 Federica Invernizzi1†
Federica Invernizzi1† Anna Ardissone2†
Anna Ardissone2† Eleonora Lamantea1
Eleonora Lamantea1 Barbara Garavaglia1
Barbara Garavaglia1 Massimo Zeviani1Laura Farina3
Massimo Zeviani1Laura Farina3 Daniele Ghezzi1*Isabella Moroni2*
Daniele Ghezzi1*Isabella Moroni2*- 1Unit of Molecular Neurogenetics, Istituto Neurologico “Carlo Besta,” Fondazione Istituto di Ricovero e Cura a Carattere Scientifico, Milan, Italy
- 2Unit of Child Neurology, Istituto Neurologico “Carlo Besta,” Fondazione Istituto di Ricovero e Cura a Carattere Scientifico, Milan, Italy
- 3Unit of Neuroradiology, Istituto Neurologico “Carlo Besta,” Fondazione Istituto di Ricovero e Cura a Carattere Scientifico, Milan, Italy
Multiple Mitochondrial Dysfunction Syndrome (MMDS) comprises a group of severe autosomal recessive diseases with onset in early infancy and characterized by a systemic disorder of energy metabolism, resulting in weakness, respiratory failure, lack of neurological development, lactic acidosis, and early death. Biochemical findings include defects of complexes I, II, and III of the mitochondrial respiratory chain and severe deficiency of Pyruvate dehydrogenase complex (PDHc). Three genes have been associated with MMDS since now: NFU1, BOLA3, and IBA57. We describe an Italian male patient presenting with severe psychomotor regression after an infectious episode, lactic acidosis, hyperglycinemia, reduction of respiratory chain complex II associated with a marked deficiency of PDHc activity. He carried two heterozygous mutations in NFU1, one novel (p.Cys210Phe) and one previously reported (p.Gly189Arg) missense change affecting highly conserved residues. A severe leukoencephalopathy with cavitations in deep white matter was disclosed at brain MRI, suggesting a peculiar neuroradiological phenotype associated with defect in this gene.
Introduction
Multiple Mitochondrial Dysfunction Syndrome (MMDS) is a severe autosomal recessive disease with onset in early infancy. It is characterized by a systemic disorder of energy metabolism resulting in marked impairment of neurologic development, with diffuse weakness, respiratory failure, lactic acidosis, and early death (Seyda et al., 2001; Roualt and Tong, 2008). MMDS has been associated with mutations in NFU1 (causing MMDS1, MIM#605711), BOLA3 (causing MMDS2, MIM#614299) and IBA57 (causing MMDS3, MIM#615330) (Cameron et al., 2011; Navarro-Sastre et al., 2011; Ajit Bolar et al., 2013), all genes coding proteins with a role in iron-sulfur (Fe-S) cluster biosynthesis. Defects in this pathway, comprising also mutations in ISCU (MIM#255125) (Mochel et al., 2008), lead to abnormal function of many proteins containing Fe-S centers, including mitochondrial respiratory chain (MRC) complexes I, II, and III, and lipoic acid synthase (LIAS). The latter enzyme is needed for the maturation of four mitochondrial lipoate-dependent enzymes: pyruvate dehydrogenase complex (PDHc), α-ketoglutarate dehydrogenase (α-KGDH), branched-chain ketoacid dehydrogenase (BCKDH) and the H protein of the glycine cleavage system (MIM*238330) (Roualt and Tong, 2008; Stehling et al., 2014). Interestingly, mutations in LIAS cause Pyruvate dehydrogenase lipoic acid synthetase deficiency (MIM#614462), a syndrome characterized by several biochemical and clinical features overlapping with MMDS (Mayr et al., 2011).
NFU1 is a Fe-S cluster scaffold, initially considered as an alternative scaffold to ISCU, while recently characterized as a late targeting factor of Fe-S clusters, specifically required for the proper assembly of only a subset of (4Fe4S) proteins including LIAS and subunits of MRC complexes I and II (Cameron et al., 2011; Navarro-Sastre et al., 2011; Stehling et al., 2014). Since now three reports have described 14 patients belonging to 11 families with NFU1 mutations (Cameron et al., 2011; Navarro-Sastre et al., 2011; Nizon et al., 2014): most cases presented with infantile encephalopathy associate with pulmonary hypertension, lactic acidosis, hyperglycinemia and severe regression leading to death before the age 15 months (Cameron et al., 2011; Navarro-Sastre et al., 2011). Only one child survived until the age of 2½ years (Nizon et al., 2014); leukoencephalopathy at brain MRI, has been illustrated only in this last patient, and reported without documentation in an additional case. Ten individuals from Spanish and French families were homozygous for p.Gly208Cys mutation, suggesting a founder effect in Europe. Other three mutations have been described in the remaining cases.
We describe the first Italian patient affected by MMDS1 associated with two heterozygous mutations in NFU1.
Background
Case Report
The proband was the first child born from unrelated Italian parents. Family history was negative; pregnancy and delivery were uneventful. Psychomotor development was referred normal until 7 months of age. During a mild febrile illness, he presented severe and rapid psychomotor regression, loosing sitting position and smiling, and developed spastic tetraparesis. Metabolic exams revealed high plasma lactate (5620 mmol/L; nv 580–2100 mmol/L) and pyruvate levels (294 mmol/L; nv 55–145), abnormal urinary excretion of lactic, succinic, fumaric, and glutaric acids; plasma amino acids analysis disclosed elevation of glycine (589 mmol/l, nv 11–360). Brain MRI showed the presence of a diffuse and symmetric involvement of hemispheric white matter with more marked abnormalities in the deep white matter that presented also cavitations (Figure 1). The cerebellum, brainstem and basal ganglia had normal appearance, while spectroscopy revealed a peak of lactic acid and decreased N-acetyl Aspartate/creatine ratio.
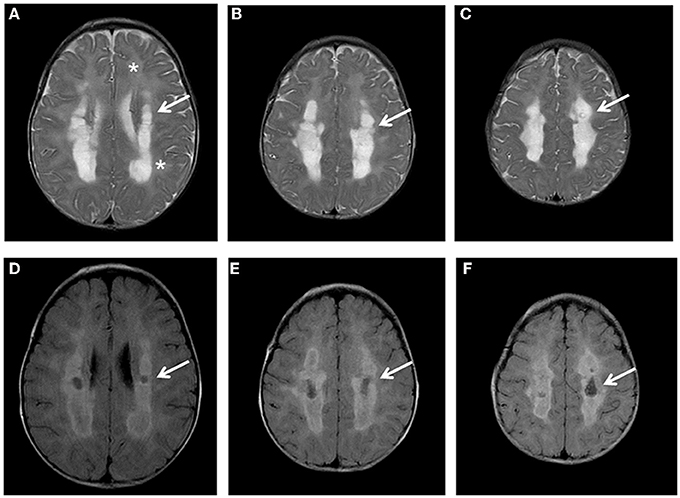
Figure 1. Brain MRI. Axial T2 (A-C) and FLAIR images (D-F) show diffuse hyper intensity of the white matter (A: asterisks) more prominent in the posterior periventricular and deep regions (A-C: arrows) with evidence of partial cystic degeneration and cavitations in FLAIR sections (D-F: arrows).
The neurological examination at 9 months revealed low body weight and height (10–25 centiles) with normal head circumference (50–75 centiles). He had good visual contact and response to sound, severe spasticity, absence of head and trunk control, dystonic postures, and frequent crying. Electroencephalogram showed a poor organization without epileptic abnormalities.
Electrocardiogram was normal and pulmonary hypertension was never detected. A mitochondrial disease was suspected and he underwent muscle and skin biopsies.
Despite the beginning of treatment with vitamin B2, B1, and Coenzyme Q the child further deteriorated during the following months: he suffered from several episodes of metabolic decompensation, developed epileptic seizures and required nasogastric feeding tube. He died at 18 months of age during a severe pneumonia; autopsy was not performed.
Muscle biopsy showed normal histological pictures. The analysis of MRC complexes activities revealed a clear reduction of complex II and SDH in muscle (43 and 48% of the control mean, respectively), and fibroblasts (30 and 55% respectively) and mild decrease of complex I in muscle (62%), and complex IV in fibroblasts (62%); the activate form of PDHc was also markedly reduced in both muscle and fibroblasts (28 and 1% respectively) (Table 1).
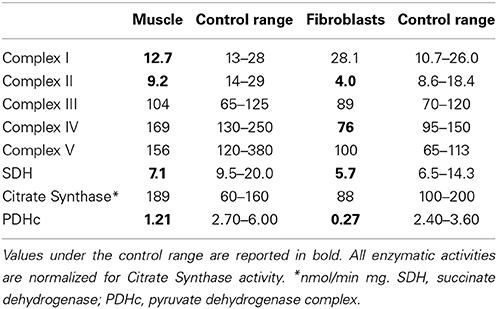
Table 1. Biochemical activity of respiratory chain complexes and PDHc activities in muscle and fibroblasts.
Material and Methods
Informed consent was obtained from patient's parents, in agreement with the Declaration of Helsinki and approved by the Ethical Committees of the Foundation IRCCS Istituto Neurologico “C. Besta,” Milan, Italy.
Biochemical studies
Skeletal muscle and skin biopsies were performed according to standard protocols. The measurement of MRC complexes activities was performed using standard spectrophotometric techniques (Bugiani et al., 2004) in both muscle homogenate and cultured skin fibroblasts, and normalized to citrate synthase (CS) activity, an index of mitochondrial content in the analyzed specimens. PDHc assay was performed with radioactive method (Uziel et al., 1988).
Molecular studies
DNA was extracted from peripheral blood of the patient and their parents using standard methods. The coding sequence and flanking intronic regions of NFU1 were analyzed by PCR amplification using the primers reported in Supplementary Table 1. Amplicons were stained with ethidium bromide on 2% agarose gels, cycle-sequenced using BigDye chemistry 3.1, and run on an ABI 3130XL automatic sequencer (Applied Biosystems).
Western-Blot analysis
Approximately 2 × 106 fibroblasts were trypsinized, pelleted, sonicated, solubilized with 10 mM phosphate pH 7.2, 10 mM EDTA, 150 mM NaCl, 2% Triton X-100 and 0.25% SDS, in presence of a mixture of protease inhibitors, and centrifuged at 100,000 g for 30 min at 4°C. A 50 μg aliquot of protein per lane was then electrophoresed through an SDS-polyacrylamide gel. The gel was then electroblotted for 90 min onto a nitrocellulose filter. The latter was incubated with 5% non-fat milk in 20 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween-20 (MTT) for 60 min at room temperature followed by incubation overnight at 4°C with primary antibody. After four washing in MTT the filter was incubated for 60 min at room temperature with a goat anti-rabbit/mouse IgG conjugated to horseradish peroxidase (HRP) (1:5000 in MTT). After four additional washing, the peroxidase reaction was revealed by autoradiography using the chemiluminescence ECL kit (GE Healthcare). The following primary antibody, diluted in MTT, were used: α-NFU1 (Biorbyt, 1:50), α-NDUFA9 (subunit of complex I; Mitoscience, 1:2000), α-SDHA (subunit of complex II; Mitoscience, 1:10,000), α-SDHB (subunit of complex II; Mitoscience, 1:200), α-UQCRFS1 (subunit of complex III; Mitoscience, 1:1000), α-MTCO2 (subunit of complex IV; Mitoscience, 1:1500), α-COX4 (subunit of complex IV; Mitoscience, 1:2000), α-Tubulin beta (Sigma, 1:2000).
Genetic and Biochemical Results
The genetic analysis of NFU1 showed the presence of two heterozygous mutations, c.565G>A and c.629G>T (NM_001002755.2), causing the p.Gly189Arg and p.Cys210Phe changes in the protein amino acid sequence (NP_001002755.1) (Figure 2A). Segregation analysis confirmed that the first mutation was inherited from the mother and the second from the father. The two identified nucleotide substitutions were not reported in public databases, including dbSNPs and NHLBI Exome Sequencing Project. The two affected amino acids are conserved in several species (Figure 2B) and the amino acid changes have high scores for pathogenicity according to different bioinformatic tools.
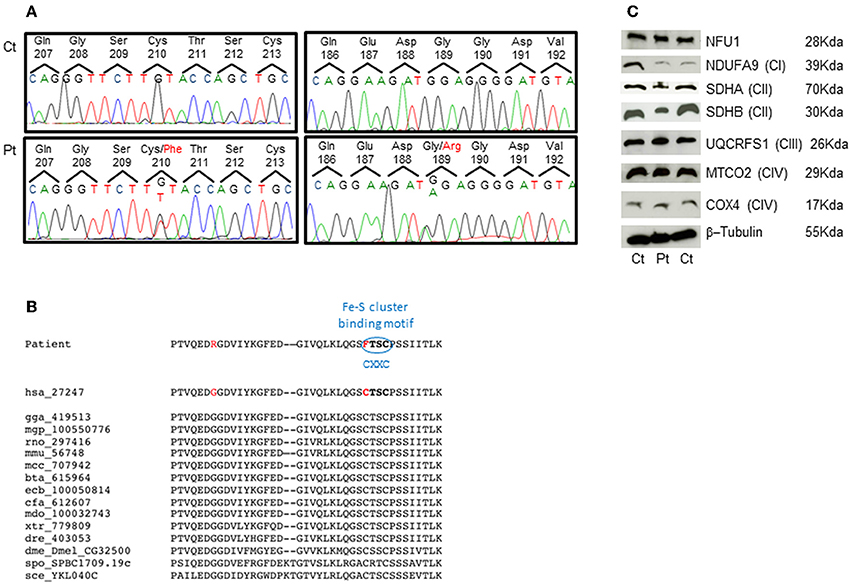
Figure 2. Genetic studies and protein characterization in fibroblasts. (A) Chromatograms of the NFU1 genomic regions comprising the identified variant in the patient (Pt, lower panel) and in a control subject (Ct, upper panel). (B) Multiple alignment of NFU1 protein domain containing mutations. Both mutations affect highly conserved amino acids and the Cys210Phe strikes the first cysteine of the CXXC motif that is a characteristic binding domain for Fe-S cluster. (C) SDS-gel electrophoresis and Western blot analysis of fibroblasts from patient (Pt) and two controls (Ct). We used antibodies against NFU1; NDUFA9 (subunit of complex I); SDHA and SDHB (subunits of complex II); UQCRFS1 (subunit of complex III); MTCO2 and COX4 (subunits of complex IV); and β-Tubulin (as loading control).
Western blot analysis performed on patient's fibroblasts showed no reduction of NFU1 protein; however, we found a partial reduction of the subunits of complex II, SDHA (70 kDa), and SDHB (30 kDa), whereas the amount of subunits belonging to other MRC complexes were similar to control levels (Figure 2C), using β-tubulin as loading control. These findings are in agreement with those recently reported by Ferrer-Cortes et al. (2013).
Discussion
Review Similar Cases
In 2001, Seyda et al. (2001) reported on three siblings, born from non-consanguineous parents of Mexican descent, affected with metabolic acidosis and high lactate levels, feeding difficulty, weakness and lethargy present since few days after birth; all three babies died within the first month of life. Biochemical studies demonstrated a combined defect of PDHc, MRC complex II, complex I + III, and complex II + III in fibroblasts from all patients. Laboratory investigations showed lactic acidosis and elevated levels of serum glycine.
Ten years later, next-generation sequencing (NGS) allowed Cameron et al. (2011) to detect a homozygous missense mutation (c.545G>A; p.Arg182Gln) in exon 6 of NFU1 in these previously reported patients. Contemporary, Navarro-Sastre et al. (2011) reported ten individuals from nine unrelated Spanish families affected with fatal MMDS and carrying mutations in NFU1 gene. In all patients the onset of symptoms started in the first months of life, and all died before the age of 16 months. They presented with failure to thrive, pulmonary hypertension, and neurological regression. For 3/13 published cases brain imaging data were available, and in only one case the presence of leukodystrophy was reported; the others were described with normal MRI pattern or unspecific and inconsistent features (e.g., cerebral atrophy, cerebellar lesions). However, white-matter necrosis with preservation of U fibers or areas of white-matter demyelination and vacuolization were found in cases in which autopsy was performed (Seyda et al., 2001; Navarro-Sastre et al., 2011). The biochemical phenotype included metabolic acidosis with variable lactic acidemia and hyperglycinemia, and high urinary excretion of 2-ketoglutaric, 2-ketoadipic, 2-hydroxyadipic, and glutaric acid. In the cases where tissues were available, activity of the glycine cleavage system in liver was low or undetectable. PDHc activity in fibroblasts was also decreased. The rates of 14C-substrate oxidation (pyruvate, leucine, and glutamate) in fibroblasts were low. Only in 3 patients MRC enzyme activities were assessed in frozen muscle, and a reduction of complex II + III activity (26–43% of control value) was detected. Genetic studies of NFU1 in these patients showed the same c.622G>T/p.Gly208Cys mutation in exon 7 (9/10 homozygous, 1/10 heterozygous) and an additional intronic mutation c.545 + 5G>A affecting the donor splice-site of exon 6 (1/10 heterozygous). Recently, a further case was reported by Nizon et al. (2014), a French female with normal psychomotor development until 14 months of age, when she showed progressively losing of standing and sitting abilities with failure to thrive and pulmonary hypertension; then she developed a severe spastic tetraparesis associated with extrapyramidal signs. Lactate and pyruvate levels resulted within normal values, except during two episodes of metabolic decompensation. Glycine levels were high in plasma, urine and CSF, and urinary organic acid analysis showed mildly increased excretion of glutaric acid, 2-hydroxyglutaric acid and α-ketoglutaric acid. Fibroblasts analysis demonstrated a defect of PDHc activity (25%) and of complex II (28%). Brain MRI revealed the presence of leukoencephalopathy involving periventricular white matter and corpus callosum, that were partially cystic.
The clinical, biochemical and genetic data of the previous and present cases are reported in Table 2.
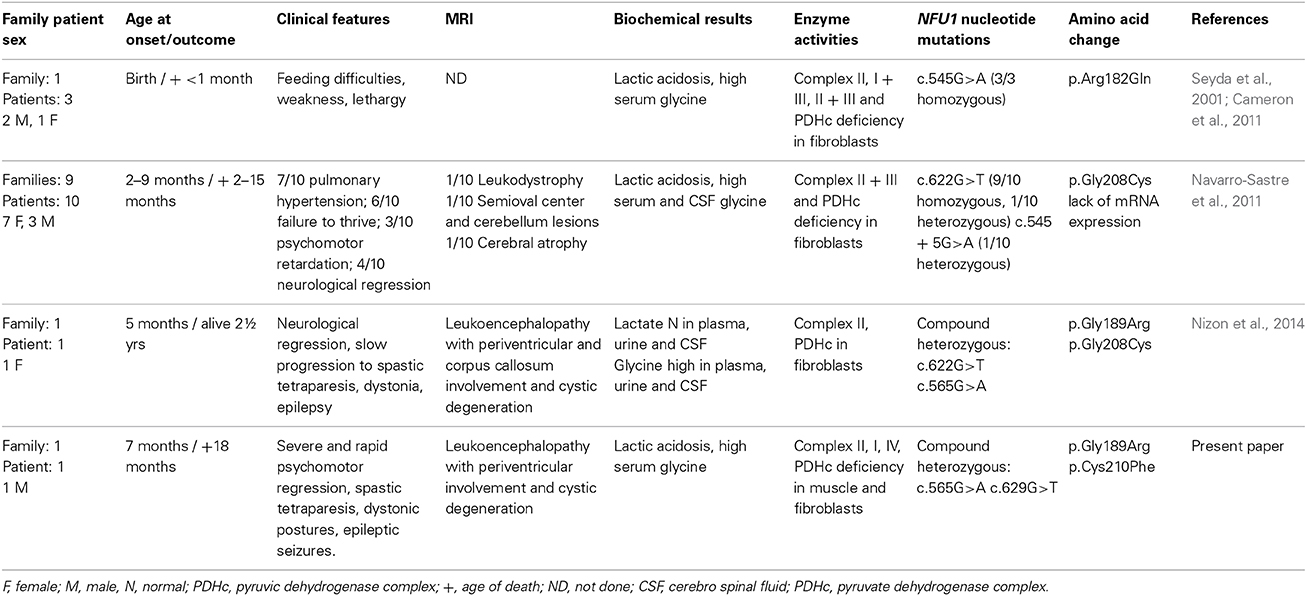
Table 2. Clinical, biochemical and genetic findings in patients with NFU1 mutations.
NFU1 is required for efficient assembly of lipoate synthase and respiratory complex II (SDH); NFU1 transiently bind the (4Fe–4S) cluster which may be transferred to the target apoprotein. The two mutations identified in our patient are both located in a highly conserved iron sulfur cluster assembly domain; moreover, the p.Cys210Phe change hits the first cysteine of a CxxC motif essential for the binding of Fe-S cluster. Although Western Blot analysis on fibroblasts failed to reveal any reduction of NFU1, the identified amino acid changes suggest a severe dysfunction of the protein, confirmed by the biochemical profile identified in our patients.
The presence of the p.Gly208Cys mutation in ten families from Spanish and French has suggested a founder effect in Europe (Navarro-Sastre et al., 2011; Nizon et al., 2014); the p.Gly189Arg mutation, found in our patient and in a previously described case (Nizon et al., 2014) can represent a second possible common European mutation.
The onset of symptoms and the clinical course in our case were similar to previously described patients, except for absence of pulmonary hypertension. The presence of abnormal urinary excretion of lactate and elevated levels of plasma glycine confirm that they represent the biochemical hallmarks of this syndrome.
Brain MRI pictures have not been fully described in the previous cases; the presence of white matter abnormalities has been reported in only one case from Navarro-Sastre series, but no MRI was available (Navarro-Sastre et al., 2011). Moreover, the evidence of a leukodystrophy associated with early development of cavitations in our patient similar to that described in one patient by Nizon et al. (2014), suggests that this may be the MRI specific pattern related to MMDS1. Interestingly, cavitating leukoencephalopathy represents an increasingly common MRI finding in mitochondrial disorders due to different genetic defects (Kashani et al., 2014; Melchionda et al., 2014).
Concluding Remarks
We have described the first Italian patient affected with MMDS1. This case contributes to delineate the phenotype associated with defects in NFU1, represented by severe early onset neurological impairment, multiple defects of respiratory chain and PDHc, high lactate and glycine levels. Moreover, the presence of leukodystrophy with cysts or cavitations is probably the commonest neuroradiological picture in MMDS1 patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Italian Ministry of Health (GR2010–2316392); Fondazione Telethon grant GGP11011; Fondazione CARIPLO grant 2011/0526; Fondazione Pierfranco e Luisa Mariani.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2014.00412/abstract
References
Ajit Bolar, N. L., Vanlander, A. V., Wilbrecht, C., Van der Aa, N., Smet, J., De Paepe, B., et al. (2013). Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum. Mol. Genet. 22, 2590–2602. doi: 10.1093/hmg/ddt107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bugiani, M., Invernizzi, F., Alberio, S., Briem, E., Lamantea, E., Carrara, F., et al. (2004). Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta 1659, 136–147. doi: 10.1016/j.bbabio.2004.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cameron, J. M., Janer, A., Levandovskiy, V., MacKay, N., Rouault, T. A., Tong, W. H., et al. (2011). Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 89, 486–495. doi: 10.1016/j.ajhg.2011.08.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrer-Cortes, X., Font, A., Bujan, N., Navarro-Sastre, A., Matalonga, L., Arranz, J. A., et al. (2013). Protein expression profiles in patients carrying NFU1 mutations. Contribution to the pathophysiology of the disease. J. Inherit. Metab. Dis. 36, 841–847. doi: 10.1007/s10545-012-9565-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kashani, A. L., Thiffault, I., Dilenge, M. E., Saint-Martin, C., Guerrero, K., Tran, L. T., et al. (2014). A homozygous mutation in the NDUFS1 gene presents with a mild cavitating leukoencephalopathy. Neurogenetics 15, 161–164. doi: 10.1007/s10048-014-0412-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayr, J. A., Zimmermann, F. A., Fauth, C., Bergheim, C., Meierhofer, D., Radmayr, D., et al. (2011). Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am. J. Hum. Genet. 89, 792–797. doi: 10.1016/j.ajhg.2011.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Melchionda, L., Haack, T. B., Hardy, S., Abbink, T. E., Fernandez-Vizarra, E., Lamantea, E., et al. (2014). Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 95, 315–325. doi: 10.1016/j.ajhg.2014.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mochel, F., Knight, M. A., Tong, W. H., Hernandez, D., Ayyad, K., Taivassalo, T., et al. (2008). Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 82, 652–660. doi: 10.1016/j.ajhg.2007.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Navarro-Sastre, A., Tort, F., Stehling, O., Uzarska, M. A., Arranz, J. A., Del Toro, M., et al. (2011). A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 89, 656–667. doi: 10.1016/j.ajhg.2011.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nizon, M., Boutron, A., Boddaert, N., Slama, A., Delpech, H., Sardet, C., et al. (2014). Leukoencephalopathy with cysts and hypeglycinemia may result from NFU1 deficiency. Mitochondrion 15, 59–64. doi: 10.1016/j.mito.2014.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roualt, T. A., and Tong, W. H. (2008). Iron-sulfur cluster biogenesis and human disease. Trends Genet. 24, 398–407. doi: 10.1016/j.tig.2008.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seyda, A., Newbold, R. F., Hudson, T. J., Verner, A., MacKay, N., Winter, S., et al. (2001). A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am. J. Hum. Genet. 68, 386–396. doi: 10.1086/318196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stehling, O., Wilbrecht, C., and Lill, R. (2014). Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie. 100, 61–77. doi: 10.1016/j.biochi.2014.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uziel, G., Garavaglia, B., and Di Donato, S. (1988). Carnitine stimulation of pyruvate dehydrogenase complex (PDHC) in isolated human skeletal muscle mitochondria. Muscle Nerve 11, 720–724. doi: 10.1002/mus.880110708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: leukoencephalopathy, multiple mitochondrial dysfunction syndrome, MMDS1, NFU1, mitochondrial disorders, brain MRI
Citation: Invernizzi F, Ardissone A, Lamantea E, Garavaglia B, Zeviani M, Farina L, Ghezzi D and Moroni I (2014) Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Front. Genet. 5:412. doi: 10.3389/fgene.2014.00412
Received: 11 October 2014; Accepted: 06 November 2014;
Published online: 20 November 2014.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Vita Dolzan, University of Ljubljana, SloveniaWladimir Bocca Vieira De Rezende Pinto, Federal University of São Paulo, Brazil
Copyright © 2014 Invernizzi, Ardissone, Lamantea, Garavaglia, Zeviani, Farina, Ghezzi and Moroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Ghezzi, Unit of Molecular Neurogenetics, Fondazione Istituto Neurologico “Carlo Besta,” via Temolo 4, 20126 Milan, Italy e-mail:ZGdoZXp6aUBpc3RpdHV0by1iZXN0YS5pdA==;
Isabella Moroni, Unit of Child Neurology, Fondazione Istituto Neurologico “Carlo Besta,” via Celoria 11, 20133 Milan, Italy e-mail:aXNhYmVsbGEubW9yb25pQGlzdGl0dXRvLWJlc3RhLml0
†These authors have contributed equally to this work.