 Petra M. Hermann
Petra M. Hermann Shawn N. Watson
Shawn N. Watson Willem C. Wildering
Willem C. Wildering- 1Department of Biological Sciences, University of Calgary, Calgary, AB, Canada
- 2Department of Physiology and Pharmacology, University of Calgary, Calgary, AB, Canada
- 3Hotchkiss Brain Institute, University of Calgary, Calgary, AB, Canada
The aging brain undergoes a range of changes varying from subtle structural and physiological changes causing only minor functional decline under healthy normal aging conditions, to severe cognitive or neurological impairment associated with extensive loss of neurons and circuits due to age-associated neurodegenerative disease conditions. Understanding how biological aging processes affect the brain and how they contribute to the onset and progress of age-associated neurodegenerative diseases is a core research goal in contemporary neuroscience. This review focuses on the idea that changes in intrinsic neuronal electrical excitability associated with (per)oxidation of membrane lipids and activation of phospholipase A2 (PLA2) enzymes are an important mechanism of learning and memory failure under normal aging conditions. Specifically, in the context of this special issue on the biology of cognitive aging we portray the opportunities offered by the identifiable neurons and behaviorally characterized neural circuits of the freshwater snail Lymnaea stagnalis in neuronal aging research and recapitulate recent insights indicating a key role of lipid peroxidation-induced PLA2 as instruments of aging, oxidative stress and inflammation in age-associated neuronal and memory impairment in this model system. The findings are discussed in view of accumulating evidence suggesting involvement of analogous mechanisms in the etiology of age-associated dysfunction and disease of the human and mammalian brain.
Introduction
Most organisms age, humans and the vast majority of other complex animals included. That is, the life functions required for their survival and reproduction weaken as they grow older due to natural processes that are still only partially understood and involve a complex combination of chemical, genetic, metabolic, molecular, physiological, ecological, and evolutionary features (see for instance Kirkwood and Austad, 2000; Ljubuncic and Reznick, 2009; Masoro and Austad, 2011).
Aging generally affects all aspects of an organism’s biology including, in the case of humans and complex multicellular animals, the nervous system’s ability to execute its control processes and support learning and memory functions. For example, declining learning and memory performance, weakening sensory, and motor functions, and slowing of reaction times are common symptoms of old age in humans and many vertebrate and invertebrate model systems (Burke and Barnes, 2006; Chawla and Barnes, 2007; Murakami, 2007; Partridge, 2008; Yeoman et al., 2012; Haddadi et al., 2014). Some of these changes, particularly in our own species, may involve pathological processes such as Alzheimer’s and Parkinson’s disease. However, even in the healthy aging brain where loss of neurons and circuits to cell death is generally not a significant factor, functional performance tends to decline with age (Peters et al., 1994; West et al., 1994; Rapp and Gallagher, 1996; Rasmussen et al., 1996; Gazzaley et al., 1997; Merrill et al., 2000, 2001; Keuker et al., 2003; Burke and Barnes, 2006). Why this happens, particularly what makes healthy aging neurons change the way in which they work and communicate with each other is the focus of this article. Specifically, we explore the idea that processes triggered by oxidative stress ensuing at the level of phospholipid bilayer membranes of cells and organelles play a central role in age-associated deterioration of the healthy aging nervous system (Note: while this review centers on non-pathological mechanisms of brain aging it should be kept in mind that many consider normal and pathological aging as two causally related extremes of a continuum in which it is not always clear where normal aging ends and pathological aging starts. See for instance Hung et al., 2010; Dai et al., 2014).
The idea that oxidative stress is a major factor in biological aging has a long history and extensive experimental support (Harman, 1956; Pérez et al., 2009; Hekimi et al., 2011; Speakman and Selman, 2011; Zimniak, 2011; Alcedo et al., 2013). Yet, despite its prominence in the conceptual framework of biological aging, understanding of how oxidative stress translates to functional decline of the aging nervous system remains incomplete. Here we examine how (per)oxidation of methylene-interrupted poly-unsaturated fatty acids1 (PUFAs) and subsequent activation of members of the phospholipase A2 (PLA2) family of fatty acylases may play a central role in declining excitability of aging neurons and age-associated memory impairment (AMI; see Figure 1 for conceptual framework; see Figure 2 for mechanisms and potential implications of PUFA peroxidation).
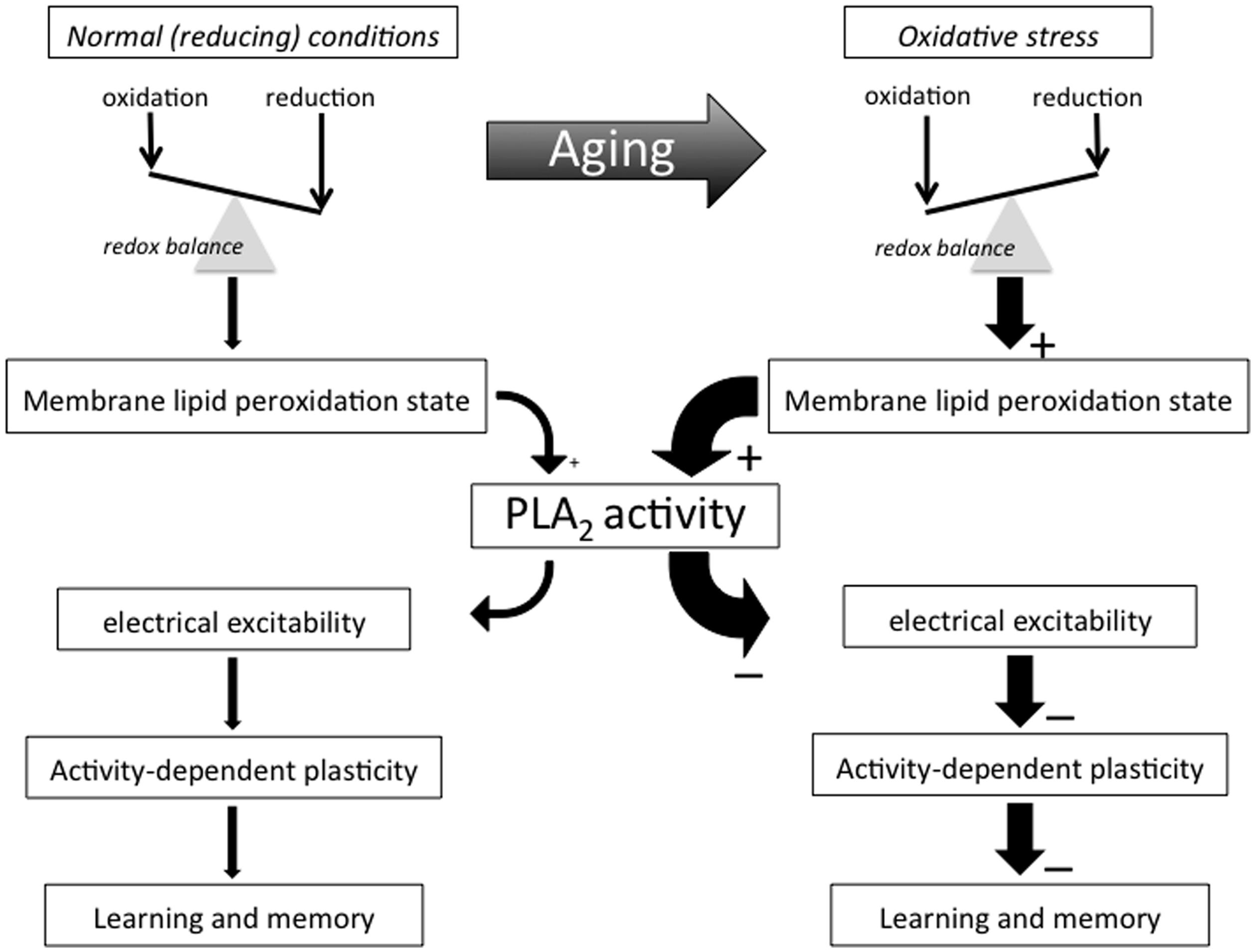
FIGURE 1. Conceptual framework. This flow diagram outlines key concepts and hypotheses addressed in this essay and illustrate the way we propose they contribute to AMI. Many important biological processes involve redox reactions and many of life’s macromolecules are sensitive to electrophilic attacks that may impair their ability to perform their normal functional or structural roles. Few of life’s building blocks are as sensitive to oxidation as the poly-unsaturated fatty acids (PUFAs) making up much of cell and organelle membranes. Organisms generate electrophiles like reactive oxygen and nitrogen species as part of many essential live processes, including metabolism, host defense, and intercellular signaling processes. Active maintenance of an appropriate redox balance is therefore of the utmost importance for organismal function and survival. Biological aging tends to be associated with shifts in redox balance away from reducing towards oxidative conditions in many of an organism’s molecular, cellular, and organismal systems and domains, including an advanced state of membrane PUFA peroxidation that recruits PLA2 and induces a (PLA2-dependent) decline in neuronal excitability and activity-dependent plasticity that manifests itself as AMI.
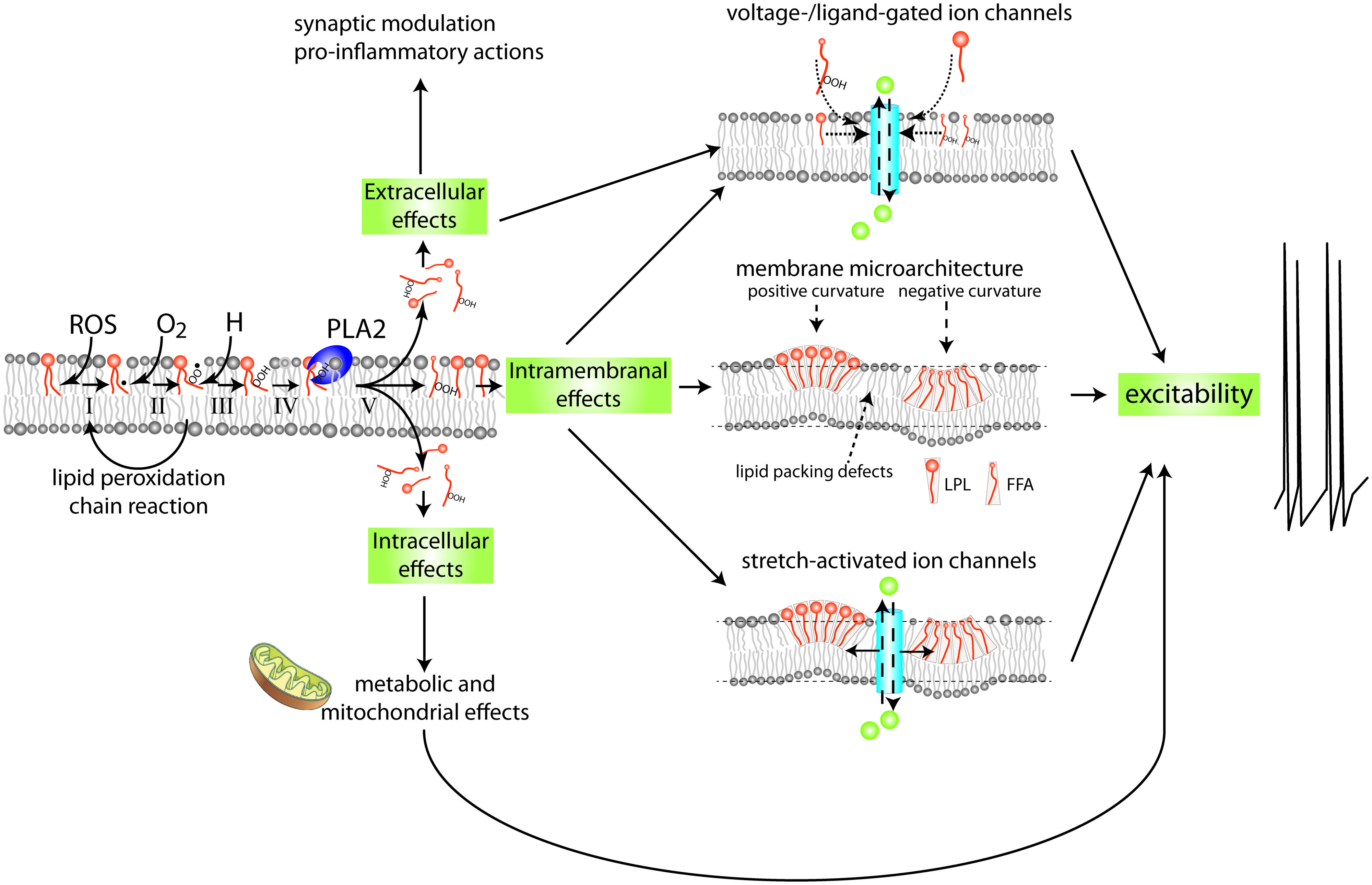
FIGURE 2. Mechanism and possible implications of membrane lipid peroxidation and phospholipase A2 activation. PUFAs are highly susceptible to (per)oxidation. Peroxidized PUFAs are prone to excision by members of the PLA2 family of enzymes. The products of PLA2’s enzymatic activity, free fatty acids (FFAs) and lysophospholipids (LPLs), have a wide range of extracellular, intracellular, and intramembranal biological activities. This highly simplified diagram indicates a few of the mechanisms and targets with special relevance to the nervous system. FFAs and LPLs can themselves act as signaling molecules and modify gating behavior of various types of ion channels. In addition they can act as substrate for other lipid-based intra- and extracellular signaling cascades, including signaling systems regulating inflammation. Moreover, through their effect on membrane microarchitecture they may change functions of integral membrane proteins such as stretch-activated ion channels, alter lipid micro domain organization, disrupt organization of transmembrane signaling complexes, or alter other cellular processes such as membrane fission and fusion that are important in the regulation of intrinsic neuronal excitability, synaptic transmission, and molecular mechanisms underlying neuronal plasticity. See Figure 6 for details on mechanisms involved in PUFA peroxidation.
Neurobiological Correlates of Normal Aging
Considerable effort has been put into identifying neurobiological correlates of normal brain aging for several decades. The picture emerging from these studies is that structural and functional changes occurring in the normal aging brain are generally much more low-key and variable than those found in pathological aging brains. The list of physiological and biochemical symptoms of neuronal aging is extensive and includes evidence for oxidative stress, impaired energy and redox metabolism, Ca2+ signaling perturbation, accumulating damage to proteins, lipids, nucleic acids and organelles, declining dendritic complexity, alterations in synaptic density, distribution and function, changes in gene regulation, and molecular processes underlying remodeling of synapses (Barnes, 1994; Burke and Barnes, 2006; Mattson and Magnus, 2006; Esiri, 2007; Shankar, 2010; Toescu and Vreugdenhil, 2010; Yeoman et al., 2012).
While cell autonomous processes likely constitute an important facet of neuronal aging, evidence for significant cell non-autonomous (i.e., organismal) dimensions of neuronal aging is growing. For instance, it is evident that brain aging has vascular, immunological and endocrinological dimensions (Joseph et al., 2005; Aïd and Bosetti, 2011; Yirmiya and Goshen, 2011; Alcedo et al., 2013; Rincon and Wright, 2013; Sadagurski and White, 2013; Oakley and Tharakan, 2014; Tarumi and Zhang, 2014). One particularly intriguing organismal dimension to normal brain aging emerging in recent decades is the concept that the brain at least in part regulates its own destiny through insulin-like peptide signaling systems coupling nutrient availability and dietary conditions to the control of cell and organismal metabolism (e.g., Alcedo et al., 2013; Sadagurski and White, 2013). Furthermore, while there seems little doubt that many of the phenomena associated with neuronal aging are of a deteriorative nature, evidence for functional preservation and compensation has also been reported (see Barnes, 1994; Burke and Barnes, 2006 for review).
The Need for Simplification: Lymnaea stagnalis as a Model for Integrative Molecule-to-Behavior Investigations of Neuronal and Behavioral Aging
How all dietary, (bio)chemical, metabolic, molecular, cellular, physiological, system-, and organismal-level processes factor into biological aging of neurons and the nervous system and contribute to AMI remains one of the most important unsolved puzzles of neuroscience. Answering fundamental questions of this kind in a system as complex, dynamic, and inherently adaptive as the nervous system is not a trivial problem. With the goal of reducing the complexity of such undertaking, we adopted the pond snail L. stagnalis as our research platform (Figure 3). Since Lymnaea is not as widely known as other model systems in neuronal aging and AMI we will briefly summarize its most relevant features before continuing with the main focus of this review.
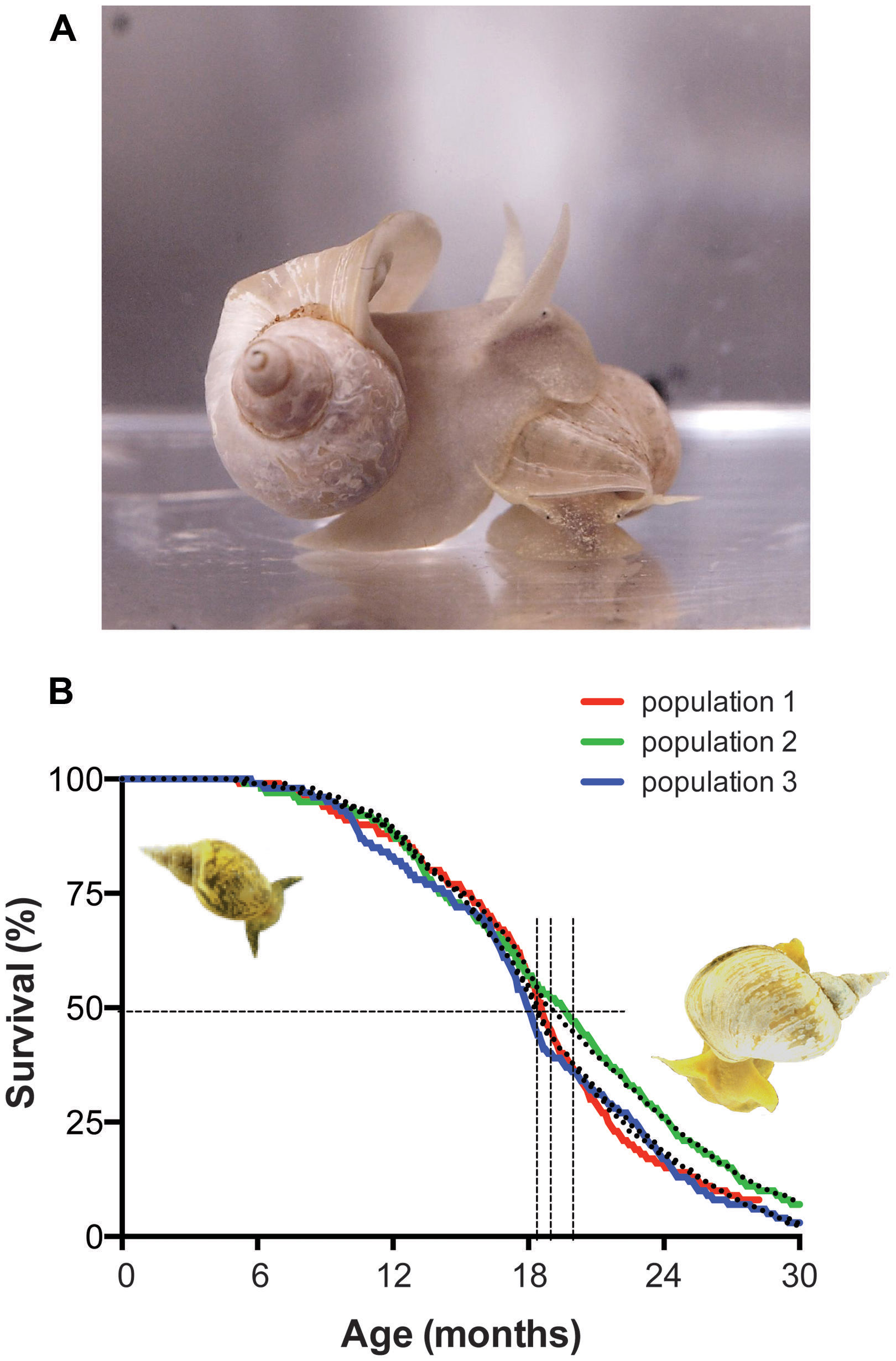
FIGURE 3. Lymnaea stagnalis, the animal and its survival characteristics. (A) L. stagnalis is an obligate freshwater aquatic gastropod of the clade Euthyneura. The animals are bimodal breathers capable of absorbing oxygen through their skin and through ventilating a simple lung cavity to air. (B) Survival characteristics of populations of Lymnaea held in our aquatic facility. Shown are the census data for three different populations, representative for normal population survival in the facility (colored lines) fitted by a Weibull power law failure model (dotted lines). Under the conditions in our facility the median population age hovers around 1.5 years (vertical dotted lines median age ± 95% confidence interval) and individual maximum age routinely exceeds 2 years. Under standard culture conditions we maintain in our in house facility (i.e., ad libitum feeding of standard diet at 17–19°C water temperature under a 12:12 light:dark regimen) survival characteristics of healthy captive populations show very little variation and reach an average median age of 593 ± 13 days and average maximal age of 880 ± 10 days (arithmetic means ± SE of the mean, n = 15; for more details on aquaculture conditions see Watson et al., 2012a).
Lymnaea stagnalis is a freshwater pulmonate snail with a long and varied track record as a model system in a wide range of basic and applied biological research, including the study of behavioral, neural, and molecular aspects of associative learning and memory. It has a relatively simple central nervous system (CNS, Figure 4) of about 20,000–25,000 neurons organized in 11 ganglia. The CNS contains many individually identifiable neurons and well-mapped, behaviorally characterized neural circuits supporting direct behavior-to-molecule inferences about the foundations of neuronal function and dysfunction (see for instance Benjamin et al., 2000; Taylor and Lukowiak, 2000; Brembs, 2003; Benjamin and Kemenes, 2010) including the biochemical, molecular, cell biological, and physiological foundations of neuronal aging and AMI.
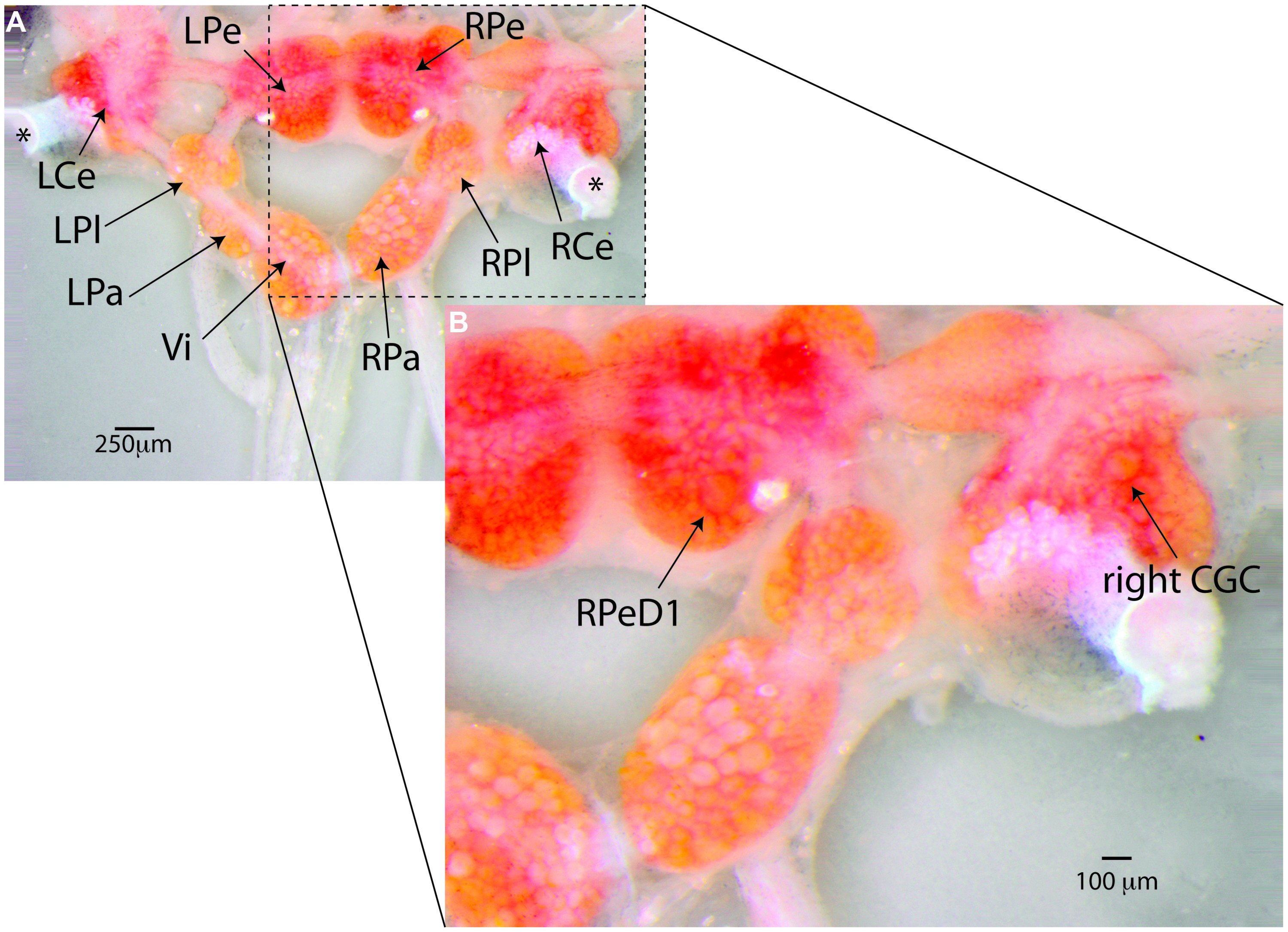
FIGURE 4. Lymnaea’s central nervous system and its identified neurons. (A) Lymnaea’s central nervous system (CNS) is comprised of a circumesophagal ganglionic ring made up of nine ganglia plus two buccal ganglia that are not part of the inner ganglionic ring (LCe, left cerebral ganglion; RCe, right cerebral ganglion; LPe, left pedal ganglion; RPe, right pedal ganglion; LPl, left pleural ganglion; RPl, right pleural ganglion; LPa, left parietal ganglion; RPa, right parietal ganglion; Vi, visceral ganglion; the paired left and right buccal ganglia are not shown in this image. To expose location of neurons discussed in this paper, the cerebral commissure connecting LCe and RCe has been cut and LCe and RCe have been folded out laterally). Inside the ganglia somata of many neurons can be recognized. Many of these neurons are individually identifiable. (B) Enlargement of the boxed area in (A) showing the location of the two identified neurons that are the main focus of the paper. Right pedal dorsal 1 (RPeD1), located on dorsal surface of the RPe ganglion is a multi-modal neuron involved in the control of Lymnaea’s aerial respiration and an important locus of operant-conditioning induced plasticity of the animals’ respiratory behavior. The cerebral giant cell (GCG) indicated here is one of a bilaterally symmetrical pair of interneurons instrumental in the control of Lymnaea’s feeding behavior. The CGCs are important loci of reward-conditioning of the animals feeding behavior. These composite images were generated from 15–20 images taken at different focal depths using DMap focus stacking algorithm in Zerene Stacker build T201404082055 (Zerene Systems, Richland, WA, USA).
Under laboratory conditions, Lymnaea can be effectively reared to ages and quantities required for aging research. Depending on variables like feeding regimen, animal density, day length and water temperature, the maximal age of the animals routinely exceeds 2.5 years with median ages of around 1.5 years under these conditions and age-dependent mortality follows Gompertz and Weibull survival characteristics indicative of aging (Figure 3B; see also Janse et al., 1988; Slob and Janse, 1988).
Lymnaea’s application in aging studies dates back to the 1980s with the publication of several neurophysiological studies (Frolkis et al., 1984; Nagy et al., 1985; Janse et al., 1986), the first report of age effects on associative memory performance (Audesirk et al., 1982) and work on the general biology and population-level aspects of the species’ aging process (Janse et al., 1988; Slob and Janse, 1988). Since then publications on various cell biological, endocrinological, neurophysiological, biophysical, behavioral, and reproductive aspects of aging in Lymnaea have appeared (Frolkis et al., 1989, 1991; Janse et al., 1989, 1999; Wildering et al., 1991; Klaassen et al., 1998; Janse and van der Roest, 2001; Arundell et al., 2006; Patel et al., 2006, 2010). Its use in research of the molecular and cellular foundations of AMI started in earnest with Hermann et al. (2007) followed by a series of other publications from our laboratory linking AMI and neuronal excitability changes to oxidative stress, inflammation, lipid peroxidation and declining glutathione (GSH) availability, and identifying PLA2 as a central player in these phenomena (Watson et al., 2012a,b, 2013, 2014; Hermann et al., 2013; Beaulieu et al., 2014).
Lymnaea has quite a broad behavioral and sensory repertoire and can be taught to change many of its behaviors in response to a variety of chemical, mechanical, and even visual cues (Audesirk et al., 1982; Lukowiak et al., 1996; Sakakibara et al., 1998; Kawai et al., 2004; reviewed in Benjamin et al., 2000; Brembs, 2003; Lukowiak et al., 2006; Benjamin, 2012). Neurobiological substrates of several behavioral paradigms of associative learning and memory have been well characterized. In recent decades most research has focused on two training paradigms: classical conditioning of the animals’ feeding behavior, a paradigm with critical involvement of a pair of interneurons called cerebral giant cells (CGCs), and operant conditioning of their aerial respiration, a paradigm with a multi-modal neuron called right pedal dorsal 1 (RPeD1) at its center (Figures 4 and 5; Spencer et al., 1999; Staras et al., 1999; for detailed review on the mechanisms and circuitry underlying these behaviors and learning paradigms see Benjamin et al., 2000; Brembs, 2003; Lukowiak et al., 2006; Benjamin, 2012). Using these two learning models, detailed insights in the molecular, cellular, neuronal network, and behavioral aspects of learning and memory have been obtained (Ribeiro et al., 2003, 2005; Kemenes et al., 2006a; Nikitin et al., 2006; Sadamoto et al., 2010; Wan et al., 2010; Dalesman and Lukowiak, 2012; Rosenegger and Lukowiak, 2013; reviewed in Benjamin et al., 2000 and Benjamin, 2012). Importantly, these studies show that, like other invertebrates, Lymnaea learning shares many key behavioral, molecular, and functional facets with learning in vertebrate model systems.
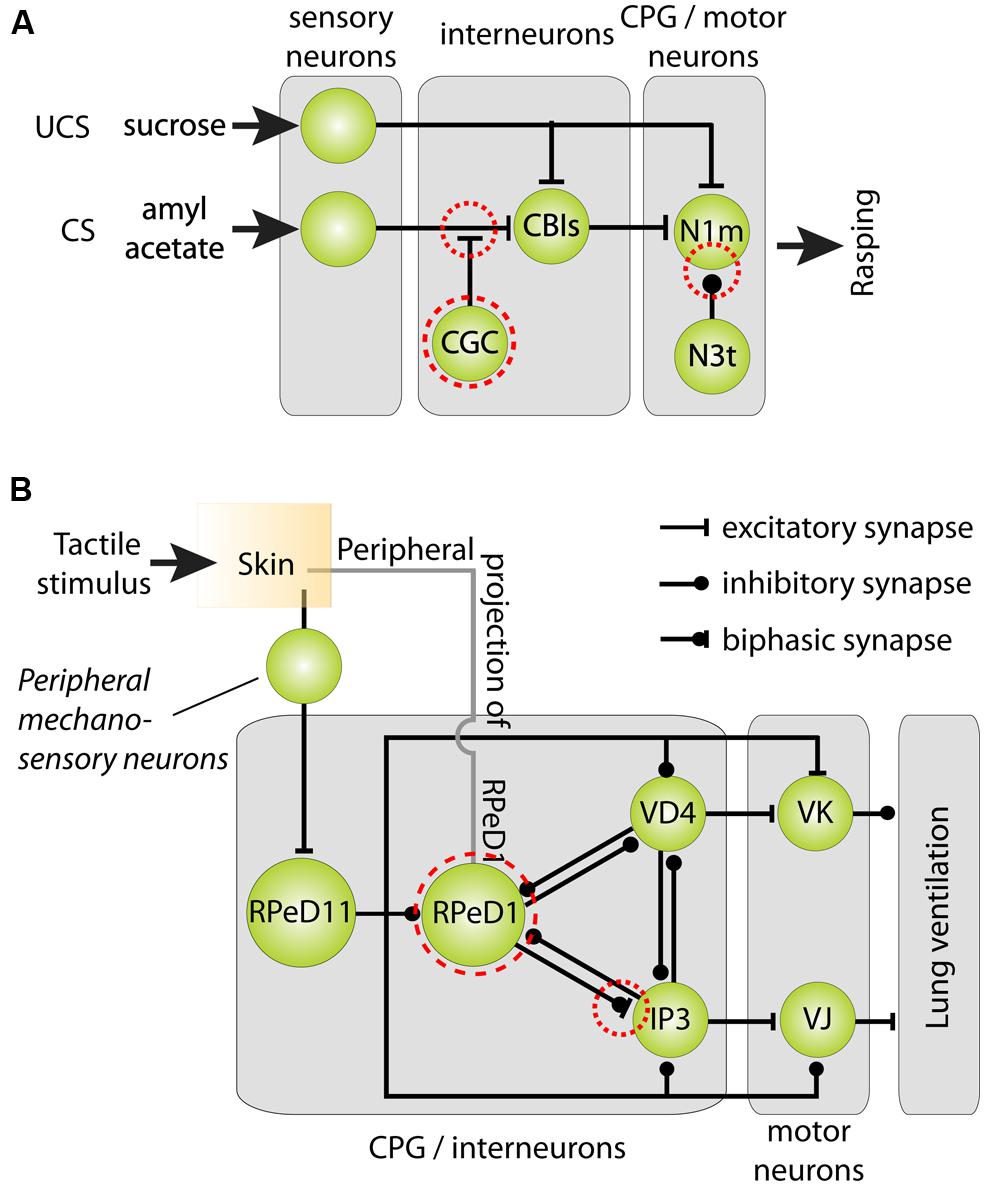
FIGURE 5. Cellular models of classical reward and operant aversive conditioning in Lymnaea. (A) Parsimonious model of the neural mechanisms underlying classical chemical reward conditioning of Lymnaea’s feeding behavior. Behavioral conditioning by peripheral application of sucrose as unconditioned stimulus (UCS) and amylacetate as conditioned stimulus (CS) induces a delayed persistent depolarization of the cerebral giant cells (CGC). This depolarization facilitates throughput of peripheral CS connections with cerebral-buccal interneurons (CBIs) thereby gating throughput of the CS to Neuron 1 medial (N1m) interneurons of the feeding circuit’s central pattern generator (CPG) that play a pivotal role in the initiation of the three phase rasping cycle. At the same time, the spike frequency of the CPG interneuron N3t is reduced after behaviorally conditioning thereby releasing N1m from its inhibition. (B) Parsimonious model of the neural mechanisms underlying aversive operant conditioning of Lymnaea’s aerial respiratory behavior. Aerial respiration involves opening and closing of an orifice giving access to the lung cavity (the pneumostome). The CPG driving the animals’ aerial respiration behavior consists of three identified interneurons: right pedal dorsal 1 (RPeD1), visceral dorsal 4 (VD4) and (wide-acting) input 3 neuron (IP3). Spiking activity in RPeD1 initiates rhythmic pattern of spiking activity in VD4 and IP3 that in turn trigger electrical activity in, respectively, Visceral K (VK) pneumostome closer motor neurons and visceral J (VJ) pneumostome opener motor neurons. Aversive operant conditioning through tactile stimulation of the pneumostome area induces a hyperpolarization of RPeD1 and reduces the latter’s ability to trigger rhythmic activity in IP3. Mechanosensory conditioning of the respiratory CPG’s electrical activity through tactile stimulation of the pneumostome area involves both afferent projections of RPeD1 (“peripheral projections of RPeD1”) and excitation of the ‘whole-body withdrawal interneuron’ RPeD11 by identified mechanosensory neurons. Dashed red circles in both circuit diagrams indicate neural loci of plasticity. Circuit diagrams shown were adapted from Benjamin et al. (2000), Taylor and Lukowiak (2000) and Benjamin and Kemenes (2010).
Lipid Membranes as a Key Theater of Aging and Age-Associated Decline of the Nervous System
A large body of evidence indicates that oxidative stress is a prominent feature of both pathological and non-pathological forms of nervous system aging (Choi, 1995; Mattson and Magnus, 2006; Torres et al., 2011; Lopez et al., 2013; Orr et al., 2013; Haddadi et al., 2014). How oxidative stress contributes to age-associated disease processes of the brain and translates to functional decline of normal healthy aging neurons is not well understood. Here we explore the theory that processes associated with the high level of unsaturation of the lipid portion of neuronal membrane constitute a key link between aging, oxidative stress, and the age-associated physiological and behavioral decline frequently observed in normal healthy aging animals and the etiology of age-associated disorders of the brain. Central to this idea are the inordinate sensitivity of methylene-interrupted PUFAs to electrophilic attacks and subsequent activation of PLA2’s, a family of enzymes involved in maintenance of membrane phospholipid integrity and membrane-associated lipid signaling processes (Figure 2).
Eukaryote cell and organelle membranes are functionally complex, dynamically regulated assemblies of mostly glycerophospholipids, fatty acids (FAs), sterols, proteins, glycoproteins, and glycolipids (Alberts et al., 2002). While most of life’s structures are sensitive to oxidation, few are as susceptible to oxidative damage as phospholipid bilayer membranes (Zimniak, 2011). This elevated susceptibility to oxidation is due to a number of factors that conspire, as a manner of speaking, to create “perfect storm” conditions. First, under normal conditions solubility of molecular oxygen and many reactive oxygen and nitrogen species (RONS) in the lipid microenvironment of membranes is quite high (Subczynski and Wisniewska, 2000). Second, as mentioned, PUFAs are inordinately sensitive to attack by RONS and other electrophiles (Hulbert et al., 2007). Third, PUFAs that have undergone free radical attack are themselves reactive free radicals and are, because of the tightly packed molecular arrangement of phospholipids in bilayer membranes, capable of propagating potentially catastrophic lipid peroxidation cascades (Forlenza et al., 2007; Ballatori et al., 2009; Currais and Maher, 2013; Sultana et al., 2013). Neuronal membranes contain high levels of PUFAs such as arachidonic acids (AA), docosahexaenoic acids (DHA), and eicosapentaenoic acids (EPA), and are therefore particularly sensitive to (per)oxidation making lipid peroxidation potentially one of the major instruments of free radical-mediated injury and cell death in the nervous system.
Parenthetically, the idea that phospholipid membranes are one of the primary molecular theaters of aging and longevity has gained substantial traction in recent decades (for reviews see Pamplona, 2008; Zimniak, 2011). In fact, the observation that the level of lipid membrane unsaturation correlates inversely with longevity in mammals, birds, and even invertebrates led to formulation of the “Membrane hypothesis of aging” which essentially sees lipid membranes as one of the weakest and therefore highest maintenance links in the biochemical armature of living organisms (Hulbert et al., 2007; Ungvari et al., 2011, 2013; Munro and Blier, 2012; Munro et al., 2013).
Lipid Peroxidation Induced Phospholipase A2 Activity and its Consequences
One characteristic cellular response triggered by lipid peroxidation is activation of one or more PLA2 family members that catalyze cleavage of FAs from the sn-2 position of glycero-phospholipids in plasma- and organelle membranes (Brown et al., 2003; Sun et al., 2004; Farooqui and Horrocks, 2006; Chen et al., 2008; Dennis et al., 2011). Although the precise underlying molecular mechanisms of lipid-peroxidation induced PLA2 activation are still a matter of debate (see for instance Greenberg et al., 2008), it is generally thought to involve peroxidation induced changes in fatty acyl chain geometry (Niki, 1990; Salgo et al., 1992; Rashba-Step et al., 1997; Greenberg et al., 2008).
Activation of PLA2 leads to the production of (peroxidized) free FAs (FFAs) and lysophospholipids (LPLs), both classes of molecules with potentially substantial biological activity (Figure 6; see below; Brown et al., 2003; Farooqui and Horrocks, 2006). For example, the molecular geometry of LPLs and FFAs is quite different from their mutual parent molecule (i.e., a phospholipid). They may therefore also cause changes in the micro-architecture and biophysical properties of phospholipid membranes and potentially affect membrane-associated signaling functions (Figure 2; Yu et al., 1992; Choi and Yu, 1995; Choe et al., 1995; Patel et al., 2001; reviewed in Piomelli et al., 2007). In addition, PLA2 mediated FFA liberation is the starting point for the generation of an extensive array of lipid-based neuromodulators. That includes FFAs themselves (e.g., AA and DHA) and LPLs (e.g., lysolecithin) but also encompasses an extensive array of biologically active molecules such as eicosanoids, isoprostanes, and neuroprostanes generated by a variety of lipid metabolizing pathways. Many of these pathways act as direct effectors of signal transduction in the nervous system through either direct interaction with ion channels or binding to G-protein-coupled or nuclear receptors (reviewed in Piomelli et al., 2007; Sultana et al., 2013).
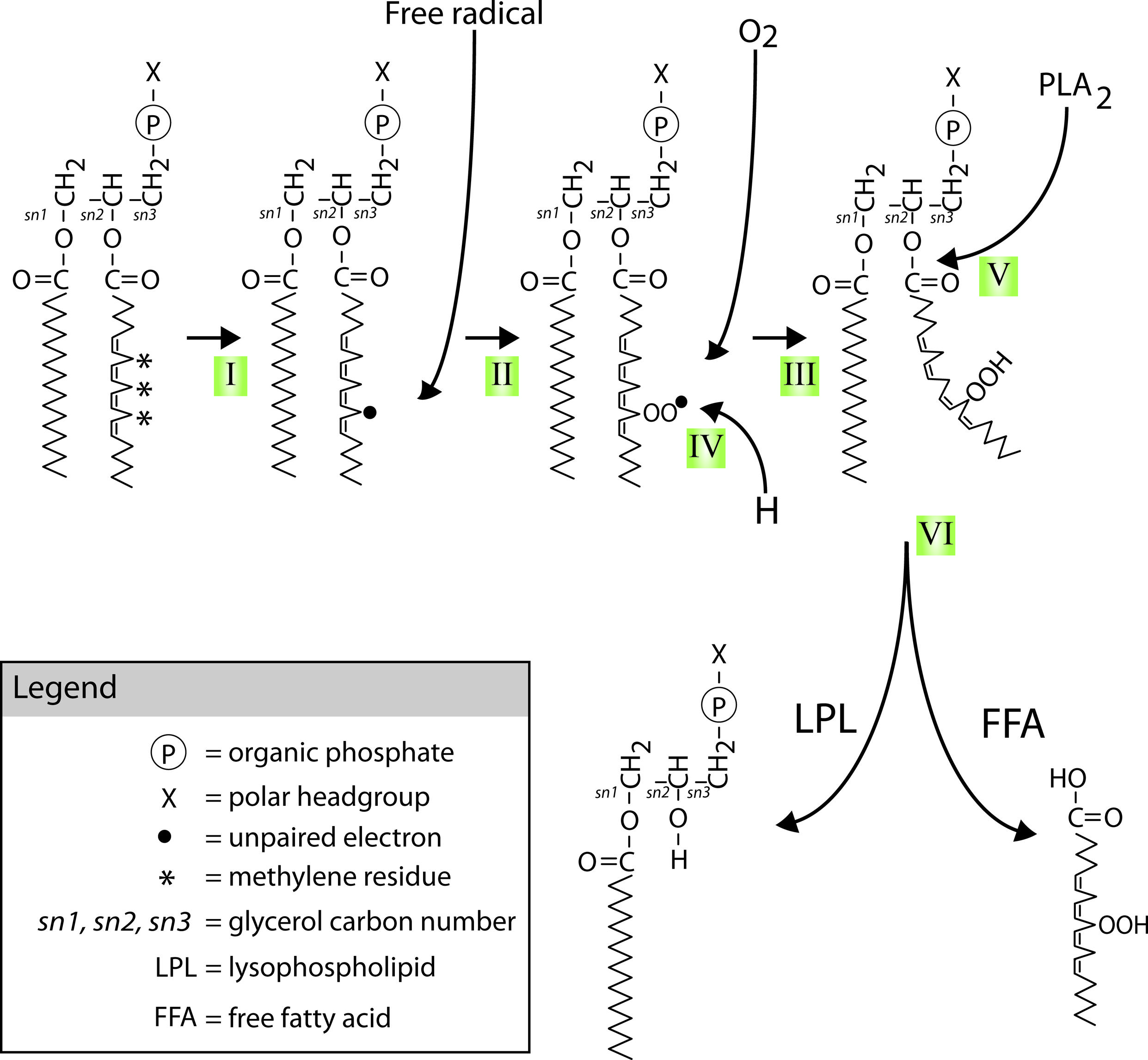
FIGURE 6. Mechanisms of PUFAs peroxidation and hydrolysis. Neuronal glycerophospholipids contain high levels of methylene-separated PUFAs such as the 20:4 omega-6 fatty acid arachidonic acid (AA) shown attached at the second carbon of the phospholipid glycerol backbone (the sn-2 position) in the diagram. These PUFAs are very sensitive to free radical attack at their bis-allylic C-H bonds (indicated by *) and subsequent (per)oxidation. This process starts with (I) the formation of a C-centered radical (indicated by •) followed by (II) recombination with molecular oxygen that is normally present in abundance in lipid bilayers. The lipid peroxide radicals formed in step II can start a peroxidation chain reaction unless (IV) defused by reduction through for instance donation of a proton by vitamin E or neutralization by other chemical mechanisms. Peroxidation of these PUFAs (V) recruits members of the PLA2 family of phospholipases. PLA2 mediated hydrolysis of peroxidized PUFAs (VI) generates peroxidized FFAs and LPLs.
Poly-unsaturated fatty acid peroxidation generally also leads to the formation of electrophilic aldehydes such as acrolein, malondialdehyde (MDA), 4-hydroxy-2-nonenal (4-HNE; Pamplona, 2008, 2011). These highly reactive aldehydes can seriously compromise molecular integrity of cells and tissues through the formation of molecular adducts with phospholipids, proteins, and DNA. However, recent evidence suggests that these lipid-derived aldehydes may also induce adaptive responses driven to decrease oxidative damage and improve antioxidant defenses (see for instance Pizzimenti et al., 2013).
Antioxidant Defenses
Organisms are not defenseless against the actions of pro-oxidants. Rather, they posses a wide and varied array of enzymatic and non-enzymatic instruments to defend their structures and functions against oxidation that includes catalases, peroxidases, and dismutases, the GSH system and various other non-enzymatic anti-oxidants like vitamin C and vitamin E.
Enzymatic Mechanisms
Because of its relatively low redox potential and high abundance, the tripeptide GSH is a versatile anti-oxidant involved as primary redox buffering of many intra- and extra-cellular compartments, including lipid membranes, and mitochondria (Mari et al., 2009; Ribas et al., 2014). In addition to its antioxidant actions, GSH also plays an important role in detoxification processes, including the detoxification of potentially harmful products of lipid peroxidation such as aforementioned aldehydes. This process is mediated by members of the glutathione-S-transferase (GST) family of enzymes (Zimniak, 2008; Ribas et al., 2014). GSH is one of the most abundant molecules in healthy cells and tissues. Its availability is a function of usage, de novo synthesis and redox regeneration. The rate-determining step in GSH synthesis is catalyzed by the enzyme γ-glutamylcysteine synthetase (GCL; Chen et al., 2005; Genovese et al., 2007). Perturbation of GCL activity has been shown to significantly reduce GSH availability. In the context of the current focus of lipid bilayer redox homeostasis as a instrument of neuronal aging it is thought provoking that several studies report age-related declines in in GSH levels and GCL activity in various cell types, including neurons (Toroser and Sohal, 2005, 2007; Zhu et al., 2006; Jacob et al., 2013; Watson et al., 2014). This suggests that anti-oxidant failure either with or without escalation of pro-oxidant activity may be a key facet of the progressive lipid peroxidation stress characteristic of aging.
Non-Enzymatic Mechanisms
Non-enzymatic antioxidants such as Vitamins A, C, and E also play key roles in the defense against oxidative stress, including the maintenance of membrane redox homeostasis. Of these three vitamin families, Vitamin E is considered the major antioxidant present in membranes. Vitamin E is comprised of two families of closely related lipid-soluble compounds, the tocopherols, and tocotrienols. Alpha-tocopherol is the most abundant Vitamin E analog and according to recent insights has a preference for non-lipid raft PUFA-containing domains (Atkinson et al., 2010; Lemaire-Ewing et al., 2010). Alpha-tocopherol associates with membranes such that its active hydroxyl moiety is located at the lipid/water interface and not within the hydrocarbon matrix (Marquardt et al., 2013). As a consequence, α-tocopherol appears to exert its anti-oxidant activity at the surface of a membrane, possibly through the interception of diffusing ROS, or by means of terminating lipid radicals (Marquardt et al., 2013).
Non-Traditional, Non-Antioxidant Actions of α-Tocopherols
In addition, several studies provide an alternative, non-antioxidant, scenario for α-tocopherol’s beneficial effects. That is, it is suggested that incorporation of α-tocopherols in membranes can structurally stabilize the membrane against lipid-peroxidation induced curvature stresses or normalize fatty acid signaling by acting as FFA trap through its ability to form hydrogen-bridges and engage in Vanderwaals interactions with FFAs (Atkinson et al., 2008; Lemaire-Ewing et al., 2010; Marquardt et al., 2013). Especially α-tocopherol’s acyl tail has been shown to play a critical role in its membrane stabilizing effect (Kagan, 1989). Methylation of this hydroxyl group disrupts α-tocopherol’s ability to engage in hydrogen-bridge formations. In this respect it is interesting to note that work from our lab shows that only non-methylated α-tocopherol is capable of reversing a decline in neuronal excitability due to age- or experimentally induced oxidative stress (Watson et al., 2012b).
Lipid Peroxidation and (Brain) Aging
Considering that neuronal membranes tend to have a high PUFA content it is not surprising that lipid peroxidation and its byproducts are increasingly implicated in brain aging. For instance, accumulation of MDA, 4-HNE, acrolein is commonly reported in aged brains of vertebrate and invertebrate model systems (Terman and Brunk, 2006; Zhu et al., 2006; Watson et al., 2012b). Moreover, PLA2s are also increasingly implicated in normal brain aging and the etiology of age-associated neurodegenerative disorders (Farooqui and Horrocks, 2006; Ong et al., 2010). How the products of PLA2 activity exert their detrimental effects in the aging brain is, however, not always clear but may involve one or more of the scenarios outlined in the preceding sections.
Intrinsic Electrical Properties as a Substrate of Plasticity
This review focuses on the role of intrinsic excitability of neurons in age-associated decline in behavioral plasticity. Decades of research on the cellular and molecular foundations of learning and memory have led to a “synapse-centric” view of learning and memory mechanisms that holds that memories are formed and stored through activity-dependent alterations of synaptic strength and neuronal circuit architecture (Bliss and Collingridge, 1993; Kandel, 2001). In this context non-synaptic mechanisms of plasticity, i.e., mechanisms involving changes in intrinsic electrical properties of neurons, have received much less attention. Yet, experimental evidence that changes in intrinsic neuronal electrical properties could be serving as part or whole of an engram beyond merely facilitating activity-dependent synaptic modulation goes back as far as the 1970s and the concept has gained substantial traction in recent years (reviewed in Benjamin et al., 2008; Mozzachiodi and Byrne, 2010; Sehgal et al., 2013; D’Angelo, 2014; for review of early work in this area see Byrne, 1987). For example, numerous studies have shown that eye blink trace conditioning in rodents induces an increase in intrinsic excitability of hippocampal CA1 and CA3 neurons through reduction of spike frequency adaptation caused by action potential afterhyperpolarizations (AHP) mediated by Ca2+-dependent outward K+ currents2 (Coulter et al., 1989; Moyer et al., 1992, 2000; Disterhoft et al., 1996; Thompson et al., 1996; Oh et al., 2010; Sehgal et al., 2014; for review, see Christian and Thompson, 2003). There is also substantial evidence of non-synaptic forms of plasticity in invertebrates, particularly in gastropod mollusks. For instance, Alkon (1974, 1979) demonstrated that associative learning in the nudibranch snail Hermissenda crasicornis involved an increase in excitability of type B photo-receptor cell associated with a decrease in the types of voltage- and Ca2+-sensitive K+ currents mediating the AHP and spike frequency adaptation. In Lymnaea, long-term memory (LTM) for classical conditioning of feeding has been associated with a persistent Na+-current dependent depolarization of the CGCs, two interneurons that act as gate-keepers to the feeding circuit and that play an essential role in chemosensory behavioral conditioning of the animals feeding behavior (Jones et al., 2003; Kemenes et al., 2006b; Nikitin et al., 2008, 2013). In the marine gastropod Aplysia californica, changes in intrinsic excitability of identified neurons in the central pattern-generating (CPG) network for feeding have been associated with the network’s behavioral conditioning induced motor output (Sieling et al., 2014).
More examples of non-synaptic plasticity can be found in the literature (see reviews by Byrne, 1987; Benjamin et al., 2008; Mozzachiodi and Byrne, 2010; Sehgal et al., 2013; D’Angelo, 2014). For the purpose of the current review it is suffice to say that the idea of intrinsic excitability regulation as a non-synaptic instrument of plasticity is now on firmer ground than ever before. The balance of this paper focuses on the idea that the mechanisms controlling intrinsic neuronal excitability, particularly those underlying the control of repetitive action potential firing, are a key target of oxidative stress-associated aging processes and a root cause of functional decline of normal aging neurons and brains.
Evidence for Age-Associated Alterations in Intrinsic Electrical Properties
Considering that neuronal electrical excitability is fundamental to brain function, substantial research on the neurobiological basis of behavioral aging has focused on the possibility that aging neurons undergo changes in intrinsic excitability. Neuronal excitability is a complex function of a neuron’s shape, the repertoire, distribution, and density of voltage- and ligand-gated ion channels in their plasma-membrane, the activity of ion pumps and various intra- and extracellular signaling systems and state variables, including intracellular Ca2+-, metabolic-, and redox homeostasis (Bean, 2007; Beck and Yaari, 2008; Vacher et al., 2008; Nusser, 2012). There is evidence for age-associated changes in many of these parameters. It is beyond the scope of this paper to review this vast and diverse literature. For a more in-depth treatment of this subject the reader is referred to many excellent reviews available in this area (see Barnes, 1994; Burke and Barnes, 2006; Mattson and Magnus, 2006; Chawla and Barnes, 2007; Thibault et al., 2007; Mattson et al., 2008; Oh et al., 2010; Toescu and Vreugdenhil, 2010; Yeoman et al., 2012). Instead in the next paragraphs we briefly review key insights gained from vertebrate and invertebrate model systems suggesting that intensification of spike frequency adaptation mechanisms is an important facet of the normal neuronal aging process.
Evidence from Vertebrate Model Systems
Most of the work on age-related intrinsic neuronal excitability changes in vertebrates has been done on the rodent hippocampus. On balance, the evidence from a large body of in vitro electrophysiological studies on different hippocampal regions of young and old mice, rats and rabbits suggest that most intrinsic neuronal electrical properties such as resting membrane potential, membrane time constant, input resistance, rheobase current, action potential threshold, and various aspects of action potential dynamics change very little across the lifespan (reviewed in Barnes, 1994; Burke and Barnes, 2006; Chawla and Barnes, 2007). However, “a number of consistent changes do occur” (Barnes, 1994). One of those changes involves alterations in voltage-gated Ca2+ currents, intracellular Ca2+-homeostasis, Ca2+-activated outward K+ currents and interactions between them enhancing the AHP and associated spike frequency adaptation of older neurons (Moyer et al., 1992; Tombaugh et al., 2005; reviewed in Oh et al., 2010). It should be noted, however, that even though the in vitro evidence for a role of these mechanisms in cognitive decline associated with normal brain aging is compelling, the idea is not undisputed (for review of this issue, see Burke and Barnes, 2006). Other studies report changes in voltage-activated Ca2+- or Na+-channel properties to age-associated decline in intrinsic excitability of various types of rodent neurons (Scott et al., 1988; Randall et al., 2012; Branch et al., 2014; for review see Annunziato et al., 2002; Foster, 2012).
Evidence from Invertebrate Model Systems
Several studies, particularly in gastropod mollusks, have looked into the question of intrinsic neuronal excitability changes in invertebrate neurons. For example, Frolkis et al. (1991, 1995) reported increased voltage-gated Ca2+ current densities and changes in other aspects of intrinsic electrical excitability in aged Lymnaea neurons. The same group reported changes in voltage-gated K+ currents amplitudes and inactivation kinetics in old Lymnaea neurons (Frolkis et al., 1989). Patel et al. (2006) reported a decline in intrinsic excitability associated with a decline in input resistance and an increase in AHP magnitude and duration in CGGs of older Lymnaea and Klaassen et al. (1998) observed a decline in excitability associated with a reduction in input resistance in aging RPeD1. We observed a decline in intrinsic excitability of CGCs in aged learning-impaired older animals and concluded that “this decline in excitability is not due to changes in passive neurophysiological properties but rather involves alterations in active properties of the cells” (Hermann et al., 2007). An age-associated decline in input resistance associated with lengthening membrane time constant was reported for at least three identified neurons in Aplysia (Rattan and Peretz, 1981).
Age-associated neurophysiological changes linked to behavioral impairment have been demonstrated in Caenorhabditis elegans, Aplysia, and Lymnaea. For instance, using a classical reward conditioning learning paradigm, we demonstrated in Lymnaea that a selective age-related impairment of appetitive LTM is associated with a decline in intrinsic excitability of the CGCs that is reflected in more pronounced spike frequency adaptation (Figure 7; Hermann et al., 2007). Along a similar vein, we also showed that age-related LTM impairment in aerial respiration operant-conditioning model is associated with enhanced spike frequency adaptation properties of RPeD1, an interneuron instrumental in the execution of this behavior and one of the cellular substrates of LTM in this learning paradigm (Watson et al., 2013). In addition, Janse et al. (1999) showed a correlation between reproductive senescence and reduced electrical excitability of the caudodorsal cells (CDCs), neuroendocrine cells producing Lymnaea’s ovulation hormone and other neuropeptides involved in the regulation of reproductive behavior (Janse et al., 1999). In Aplysia, studies from the Peretz lab and a recent study by Kempsell and Fieber (2014) have linked age-associated behavioral and homeostatic deficiencies to declining neuronal excitability (Rattan and Peretz, 1981; Peretz, 1988; Skinner and Peretz, 1989). Interestingly, in the nematode C. elegans age-associated decline in chemotactic behavior is associated with a decline in sensory neuron excitability due to oxidation-induced enhancement of voltage-gated K+ currents (Cai and Sesti, 2009; Sesti et al., 2010).
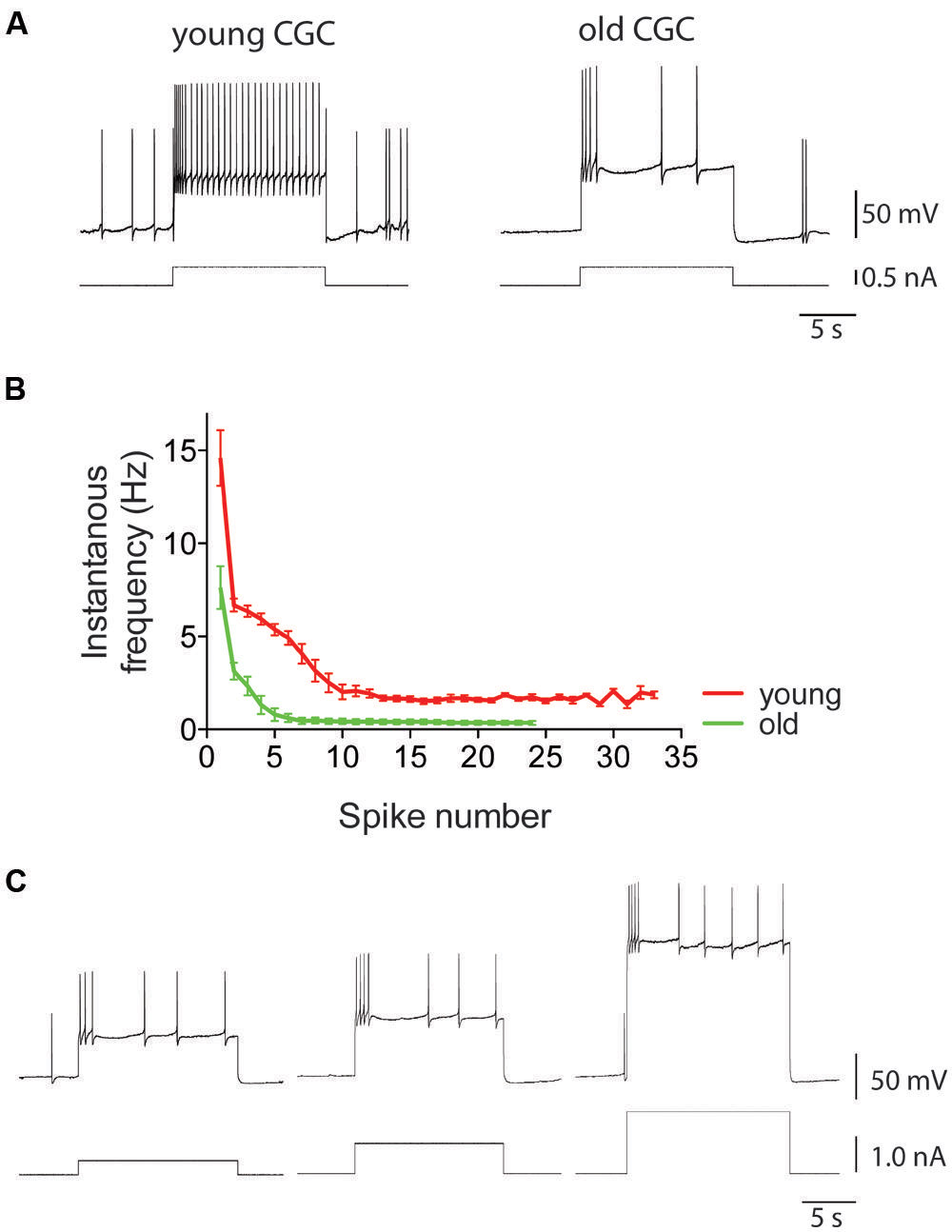
FIGURE 7. Intrinsic electrical excitability of Lymnaea neurons declines with age. Using CGCs as example, this figure illustrates the characteristic age-associated enhancement of spike frequency adaptation that is also observed in other identified Lymnaea neurons, including RPeD1 (see for instance Watson et al., 2012b, 2013). (A) This figure shows examples of intracellular recordings of spiking activity evoked in young and old CGCs by injection of 0.5 nA depolarizing current. Identical current injection results in a non-accommodating response in young CGCs but a rapidly accommodating response in old CGCs. (B) Average instantaneous frequency response of young and old CGCs. Current injected in young CGCs evokes a high-frequency action potential burst followed by a steady non accommodating firing response. In contrast, current injected in old CGCs evokes a few action potentials and a rapidly accommodating response. (C) Example of reduced excitability in old CGCs. Increasing stimulus strength fails to evoke a high-frequency non-accommodating response in old CGCs. Adapted from Hermann et al. (2007) with permission from American Psychological Association (APA).
How Does Oxidative Stress Contribute to Declining Excitability of Aging Neurons?
How oxidative stress translates into functional decline and increased vulnerability to disease of the aging brain is only partly understood (Mattson and Magnus, 2006). First it should be noted that in the context of redox modulation of neuronal functions many pro-oxidants have both “good” and a “bad” aspects. For instance, under normal conditions pro-oxidants like RONS play important physiological roles in synaptic plasticity and memory formation (reviewed in Massaad and Klann, 2011; Milton and Sweeney, 2011). Yet, under conditions of oxidative stress the same mechanisms may affect neuronal plasticity and memory function in a negative manner (Hu et al., 2006; Negre-Salvayre et al., 2010; Massaad and Klann, 2011; Niki, 2012).
Second, many of the molecular instruments of neuronal excitability (i.e., membrane ion channels, receptors, vesicle fusion processes) are susceptible to oxidation or sensitive to alterations in cellular redox status (Annunziato et al., 2002; Dukoff et al., 2014). For instance, a particularly salient example concerns the so-called A-current (IA) family, a family of outward transient voltage-gated K+ currents (i.e., they are self-inactivating) that plays important roles in the regulation of neuronal excitability (Adams and Galvan, 1986; Bean, 2007). It has long been known that IA’s inactivation properties are modulated by changes in intracellular redox conditions (Ruppersberg et al., 1991; Bähring et al., 2001). That is, under cytosolic reducing conditions the current displays its characteristic fast N-type inactivation behavior whereas under oxidizing conditions this inactivation process is slowed down. In a manner of speaking, this process turns IA from a “temporary brake” into a “permanent brake” on a neuron’s spiking ability. It has been suggested that these mechanisms may operate as a safety switch against metabolic overload by matching a neuron’s energy demanding electrical activity to its metabolic capacity (Gulbis, 2002). The IA family of ion channels is not the only type of ion channels susceptible to changes in cellular redox homeostasis. Many other types of ion channels playing significant roles in the control and execution of neuronal excitability, including voltage-gated Na+ and Ca2+, are subject to redox modulation (for review see Annunziato et al., 2002).
In addition, over the last number of decades it has become clear that many of the molecular correlates of neuronal excitability and processes are sensitive to the composition and micro-architecture of their membrane environment and/or are modulated by various membrane-derived lipid signaling moieties (for review see Piomelli et al., 2007; Dart, 2010; Rosenhouse-Dantsker et al., 2012). Maintaining lipid membrane homeostasis is therefore a crucial facet of the maintenance of functional integrity of neurons and the brain. Below we illustrate these ideas with some of the most salient examples.
Membrane-Associated Correlates of Neuronal Excitability Control
There are numerous examples of lipid-mediated signaling in the nervous system. For instance, although there is still some debate whether its actions arise from direct or indirect interactions with ion channel proteins it is evident that AA, one of the most common PUFAs in the nervous system, can modulate a variety of ion channels, including voltage-gated Ca2+ channels, mechano-gated K+ channels like TRAAK, TREK-1, and other members of the four-transmembrane/two-pore domain (4TM2P) family of background K+ channels (Patel et al., 1998; Maingret et al., 1999; Roberts-Crowley et al., 2009). Alternatively, PUFAs like AA or EPA liberated from their plasma membrane environment can act as metabolic precursors for several signaling pathways active in the nervous system. This includes the eicosanoid pathway that generates a collection of signaling molecules (i.e., prostaglandins, prostacyclins, thromboxanes, lipoxins, and leukotrienes). Many eicosanoids regulate immune functions and inflammation but also double as neuromodulators (for review see Nigam and Schewe, 2000; Liang et al., 2007). Intriguingly, one of those eicosanoid neuromodulators, the 12-lipoxygenase (12-LOX) metabolite 12-HPETE has long been known as an activator of Aplysia’s S-type K+-channels (Piomelli et al., 1987). These K+ channels, later identified to belong to the TREK-1 family, are instrumental in non-synaptic forms of plasticity underlying behavioral modification of Aplysia’s gill withdrawal reflex (Hawkins, 1984) Similar PLA2- and/or LOX-dependent arachidonic-acid modulated background K+ channels have been described in Lymnaea (Kits et al., 1997; Lopes et al., 1998). Opening of these 4TM2P channels generates an outward current. It is, therefore, conceivable that elevated levels of PLA2-activity associated with inflammation, oxidative stress, and aging induce a decline in neuronal excitability through activation of these 4TM2P background K+ channels. An alternative route linking membrane oxidative stress to a decline in neuronal excitability involves aforementioned aldehyde byproducts of lipid peroxidation. As mentioned MDA, 4-HNE and acrolein can cause irreversible damage to many of the cells macromolecules through covalent conjugation (Farooqui and Horrocks, 2006) or engage in various signaling activities including pathways regulating mitochondrial function and other aspects of cell metabolism (Riahi et al., 2010; Ayala et al., 2014).
Yet another intriguing plausible explanation for the effects of lipid peroxidation on neuronal excitability involves structural rather than signaling arguments. Specifically, as noted before peroxidation very likely incites considerable changes in the molecular geometry of affected PUFAs that affect the way they “fit” in the membrane and may introduce considerable changes in the micro-architecture and biophysical properties of bilayer membranes including parameters such as membrane curvature, membrane viscosity, lipid composition, and distribution. These changes may have various implications for neuronal signaling functions, including the organization of transmembrane signaling complexes, vesicle fusion processes, receptor trafficking, and ion channel functions (Yu et al., 1992; Choi and Yu, 1995; Choe et al., 1995; Patel et al., 2001; reviewed in Piomelli et al., 2007). For example, membrane curvature stresses have been found an important factor in the regulation of neuronal electrical activity through the activation of stretch/mechano sensitive background K+ channels known to contribute to the regulation of neuronal excitability (Figure 2; Patel et al., 2001). Also, the lipid architecture of presynaptic terminal membranes does play a vital role in synaptic Ca2+-triggered exocytosis process raising the possibility that PUFA peroxidation interferes with synaptic functions (reviewed in Davletov and Montecucco, 2010).
Taken together, even though there are still a lot of loose ends in the conceptual framework we sketched here it seems evident that proper management of membrane redox status, lipid peroxidation and phospholipid lysis products generated by or with the aid of PLA2 is likely critical for functional homeostasis of neurons and by extension the nervous system. Below we summarize and discuss evidence from recent work done in our laboratory trying to fit the pieces of this puzzle together.
PLA2 – A Lipase at the Nexus of Aging, Oxidative Stress, Neuronal Electrical Excitability Control and Functional Decline of the Aging Nervous System?
In the preceding paragraphs we explored the prospect for interplay between oxidative stress, lipid membrane factors, PLA2, and changes in intrinsic neuronal excitability as a factor in functional decline of normal aging neurons. In this section we review the evidence that lipid peroxidation-induced PLA2 activity lays at the root of inflammation-, age-, and oxidative stress-associated functional impairment of the nervous system in Lymnaea and put our findings in the context of a growing literature suggesting that we are dealing with an evolutionary widely conserved mechanism.
As described above, aging Lymnaea display associative LTM impairment in two functionally and anatomically distinct (i.e., appetitive and respiratory) learning paradigms (Hermann et al., 2007, 2013; Watson et al., 2012a, 2013, 2014; Beaulieu et al., 2014). In both instances, LTM failure was associated with reduced spontaneous action potential activity and enhanced spike frequency adaptation of the two key interneurons underlying both execution and plasticity of the respective behaviors (i.e., CGCs and RPeD1). Both behavioral and electrophysiological facets of age-associated respiratory and appetitive LTM impairment could be reproduced in young animals through treatment with 2,2′- azobis-2-methyl-propanimidamide (AAPH), a water-soluble free radical generator commonly used to induce PUFA peroxidation, furthering the notion of plasmamembrane lipid peroxidation as a significant agent of neuronal and behavioral aging in Lymnaea (Figure 8). Evidence of increased FFA mobilization from brains and other tissues was found in both old and AAPH-treated young animals (Watson et al., 2013; Beaulieu et al., 2014). Moreover, treatment with the lipid-domain anti-oxidant α-tocopherol reversed decline in neuronal excitability in both experimental oxidative stressed and naturally aged neurons (Watson et al., 2012a,b). Remarkably, all these behavioral, electrophysiological, and biochemical symptoms of aging and experimental oxidative stress could be reversed by treatment with aristolochic acid, a broad spectrum PLA2 inhibitor that inhibits both Ca2+-dependent cPLA2 and Ca2+-independent iPLA2 (Figure 8; Vishwanath et al., 1988; Lopes et al., 1998; Carnevale and Cathcart, 2001).
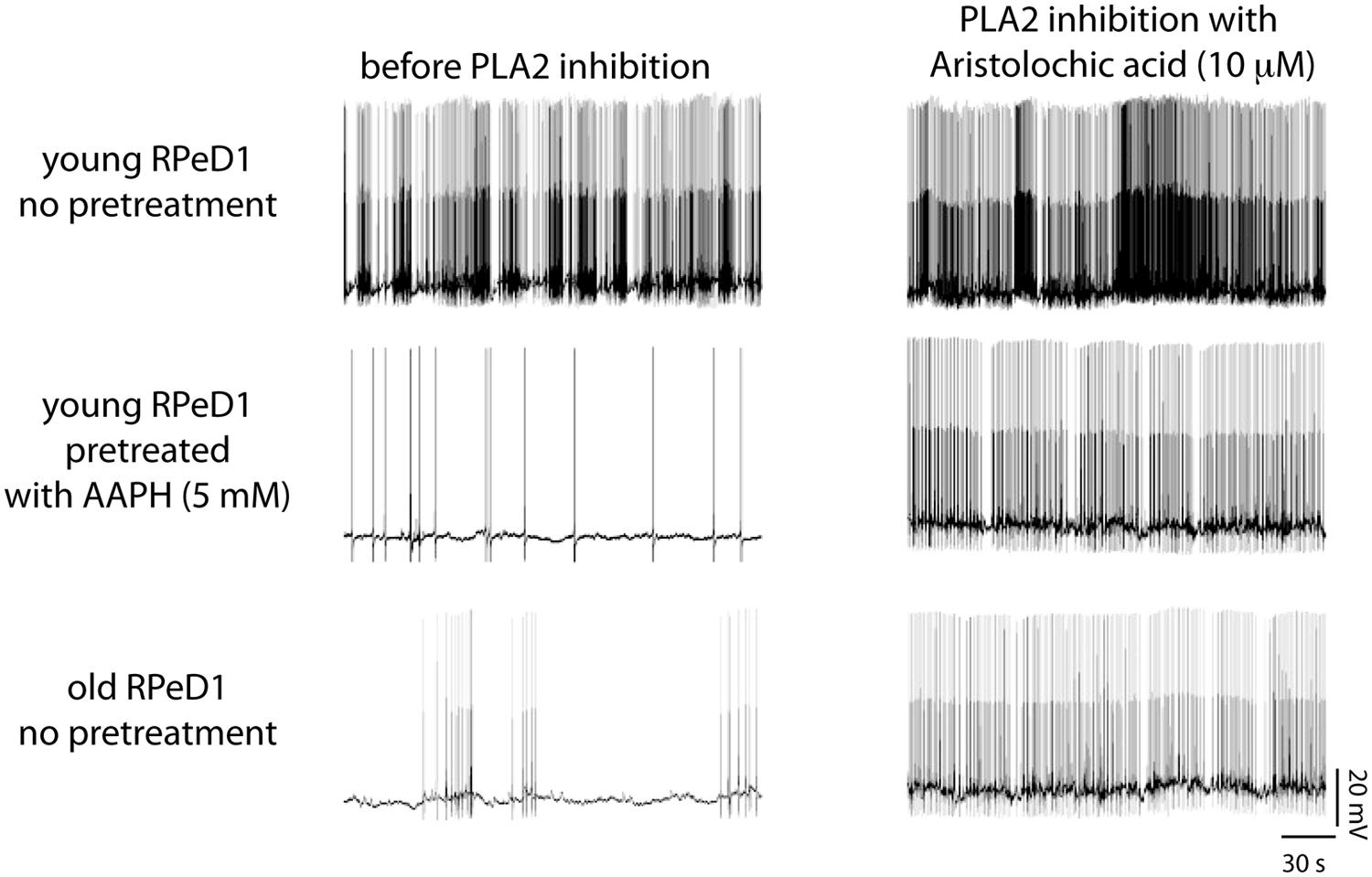
FIGURE 8. Phospholipase A2 (PLA2) contributes to regulation of respiratory neural circuit activity in old and oxidative stressed young brains. Examples of action potential activity recorded from RPeD1 in isolated young central nervous systems (CNSs) pretreated for 30 min with either vehicle-only (top row) or the pro-oxidant 2,2-azobis (2-methylpropion-amidine) dihydrochloride (AAPH; middle row), and old CNSs maintained in vehicle-only (bottom row), before (left column), and after 30 min of treatment with the PLA2 inhibitor aristolochic acid (right column). Reprinted from Watson et al. (2013) with permission from Elsevier.
No evidence was found of significant age-associated impairment or experimental oxidative stress-induced repression of transcription-independent short/intermediate term memory (S/ITM) in either of the two behavioral conditioning paradigms, suggesting that transcription-independent forms of memory appear impervious to aging or oxidative stress. As yet, we have no definitive answer to the question why LTM is selectively susceptible. However, preliminary evidence from our laboratory suggests that both aging and AAPH-induced oxidative stress in participation with PLA2 alters aspects of mitochondrial functions that may affect their ability to contribute to intracellular Ca2+-buffering. The molecular pathways underlying LTM formation in Lymnaea involve “traditional” conserved cAMP- and Ca2+-sensitive CREB- and MAPK-dependent pathways (Ribeiro et al., 2003; Wagatsuma et al., 2006; Guo et al., 2010). It is therefore conceivable that dysregulation of intracellular Ca2+ homeostasis disrupts transcription-regulation pathways involved in LTM expression. Furthermore, the observation that PLA2 inhibition rescues LTM is particularly intriguing in view of evidence implicating FFAs and PLA2 in mitochondrial uncoupling in pancreatic β-cells (Ježek et al., 2010, 2014). Although speculative at this point in time, it is not unreasonable to postulate that a similar mechanism may underlie the PLA2-associated LTM impairment in Lymnaea.
Intriguingly, selective appetitive LTM impairment could also be induced in young Lymnaea through systemic activation of their cellular immune system (Hermann et al., 2013). As before in naturally aged and experimentally oxidation-stressed young animals, inflammation-associated LTM impairment could be rescued by means of PLA2 inhibition (Hermann et al., 2013). Together, our data indicate that the processes underlying Lymnaea’s transcription-dependent forms of memory are particularly vulnerable to aging, oxidative stress, and inflammation and that PLA2 plays a pivotal role in each of these models of associative LTM failure.
PLA2’s as Therapeutic Targets?
PLA2’s and deregulation of lipid metabolism are increasingly implicated in human age-, oxidative stress-, and inflammation-associated conditions including cardiovascular diseases and age-associated neurodegenerative diseases such as Alzheimer’s disease (Sun et al., 2004; Farooqui and Horrocks, 2006; Adibhatla and Hatcher, 2008; Sanchez-Mejia et al., 2008; Chalimoniuk et al., 2009; Sanchez-Mejia and Mucke, 2010; Desbene et al., 2012; Gentile et al., 2012; Hui, 2012; reviewed in Sun et al., 2014). They are therefore, not surprisingly, increasingly targeted or considered as targets for therapeutic intervention (see for instance, Garcia-Garcia and Serruys, 2009; Quach et al., 2014). Particularly, it is intriguing that lipoprotein-associated phospholipase A2 (Lp-PLA2), also known as platelet-activating factor acetylhydrolase (PAF-AH) was identified as a potential factor in cerebrovascular oxidative stress and inflammation and risk factor for human dementia and subsequently drew considerable interest as a therapeutic target in Alzheimer’s disease (Davidson et al., 2012; Acharya et al., 2013; Fitzpatrick et al., 2014). However, there are still a lot of unknowns surrounding the pathways, processes and conditions leading to PLA2 activation in different cell types that should be resolved before effective and safe therapeutic targets and agents for the treatment and prevention of these increasingly germane age-associated public health concerns (Sun et al., 2014). For instance, whereas many studies implicate PLA2 hyperactivity as a factor in the etiology of Alzheimer’s disease (Sanchez-Mejia and Mucke, 2010; Gentile et al., 2012; Hui, 2012), other studies suggest that hypoactivity of intracellular PLA2s (cPLA2 and iPLA2) are a contributing factor to memory impairment and Alzheimer’s neuropathology (reviewed by Schaeffer et al., 2009).
Relevance and Future Directions
The evidence reviewed here infers close ties between inflammation, oxidative stress, lipid-peroxidation, PLA2-activation, neuronal excitability, and AMI in Lymnaea and echo a rapidly growing evidence from basic, clinical, and epidemiological research that PLA2s, inflammation and oxidative stress are important factors in age-related cognitive decline and impairment in humans and other mammals (Sun et al., 2004, 2014; Phillis et al., 2006; Adibhatla and Hatcher, 2008; Lee et al., 2011). Considering this remarkable conservation we believe that Lymnaea’s extraordinary suitability for detailed, direct, and tightly integrated molecule-to-behavior dissection of the neurobiological substrates of AMI will in addition to furthering understanding of the biological foundations and parameters of aging potentially also generate valuable insights into mechanisms of and possible solutions for human AMI and other age-associated afflictions of our own nervous system. Before that goal can be achieved many questions need to be answered.
Among the unanswered questions requiring further attention is the identity of the ionic mechanisms through which the products of PLA2-mediated fatty acyl hydrolysis exert their inhibitory influence on neuronal excitability. As outlined in the preceding pages, there are many possibilities to test ranging from FA dependent modulation of ion channels to alterations in autacoid modulation of synaptic functions and hypotheses encompassing elements of membrane architecture dependent regulation of neuronal electrical properties.
Another important puzzle to solve is what inhibition of PLA2, one of the main tools of membrane homeostasis and repair, does to long-term cell/tissues homeostatic integrity. Considering the impact lipid peroxidation is expected to have on membrane structure and function, our finding that PLA2 inhibition restored rather than aggravated neuro-physiological and behavioral correlates of aging, experimental oxidative stress, and inflammation is puzzling and urges further study.
Yet another intriguing perspective we are currently exploring is mitochondrial involvement in the lipid peroxidation- and PLA2-dependent declining excitability and plasticity of aging Lymnaea neurons. Neurons critically depend on mitochondria to provide the energy to establish and maintain membrane excitability and engage in their quintessential and metabolically costly electrochemical signaling activities (Kann and Kovács, 2007; Mattson et al., 2008). Mitochondria are also a critical partner in intracellular free Ca2+ homeostasis and are one of the main sources of RONS and other pro-oxidants. There are many tight reciprocal links between neuronal electrical and mitochondrial activities and many neuronal excitability disorders appear to be associated with mitochondrial aberrancies (Kann and Kovács, 2007; Mattson et al., 2008; Surmeier et al., 2012). Moreover, there is also a substantial body of evidence indicating involvement of mitochondria-associated oxidative stress, lipid signaling, Ca2+ dysregulation in normal aging, and the pathogenesis of common age-associated neurological diseases including AD and PD in which lipid peroxidation and PLA2 may be key partners (reviewed in Thibault et al., 2007; Mattson et al., 2008; Toescu and Vreugdenhil, 2010; Waldbaum and Patel, 2010). The Lymnaea model system we have developed provides an excellent platform to investigate these interactions between neuronal energy metabolism, electrical excitability, and plasticity in great detail.
The Lymnaea research platform we portrayed here will also be a great tool to investigate fundamental mechanisms of neuronal and behavioral aging and test other existing and emerging aging theories. For example, there is growing evidence that insulin and other insulin-like peptides (ILPs) modulate aspects of plasticity in the Lymnaea CNS and enhance learning abilities in older learning-impaired snails (Murakami et al., 2013; Pirger et al., 2014). These findings resonate very well with a rapidly expanding literature that ILPs and insulin resistance are a major endocrine factor in aging in general and aging of the nervous system in particular (Kim and Feldman, 2012; Alcedo et al., 2013). Moreover, because of its identified neurons the model system will allow us to explore many important fundamental scientific questions about aging neurons and brains like: do neurons age according to “plan” or do they fail at random? Are some neuronal physiological phenotypes more vulnerable to aging than others? Can we reverse the effects of aging without repercussion? What are the principles and mechanisms underlying robust and reliable complex brains? Are all changes observed in aging neurons mal-adaptive or are some perhaps compensatory and adaptive? Are neurons and brains life cycles subject to the same resource constraints and trade-offs as other types of cells and tissues in the animal body?
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Discovery Grant from the National Science and Engineering Research Council (NSERC) of Canada to Willem C. Wildering.
Footnotes
- ^ Key concept 1 | Methylene-interrupted PUFAs. Poly-unsaturated fatty acids with one or more pairs of hydrogen unsaturated carbons in cis-arrangement separated by a hydrogen-saturated (bis-allylic) methylene bridge. Because the methylene moiety’s bis-allylic C–H bonds are weaker than ordinary C–H bonds they are prone to free radical attack. The main PUFAs with more than one bis-allylic methylene site in the plasmamembranes of neurons are arachidonic acid (AA), docosahexanoic acid (DHA), and eicosapentaenoic acid (EPA).
- ^ Spike frequency adaptation, Action potential afterhyperpolarization (AHP), Ca2+-activated K+ currents – Spike frequency adaptation is a process by which the action potential firing rate of a neuron declines with time despite the maintenance of a constant excitatory stimulus. The action potential of most neurons is followed by an HP, a period of varying length of time during which the cells’ membrane potential is lower than the resting potential preceding the action potential and during which the cell is less excitable than before. Various ionic mechanisms may contribute to the AHP including a class of outward K+ currents activated either exclusively or in part by increase of intracellular free Ca2+ levels (i.e., Ca2+-activated K+ currents). The AHP as part of a negative feedback loop linking spiking frequency and opening of voltage-gated Ca2+ channels to activation of inhibitory Ca2+-activated K+ currents is a key component of spike frequency adaptation in most neurons (see Lewis et al., 1986; Sah, 1996; Sah and Davies, 2000 for more detail).
References
Acharya, N. K., Levin, E. C., Clifford, P. M., Han, M., Tourtellotte, R., Chamberlain, D.,et al. (2013). Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J. Alzheimers Dis. 35, 179–198. doi: 10.3233/JAD-122254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adams, P. R., and Galvan, M. (1986). Voltage-dependent currents of vertebrate neurons and their role in membrane excitability. Adv. Neurol. 44, 137–170.
Adibhatla, R. M., and Hatcher, J. F. (2008). Phospholipase A2, reactive oxygen species and lipid peroxidation in CNS pathologies. BMB Rep. 41, 560–567. doi: 10.5483/BMBRep.2008.41.8.560
Aïd, S., and Bosetti, F. (2011). Targeting cyclooxygenases-1 and -2 in neuroinflammation: therapeutic implications. Biochimie 93, 46–51. doi: 10.1016/j.biochi.2010.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002). Molecular Biology of the Cell, 4th Edn. New York: Garland Science.
Alcedo, J., Flatt, T., and Pasyukova, E. G. (2013). Neuronal inputs and outputs of aging and longevity. Front. Genet. 4:71. doi: 10.3389/fgene.2013.00071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alkon, D. L. (1974). Associative training of Hermissenda. J. Gen. Physiol. 64, 70–84. doi: 10.1085/jgp.64.1.70
Alkon, D. L. (1979). Voltage-dependent calcium and potassium ion conductances: a contingency mechanism for an associative learning model. Science 205, 810–816. doi: 10.1126/science.223244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Annunziato, L., Pannaccione, A., Cataldi, M., Secondo, A., Castaldo, P., Di Renzo, G.,et al. (2002). Modulation of ion channels by reactive oxygen and nitrogen species: a pathophysiological role in brain aging? Neurobiol. Aging 23, 819–834. doi: 10.1016/S0197-4580(02)00069-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arundell, M., Patel, B. A., Straub, V., Allen, M. C., Janse, C. K., O’Hare, D.,et al. (2006). Effects of age on feeding behavior and chemosensory processing in the pond snail, Lymnaea stagnalis. Neurobiol. Aging 27, 1880–1891. doi: 10.1016/j.neurobiolaging.2005.09.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atkinson, J., Epand, R. F., and Epand, R. M. (2008). Tocopherols and tocotrienols in membranes: a critical review. Free Rad. Biol. Med. 44, 739–764. doi: 10.1016/j.freeradbiomed.2007.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atkinson, J., Harroun, T., Wassal, S. R., Stillwell, W., and Katsaras, J. (2010). The location and behavior of alpha -tocopherol in membranes. Mol. Nutr. Food Res. 54, 641–651. doi: 10.1002/mnfr.200900439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Audesirk, T. E., Alexander, J. E., Audesirk, G. J., and Moyer, C. M. (1982). Rapid, nonaversive conditioning in a freshwater gastropod. I. Effects of age and motivation. Behav. Neural Biol. 36, 379–390. doi: 10.1016/S0163-1047(82)90782-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ayala, A., Muñoz, M. F., and Argüelles, S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 1–31. doi: 10.1155/2014/360438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bähring, R., Milligan, C. J., Vardanyan, V., Engeland, B., Young, B. A., Dannenberg, J.,et al. (2001). Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvbeta subunits. J. Biol. Chem. 276, 22923–22929. doi: 10.1074/jbc.M100483200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ballatori, N., Krance, S. M., Notenboom, S., Shi, S., Tieu, K., and Hammond, C. L. (2009). Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 390, 191–214. doi: 10.1515/BC.2009.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barnes, C. A. (1994). Normal aging: regionally specific changes in hippocampal synaptic transmission. Trends Neurosci. 17, 13–18. doi: 10.1016/0166-2236(94)90029-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bean, B. P. (2007). The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465. doi: 10.1038/nrn2148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beaulieu, E., Ioffe, J., Watson, S. N., Hermann, P. M., and Wildering, W. C. (2014). Oxidative-stress induced increase in circulating fatty acids does not contribute to phospholipase A2-dependent appetitive long-term memory failure in the pond snail Lymnaea stagnalis. BMC Neurosci. 15:56. doi: 10.1186/1471-2202-15-56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beck, H., and Yaari, Y. (2008). Plasticity of intrinsic neuronal properties in CNS disorders. Nat. Rev. Neurosci. 9, 357–369. doi: 10.1038/nrn2371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benjamin, P. R. (2012). Distributed network organization underlying feeding behavior in the mollusk Lymnaea. Neural Syst. Circuits 2:4. doi: 10.1186/2042-1001-2-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benjamin, P. R., and Kemenes, G. (2010). Lymnaea learning and memory. Scholarpedia 5, 4247. doi: 10.4249/scholarpedia.4247
Benjamin, P. R., Kemenes, G., and Kemenes, I. (2008). Non-synaptic neuronal mechanisms of learning and memory in gastropod molluscs. Front. Biosci. 13:4051–4057.
Benjamin, P. R., Staras, K., and Kemenes, G. (2000). A systems approach to the cellular analysis of associative learning in the pond snail Lymnaea. Learn. Mem. 7, 124–131. doi: 10.1101/lm.7.3.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bliss, T. V. P., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Branch, S. Y., Sharma, R., and Beckstead, M. J. (2014). Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J. Neurosci. 34, 9310–9318. doi: 10.1523/JNEUROSCI.4228-13.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brembs, B. (2003). Operant conditioning in invertebrates. Curr. Opin. Neurobiol. 13, 710–717. doi: 10.1016/j.conb.2003.10.002
Brown, W. J., Chambers, K., and Doody, A. (2003). Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 4, 214–221. doi: 10.1034/j.1600-0854.2003.00078.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burke, S. N., and Barnes, C. A. (2006). Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 7, 30–40. doi: 10.1038/nrn1809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cai, S.-Q., and Sesti, F. (2009). Oxidation of a potassium channel causes progressive sensory function loss during aging. Nat. Neurosci. 12, 611–617. doi: 10.1038/nn.2291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carnevale, K. A., and Cathcart, M. K. (2001). Calcium-independent phospholipase A(2) is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J. Immunol. 167, 3414–3421. doi: 10.4049/jimmunol.167.6.3414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chalimoniuk, M., Stolecka, A., Ziemiñska, E., Stepieñ, A., Langfort, J., and Strosznajder, J. B. (2009). Involvement of multiple protein kinases in cPLA2 phosphorylation, arachidonic acid release, and cell death in in vivo and in vitro models of 1-methyl-4-phenylpyridinium-induced parkinsonism - the possible key role of PKG. J. Neurochem. 110, 307–317. doi: 10.1111/j.1471-4159.2009.06147.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chawla, M. K., and Barnes, C. A. (2007). Hippocampal granule cells in normal aging: insights from electrophysiological and functional imaging experiments. Progr. Brain Res. 163, 661–821. doi: 10.1016/S0079-6123(07)63036-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, C. T., Green, J. T., Orr, S. K., and Bazinet, R. P. (2008). Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins Leukot. Essent. Fatty Acids 79, 85–91. doi: 10.1016/j.plefa.2008.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J., Small-Howard, A., Yin, A., and Berry, M. J. (2005). The responses of Ht22 cells to oxidative stress induced by buthionine sulfoximine (BSO). BMC Neurosci. 6:10. doi: 10.1186/1471-2202-6-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choe, M., Jackson, C., and Yu, B. P. (1995). Lipid peroxidation contributes to age-related membrane rigidity. Free Radic. Biol. Med. 18, 977–984. doi: 10.1016/0891-5849(94)00217-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, B. H. (1995). Oxidative stress and Alzheimer’s disease. Neurobiol. Aging 16, 675–678. doi: 10.1016/0197-4580(95)00065-M
Choi, J. H., and Yu, B. P. (1995). Brain synaptosomal aging: free radicals and membrane fluidity. Free Radic. Biol. Med. 18, 133–139. doi: 10.1016/0891-5849(94)00106-T
Christian, K. M., and Thompson, R. F. (2003). Neural substrates of eyeblink conditioning: acquisition and retention. Learn. Mem. 10, 427–455. doi: 10.1101/lm.59603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coulter, D. A., Lo Turco, J. J., Kubota, M., Disterhoft, J. F., Moore, J. W., and Alkon, D. L. (1989). Classical conditioning reduces amplitude and duration of calcium-dependent afterhyperpolarization in rabbit hippocampal pyramidal cells. J. Neurophysiol. 61, 971–981.
Currais, A., and Maher, P. (2013). Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox Signal. 19, 813–822. doi: 10.1089/ars.2012.4996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dai, D.-F., Chiao, Y. A., Marcinek, D. J., Szeto, H. H., and Rabinovitch, P. S. (2014). Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 3, 6. doi: 10.1186/2046-2395-3-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalesman, S., and Lukowiak, K. (2012). How stress alters memory in ’smart’ snails. PLoS ONE 7:e32334. doi: 10.1371/journal.pone.0032334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Angelo, E. (2014). The Organization of Plasticity in the Cerebellar Cortex: From Synapses to Control, Vol. 210 (Amsterdam: Elsevier), 31–58.
Dart, C. (2010). Lipid microdomains and the regulation of ion channel function. J. Physiol. 588, 3169–3178. doi: 10.1113/jphysiol.2010.191585
Davidson, J. E., Lockhart, A., Amos, L., Stirnadel-Farrant, H. A., Mooser, V., Sollberger, M.,et al. (2012). Plasma lipoprotein-associated phospholipase A2 activity in Alzheimer’s disease, amnestic mild cognitive impairment, and cognitively healthy elderly subjects: a cross-sectional study. Alzheimers Res. Ther. 4, 51. doi: 10.1186/alzrt154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davletov, B., and Montecucco, C. (2010). Lipid function at synapses. Curr. Opin. Neurobiol. 20, 543–549. doi: 10.1016/j.conb.2010.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dennis, E. A., Cao, J., Hsu, Y. H., Magrioti, V., and Kokotos, G. (2011). Phospholipase A2 enzymes: physical structure. Biological function, disease implications, chemical inhibition, and therapeutic intervention. Chem. Rev. 111, 6130–6185. doi: 10.1021/cr200085w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Desbene, C., Malaplate-Armand, C., Youssef, I., Garcia, P., Stenger, C., Sauvée, M.,et al. (2012). Critical role of cPLA2 in Abeta oligomer- induced neurodegeneration and memory deficit. Neurobiol. Aging 33, 1123.e17–1123.e29. doi: 10.1016/j.neurobiolaging.2011.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Disterhoft, J. F., Thompson, L. T., Moyer, J. R., and Mogul, D. J. (1996). Calcium-dependent after hyperpolarization and learning in young and aging hippocampus. Life Sci. 59, 413–420. doi: 10.1016/0024-3205(96)00320-7
Dukoff, D. J., Hogg, D. W., Hawrsyh, P. J., and Buck, L. T. (2014). Scavenging ROS dramatically increases NMDA receptor whole cell currents in painted turtle cortical neurons. J. Exp. Biol. 217, 3346–3355. doi: 10.1242/jeb.105825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Esiri, M. M. (2007). Ageing and the brain. J. Pathol. 211, 181–187. doi: 10.1002/path.2089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farooqui, A. A., and Horrocks, L. A. (2006). Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly. Neuroscientist 12, 245–260. doi: 10.1177/1073858405285923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fitzpatrick, A. L., Irizarry, M. C., Cushman, M., Jenny, N. S., Chi, G. C., and Koro, C. (2014). Lipoprotein-associated phospholipase A2 and risk of dementia in the Cardiovascular Health Study. Atherosclerosis 235, 384–391. doi: 10.1016/j.atherosclerosis.2014.04.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forlenza, O. V., Schaeffer, E. L., and Gattaz, W. F. (2007). The role of phospholipase A2 in neuronal homeostasis and memory formation: implications for the pathogenesis of Alzheimer’s disease. J. Neural Transm. 114, 231–238. doi: 10.1007/s00702-006-0597-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Foster, T. C. (2012). Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Prog. Neurobiol. 96, 283–303. doi: 10.1016/j.pneurobio.2012.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frolkis, V. V., Kvitnitskaya-Ryzhova, T. Yu., and Martynenko, O. A. (1995). Aging of neurons in the mollusc Lymnaea stagnalis small parietal ganglion: a morpho-functional comparison in the same neuron. Exp. Gerontol. 30, 533–544. doi: 10.1016/0531-5565(95)00008-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frolkis, V. V., Martynenko, O. A., and Timchenko, A. N. (1989). Age-related changes in the function of somatic membrane potassium channels of neurons in the mollusc Lymnaea stagnalis. Mech. Ageing Dev. 47, 47–54. doi: 10.1016/0047-6374(89)90006-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frolkis, V. V., Martynenko, O. A., and Timchenko, A. N. (1991). Potential-dependent Ca channels of neurons in the mollusc Lymnaea stagnalis in aging: effect of norepinephrine. Mech. Ageing Dev. 58, 75–83. doi: 10.1016/0047-6374(91)90121-F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frolkis, V. V., Stupina, A. S., Martinenko, O. A., Tòth, S., and Timchenko, A. I. (1984). Aging of neurons in the mollusc Lymnaea stagnalis. Structure, function and sensitivity to transmitters. Mech. Ageing Dev. 25, 91–102. doi: 10.1016/0047-6374(84)90132-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia-Garcia, H. M., and Serruys, P. W. (2009). Phospholipase A2 inhibitors. Curr. Opin. Lipidol. 20, 327–332. doi: 10.1097/MOL.0b013e32832dd4c7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gazzaley, A. H., Thakker, M. M., Hof, P. R., and Morrison, J. H. (1997). Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neurobiol. Aging 18, 549–553. doi: 10.1016/S0197-4580(97)00112-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Genovese, T., Mazzon, E., Esposito, E., Muia, C., Di Paola, R., Di Bella, P.,et al. (2007). Role of endogenous glutathione in the secondary damage in experimental spinal cord injury in mice. Neurosci. Lett. 423, 41–46. doi: 10.1016/j.neulet.2007.05.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gentile, M. T., Reccia, M. G., Sorrentino, P. P., Vitale, E., Sorrentino, G., Puca, A. A.,et al. (2012). Role of cytosolic calcium-dependent phospholipase A2 in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 45, 596–604. doi: 10.1007/s12035-012-8279-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greenberg, M. E., Li, X. M., Gugiu, B. G., Gu, X., Qin, J., Salomon, R. G.,et al. (2008). The lipid whisker model of the structure of oxidized cell membranes. J. Biol. Chem. 283, 2385–2396. doi: 10.1074/jbc.M707348200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gulbis, J. M. (2002). The beta subunit of Kv1 channels: voltage-gated enzyme or safety switch? Novartis Found. Symp. 245, 127–141. doi: 10.1002/0470868759.ch9
Guo, C. H., Senzel, A., Li, K., and Feng, Z. P. (2010). De novo protein synthesis of syntaxin-1 and dynamin-1 in long-term memory formation requires CREB1 gene transcription in Lymnaea stagnalis. Behav. Genet. 40, 680–693. doi: 10.1007/s10519-010-9374-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haddadi, M., Jahromi, S. R., Sagar, B. K., Patil, R. K., Shivanandappa, T., and Ramesh, S. R. (2014). Brain aging, memory impairment and oxidative stress: a study in Drosophila melanogaster. Behav. Brain Res. 259, 60–69. doi: 10.1016/j.bbr.2013.10.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. doi: 10.1093/geronj/11.3.298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hawkins, R. D. (1984). A cellular mechanism of classical conditioning in Aplysia. J. Exp. Biol. 112, 113–128.
Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. doi: 10.1016/j.tcb.2011.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hermann, P. M., Lee, A., Hulliger, S., Minvielle, M., Ma, B., and Wildering, W. C. (2007). Impairment of long-term associative memory in aging snails (Lymnaea stagnalis). Behav. Neurosci. 121, 1400–1414. doi: 10.1037/0735-7044.121.6.1400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hermann, P. M., Park, D., Beaulieu, E., and Wildering, W. C. (2013). Evidence for inflammation-mediated memory dysfunction in gastropods: putative PLA2 and COX inhibitors abolish long-term memory failure induced by systemic immune challenges. BMC Neurosci. 14:83. doi: 10.1186/1471-2202-14-83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, D., Serrano, F., Oury, T. D., and Klann, E. (2006). Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J. Neurosci. 26, 3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hui, D. Y. (2012). Phospholipase A(2) enzymes in metabolic and cardiovascular diseases. Curr. Opin. Lipidol. 23, 235–240.
Hulbert, A. J., Pamplona, R., Buffenstein, R., and Buttemer, W. A. (2007). Life and death: metabolic rate, membrane composition, and life span of animals. Physiol. Rev. 87, 1175–1213. doi: 10.1152/physrev.00047.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hung, C.-W., Chen, Y.-C., Hsieh, W.-L., Chiou, S.-H., and Kao, C.-L. (2010). Ageing and neurodegenerative diseases. Ageing Res. Rev. 9(Suppl. )1, S36–S46. doi: 10.1016/j.arr.2010.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacob, K. D., Hooten, N. N., Trzeciak, A. R., and Evans, M. K. (2013). Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech. Ageing Dev. 134, 139–157. doi: 10.1016/j.mad.2013.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janse, C., and van der Roest, M. (2001). cAMP and excitability in neuroendocrine cells during reproductive senescence. Neurobiol. Aging 22, 503–514. doi: 10.1016/S0197-4580(01)00211-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janse, C., van der Roest, M., and Slob, W. (1986). Age-related decrease in electrical coupling of two identified neurons in the mollusk Lymnaea stagnalis. Brain Res. 376, 208–212. doi: 10.1016/0006-8993(86)90920-0
Janse, C. K., Peretz, B., van der Roest, M., and Dubelaar, E. J. (1999). Excitability and branching of neuroendocrine cells during reproductive senescence. Neurobiol. Aging 20, 675–683. doi: 10.1016/S0197-4580(99)00021-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janse, C. K., Slob, W., Popelier, C. M., and Vogelaar, J. W. (1988). Survival characteristics of the mollusc Lymnaea stagnalis under constant culture conditions: effects of aging and disease. Mech. Ageing Dev. 42, 263–274. doi: 10.1016/0047-6374(88)90052-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janse, C. K., Wildering, W. C., and Popelier, C. M. (1989). Age-related changes in female reproductive activity and growth in the mollusc Lymnaea stagnalis. J. Gerontol. 44, B148–B155. doi: 10.1093/geronj/44.6.B148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ježek, J., Jabùrek, M., Zelenka, J., and Ježek, P. (2010). Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 59, 737–747.
Ježek, P., Olejár, T., Smolková, K., Ježek, J., Dlasková, A., Plecitá-Hlavatá, L.,et al. (2014). Antioxidant and regulatory role of mitochondrial uncoupling protein UCP2 in pancreatic beta-cells. Physiol. Res. 63(Suppl. 1), S73–S91.
Jones, N. G., Kemenes, I., Kemenes, G., and Benjamin, P. R. (2003). A persistent cellular change in a single modulatory neuron contributes to associative long-term memory. Curr. Biol. 13, 1064–1069. doi: 10.1016/S0960-9822(03)00380-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joseph, J. A., Shukitt-Hale, B., Casadesus, G., and Fisher, D. (2005). Oxidative stress and inflammation in brain aging: nutritional considerations. Neurochem. Res. 30, 927–935. doi: 10.1007/s11064-005-6967-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kagan, V. E. (1989). Tocopherol stabilizes membrane against phospholipase A, free fatty acids and lysophospholipids. Ann. N. Y. Acad. Sci. 570, 121–135. doi: 10.1111/j.1749-6632.1989.tb14913.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kann, O., and Kovács, R. (2007). Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 292, C641–C657. doi: 10.1152/ajpcell.00222.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawai, R., Sunada, H., Horikoshi, T., and Sakakibara, M. (2004). Conditioned taste aversion with sucrose and tactile stimuli in the pond snail Lymnaea stagnalis. Neurobiol. Learn. Mem. 82, 164–168. doi: 10.1016/j.nlm.2004.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kemenes, G., Kemenes, I., Michel, M., Papp, A., and Müller, U. (2006a). Phase-dependent molecular requirements for memory reconsolidation: differential roles for protein synthesis and protein kinase A activity. J. Neurosci. 26, 6298–6302. doi: 10.1523/JNEUROSCI.0890-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kemenes, I., Straub, V. A., Nikitin, E. S., Staras, K., O’Shea, M., Kemenes, G.,et al. (2006b). Role of delayed nonsynaptic neuronal plasticity in long-term associative memory. Curr. Biol. 16, 1269–1279. doi: 10.1016/j.cub.2006.05.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kempsell, A. T., and Fieber, L. A. (2014). Behavioral aging is associated with reduced sensory neuron excitability in Aplysia californica. Front. Aging Neurosci. 6:84. doi: 10.3389/fnagi.2014.00084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keuker, J. I., Luiten, P. G., and Fuchs, E. (2003). Preservation of hippocampal neuron numbers in aged rhesus monkeys. Neurobiol. Aging 24, 157–165. doi: 10.1016/S0197-4580(02)00062-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, B., and Feldman, E. L. (2012). Insulin resistance in the nervous system. Trends Endocrinol. Metabol. 23, 133–141. doi: 10.1016/j.tem.2011.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kirkwood, T. B. L., and Austad, S. N. (2000). Why do we age? Nature, 408, 233–238. doi: 10.1038/35041682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kits, K. S., Lodder, J. C., and Veerman, M. J. (1997). Phe-Met-Arg-Phe-amide activates a novel voltage-dependent K+ current through a lipoxygenase pathway in molluscan neurones. J. Gen. Physiol. 110, 611–628. doi: 10.1085/jgp.110.5.611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klaassen, L. J., Janse, C. K., and van der Roest, M. (1998). Multiple synaptic connections of a single neuron change differentially with age. Neurobiol. Aging 19, 341–349. doi: 10.1016/S0197-4580(98)00065-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J., Kosaras, B., Del Signore, S. J., Cormier, K., McKee, A., Ratan, R. R.,et al. (2011). Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropath. 121, 487–498. doi: 10.1007/s00401-010-0788-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemaire-Ewing, S., Desrumaux, C., Neel, D., and Laurent, L. (2010). Vitamin E transport, membrane incorporation and cell metabolism: is α-tocopherol in lipid rafts an oar in the lifeboat? Mol. Nutr. Food Res. 54, 631–640. doi: 10.1002/mnfr.200900445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewis, D. V., Huguenard, J. R., Anderson, W. W., and Wilson, W. A. (1986). Membrane currents underlying bursting pacemaker activity and spike frequency adaptation in invertebrates. Adv. Neurol. 44, 235–261.
Liang, X., Wu, L., Wang, Q., Hand, T., Bilak, M., McCullough, L.,et al. (2007). Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 33, 94–99. doi: 10.1007/s12031-007-0058-8
Ljubuncic, P., and Reznick, A. Z. (2009). The evolutionary theories of aging revisited- a mini review. Gerontology 55, 205–216. doi: 10.1159/000200772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopes, C. M. B., Franks, N. P., and Lieb, W. R. (1998). Actions of general anaesthetics and arachidonic acid pathway inhibitors on K +currents activated by volatile anaesthetics and FMRFamide in molluscan neurones. Br. J. Pharmacol. 125, 309–318. doi: 10.1038/sj.bjp.0702069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopez, N., Tormo, C., De Blas, I., Llinares, I., and Alom, J. (2013). Oxidative stress in Alzheimer’s disease and mild cognitive impairment with high sensitivity and specificity. J. Alzheimers Dis. 33, 823–829. doi: 10.3233/JAD-2012-121528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lukowiak, K., Martens, K., Orr, M., Parvez, K., Rosenegger, D., and Sangha, S. (2006). Modulation of aerial respiratory behaviour in a pond snail. Respir. Physiol. Neurobiol. 154, 61–72. doi: 10.1016/j.resp.2006.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lukowiak, K., Ringseis, E., Spencer, G., Wildering, W. C., and Syed, N. I. (1996). Operant conditioning of aerial respiratory behaviour in Lymnaea stagnalis. J. Exp. Biol. 199, 683–691.
Maingret, F., Fosset, M., Lesage, F., Lazdunski, M., and Honoré, E. (1999). TRAAK is a mammalian neuronal mechano-gated K+ channel. J. Biol. Chem. 274, 1381–1387. doi: 10.1074/jbc.274.3.1381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mari, M., Morales, A., Colell, A., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2009). Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 11, 2685–2700. doi: 10.1089/ars.2009.2695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marquardt, D., Williams, J. A., Kucerka, N., Atkinson, J., Wassal, S. R., Katsaras, J.,et al. (2013). Tocopherol activity correlates with its location in a membrane: a new perspective on the antioxidant Vitamin E. J. Am. Chem. Soc. 135, 7523–7533. doi: 10.1021/ja312665r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masoro E. J., and Austad, S. N. (2011). Handbook of the Biology of Aging. Amsterdam: Elsevier Academic Press.
Massaad, C. A., and Klann, E. (2011). Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 14, 2013–2053. doi: 10.1089/ars.2010.3208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattson, M. P., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294. doi: 10.1038/nrn1886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merrill, D. A., Chiba, A. A., and Tuszynski, M. H. (2001). Conservation of neuronal number and size in the entorhinal cortex of behaviorally characterized aged rats. J. Comp. Neurol. 438, 445–456. doi: 10.1002/cne.1327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merrill, D. A., Roberts, J. A., and Tuszynski, M. H. (2000). Conservation of neuron number and size in entorhinal cortex layers II, III, and V/VI of aged primates. J. Comp. Neurol. 422, 396–401. doi: 10.1002/1096-9861(20000703)422:3<396::AID-CNE6>3.0.CO;2-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Milton, V. J., and Sweeney, S. T. (2011). Oxidative stress in synapse development and function. Dev. Neurobiol. 72, 100–110. doi: 10.1002/dneu.20957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moyer, J. R., Power, J. M., Thompson, L. T., and Disterhoft, J. F. (2000). Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. J. Neurosci. 20, 5476–5482.
Moyer, J. R., Thompson, L. T., Black, J. P., and Disterhoft, J. F. (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J. Neurophysiol. 68, 2100–2109.
Mozzachiodi, R., and Byrne, J. H. (2010). More than synaptic plasticity: role of nonsynaptic plasticity in learning and memory. Trends Neurosci. 33, 17–26. doi: 10.1016/j.tins.2009.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munro, D., and Blier, P. U. (2012). The extreme longevity of Arctica islandicais associated with increased peroxidation resistance in mitochondrial membranes. Aging Cell 11, 845–855. doi: 10.1111/j.1474-9726.2012.00847.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munro, D., Pichaud, N., Paquin, F., Kemeid, V., and Blier, P. U. (2013). Low hydrogen peroxide production in mitochondria of the long-lived Arctica islandica: underlying mechanisms for slow aging. Aging Cell 12, 584–592. doi: 10.1111/acel.12082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murakami, S. (2007). Caenorhabditis elegans as a model system to study aging of learning and memory. Mol. Neurobiol. 35, 85–94. doi: 10.1007/BF02700625
Murakami, J., Okada, R., Sadamoto, H., Kobayashi, S., Mita, K., Sakamoto, Y.,et al. (2013). Involvement of insulin-like peptide in long-term synaptic plasticity and long-term memory of the pond snail Lymnaea stagnalis. J. Neurosci. 33, 371–383. doi: 10.1523/JNEUROSCI.0679-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagy, I., Toth, S., and Lustyik, G. (1985). Verification of the membrane hypothesis of aging on the identified giant neurons of the snail Lymnaea stagnalis L. (Gastropoda, Pulmonata) by a combined application of intracellular electrophysiology and X-ray microanalysis. Arch. Gerontol. Geriatr. 4, 53–66. doi: 10.1016/0167-4943(85)90018-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Negre-Salvayre, A., Auge, N., Ayala, V., Basaga, H., Boada, J., Brenke, R.,et al. (2010). Pathological aspects of lipid peroxidation. Free Radic. Res. 44, 1125–1171. doi: 10.3109/10715762.2010.498478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nigam, S., and Schewe, T. (2000). Phospholipase A(2)s and lipid peroxidation. Biochim. Biophys. Acta 1488, 167–181. doi: 10.1016/S1388-1981(00)00119-0
Niki, E. (1990). Free-radical initiators as source of water-soluble or lipid-soluble peroxyl radicals. Methods Enzymol. 186, 100–108. doi: 10.1016/0076-6879(90)86095-D
Niki, E. (2012). Do antioxidants impair signaling by reactive oxygen species and lipid peroxidation products. FEBS Lett. 586, 3767–3770. doi: 10.1016/j.febslet.2012.09.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikitin, E. S., Balaban, P. M., and Kemenes, G. (2013). Nonsynaptic plasticity underlies a compartmentalized increase in synaptic efficacy after classical conditioning. Curr. Biol. 23, 614–619. doi: 10.1016/j.cub.2013.02.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikitin, E. S., Kiss, T., Staras, K., O’Shea, M., Benjamin, P. R., and Kemenes, G. (2006). Persistent sodium current is a target for cAMP-induced neuronal plasticity in a state-setting modulatory interneuron. J. Neurophysiol. 95, 453–463. doi: 10.1152/jn.00785.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikitin, E. S., Vavoulis, D. V., Kemenes, I., Marra, V., Pirger, Z., Michel, M.,et al. (2008). Persistent sodium current is a nonsynaptic substrate for long-term associative memory. Curr. Biol. 18, 1221–1226. doi: 10.1016/j.cub.2008.07.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nusser, Z. (2012). Differential subcellular distribution of ion channels and the diversity of neuronal function. Curr. Opin. Neurobiol. 22, 366–371. doi: 10.1016/j.conb.2011.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oakley, R., and Tharakan, B. (2014). Vascular hyperpermeability and aging. Aging Dis. 5, 114–125. doi: 10.14336/AD.2014.0500114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oh, M. M., Oliveira, F. A., and Disterhoft, J. F. (2010). Learning and aging related changes in intrinsic neuronal excitability. Front. Aging Neurosci. 2:2. doi: 10.3389/neuro.24.002.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ong, W. Y., Farooqui, T., and Farooqui A. A. (2010). Involvement of cytosolic phospholipase A2, calcium independent phospholipase A2 and plasmalogen selective phospholipase A2 in neurodegenerative and neuropsychiatric conditions. Curr. Med. Chem. 17, 2746–2763. doi: 10.2174/092986710791859289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orr, W. C., Radyuk, S. N., and Sohal, R. S. (2013). Involvement of redox state in the aging of Drosophila melanogaster. Antioxid. Redox Signal. 19, 788–803. doi: 10.1089/ars.2012.5002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pamplona, R. (2008). Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim. Biophys. Acta 1777, 1249–1262. doi: 10.1016/j.bbabio.2008.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pamplona, R. (2011). Advanced lipoxidation end-products. Chem. Biol. Interact 192, 14–20. doi: 10.1016/j.cbi.2011.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Partridge, L. (2008). Some highlights of research on aging with invertebrates. Aging Cell 7, 605–608. doi: 10.1111/j.1474-9726.2008.00415.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patel, A. J., Honoré, E., Maingret, F., Lesage, F., Fink, M., Duprat, F.,et al. (1998). A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 17, 4283–4290. doi: 10.1093/emboj/17.15.4283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patel, A. J., Lazdunski, M., and Honoré, E. (2001). Lipid and mechano-gated 2P domain K(+) channels. Curr. Opin. Cell Biol. 13, 422–428. doi: 10.1016/S0955-0674(00)00231-3
Patel, B. A., Arundell, M., Allen, M. C., Gard, P., O’Hare, D., Parker, K.,et al. (2006). Changes in the properties of the modulatory cerebral giant cells contribute to aging in the feeding system of Lymnaea. Neurobiol. Aging 27, 1892–1901. doi: 10.1016/j.neurobiolaging.2005.09.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patel, B. A., Arundell, M., Parker, K. H., Yeoman, M. S., and O’Hare, D. (2010). Microelectrode investigation of neuronal ageing from a single identified neurone. Phys. Chem. Chem. Phys. 12, 10065–10072. doi: 10.1039/c0cp00310g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peretz, B. (1988). Long-term change of viability of neuron functioning and its possible behavioral consequences in the adult Aplysia. J. Physiol. 83, 217–223.
Pérez, V. I., Bokov, A., Remmen, H. V., Mele, J., Ran, Q., Ikeno, Y.,et al. (2009). Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790, 1005–1014. doi: 10.1016/j.bbagen.2009.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peters, A., Leahu, D., Moss, M. B., and McNally, K. J. (1994). The effects of aging on area 46 of the frontal cortex of the rhesus monkey. Cereb. Cortex 4, 621–635. doi: 10.1093/cercor/4.6.621
Phillis, J. W., Horrocks, L. A., and Farooqui, A. A. (2006). Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res. Rev. 52, 201–243. doi: 10.1016/j.brainresrev.2006.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piomelli, D., Astarita, G., and Rapaka, R. (2007). A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 8, 743–754. doi: 10.1038/nrn2233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piomelli, D., Volterra, A., Dale, N., Siegelbaum, S. A., Kandel, E. R., Schwartz, J. H.,et al. (1987). Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature 328, 38–43. doi: 10.1038/328038a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pirger, Z., Naskar, S., László, Z., Kemenes, G., Reglõdi, D., and Kemenes, I. (2014). Reversal of age-related learning deficiency by the vertebrate PACAP and IGF-1 in a novel invertebrate model of aging: the pond snail (Lymnaea stagnalis). J. Gerontol. A Biol. Sci. Med. Sci. 69, 1331–1338. doi: 10.1093/gerona/glu068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pizzimenti, S., Ciamporcero, E. S., Daga, M., Pettazzoni, P., Arcaro, A., Cetrangolo, G.,et al. (2013). Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 4:242. doi: 10.3389/fphys.2013.00242
Quach, N. D., Arnold, R. D., and Cummings, B. S. (2014). Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem. Pharmacol. 90, 338–348. doi: 10.1016/j.bcp.2014.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Randall, A. D., Booth, C., and Brown, J. T. (2012). Age-related changes to Na+ channel gating contribute to modified intrinsic neuronal excitability. Neurobiol. Aging 33, 2715–2720. doi: 10.1016/j.neurobiolaging.2011.12.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rapp, P. R., and Gallagher, M. (1996). Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc. Natl. Acad. Sci. U.S.A. 93, 9926–9930. doi: 10.1073/pnas.93.18.9926
Rashba-Step, J., Tatoyan, A., Duncan, R., Ann, D., Pushpa-Rehka, T. R., and Sevanian, A. (1997). Phospholipid peroxidation induces cytosolic phospholipase A2 activity: membrane effects versus enzyme phosphorylation. Arch. Biochem. Biophys. 343, 44–54. doi: 10.1006/abbi.1997.0134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rasmussen, T., Schliemann, T., Sorensen, J. C., Zimmer, J., and West, M. J. (1996). Memory impaired aged rats: no loss of principal hippocampal and subicular neurons. Neurobiol. Aging 17, 143–147. doi: 10.1016/0197-4580(95)02032-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rattan, K. S., and Peretz, B. (1981). Age-dependent behavioral changes and physiological changes in identified neurons in Aplysia californica. J. Neurobiol. 12, 469–478. doi: 10.1002/neu.480120506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Riahi, Y., Cohen, G., Shamni, O., and Sasson, S. (2010). Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 299, E879–E886. doi: 10.1152/ajpendo.00508.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ribas, V., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2014). Glutathione and mitochondria. Front. Pharmacol. 5:151. doi: 10.3389/fphar.2014.00151
Ribeiro, M. J., Schofield, M. G., Kemenes, I., O’Shea, M., Kemenes, G., and Benjamin, P. R. (2005). Activation of MAPK is necessary for long-term memory consolidation following food-reward conditioning. Learn. Mem. 12, 538–545. doi: 10.1101/lm.8305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ribeiro, M. J., Serfozo, Z., Papp, A., Kemenes, I., O’Shea, M., Yin, J. C.,et al. (2003). Cyclic AMP response element-binding (CREB)-like proteins in a molluscan brain: cellular localization and learning-induced phosphorylation. Eur. J. Neurosci. 18, 1223–1234. doi: 10.1046/j.1460-9568.2003.02856.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rincon, F., and Wright, C. B. (2013). Vascular cognitive impairment. Curr. Opin. Neurol. 26, 29–36. doi: 10.1097/WCO.0b013e32835c4f04
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roberts-Crowley, M. L., Mitra-Ganguli, T., Liu, L., and Rittenhouse, A. R. (2009). Regulation of voltage-gated Ca2+ channels by lipids. Cell Calcium 45, 589–601. doi: 10.1016/j.ceca.2009.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosenegger, D., and Lukowiak, K. (2013). The participation of NMDA receptors, PKC, and MAPK in Lymnaea memory extinction. Neurobiol. Learn. Mem. 100, 64–69. doi: 10.1016/j.nlm.2012.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosenhouse-Dantsker, A., Mehta, D., and Levitan, I. (2012). Regulation of ion channels by membrane lipids. Compr. Physiol. 2, 31–68. doi: 10.1002/cphy.c110001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruppersberg, J. P., Stacker, M., Pongs, O., Heinemann, S. H., Frank, R., and Koenen, M. (1991). Regulation of fast inactivation of cloned mammalian IK (A) channels by cysteine oxidation. Nature 352, 711–714. doi: 10.1038/352711a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sadagurski, M., and White, M. F. (2013). Integrating metabolism and longevity through insulin and IGF1 signaling. Endocrinol. Metab. Clin. North Am. 42, 127–148. doi: 10.1016/j.ecl.2012.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sadamoto, H., Kitahashi, T., Fujito, Y., and Ito, E. (2010). Learning-dependent gene expression of CREB1 isoforms in the molluscan brain. Front. Behav. Neurosci. 4:25. doi: 10.3389/fnbeh.2010.00025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sah, P. (1996). Ca (2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 19, 150–154. doi: 10.1016/S0166-2236(96)80026-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sah, P., and Davies, P. (2000). Calcium-activated potassium currents in mammalian neurons. Clin. Exp. Pharm. Physiol. 27, 657–663. doi: 10.1046/j.1440-1681.2000.03317.x
Sakakibara, M., Kawai, R., Kobayashi, S., and Horikoshi, T. (1998). Associative learning of visual and vestibular stimuli in Lymnaea. Neurobiol. Learn. Mem. 69, 1–12. doi: 10.1006/nlme.1997.3805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salgo, M. G., Corongiu, F. P., and Sevanian, A. (1992). Peroxidation and phospholipase A2 hydrolytic susceptibility of liposomes consisting of mixed species of phosphatidylcholine and phosphatidylethanolamine. Biochim. Biophys. Acta 1127, 131–140. doi: 10.1016/0005-2760(92)90268-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanchez-Mejia, R. O., and Mucke, L. (2010). Phospholipase A2 and arachidonic acid in Alzheimer’s disease. Biochim. Biophys. Acta 1801, 784–790. doi: 10.1016/j.bbalip.2010.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanchez-Mejia, R. O., Newman, J. W., Toh, S., Yu, G. Q., Zhou, Y., Halabisky, B.,et al. (2008). Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 11, 1311–1318. doi: 10.1038/nn.2213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaeffer, E. L., Forlenza, O. V., and Gattaz, W. F. (2009). Phospholipase A2 activation as a therapeutic approach for cognitive enhancement in early-stage Alzheimer disease. Psychopharmacol. (Berl). 202, 37–51. doi: 10.1007/s00213-008-1351-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scott, B., Leu, J., and Cinader, B. (1988). Effects of aging on neuronal electrical membrane properties. Mech. Ageing Dev. 44, 203–214. doi: 10.1016/0047-6374(88)90022-X
Sehgal, M., Ehlers, V. L., and Moyer, J. R. (2014). Learning enhances intrinsic excitability in a subset of lateral amygdala neurons. Learn. Mem. 21, 161–170. doi: 10.1101/lm.032730.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sehgal, M., Song, C., Ehlers, V. L., and Moyer, J. R. (2013). Learning to learn - intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol. Learn. Mem. 105, 186–199. doi: 10.1016/j.nlm.2013.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sesti, F., Liu, S., and Cai, S. Q. (2010). Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration? Trends Cell Biol. 20, 45–51. doi: 10.1016/j.tcb.2009.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shankar, S. K. (2010). Biology of aging brain. Indian J. Pathol. Microbiol. 53, 595. doi: 10.4103/0377-4929.71995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sieling, F., Bédécarrats, A., Simmers, J., Prinz, A. A., and Nargeot, R. (2014). Differential roles of nonsynaptic and synaptic plasticity in operant reward learning-induced compulsive behavior. Curr. Biol. 24, 941–950. doi: 10.1016/j.cub.2014.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Skinner, T. L., and Peretz, B. (1989). Age sensitivity of osmoregulation and of its neural correlates in Aplysia. Am. J. Physiol. 256, R989–R996.
Slob, W., and Janse, C. K. (1988). A quantitative method to evaluate the quality of interrupted animal cultures in aging studies. Mech. Ageing Dev. 42, 275–290. doi: 10.1016/0047-6374(88)90053-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Speakman, J. R., and Selman, C. (2011). The free-radical damage theory: accumulating evidence against a simple link of oxidative stress to ageing and lifespan. BioEssays 33, 255–259. doi: 10.1002/bies.201000132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spencer, G. E., Syed, N. I., and Lukowiak, K. (1999). Neural changes after operant conditioning of the aerial respiratory behavior in Lymnaea stagnalis. J. Neurosci. 19, 1836–1843.
Staras, K., Kemenes, G., and Benjamin, P. R. (1999). Cellular traces of behavioral classical conditioning can be recorded at several specific sites in a simple nervous system. J. Neurosci. 19, 347–357.
Subczynski, W. K., and Wisniewska, A. (2000). Physical properties of lipid bilayer membranes: relevance to membrane biological functions. Acta Biochim. Pol. 47, 613–625.
Sultana, R., Perluigi, M., and Butterfield, D. A. (2013). Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 62, 157–169. doi: 10.1016/j.freeradbiomed.2012.09.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, G. Y., Chuang, D. Y., Zong, Y., Jiang, J., Lee, J. C.-M., Gu, Z.,et al. (2014). Role of cytosolic phospholipase A2 in oxidative and inflammatory signaling pathways in different cell types in the central nervous system. Mol. Neurobiol. 50, 6–14. doi: 10.1007/s12035-014-8662-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, G. Y., Xu, J., Jensen, M. D., and Simonyi, A. (2004). Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. J. Lipid Res. 45, 205–213. doi: 10.1194/jlr.R300016-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Surmeier, D. J., Guzman, J. N., Sanchez, J., and Schumacker, P. T. (2012). Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009290. doi: 10.1101/cshperspect.a009290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tarumi, T., and Zhang, R. (2014). Cerebral hemodynamics of the aging brain: risk of Alzheimer disease and benefit of aerobic exercise. Front. Physiol. 5:6. doi: 10.3389/fphys.2014.00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor B. E., and Lukowiak K. (2000). The respiratory central pattern generator of Lymnaea: a model, measured and malleable. Respir. Physiol. 122, 197–207. doi: 10.1016/S0034-5687(00)00159-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terman, A., and Brunk, U. T. (2006). Oxidative stress, accumulation of biological ‘garbage’, and aging. Antiox. Redox Signal. 8, 197–204. doi: 10.1089/ars.2006.8.197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thibault, O., Gant, J. C., and Landfield, P. W. (2007). Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 6, 307–317. doi: 10.1111/j.1474-9726.2007.00295.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thompson, L. T., Moyer, J. R., and Disterhoft, J. F. (1996). Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J. Neurophysiol. 76, 1836–1849.
Toescu, E. C., and Vreugdenhil, M. (2010). Calcium and normal brain ageing. Cell Calcium 47, 158–164. doi: 10.1016/j.ceca.2009.11.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tombaugh, G. C., Rowe, W. B., and Rose, G. M. (2005). The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J. Neurosci. 25, 2609–2616. doi: 10.1523/JNEUROSCI.5023-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toroser, D., and Sohal, R. S. (2005). Kinetic characteristics of native gamma-glutamylcysteine ligase in the aging housefly, Musca domestica L. Biochem. Biophys. Res. Commun. 326, 586–593. doi: 10.1016/j.bbrc.2004.11.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toroser, D., and Sohal, R. S. (2007). Age-associated perturbations in glutathione synthesis in mouse liver. Biochem. J. 405, 583–589. doi: 10.1042/BJ20061868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torres, L. L., Quaglio, N. B., de Souza, G. T., Garcia, R. T., Dati, L. M., Moreira, W. L.,et al. (2011). Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 6, 59–68. doi: 10.3233/JAD-2011-110284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ungvari, Z., Csiszar, A., Sosnowska, D., Philipp, E. E., Campbell, C. M., McQuary, P. R.,et al. (2013). Testing predictions of the oxidative stress hypothesis of aging using a novel invertebrate model of longevity: the giant clam (Tridacna derasa). J. Gerontol. A Biol. Sci. Med. Sci. 68, 359–367. doi: 10.1093/gerona/gls159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ungvari, Z., Ridgway, I., Philipp, E. E. R., Campbell, C. M., McQuary, P., Chow, T.,et al. (2011). Extreme longevity is associated with increased resistance to oxidative stress in Arctica islandica, the longest-living non-colonial animal. J. Gerontol. A Biol. Sci. Med. Sci. 66, 741–750. doi: 10.1093/gerona/glr044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vacher, H., Mohapatra, D. P., and Trimmer, J. S. (2008). Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88, 1407–1447. doi: 10.1152/physrev.00002.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vishwanath, B. S., Fawzy, A. A., and Franson, R. C. (1988). Edema-inducing activity of phospholipase A2 purified from human synovial fluid and inhibition by aristolochic acid. Inflammation 12, 549–561. doi: 10.1007/BF00914317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagatsuma, A., Azami, S., Sakura, M., Hatakeyama, D., Aonuma, H., and Ito, E. (2006). De Novo synthesis of CREB in a presynaptic neuron is required for synaptic enhancement involved in memory consolidation. J. Neurosci. Res. 84, 954–960. doi: 10.1002/jnr.21012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waldbaum, S., and Patel, M. (2010). Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 42, 449–455. doi: 10.1007/s10863-010-9320-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wan, H., Mackay, B., Iqbal, H., Naskar, S., and Kemenes, G. (2010). Delayed intrinsic activation of an NMDA-independent CaM-kinase II in a critical time window is necessary for late consolidation of an associative memory. J. Neurosci. 30, 56–63. doi: 10.1523/JNEUROSCI.2577-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watson, S. N., Lee, J. R., Risling, T. E., Hermann, P. M., and Wildering, W. C. (2014). Diminishing glutathione availability and age-associated decline in neuronal excitability. Neurobiol. Aging 35, 1074–1085. doi: 10.1016/j.neurobiolaging.2013.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watson, S. N., Risling, T. E., Hermann, P. M., and Wildering, W. C. (2012a). Failure of delayed nonsynaptic neuronal plasticity underlies age-associated long-term associative memory impairment. BMC Neurosci. 13:103. doi: 10.1186/1471-2202-13-103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watson, S. N., Nelson, M. A., and Wildering, W. C. (2012b). Redox agents modulate neuronal activity and reproduce physiological aspects of neuronal aging. Neurobiol. Aging 33, 149–161. doi: 10.1016/j.neurobiolaging.2010.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watson, S. N., Wright, N., Hermann, P. M., and Wildering, W. C. (2013). Phospholipase A2: the key to reversing long-term memory impairment in a gastropod model of aging. Neurobiol. Aging 34, 610–620. doi: 10.1016/j.neurobiolaging.2012.02.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
West, M. J., Coleman, P. D., Flood, D. G., and Troncoso, J. C. (1994). Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344, 769–772. doi: 10.1016/S0140-6736(94)92338-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wildering, W. C., van der Roest, M., de Vlieger, T. A., and Janse, C. K. (1991). Age-related changes in junctional and non-junctional conductances in two electrically coupled peptidergic neurons of the mollusc Lymnaea stagnalis. Brain Res. 547, 89–98. doi: 10.1016/0006-8993(91)90578-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeoman, M. S., Scutt, G., and Faragher, R. (2012). Insights into CNS ageing from animal models of senescence. Nat. Rev. Neurosci. 13, 435–445. doi: 10.1038/nrn3230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213. doi: 10.1016/j.bbi.2010.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, B. P., Suescun, E. A., and Yang, S. Y. (1992). Effect of age-related lipid peroxidation on membrane fluidity and phospholipase A2: modulation by dietary restriction. Mech. Ageing Dev. 65, 17–33. doi: 10.1016/0047-6374(92)90123-U
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, Y., Carvey, P. M., and Ling, Z. (2006). Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 1090, 35–44. doi: 10.1016/j.brainres.2006.03.063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zimniak, P. (2008). Detoxification reactions: relevance to aging. Ageing Res. Rev. 7, 281–300. doi: 10.1016/j.arr.2008.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zimniak, P. (2011). Relationship of electrophilic stress to aging. Free Radic. Biol. Med. 51, 1087–1105. doi: 10.1016/j.freeradbiomed.2011.05.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: aging, age-associated memory impairment (AMI), lipid peroxidation, phospholipases A2, neuronal excitability, neuronal plasticity, Lymnaea stagnalis
Citation: Hermann PM, Watson SN and Wildering WC (2014) Phospholipase A2 – nexus of aging, oxidative stress, neuronal excitability, and functional decline of the aging nervous system? Insights from a snail model system of neuronal aging and age-associated memory impairment. Front. Genet. 5:419. doi: 10.3389/fgene.2014.00419
Received: 19 April 2014; Accepted: 13 November 2014;
Published online: 04 December 2014.
Edited by:
Shin Murakami, Touro University-California, USAReviewed by:
Deborah Baro, Georgia State University, USASrinivas Ayyadevara, Central Arkansas Veterans Healthcare System, USA
Copyright © 2014 Hermann, Watson and Wildering. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Willem C. Wildering, Department of Biological Sciences, University of Calgary, 2500 University Drive NW, Calgary, AB T2N 1N4, Canada e-mail:d2lsZGVyaW5AdWNhbGdhcnkuY2E=