiol. 2:24. doi: 10.3389/fmicb.2011.00024
Luo, T., Kuriakose, J. A., Zhu, B., Wakeel, A., and McBride, J. W. (2011). Ehrlichia chaffeensis TRP120 interacts with a diverse array of eukaryotic proteins involved in transcription, signaling, and cytoskeleton organization. Infect. Immun. 79, 4382–4391. doi: 10.1128/IAI.05608-11
Luo, T., and McBride, J. W. (2012). Ehrlichia chaffeensis TRP32 interacts with host cell targets that influence intracellular survival. Infect. Immun. 80, 2297–2306. doi: 10.1128/IAI.00154-12
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N.,et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C. A., Turner, B. M., Erdjument-Bromage, H.,et al. (1999). MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 23, 58–61. doi: 10.1038/12659
Ohashi, N., Zhi, N., Lin, Q., and Rikihisa, Y. (2002). Characterization and transcriptional analysis of gene clusters for a type IV secretion machinery in human granulocytic and monocytic ehrlichiosis agents. Infect. Immun. 70, 2128–2138. doi: 10.1128/IAI.70.4.2128-2138.2002
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
Ottaviani, D., Lever, E., Mitter, R., Jones, T., Forshew, T., Christova, R.,et al. (2008). Reconfiguration of genomic anchors upon transcriptional activation of the human major histocompatibility complex. Genome Res. 18, 1778–1786. doi: 10.1101/gr.082313.108
Park, J., Kim, K. J., Choi, K. S., Grab, D. J., and Dumler, J. S. (2004). Anaplasma phagocytophilum AnkA binds to granulocyte DNA and nuclear proteins. Cell Microbiol. 6, 743–751. doi: 10.1111/j.1462-5822.2004.00400.x
Paschos, K., and Allday, M. J. (2010). Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 18, 439–447. doi: 10.1016/j.tim.2010.07.003
Pennini, M. E., Perrinet, S., Dautry-Varsat, A., and Subtil, A. (2010). Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis. PLoS Pathog. 6:e1000995. doi: 10.1371/journal.ppat.1000995
Reddington, J. P., Pennings, S., and Meehan, R. R. (2013). Non-canonical functions of the DNA methylome in gene regulation. Biochem. J. 451, 13–23. doi: 10.1042/BJ20121585
Rennoll-Bankert, K. E., and Dumler, J. S. (2012). Lessons from Anaplasma phagocytophilum: chromatin remodeling by bacterial effectors. Infect. Disord. Drug Targets 12, 380–387. doi: 10.2174/187152612804142242
Rohde, J. R. (2011). Microbiology. Listeria unwinds host DNA. Science (New York, N.Y.) 331, 1271–1272. doi: 10.1126/science.1203271
Rolando, M., Sanulli, S., Rusniok, C., Gomez-Valero, L., Bertholet, C., Sahr, T.,et al. (2013). Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 13, 395–405. doi: 10.1016/j.chom.2013.03.004
Scorpio, D. G., Akkoyunlu, M., Fikrig, E., and Dumler, J. S. (2004). CXCR2 blockade influences Anaplasma phagocytophilum propagation but not histopathology in the mouse model of human granulocytic anaplasmosis. Clin. Diagn. Lab. Immunol. 11, 963–968. doi: 10.1128/CDLI.11.5.963-968.2004
Silmon de Monerri, N. C., and Kim, K. (2014). Pathogens hijack the epigenome: a new twist on host–pathogen interactions. Am. J. Pathol. 184, 897–911. doi: 10.1016/j.ajpath.2013.12.022
Spector, D. L. (2003). The dynamics of chromosome organization and gene regulation. Annu. Rev. Biochem. 72, 573–608. doi: 10.1146/annurev.biochem.72.121801.161724
Srivastava, S., Mishra, R. K., and Dhawan, J. (2010). Regulation of cellular chromatin state: insights from quiescence and differentiation. Organogenesis 6, 37–47. doi: 10.4161/org.6.1.11337
Stephens, R. S., Kalman, S., Lammel, C., Fan, J., Marathe, R., Aravind, L. W.,et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science (New York, N.Y.) 282, 754–759. doi: 10.1126/science.282.5389.754
Stirzaker, C., Song, J. Z., Davidson, B., and Clark, S. J. (2004). Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res. 64, 3871–3877. doi: 10.1158/0008-5472.CAN-03-3690
Sugio, A., Yang, B., Zhu, T., and White, F. F. (2007). Two type III effector genes of Xanthomonas oryzae Pv. oryzae control the induction of the host genes OsTFIIAγ1 and OsTFX1 during bacterial blight of rice. Proc. Natl. Acad. Sci. U.S.A. 104, 10720–10725. doi: 10.1073/pnas.0701742104
Thomas, V., Samanta, S., Wu, C., Berliner, N., and Fikrig, E. (2005). Anaplasma phagocytophilum modulates gp91phox gene expression through altered Interferon Regulatory Factor 1 and PU.1 levels and binding of CCAAT displacement protein. Infect. Immun. 73, 208–218. doi: 10.1128/IAI.73.1.208-218.2005
Yasui, D., Miyano, M., Cai, S., Varga-Weisz, P., and Kohwi-Shigematsu, T. (2002). SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 419, 641–645. doi: 10.1038/nature01084
Keywords: epigenetics, nucleomodulin, DNA methylation, histone deacetylase, chromatin, Anaplasma phagocytophilum
Citation: Sinclair SH, Rennoll-Bankert KE and Dumler JS (2014) Effector bottleneck: microbial reprogramming of parasitized host cell transcription by epigenetic remodeling of chromatin structure. Front. Genet. 5:274. doi: 10.3389/fgene.2014.00274
Received: 17 June 2014; Paper pending published: 23 July 2014;
Accepted: 26 July 2014; Published online: 14 August 2014.
Edited by:
Ilaria Negri, University of Turin, ItalyCopyright © 2014 Sinclair, Rennoll-Bankert and Dumler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. S. Dumler, Departments of Pathology and Department of Microbiology and Immunology, School of Medicine, University of Maryland Baltimore, 685 West Baltimore Street, Health Sciences Facility-1 Room 322D, Baltimore, MD 21201, USA e-mail:c2R1bWxlckBzb20udW1hcnlsYW5kLmVkdQ==