 Abigail J. Renoux
Abigail J. Renoux- 1Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI, USA
- 2Department of Neurology, University of Michigan, Ann Arbor, MI, USA
The FMR1 protein product, FMRP, is an mRNA binding protein associated with translational inhibition of target transcripts. One FMRP target is the amyloid precursor protein (APP) mRNA, and APP levels are elevated in Fmr1 KO mice. Given that elevated APP protein expression can elicit Alzheimer’s disease (AD) in patients and model systems, we evaluated whether FMRP expression might be altered in Alzheimer’s autopsy brain samples and mouse models compared to controls. In a double transgenic mouse model of AD (APP/PS1), we found no difference in FMRP expression in aged AD model mice compared to littermate controls. FMRP expression was also similar in AD and control patient frontal cortex and cerebellum samples. Fragile X-associated tremor/ataxia syndrome (FXTAS) is an age-related neurodegenerative disorder caused by expanded CGG repeats in the 5′ untranslated region of the FMR1 gene. Patients experience cognitive impairment and dementia in addition to motor symptoms. In parallel studies, we measured FMRP expression in cortex and cerebellum from three FXTAS patients and found reduced expression compared to both controls and Alzheimer’s patient brains, consistent with animal models. We also find increased APP levels in cerebellar, but not cortical, samples of FXTAS patients compared to controls. Taken together, these data suggest that a decrease in FMRP expression is unlikely to be a primary contributor to Alzheimer’s disease pathogenesis.
Introduction
Fragile X-associated disorders result from intergenerational instability in a CGG microsatellite repeat expansion located in the 5′ untranslated region (UTR) of the fragile X mental retardation (FMR1) gene. In the general population, the mean repeat length is 30 CGG repeats with a range from 4 to 55 (Strom et al., 2007). When this repeat expands beyond this normal range, it can cause symptoms associated with fragile X spectrum disorders (Renoux and Todd, 2012; Nelson et al., 2013). Greater than 200 CGG repeats elicits transcriptional silencing of the FMR1 locus, with absent or markedly reduced FMR1 mRNA and FMR protein (FMRP) production (Bagni and Oostra, 2013). These large expansions result clinically in fragile X syndrome (FXS), which is the most common monogenic cause of autism and intellectual disability (Rogers et al., 2001; Hernandez et al., 2009).
In contrast, repeat expansions in the “premutation” range between 55 and 200 CGG repeats cause a distinct set of human disorders, including fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated Premature Ovarian Insufficiency (FXPOI; Berry-Kravis et al., 2007; Hagerman, 2013; Nelson et al., 2013). FXTAS is an age-related neurodegenerative disorder characterized by gait difficulties, action tremor, and variably present Parkinsonism, dysautonomia, and dementia (Berry-Kravis et al., 2007). Unlike the scenario in FXS, premutation sized CGG repeats elicit enhanced FMR1 transcription through alterations in the local chromatin structure (Tassone et al., 2000a, 2007; Todd et al., 2010; Hagerman and Hagerman, 2013). However, this increase in FMR1 mRNA is paradoxically associated with a reduction in total and activity-dependent FMRP expression (Tassone et al., 2000b; Kenneson et al., 2001; Entezam et al., 2007; Qin et al., 2011; Iliff et al., 2013; Ludwig et al., 2014; Pretto et al., 2014). This decrease in FMRP likely derives from an alteration in FMR1 mRNA translational efficiency, where the repeat forms a hairpin secondary structure that impairs ribosomal scanning (Feng et al., 1995; Kenneson et al., 2001; Primerano et al., 2002; Chen et al., 2003; Ludwig et al., 2011). Research into the mechanisms of neurodegeneration in FXTAS has largely focused on how the FMR1 mRNA might elicit a gain of function toxicity, either through sequestration of RNA binding proteins or through triggered aberrant translation through the repeat of aggregate prone proteins that underlie the intranuclear inclusions observed in patients (Jin et al., 2003, 2007; Hagerman and Hagerman, 2004, 2013; Iwahashi et al., 2006; Sofola et al., 2007; Sellier et al., 2010, 2013; Todd and Paulson, 2010; Renoux and Todd, 2012; Todd et al., 2013). However, more recent work has begun to explore whether a reduction in FMRP expression might contribute to aspects of disease pathogenesis in these individuals (Iliff et al., 2013; Ludwig et al., 2014; Pretto et al., 2014; Renoux et al., 2014; von Leden et al., 2014).
The absence of FMRP causes the cognitive impairment seen in FXS and may contribute to some of the symptoms observed in FXTAS. A significant body of work has explored the normal functions of FMRP (O’Donnell and Warren, 2002; Santoro et al., 2012; Wang et al., 2012). FMRP is an RNA binding protein found associated with poly-ribosome complexes in soma and synapses (Tamanini et al., 1996; Willemsen et al., 1996; Feng et al., 1997; Zalfa et al., 2003; Darnell et al., 2005, 2011; Ascano et al., 2012; Chen et al., 2014). It is normally phosphorylated, and upon metabotropic glutamate receptor (mGluR) activation is dephosphorylated to allow its associated transcripts to be translated (Ceman et al., 2003; Bear et al., 2004; Weiler et al., 2004; Muddashetty et al., 2007; Narayanan et al., 2008; Nalavadi et al., 2012). This allows FMRP to participate in temporal and spatial control of activity-dependent translation. In an effort to understand how reduced levels of FMRP may alter synaptic function, many groups have identified possible FMRP target transcripts. One transcript associated with FMRP is the amyloid precursor protein (APP) mRNA (Westmark and Malter, 2007; Lee et al., 2010). High-throughput sequencing of RNAs isolated by cross-linking immunoprecipitation (HITS-CLIP and PAR-CLIP) analysis on FMRP-associated transcripts identified APP mRNA (Darnell et al., 2011; Ascano et al., 2012). This interaction appears to play a role in regulating APP translation, as APP synthesis in response to mGluR activation is increased in control mice (Westmark and Malter, 2007). Moreover, a mouse model of FXS which lacks FMRP (Fmr1 KO) exhibit higher basal levels of APP, and of the pathogenic product of APP cleavage, β-amyloid (Aβ), and FXS patients show abnormal Aβ levels in plasma and brain tissues (Westmark and Malter, 2007; Westmark et al., 2011). Overexpression of APP in Fmr1 KO mice increases mortality and seizure susceptibility (Westmark et al., 2008). Conversely, genetic reduction of APP in the Fmr1 KO mouse improved seizure activity, anxiety-associated behavior, spine morphology, and altered mGluR-dependent long-term depression (LTD), indicating a role for Aβ expression levels in FXS pathology (Westmark et al., 2011).
As there is evidence indicating FMRP participates in regulating APP production and that FMRP and APP interact genetically, we sought to explore the possibility that reduced FMRP levels may contribute to increased APP and Aβ levels in AD mouse models and spontaneous cases of AD. This is especially relevant given evidence for decreased FMRP expression with age in mouse models (Singh et al., 2007; Iliff et al., 2013; Gaur and Prasad, 2014; Ludwig et al., 2014). Impaired FMRP expression in older individuals could lead to increased basal APP translation, increasing the amyloidogenic burden and thus serving as a contributor to AD pathogenesis. We sought to test this hypothesis by measuring cortical FMRP levels by western blot and immunohistochemistry in a double transgenic AD mouse model (APP/PS1; Haass et al., 1995; Prihar et al., 1999). We also measured FMRP immunoreactivity in human cortex and cerebellum from control and confirmed AD samples. Concurrently, we included samples from three FXTAS patients who exhibited reduced FMRP levels. We found similar FMRP expression in AD model mice and AD human samples. We further examined APP expression in FXTAS patient samples, and found a selective increase in cerebellar lysates, but not in frontal cortex or in CGG KI model mice. Taken together, our data suggest that impaired FMRP expression is unlikely to contribute significantly to end-stage Alzheimer’s disease (AD) pathogenesis.
Materials and Methods
Mice
Animal use followed NIH guidelines and was in compliance with the University of Michigan Committee on Use and Care of Animals. Fmr1 KO (Bakker et al., 1994) and CGG KI (Entezam et al., 2007) mice were genotyped as described previously (Iliff et al., 2013; Renoux et al., 2014). APP/PS1 mouse (Haass et al., 1995; Prihar et al., 1999) genotypes were confirmed with western analysis for the human APP transgene (clone 6E10 1:2000; Millipore).
Patient Donor Samples
All human tissues were obtained and distributed under oversight by appropriate institution specific review boards. Frontal cortex and cerebellar tissue from 10 control and 10 clinically probable AD patients were obtained from the University of Michigan Alzheimer’s Disease Brain Bank. All AD cases were confirmed at autopsy. Brain tissues from two previously described FXTAS patients (Louis et al., 2006; Todd et al., 2013) and an additional clinically definite FXTAS patient were used as controls for reduced FMRP expression. CGG repeat size was determined in FXTAS patients by DNA isolation followed by PCR using C and F primers. See Table 1 for post-mortem interval (PMI), age, and sex of each individual.
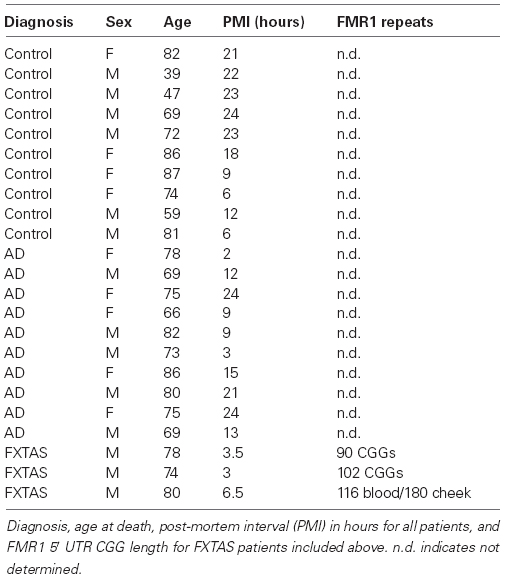
Table 1. Patient donor information.
Western Blot Analysis
Western blotting was performed as described previously (Iliff et al., 2013). Briefly, brain tissue samples (cerebral cortex and subcortical regions from mice, or frontal cortex from human, and cerebellum) were homogenized in RIPA buffer containing Complete Mini protease inhibitor cocktail (Roche). Samples were sonicated and centrifuged, and total protein content of the supernatant measured using a DC Protein assay (Bio-Rad). Equal amounts of protein were mixed with 6 × Laemmli buffer and boiled for 5 min before separation on 8% polyacrylamide gels. Gels were transferred and blocked with Tris-buffered saline containing 0.1% Triton-X (TBST) and 5% non-fat milk for 60 min at room temperature (RT), and incubated with an antibody against FMRP (Millipore mouse monoclonal 1C3 1:1000 or Abcam rabbit polyclonal 17722, 1:1000), or against the C-terminus of APP (Invitrogen 51-2700, 1:500) and β-tubulin (University of Iowa’s Developmental Studies Hybridoma Bank E7, 1:5000) overnight at 4°C. Blots were incubated with corresponding fluorescent secondary antibody (1:15000; IRDye® 680RD or 800CW, LI-COR) for 60 min at RT, and imaged with the Odyssey® Imaging System (LI-COR).
Band intensity was quantified in the linear range with densitometry using LI-COR Image StudioTM Software. Experiments were performed in technical triplicate, and FMRP/tubulin or APP/tubulin ratios to two control samples included in every experiment were combined. These ratios were averaged, normalized to control values for each experiment, and expressed as %control in experiments.
Immunohistochemistry
Antibody control experiments were performed on mice aged 50–75 weeks (n = 6 WT, n = 5 CGG KI, n = 1 Fmr1 KO), and experimental analysis on 80- to 90-week-old mice (n = 2 WT, n = 3 APP/PS1) which were anesthetized with 0.2 mg ketamine/20 µg xylazine per kilogram prior to transcardial perfusion with 15–20 mL phosphate buffered saline (PBS) and 15–20 mL 4% paraformaldehyde (PFA) in PBS. Brains were dissected, fixed overnight in 4% PFA, and then sunk in 30% sucrose in PBS at 4°C. Brains were sectioned at 30 µm and placed in a cryostorage solution of 30% sucrose/33.33 % ethylene glycol/0.05 M PBS until needed.
Prior to staining, slices were rotated in PBS overnight at 4°C to remove cryostorage solution, then basic antigen retrieval was performed by placing the slices in 0.01 M sodium citrate (pH 8.5) at 80°C for 10 min followed by three 5-min washes in PBS. The slices were then placed in 1% H2O2 in Tris to block endogenous peroxidases. Slices were permeabilized in 0.1% Triton-X/.05% bovine serum albumin (BSA) in Tris for 30 min at RT and blocked in 5% normal goat serum (NGS) for 1 h at RT. Slices were then incubated overnight in anti-FMRP antibody (1:3500). Following two washes, slices were incubated in horseradish peroxidase-conjugated secondary antibody (1:1000) in 5% NGS in Tris. Prior to peroxidase development, slices were treated with the Vectastain ABC kit (Vector) to increase diaminobenzidine (DAB) visibility. Following washes, slices from all genotypes were placed simultaneously in ImmPACT DAB solution (Vector) for 10–15 s until they just started to turn brown. Following washes, slices were mounted, allowed to dry overnight then either counterstained in Gill’s 1:2 hematoxylin for 45–60 s, or immediately dehydrated in an alcohol gradient and mounted.
Microscopy
A slide scanning microscope (Zeiss) was used to image all DAB stained tissue. Fields of view were selected in the hippocampus and cortex to be easily reproducible across multiple sections. Images were taken using the same exposure settings for all genotypes.
Statistical Analysis
All values are expressed as the mean ± standard error of the mean. Western blot immunoreactivity for each sample was measured in technical triplicate, and combined as a ratio of %control for each blot. These combined values were averaged, and compared using a Student’s t-test, with significance indicated by a P-value < 0.05.
FMRP expression compared to age, PMI, and pH were evaluated using a Pearson’s correlation coefficient on combined cortical and cerebellar FMRP values from either control and AD samples, or combined control and FXTAS cortical and cerebellar values, with significance indicated by P < 0.05. Comparison of donor gender in FMRP expression was performed using a two-way ANOVA with post hoc Sidak’s multiple comparison test.
Results
To accurately evaluate the expression of FMRP in our experiments, we compared two commonly used antibodies for their specificity by western blot and immunohistochemistry. To assess the sensitivity of each antibody, we compared FMRP levels in WT, littermate CGG KI, and Fmr1 KO mouse cortical lysates (Figure 1). Both the 1C3 mouse anti-FMRP (Millipore) and the 17722 rabbit anti-FMRP (Abcam) show reactivity in the Fmr1 KO samples (Figures 1A,B). The largest band at ∼75 kD corresponds to FMRP and is absent in Fmr1 KO lysates with both antibodies. This band was used for all measurements in subsequent figures. However, a smaller band at ∼71 kD which results at least partially from cross-reactivity with the related protein, FXR1 (Tamanini et al., 1997; Ceman et al., 1999), is still reactive with both antibodies tested. Similarly, we examined specificity of the 17722 anti-FMRP antibody in WT, CGG KI and Fmr1 KO coronal brain sections by immunohistochemistry (WT n = 3, CGG KI n = 3, Fmr1 KO n = 4; representative images; Figures 1C,D). Conditions were optimized to minimize DAB reactivity in Fmr1 KO tissue. We reliably observed reduced FMRP levels in CGG KI mice both by western analysis and immunohistochemistry, consistent with previous reports (Tassone et al., 2000a; Kenneson et al., 2001; Entezam et al., 2007; Qin et al., 2011; Iliff et al., 2013; Ludwig et al., 2014; Pretto et al., 2014; Figure 1).
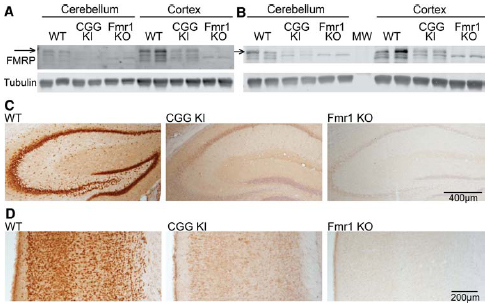
Figure 1. FMRP antibody specificity. (A) Mouse α-FMRP 1C3 (Millipore) 1:250 on cerebellar and combined cerebral cortex and subcortical brain lysates of WT (n = 2), premutation model CGG KI (n = 2), and Fmr1 KO mice (n = 2). Arrow indicates the FMRP-specific band which is absent in Fmr1 KO lysates. (B) Rabbit α-FMRP 17722 (Abcam) 1:1000 on the same brain lysates as in (A). (C) Immunohistochemistry with the 17722 α-FMRP antibody in WT (n = 3), CGG KI (n = 3) and Fmr1 KO (n = 4) hippocampus. (D) Cortical FMRP expression from identical animals as in (C) stained with the 17722 antibody. Sections were counter-stained with hematoxylin to label nuclei.
In an effort to probe a potential role of altered FMRP expression in AD pathogenesis, we evaluated FMRP in a double transgenic model of AD which contains an additional copy of the human APP gene carrying the familial Swedish (K670N/M671L) missense mutation (Haass et al., 1995), and a deletion of exon 9 in the presenilin1 gene (Prihar et al., 1999). These double transgenic mice (APP/PS1; n = 6: 6 females) and age-matched control littermates (n = 8: 4 males, 4 females) were compared at 80–90 weeks of age for combined cortical and subcortical FMRP levels. We performed western blot analysis in triplicate, averaging the percent control FMRP for each animal across blots to minimize error between experiments. We find no difference in FMRP levels in APP/PS1 mice compared to controls (t = 0.358, df = 12, NS; Figure 2A). Cerebellar samples from the same animals demonstrated no significant difference in FMRP expression (t = 1.300, df = 12, NS; Figure 2A). Any contribution of sex was evaluated in the control animals (as the APP/PS1 mice were all female), and FMRP expression was not significantly different between male and female control cortical values (male n = 4, female n = 4, t = 0.717, df = 6, NS; data not shown). As several groups have found FMRP levels change with age (Singh et al., 2007; Iliff et al., 2013; Gaur and Prasad, 2014; Ludwig et al., 2014), we explored expression in young (8-week-old) APP/PS1 mice (WT n = 3: 3 females; APP/PS1 n = 3: 3 females). We find no difference in cortical FMRP expression at this age (t = 0.919, df = 4, NS). Cerebellar FMRP expression demonstrated a non-significant increase in AD model mice (t = 2.627, df = 4, NS; Figure 2B). We also compared age-dependent FMRP expression of the 8-week-old and the 80-week-old animals, and did not find a significant difference in the cortex or the cerebellum (cortex: 8 weeks, n = 3 females; 80 weeks n = 8, 4 males, 4 females; t = 0.342, df = 9, NS; cerebellum: 8 weeks n = 3 females; 80 weeks n = 8, 4 males, 4 females; t = 1.188, df = 9, NS; Figure 2C). There was no significant difference between same sex 8- and 80-week-old female FMRP expression (cortex: 8 weeks female n = 3, 80 weeks female n = 4, t = 1.034, df = 5, NS; cerebellum: 8 weeks female n = 3, 80 weeks female n = 4, t = 1.136, df = 5, NS; data not shown). We went on to probe FMRP levels by immunohistochemistry in 80-week-old control and APP/PS1 mice. Comparing cortex, hippocampus, and subcortical regions using the 1C3 α-FMRP antibody, there were no differences detected (WT n = 2, APP/PS1 n = 3; Figures 2D,E).
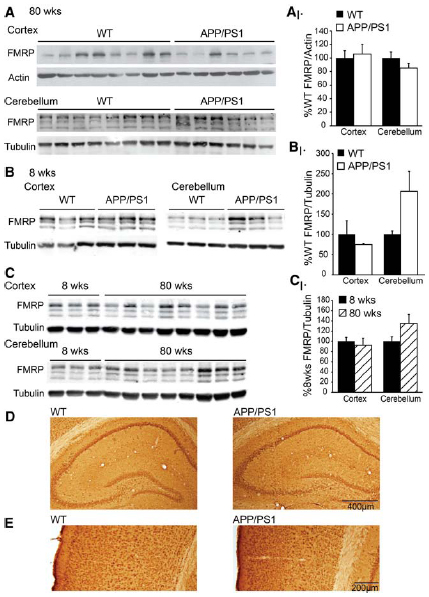
Figure 2. FMRP expression in AD model APP/PS1 mice. (A) Cortical and subcortical, and cerebellar lysates from 80-week-old APP/PS1 (n = 6) and age-matched controls (n = 8) probed with the 17722 α-FMRP antibody and actin or tubulin. (A1) FMRP/actin values were calculated and normalized to WT levels, and expressed as %WT in the quantification. Results are the summary of values calculated in technical triplicate. (B) Eight-week-old APP/PS1 (n = 3) and WT littermate controls (n = 3) were compared for FMRP expression in cortical and subcortical and cerebellar lysates. (B1) FMRP/tubulin values are expressed as %WT, and data are the summary of experiments performed in technical triplicate. (C) Eight-week-old WT (n = 3) and 80-week-old WT (n = 8) cortical and cerebellar lysates compared for FMRP expression. (C1) FMRP/tubulin values are expressed as percentage of mean 8-week-old samples, and data are the summary of experiments performed in duplicate. (D) Hippocampal FMRP expression using the 1C3 α-FMRP antibody in WT and APP/PS1 mice. (E) Cortical FMRP in the same mice as in (D).
As the mouse model we used was genetically modified to mimic some AD phenotypes, it may not recapitulate proximal pathogenic events that contribute to spontaneous cases of AD. In an attempt to better answer the question of a possible role for FMRP in AD development, we obtained frontal cortex and cerebellar autopsy samples from control and AD patients (details included in Table 1). As with our murine samples, we performed western blot analysis in technical triplicate to minimize variability in our measurements. Using this technique, we found no difference in FMRP expression level in the frontal cortex (t = 0.2836, df = 18, NS; control n = 10, AD n = 10; Figures 3A,C). As two of the control individuals were younger than the majority of the other donors (39 and 47 years old), we performed analysis excluding those values, and found no change in the result (t = 0.3915, df = 16, NS). Similarly there was no difference in FMRP expression found in the cerebellar samples analyzed (t = 0.2837, df = 18, NS; control n = 10, AD n = 10; Figures 3B,C). Again, excluding the youngest individuals did not impact the finding (t = 0.3618, df = 16; NS). As there was a wide range in PMI and age of the samples tested, we compared normalized values against these two variables, in addition to tissue pH (where available), and found a significant impact on FMRP levels with longer PMI (age: r = –0.111, df = 38, NS; PMI: r = –0.371, df = 38, P < 0.05; pH: r = 0.277, df = 22, NS; Figure 3D, data not shown). We also evaluated the impact of gender in this experiment by comparing the data with a two-way ANOVA, and found no significant difference of FMRP expression in any group in the cortex [gender: F(1,16) = 0.3187, NS; diagnosis: F(1,16) = 0.05139, NS; interaction: F(1,16) = 0.02455, NS; data not shown]. Cerebellar samples similarly showed no significant impact of gender [gender: F(1,16) = 0.1176, NS; diagnosis: F(1,16) = 0.09488, NS; interaction: F(1,16) = 0.006407, NS; data not shown]. While these analyses only showed significant contributions of PMI on FMRP levels, the negative trends of decreasing FMRP with age and lower pH suggest that these variables should also be controlled for in future studies.
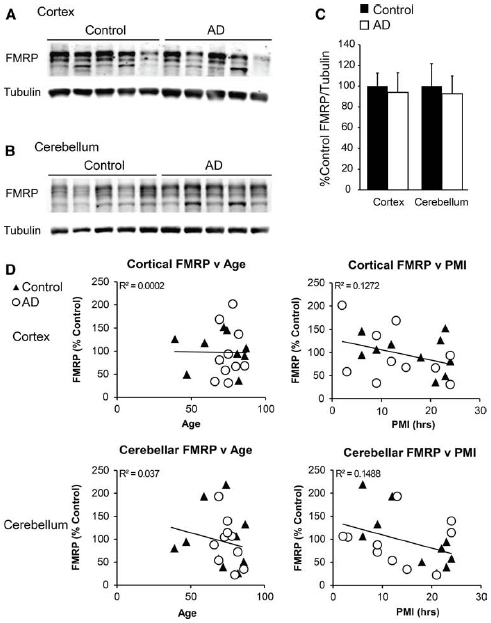
Figure 3. FMRP expression in Alzheimer’s disease cortex and cerebellum. (A) Frontal cortex lysates from control (n = 10) and autopsy-confirmed AD patients (n = 10) probed with α-FMRP 17722. (B) Cerebellar lysates from the same patients as in (A) were evaluated concurrently. (C) Quantification of cortical and cerebellar FMRP/tubulin values performed in technical triplicate. (D) Normalized FMRP values were compared to individual patient ages and post-mortem indices. Diagnosis-independent best fit curves display overall trends in FMRP expression.
We obtained samples from FXTAS patient brains, and compared FMRP levels to the controls, finding a decrement in FMRP immunoreactivity in two of the three samples tested, though the number of samples evaluated was not sufficient to discern a significant difference when the two youngest control samples were excluded (cortex: t = 2.161, df = 9, NS; cerebellum: t = 1.793, df = 9, NS; control n = 8, FXTAS n = 3; Figures 4A–C). Similarly, we compared the age and PMI as a factor which may alter FMRP levels, and found no significant difference of either variable (age: r = –0.180, df = 24, NS; PMI: r = 0.007, df = 24, NS; data not shown).
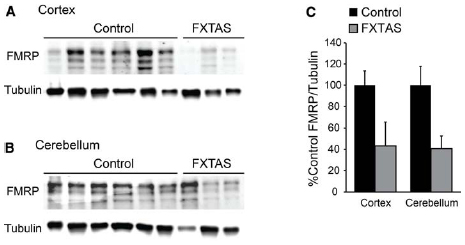
Figure 4. Cortical and cerebellar FMRP expression in FXTAS patients. (A) Frontal cortex lysates from FXTAS patients (n = 3) and the same control tissues (n = 8) probed with α-FMRP 17722. (B) Cerebellar lysates from the same individuals as in (A). (C) Normalized FMRP expression as a percent of controls, performed in technical triplicate.
To further test the hypothesis that reduced FMRP might enhance APP synthesis, we evaluated total full-length APP expression in the same FXTAS patient samples. Despite the small sample size, we did find a selective increase in cerebellar APP expression in FXTAS samples (t = 4.704, df = 11, P < 0.05; control n = 10, FXTAS n = 3; Figures 5A–C). However, APP levels were unchanged in frontal cortex samples compared to controls (t = 0.4603, df = 11, NS; control n = 10, FXTAS n = 3; Figures 5A–C). These results were not affected by the inclusion of the two youngest control samples (cortex: t = 0.2636, df = 9, NS; cerebellum: t = 4.844, df = 9, P < 0.05). This finding was not recapitulated in 12-month-old CGG KI mice, which showed unchanged levels of APP in both the cortex and cerebellum (cortex: t = 1.877, df = 5, NS; cerebellum: t = 1.131, df = 5, NS; WT n = 3, CGG KI n = 4; Figures 5D–F). To assess any age-dependent effects of APP expression we compared CGG KI and WT cortical and cerebellar lysates at 2 and 16–18 months of age, and found no significant difference between genotypes (2 months cortex: WT = 100 ± 11.66, CGG KI = 99.86 ± 21.63; t = 0.006, df = 8, NS; WT n = 5, CGG KI n = 5; 16–18 months cortex: WT = 100 ± 15.31, CGG KI = 93.09 ± 6.1; t = 0.367, df = 5, NS; WT n = 4, CGG KI n = 3; 16–18 months cerebellum: WT = 100 ± 10.84, CGG KI = 75.75 ± 7.70; t = 1.690, df = 5, NS; WT n = 3, CGG KI n = 3; data not shown). The C-terminal APP antibody used cannot detect Aβ fragments, so while total APP levels are unchanged, the relative processing or cleavage events may be altered.
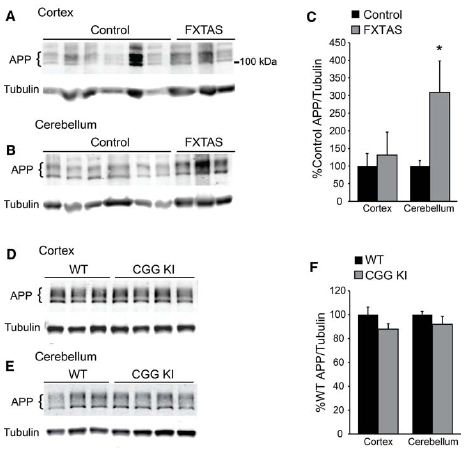
Figure 5. Amyloid precursor protein expression in FXTAS patients and mouse models. (A) Frontal cortex lysates from control (n = 10) and FXTAS patients (n = 3) probed with a C-terminal αAPP antibody which detects the three primary isoforms of full-length APP (100–125 kDa). (B) Cerebellar lysates from the same individuals as in (A). (C) Normalized APP expression relative to tubulin expressed as a percent of controls, performed in technical triplicate. (D) Twelve-month-old CGG KI (n = 4) and WT littermate control (n = 3) cortex and subcortical lysates probed with αAPP. (E) Cerebellar lysates from the same animals as in (D). (F) Normalized APP expression in CGG KI mice expressed as a percent of WT controls, performed in technical triplicate. *P < 0.05 Student’s t-test.
Discussion
FMRP is a synaptic mRNA binding protein implicated in fragile X spectrum disorders. FMRP normally acts to inhibit translation of target transcripts until synaptic transmission and mGluR activation cause its dissociation, allowing for a burst of rapid local translation. One FMRP target is APP, the precursor to the amyloidogenic Aβ peptide (Westmark and Malter, 2007). APP expression is enhanced in mouse models that lack FMRP (Westmark and Malter, 2007). FMRP expression is reported to decline with age in mouse models and might follow a similar trajectory in humans (Singh et al., 2007; Gaur and Prasad, 2014; Ludwig et al., 2014). We hypothesized that a natural age-related decline in FMRP might trigger an increase in basal APP synthesis in neurons, leading to enhanced Aβ-42 production and possibly serving as a proximal trigger of AD pathogenesis. Moreover, as AD pathogenesis progresses, we hypothesized that neuronal dysfunction and toxicity might further impair FMRP expression and hence enhance APP translation, creating a feed-forward loop that could drive AD pathogenesis and lower FMRP expression in AD models and patient tissues. To test this hypothesis, we examined FMRP expression in a double transgenic mouse model of AD (APP/PS1), and found no significant difference between AD model and control animals. Furthermore, we tested control and AD patient samples, and found no difference in cortical or cerebellar FMRP expression. In contrast, we observed a decrease in FMRP levels in the two of the three FXTAS patients tested, consistent with published results (Ludwig et al., 2014; Pretto et al., 2014), and found increased cerebellar levels of APP in these same FXTAS patients, suggesting an impact of lower FMRP on APP expression in humans. Taken together, these data indicate that decreased FMRP expression is not a common finding in AD and suggests that a primary deficiency in FMRP expression is unlikely to play a proximal role in most cases of AD. However, manipulation of FMRP expression and activity retains the potential to influence APP expression and aspects of AD pathogenesis.
FMRP’s regulatory function is dependent on phosphorylation by S6K, and dephosphorylation by PP2A (Narayanan et al., 2007, 2008). Recent work has described a function for FMRP degradation upon mGluR activation, and dephosphorylation by PP2A (Nalavadi et al., 2012). While the goal of this study was to examine total FMRP expression levels, alterations in FMRP phosphorylation would impact its regulatory function, and therefore the control of target protein translation. While we did not observe large alterations in FMRP levels, it is possible that relative phosphorylation state could play a role in AD pathogenesis. Further studies comparing phospho-FMRP would be required to examine this possibility.
Our group and others have observed decreases in FMRP expression with age (Singh et al., 2007; Iliff et al., 2013; Gaur and Prasad, 2014; Ludwig et al., 2014). However, we observed no significant difference in FMRP levels between 8- and 80-week-old WT mice evaluated in this study (Figure 2C). The reason for this difference is not immediately clear, although the smaller number of young animals evaluated and the significant variance observed in older animals may explain the discrepancy. Other groups have evaluated various time points ranging from neonate to adult (20 weeks of age; Singh et al., 2007; Gaur and Prasad, 2014; Ludwig et al., 2014), and as old as 60–70 weeks of age (Singh et al., 2007; Gaur and Prasad, 2014). Both groups have observed cerebellar expression to steadily decrease in older animals (Gaur and Prasad, 2014; Ludwig et al., 2014), though cortical FMRP expression may be more complex during postnatal development into adulthood (Ludwig et al., 2014). While we and others have studied animals near 4–6 weeks of age (Singh et al., 2007; Iliff et al., 2013; Gaur and Prasad, 2014; Ludwig et al., 2014), the specific 8-week time point utilized here has not been compared directly to old animals previously.
Our study is likely underpowered to detect subtle changes in human FMRP expression. We observed a wide range of FMRP expression levels in both the control and patient samples, consistent with published results (Ludwig et al., 2014; Pretto et al., 2014). This variance was not completely explained by patient age, tissue pH, or PMI (Figure 3), although all of these variables demonstrated a trend toward lower FMRP. This leaves open the possibility that lower basal or activity dependent FMRP expression could still contribute to altered APP expression in a subset of patients or in certain brain regions.
Impaired FMRP expression in FXTAS model mice and patients have been described previously (Tassone et al., 2000a; Kenneson et al., 2001; Entezam et al., 2007; Qin et al., 2011; Iliff et al., 2013; Ludwig et al., 2014; Pretto et al., 2014). Similarly, we find reduced FMRP levels in two of the three FXTAS patient samples tested (Figure 4). Symptomatic FXTAS patients can develop cognitive impairment in addition to motor behavior symptoms (Berry-Kravis et al., 2007; Leehey, 2009). While early studies suggested that pathologic hallmarks of AD are rare in FXTAS (Greco et al., 2002), recent studies in female premutation carriers with dementia demonstrated plaque and neurofibrillary tangle development consistent with AD pathology (Tassone et al., 2012). However, a recent study examining AD patient populations for FMR1 CGG expansions did not find a significant association (Hall et al., 2014).
To test whether FMRP insufficiency might lead to increased APP synthesis in FXTAS, we evaluated APP levels in our patient samples (Figure 5). We detected an increase in cerebellar, but not cortical APP expression in FXTAS patients and no differences in a mouse model of FXTAS (Figure 5). The C-terminal antibody used for this study does not detect Aβ fragments, so it remains possible that APP processing is altered in FXTAS or CGG premutation models. However, in the context of the published studies noted above, a strong direct relationship between CGG repeats and induction of amyloid pathology appears unlikely.
The group 1 mGluR, mGluR5, has been identified as a receptor of the APP peptide Aβ42 (Renner et al., 2010; Sokol et al., 2011; Um et al., 2013; Hamilton et al., 2014). A recent report explored the role of increased mGluR5 signaling in the double transgenic APP/PS1 model of AD (Hamilton et al., 2014). Genetic modulation of mGluR5 improved behavioral learning performance, and decreased amyloid plaques found in the APP/PS1 mice (Hamilton et al., 2014). Furthermore, cortical FMRP expression was increased in 12-month-old APP/PS1 mice compared to WT controls, presumably due to increased mGluR5 signaling, which generates a positive feedback loop leading to increased APP production and cleavage (Hamilton et al., 2014). The reason for differing results in our study is not entirely clear, however, the animals examined in this report were significantly older (18 months old compared to 12 months old), and the procedures for tissue isolation and lysate generation were different (Hamilton et al., 2014). Of note, the increase observed in Hamilton et al. (2014) is consistent with our observations of human AD cerebellar samples which showed increased FMRP expression compared to controls (Figure 3).
While reduced FMRP might contribute to neuronal dysfunction in FXTAS patients, it is unlikely to be a primary cause of neurodegeneration in FXTAS. Expression of CGG repeats as RNA in heterologous contexts is sufficient to elicit toxicity in Drosophila, cells, and mice (Jin et al., 2003, 2007; Hagerman and Hagerman, 2004; Iwahashi et al., 2006; Sofola et al., 2007; Hashem et al., 2009; Sellier et al., 2010, 2013; Todd and Paulson, 2010; Renoux and Todd, 2012; Hagerman, 2013). However, FMRP insufficiency may nonetheless contribute to the cognitive decline observed in mouse models and patients with FXTAS. In addition to the potential differential amyloidogenic burden, reduced FMRP likely impacts synaptic function in premutation mouse models (Hunsaker et al., 2012; Iliff et al., 2013; von Leden et al., 2014). Synaptic dysregulation is thought to precede neurodegeneration, and contribute to the onset of symptoms prior to gross neuronal loss (Dong et al., 2009; Milnerwood and Raymond, 2010). Delineating the role of FMRP insufficiency in FXTAS thus remains an important objective going forward given the implications for therapeutic development in patients.
In summary, we find no evidence to support a direct link between lower basal FMRP expression and AD pathogenesis. Future work will be needed to define whether changes in FMRP activity influence AD development, given the known roles of FMRP in APP processing and neuronal function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Svetlana Fischer and Edgar Rodriguez for technical assistance and helpful discussions. We thank all the reviewers for their helpful input and time on this manuscript. APP/PS1 mice were a kind gift from Henry Paulson (University of Michigan ADRC). CGG KI mice were obtained from Karen Usdin (NIH), and Fmr1 KO animals donated by Cara Westmark (University of Wisconsin), and Jim Malter (UT Southwestern). Nicholas M. Carducci received financial support for his role on this project from Michael Sutton (University of Michigan). This work was supported by the National Institutes of Mental Health (F31MH097451) to Abigail J. Renoux, and by the Veterans Administration (BLRD #1I01BX001689), NIH (1K08NS069809 and 1R01NS086810-01), and Harris Professorship to Peter K. Todd.
References
Ascano, M. Jr., Mukherjee, N., Bandaru, P., Miller, J. B., Nusbaum, J. D., et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. doi: 10.1038/nature11737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bagni, C., and Oostra, B. A. (2013). Fragile X syndrome: from protein function to therapy. Am. J. Med. Genet. A 161A, 2809–2821. doi: 10.1002/ajmg.a.36241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bakker, C. E., Verheij, C., Willemsen, R., Vanderhelm, R., Oerlemans, F., Vermey, M., et al. (1994). Fmr1 knockout mice—a model to study fragile-X mental-retardation. Cell 78, 23–33.
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E., Abrams, L., Coffey, S. M., Hall, D. A., Greco, C., Gane, L. W., et al. (2007). Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov. Disord. 22, 2018–2030, quiz 2140. doi: 10.1002/mds.21493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ceman, S., Brown, V., and Warren, S. T. (1999). Isolation of an FMRP-associated messenger ribonucleoprotein particle and identification of nucleolin and the fragile X-related proteins as components of the complex. Mol. Cell. Biol. 19, 7925–7932.
Ceman, S., O’Donnell, W. T., Reed, M., Patton, S., Pohl, J., and Warren, S. T. (2003). Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum. Mol. Genet. 12, 3295–3305. doi: 10.1093/hmg/ddg350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, E., Sharma, M. R., Shi, X., Agrawal, R. K., and Joseph, S. (2014). Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 54, 407–417. doi: 10.1016/j.molcel.2014.03.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L. S., Tassone, F., Sahota, P., and Hagerman, P. J. (2003). The (CGG)n repeat element within the 5′ untranslated region of the FMR1 message provides both positive and negative cis effects on in vivo translation of a downstream reporter. Hum. Mol. Genet. 12, 3067–3074. doi: 10.1093/hmg/ddg331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darnell, J. C., Fraser, C. E., Mostovetsky, O., Stefani, G., Jones, T. A., Eddy, S. R., et al. (2005). Kissing complex RNAs mediate interaction between the fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 19, 903–918. doi: 10.1101/gad.1276805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, X. X., Wang, Y., and Qin, Z. H. (2009). Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 30, 379–387. doi: 10.1038/aps.2009.24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Entezam, A., Biacsi, R., Orrison, B., Saha, T., Hoffman, G. E., Grabczyk, E., et al. (2007). Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene 395, 125–134. doi: 10.1016/j.gene.2007.02.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, Y., Gutekunst, C. A., Eberhart, D. E., Yi, H., Warren, S. T., and Hersch, S. M. (1997). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 17, 1539–1547.
Feng, Y., Zhang, F., Lokey, L. K., Chastain, J. L., Lakkis, L., Eberhart, D., et al. (1995). Translational suppression by trinucleotide repeat expansion at FMR1. Science 268, 731–734. doi: 10.1126/science.7732383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gaur, P., and Prasad, S. (2014). Alterations in the Sp1 binding and Fmr-1 gene expression in the cortex of the brain during maturation and aging of mouse. Mol. Biol. Rep. doi: 10.1007/s11033-014-3571-1 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greco, C. M., Hagerman, R. J., Tassone, F., Chudley, A. E., Del Bigio, M. R., Jacquemont, S., et al. (2002). Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 125, 1760–1771.
Haass, C., Lemere, C. A., Capell, A., Citron, M., Seubert, P., Schenk, D., et al. (1995). The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1, 1291–1296.
Hagerman, P. (2013). Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 126, 1–19. doi: 10.1007/s00401-013-1138-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagerman, P. J., and Hagerman, R. J. (2004). Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment. Retard. Dev. Disabil. Res. Rev. 10, 25–30. doi: 10.1002/mrdd.20005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagerman, R., and Hagerman, P. (2013). Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 12, 786–798. doi: 10.1016/S1474-4422(13)70125-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, D. A., Bennett, D. A., Filley, C. M., Shah, R. C., Kluger, B., Ouyang, B., et al. (2014). Fragile X gene expansions are not associated with dementia. Neurobiol. Aging 35, 2637–2638. doi: 10.1016/j.neurobiolaging.2014.04.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamilton, A., Esseltine, J. L., Devries, R. A., Cregan, S. P., and Ferguson, S. S. (2014). Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 7, 40. doi: 10.1186/1756-6606-7-40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashem, V., Galloway, J. N., Mori, M., Willemsen, R., Oostra, B. A., Paylor, R., et al. (2009). Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet. 18, 2443–2451. doi: 10.1093/hmg/ddp182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernandez, R. N., Feinberg, R. L., Vaurio, R., Passanante, N. M., Thompson, R. E., and Kaufmann, W. E. (2009). Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am. J. Med. Genet. A 149A, 1125–1137. doi: 10.1002/ajmg.a.32848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hunsaker, M. R., Kim, K., Willemsen, R., and Berman, R. F. (2012). CGG trinucleotide repeat length modulates neural plasticity and spatiotemporal processing in a mouse model of the fragile X premutation. Hippocampus 22, 2260–2275. doi: 10.1002/hipo.22043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iliff, A. J., Renoux, A. J., Krans, A., Usdin, K., Sutton, M. A., and Todd, P. K. (2013). Impaired activity-dependent FMRP translation and enhanced mGluR-dependent LTD in fragile X premutation mice. Hum. Mol. Genet. 22, 1180–1192. doi: 10.1093/hmg/dds525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwahashi, C. K., Yasui, D. H., An, H. J., Greco, C. M., Tassone, F., Nannen, K., et al. (2006). Protein composition of the intranuclear inclusions of FXTAS. Brain 129, 256–271. doi: 10.1093/brain/awh650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, P., Duan, R., Qurashi, A., Qin, Y., Tian, D., Rosser, T. C., et al. (2007). Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55, 556–564. doi: 10.1016/j.neuron.2007.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, P., Zarnescu, D. C., Zhang, F., Pearson, C. E., Lucchesi, J. C., Moses, K., et al. (2003). RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 39, 739–747. doi: 10.1016/S0896-6273(03)00533-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kenneson, A., Zhang, F., Hagedorn, C. H., and Warren, S. T. (2001). Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 10, 1449–1454.
Lee, E. K., Kim, H. H., Kuwano, Y., Abdelmohsen, K., Srikantan, S., Subaran, S. S., et al. (2010). hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat. Struct. Mol. Biol. 17, 732–739. doi: 10.1038/nsmb.1815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leehey, M. A. (2009). Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J. Invest. Med. 57, 830–836. doi: 10.231/JIM.0b013e3181af59c4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Louis, E., Moskowitz, C., Friez, M., Amaya, M., and Vonsattel, J. P. (2006). Parkinsonism, dysautonomia, and intranuclear inclusions in a fragile X carrier: a clinical-pathological study. Mov. Disord. 21, 420–425. doi: 10.1002/mds.20753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ludwig, A. L., Espinal, G. M., Pretto, D., Jamal, A. L., Arque, G., Tassone, F., et al. (2014). CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum. Mol. Genet. 23, 3228–3238. doi: 10.1093/hmg/ddu032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ludwig, A. L., Hershey, J. W., and Hagerman, P. J. (2011). Initiation of translation of the FMR1 mRNA Occurs predominantly through 5′-end-dependent ribosomal scanning. J. Mol. Biol. 407, 21–34. doi: 10.1016/j.jmb.2011.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Milnerwood, A. J., and Raymond, L. A. (2010). Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 33, 513–523. doi: 10.1016/j.tins.2010.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muddashetty, R. S., Kelic, S., Gross, C., Xu, M., and Bassell, G. J. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nalavadi, V. C., Muddashetty, R. S., Gross, C., and Bassell, G. J. (2012). Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J. Neurosci. 32, 2582–2587. doi: 10.1523/JNEUROSCI.5057-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narayanan, U., Nalavadi, V., Nakamoto, M., Pallas, D. C., Ceman, S., Bassell, G. J., et al. (2007). FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J. Neurosci. 27, 14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narayanan, U., Nalavadi, V., Nakamoto, M., Thomas, G., Ceman, S., Bassell, G. J., et al. (2008). S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 283, 18478–18482. doi: 10.1074/jbc.C800055200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nelson, D. L., Orr, H. T., and Warren, S. T. (2013). The unstable repeats—three evolving faces of neurological disease. Neuron 77, 825–843. doi: 10.1016/j.neuron.2013.02.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Donnell, W. T., and Warren, S. T. (2002). A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 25, 315–338. doi: 10.1146/annurev.neuro.25.112701.142909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pretto, D. I., Kumar, M., Cao, Z., Cunningham, C. L., Durbin-Johnson, B., Qi, L., et al. (2014). Reduced excitatory amino acid transporter 1 and metabotropic glutamate receptor 5 expression in the cerebellum of fragile X mental retardation gene 1 premutation carriers with fragile X-associated tremor/ataxia syndrome. Neurobiol. Aging 35, 1189–1197. doi: 10.1016/j.neurobiolaging.2013.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prihar, G., Verkkoniem, A., Perez-Tur, J., Crook, R., Lincoln, S., Houlden, H., et al. (1999). Alzheimer disease PS-1 exon 9 deletion defined. Nat. Med. 5, 1090. doi: 10.1038/13383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Primerano, B., Tassone, F., Hagerman, R. J., Hagerman, P., Amaldi, F., and Bagni, C. (2002). Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA 8, 1482–1488.
Qin, M., Entezam, A., Usdin, K., Huang, T., Liu, Z. H., Hoffman, G. E., et al. (2011). A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol. Dis. 42, 85–98. doi: 10.1016/j.nbd.2011.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Renner, M., Lacor, P. N., Velasco, P. T., Xu, J., Contractor, A., Klein, W. L., et al. (2010). Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754. doi: 10.1016/j.neuron.2010.04.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Renoux, A. J., Sala-Hamrick, K. J., Carducci, N. M., Frazer, M., Halsey, K. E., Sutton, M. A., et al. (2014). Impaired sensorimotor gating in Fmr1 knock out and fragile X premutation model mice. Behav. Brain Res. 267C, 42–45. doi: 10.1016/j.bbr.2014.03.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Renoux, A. J., and Todd, P. K. (2012). Neurodegeneration the RNA way. Prog. Neurobiol. 97, 173–189. doi: 10.1016/j.pneurobio.2011.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogers, S. J., Wehner, D. E., and Hagerman, R. (2001). The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 22, 409–417.
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sellier, C., Freyermuth, F., Tabet, R., Tran, T., He, F., Ruffenach, F., et al. (2013). Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 3, 869–880. doi: 10.1016/j.celrep.2013.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sellier, C., Rau, F., Liu, Y., Tassone, F., Hukema, R. K., Gattoni, R., et al. (2010). Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 29, 1248–1261. doi: 10.1038/emboj.2010.21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, K., Gaur, P., and Prasad, S. (2007). Fragile x mental retardation (Fmr-1) gene expression is down regulated in brain of mice during aging. Mol. Biol. Rep. 34, 173–181. doi: 10.1007/s11033-006-9032-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sofola, O. A., Jin, P., Qin, Y., Duan, R., Liu, H., De Haro, M., et al. (2007). RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55, 565–571. doi: 10.1016/j.neuron.2007.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sokol, D. K., Maloney, B., Long, J. M., Ray, B., and Lahiri, D. K. (2011). Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology 76, 1344–1352. doi: 10.1212/WNL.0b013e3182166dc7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strom, C. M., Crossley, B., Redman, J. B., Buller, A., Quan, F., Peng, M., et al. (2007). Molecular testing for fragile X syndrome: lessons learned from 119,232 tests performed in a clinical laboratory. Genet. Med. 9, 46–51. doi: 10.1097/GIM.0b013e31802d833c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamanini, F., Meijer, N., Verheij, C., Willems, P. J., Galjaard, H., Oostra, B. A., et al. (1996). FMRP is associated to the ribosomes via RNA. Hum. Mol. Genet. 5, 809–813. doi: 10.1093/hmg/5.6.809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamanini, F., Willemsen, R., Van Unen, L., Bontekoe, C., Galjaard, H., Oostra, B. A., et al. (1997). Differential expression of FMR1, FXR1 and FXR2 proteins in human brain and testis. Hum. Mol. Genet. 6, 1315–1322.
Tassone, F., Beilina, A., Carosi, C., Albertosi, S., Bagni, C., Li, L., et al. (2007). Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 13, 555–562. doi: 10.1261/rna.280807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tassone, F., Greco, C. M., Hunsaker, M. R., Seritan, A. L., Berman, R. F., Gane, L. W., et al. (2012). Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 11, 577–585. doi: 10.1111/j.1601-183X.2012.00779.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tassone, F., Hagerman, R. J., Chamberlain, W. D., and Hagerman, P. J. (2000a). Transcription of the FMR1 gene in individuals with fragile X syndrome. Am. J. Med. Genet. 97, 195–203. doi: 10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tassone, F., Hagerman, R. J., Loesch, D. Z., Lachiewicz, A., Taylor, A. K., and Hagerman, P. J. (2000b). Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am. J. Med. Genet. 94, 232–236. doi: 10.1002/1096-8628(20000918)94:3<232::AID-AJMG9>3.0.CO;2-H
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Todd, P. K., Oh, S. Y., Krans, A., He, F., Sellier, C., Frazer, M., et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. doi: 10.1016/j.neuron.2013.03.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Todd, P. K., Oh, S. Y., Krans, A., Pandey, U. B., Di Prospero, N. A., Min, K. T., et al. (2010). Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 6:e1001240. doi: 10.1371/journal.pgen.1001240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Todd, P. K., and Paulson, H. L. (2010). RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 67, 291–300. doi: 10.1002/ana.21948
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer αβ oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
von Leden, R. E., Curley, L. C., Greenberg, G. D., Hunsaker, M. R., Willemsen, R., and Berman, R. F. (2014). Reduced activity-dependent protein levels in a mouse model of the fragile X premutation. Neurobiol. Learn. Mem. 109, 160–168. doi: 10.1016/j.nlm.2014.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T., Bray, S. M., and Warren, S. T. (2012). New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 22, 256–263. doi: 10.1016/j.gde.2012.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiler, I. J., Spangler, C. C., Klintsova, A. Y., Grossman, A. W., Kim, S. H., Bertaina-Anglade, V., et al. (2004). Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc. Natl. Acad. Sci. U.S.A. 101, 17504–17509. doi: 10.1073/pnas.0407533101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., and Malter, J. S. (2007). FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 5:e52. doi: 10.1371/journal.pbio.0050052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., Westmark, P. R., Beard, A. M., Hildebrandt, S. M., and Malter, J. S. (2008). Seizure susceptibility and mortality in mice that over-express amyloid precursor protein. Int. J. Clin. Exp. Pathol. 1, 157–168.
Westmark, C. J., Westmark, P. R., O’Riordan, K. J., Ray, B. C., Hervey, C. M., Salamat, M. S., et al. (2011). Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS ONE 6:e26549. doi: 10.1371/journal.pone.0026549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Willemsen, R., Bontekoe, C., Tamanini, F., Galjaard, H., Hoogeveen, A., and Oostra, B. (1996). Association of FMRP with ribosomal precursor particles in the nucleolus. Biochem. Biophys. Res. Commun. 225, 27–33. doi: 10.1006/bbrc.1996.1126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: fragile X, FMRP, Alzheimer’s disease, APP, mouse models
Citation: Renoux AJ, Carducci NM, Ahmady AA and Todd PK (2014) Fragile X mental retardation protein expression in Alzheimer’s disease. Front. Genet. 5:360. doi: 10.3389/fgene.2014.00360
Received: 30 May 2014; Paper pending published: 03 July 2014;
Accepted: 27 September 2014; Published online: 21 October 2014.
Edited by:
Babajan Banaganapalli, King Abdulaziz University, Saudi ArabiaReviewed by:
Peter Turnpenny, Royal Devon & Exeter NHS Foundation Trust, UKCara J. Westmark, University of Wisconsin, USA
Copyright © 2014 Renoux, Carducci, Ahmady and Todd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter K. Todd, Department of Neurology, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, MI 48109, USA e-mail:cGV0ZXJ0b2RAbWVkLnVtaWNoLmVkdQ==