 Krassimira Botcheva
Krassimira Botcheva- Cell and Molecular Biology Department, Life Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
p53 is the most studied human protein because of its role in maintaining genomic stability. Binding to genomic targets is essential for transcription-dependent p53 tumor suppression, but how p53 selects targets remains unclear. Here, the impact of chromatin context on p53 genome-wide binding and targets selection is discussed. It is proposed that p53 genomic binding serves not only to regulate transcription, but to sense epigenomic changes threatening the genomic integrity. The problem of p53 navigating the human genome is discussed with respect to the degenerate p53 binding motif. This discussion relates to the fundamental problem of DNA binding factors navigating large genomes in search for cognate binding sites.
35 Years p53
p53 guards the genome by mobilizing a complex network of target genes and coordinating the cellular stress responses to prevent propagation of damaged DNA (Vogelstein et al., 2000). DNA binding is important for the p53 tumor suppression; 90% of the cancer-associated p53 mutations reside in the DNA binding domain (Hollstein et al., 1991). It is commonly viewed that in absence of stress cells keep low amount of p53, bound to some genomic targets without affecting transcription, while upon DNA damage activation, the genomic binding is accompanied by transcription regulation. Depending on the cell type and damage, p53 regulates different sets of genes, triggering cell cycle arrest, DNA repair, senescence, or apoptosis, steering the cells toward life or death (Vousden and Lu, 2002). How p53 selects its targets remains one of the foremost questions in the field.
Binding to DNA and transcription regulation are affected by the p53 mutational load (Leroy et al., 2014), extensive posttranslational modifications (Meek and Anderson, 2009), tertiary structure and oligomerization state (Joerger and Fersht, 2010), binding partners and cooperativity (Braithwaite et al., 2006; Brandt et al., 2012), basal transcription machinery (Espinosa, 2008), and genomic binding sites (Horvath et al., 2007; Riley et al., 2008; Wang et al., 2009). Adding to this complexity, the family members p63 and p73 (Belyi et al., 2010) and the isoforms expression patterns in the family (Bourdon et al., 2005; Marcel et al., 2014) have a major impact on the p53 properties, collectively defining functions previously ascribed to a single full length protein. While these components are undoubtedly important for the p53 genomic binding and transcription regulation, they function in the context of chromatin, which plays an active role in transcription and moderates the interactions with DNA (Li et al., 2007).
p53 Network Complexity
The p53 ability to load histone modifiers and chromatin remodelers upon binding to genomic targets is essential for the transcription regulation (Figure 1) and has been well documented (Beckerman and Prives, 2010; Rinn and Huarte, 2011). The opposite, the effect of the chromatin context on p53 targets selectivity is far less understood. Just as transcription factor binding affects the local chromatin, the chromatin context affects the transcription factor binding (Guertin and Lis, 2013). Genomic binding dependent on the chromatin context has been reported for proteins such as Myc (Guccione et al., 2006) and NFκB (Natoli, 2009). The chromatin state matters because it is plastic and dynamic, changing during normal development (Mikkelsen et al., 2007) and in cancer (Baylin and Jones, 2011). The chromatin context introduces a level of complexity in the p53 network, which needs to be considered to understand the p53 functions dependent on DNA binding. Even in a “competent state” for sequence-specific DNA binding (without mutations, activated by posttranslational modifications), p53 may not bind to sites buried in inaccessible chromatin, and bind if they become available. Despite the proposed and observed chromatin impact on p53 binding (Lidor Nili et al., 2010; Millau et al., 2011), there is no good understanding of how p53 binding sites availability is defined and how it affects the genomic targets selection. Analysis of the binding context relies on the binding sites detection.
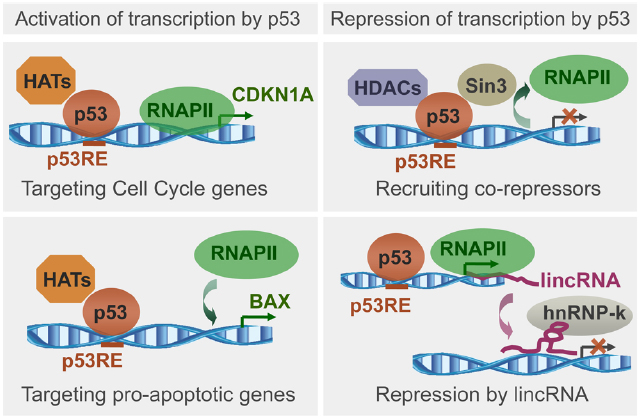
Figure 1. Schematic representation of p53 ability to load histone modifiers upon binding to genomic targets, followed by recruitment or exclusion of RNA polymerase II (RNAPII), and transcription regulation. Illustrated is transcription activation upon loading of histone acetyltransferases (HATs) to promoters of genes regulating cell cycle or apoptosis, and transcription repression upon loading of histone deacetylases (HDACs), or through lincRNAs. Based on recent reviews (Beckerman and Prives, 2010; Rinn and Huarte, 2011).
p53 Genomic Binding Sites
Broadly, two types of experiments provide binding sites information, individual gene studies and genome-wide studies. Evidence for p53-occupied genomic sites came first from individual gene studies. About 200 such sites were reported (Horvath et al., 2007; Riley et al., 2008; Wang et al., 2009). Hereafter these are called reference REs (response elements), since p53 binding at them was demonstrated to evoke transcriptional response. Development of the sequencing technologies enabled genome-wide studies at unprecedented depth, allowing mapping of global binding patterns and chromatin state analysis (Park, 2009; Landt et al., 2012), including large scale p53 binding studies.
Since the discovery of the p53 consensus motif RRRCWWGYYY-N(0–13)-RRRCWWGYYY (El-Deiry et al., 1992; Funk et al., 1992), efforts were focused on designing methods for robust detection of p53 occupied sites. Early work (Tokino et al., 1994), followed by limited scale studies (Cawley et al., 2004; Chen and Sadowski, 2005; Hearnes et al., 2005; Kaneshiro et al., 2007), generated data allowing extrapolation to the whole human genome. The estimated 1,500–3,500 sites occupied genome-wide, were in agreement with predictions by computational models (Veprintsev and Fersht, 2008). The first high-throughput p53 ChIP-PET approach based on Sanger and 454 sequencing (Ng et al., 2006; Wei et al., 2006) paved the road for the next-generation sequencing p53 studies. Most of them used ChIP-seq to map endogenous wild type p53 genomic binding in human cell lines (Botcheva et al., 2011; Smeenk et al., 2011; Nikulenkov et al., 2012; Menendez et al., 2013; Akdemir et al., 2014; McDade et al., 2014; Rashi-Elkeles et al., 2014); some mapped p53 family members, p53 mutants, or p53 variants (Kouwenhoven et al., 2010; Koeppel et al., 2011; Martynova et al., 2012; Schlereth et al., 2013; Wang et al., 2014); others looked at p53 binding in the mouse genome (Li et al., 2012; Kenzelmann Broz et al., 2013), and recently p53 binding data were generated by ChIP-exo (Chang et al., 2014).
Default and Distinct Binding
What these studies revealed about the endogenous wild type p53 binding to the human genome? Genomic binding is not always followed by transcription changes; p53 exhibits both “default” and “distinct” binding. The studies coupled with expression analysis agreed that many p53 occupied sites are not associated with transcription changes (Li et al., 2012; Nikulenkov et al., 2012; Kenzelmann Broz et al., 2013; Menendez et al., 2013; Akdemir et al., 2014; McDade et al., 2014; Rashi-Elkeles et al., 2014), which could be interpreted as absence of transcription regulation at these sites. Since p53 has induced and constitutive functions, and binding may serve to induce transcription, but also to maintain basal transcription levels (Zheltukhin and Chumakov, 2010), p53 occupancy at some of these sites may support the basal expression of the nearby genes. Although not manifested by impressive transcription changes, maintaining global gene expression patterns, ensuring functional homeostasis under stress, in the damaged genome, may represent important p53 function. Since p53 binds to enhancers (Melo et al., 2013), occupancy at some of these sites may exert long distance effect on gene expression.
p53 ability to induce stress and cell type specific responses is well recognized, but whether p53 binds to its targets in such selective manner has been a subject of debate. Recent genome-wide binding studies revealed common and distinct binding patterns. Interestingly, treatment with Nutlin (small molecule activator of p53) leads to more p53 binding, but less effect on transcription, compared to DNA damage (Nikulenkov et al., 2012; Menendez et al., 2013). A “p53 default program” was proposed to explain the observation that the most frequently occupied p53 sites were the same after different treatments (Nikulenkov et al., 2012). Besides the detected p53 at common sites, distinct binding patterns were observed as well (Botcheva et al., 2011; Menendez et al., 2013; Akdemir et al., 2014).
The proposed “p53 default program” can explain the occupancy at canonical targets (such as CDKN1A and MDM2), consistently reported bound under many stress conditions and cell types analyzed. On the other hand, significant fraction of the binding sites displays occupancy dependent on the type of treatment (Menendez et al., 2013; Akdemir et al., 2014), or on the cell context (Botcheva et al., 2011; Botcheva and McCorkle, 2014). If the “default” binding sites are strong, unambiguously detected by many, and reported “functional,” therefore clearly important for the transcription regulation, why obsessing with the “distinct” binding patterns, which despite being shaped by a large fraction of the data, may be composed by sites with less well defined individual contribution to direct transcription regulation? The “default” binding sites, being consistently occupied, may reflect fundamental p53 functions important in any cell context or stress type, while the distinct binding patterns may reveal more about the stress- and cell type- specific p53 functions. Patterns matter in nature. For example, despite the importance of individual populations for biodiversity, gross changes in tropical forests have major impact on earth climate (Lewis, 2006). In order not to lose the forest for the trees, it may be worth studying the global patterns associated with p53 binding, since these may reflect global trends in the binding context, affecting the way p53 network is engaged in response to stress.
The Chromatin Context
What causes the distinct genomic binding patterns of the endogenous wild type p53? Cancer-derived human cell lines have been widely used for p53 research (Millau et al., 2010). When high-resolution p53 binding map was generated for the first time in normal, not immortalized human cell line (Botcheva et al., 2011) and compared to previous studies in cancer cell lines (Wei et al., 2006; Smeenk et al., 2008, 2011), distinct p53 binding patterns were observed. Only in the normal cells p53 was strongly enriched within 2 kb of transcription start sites and at CpG islands (CGIs); distribution typical for the functional reference REs, but not for the sites mapped in the cancer cell lines, where p53 was depleted from CGIs. The chromatin structure at CGIs is important for the regulation of transcription; these CG rich sequences are kept hypomethylated in otherwise methylated genome, and subjected to epigenetic control (Deaton and Bird, 2011). Correlating p53 binding with high-resolution methylome generated in the same cell line (Lister et al., 2009), revealed enrichment of p53 binding sites at hypomethylated DNA. Importantly, that was true not only for sites in CGIs (generally kept hypomethylated), but for sites out of CGIs as well (Botcheva et al., 2011). Notably, when p53 DNA binding in methylation-dependent mode was first considered and examined, no specific p53 affinity was detected for particular methylation state of the binding sites on naked DNA (Petrovich and Veprintsev, 2009). Thus, p53 propensity to bind at hypomethylated regions (not just CGIs) was likely due to the chromatin structure modulated by the DNA hypomethylation, rather than affinity to hypomethylated binding site (Botcheva et al., 2011), although at present that could not be excluded.
Due to the nature of the distinct p53 binding patterns (enrichment at hypomethylated DNA and CGIs in the normal cell line; depletion from CGIs and higher enrichment at repeats in the cancer cell lines), it was proposed that epigenetic changes accompanying cancer progression (local CGIs hypermethylation and global genomic hypomethylation) modulate p53 binding to the genome (Botcheva et al., 2011). Another possibility suggested that at certain sites, the methylated CpGs may undergo tumorogenesis-dependent deamination, to eliminate p53 responsiveness (Freed-Pastor and Prives, 2011). Subsequently, a key study in mouse fibroblasts revealed that in absence of wild type p53, DNA demethylation triggers repeats instability, followed by massive apoptotic response (Leonova et al., 2013), commented in details (Levine and Greenbaum, 2012; Koonin, 2013; Tavana and Gu, 2013). These findings have implications beyond gene promoters and repeats, because cancer is accompanied by global epigenetic alterations involving enhancers and insulator elements as well (Taberlay et al., 2014), and binding at enhancers is important for the p53-dependent transcription regulation.
The datasets analyzed by Botcheva et al. (2011) differed not only by cell context (normal and cancer-derived cell lines), but by treatment and/or experimental approach, potentially contributing to the observed p53 binding differences. Recently, the distinct p53 binding patterns were confirmed under same treatment, experimental conditions and ChIP-seq approach, evidence for cell context-dependent p53 genomic binding (Botcheva and McCorkle, 2014). Moreover, the analysis was extended to examine the differentially bound types of repeats and p53 was found enriched at LINE (long interspersed nuclear elements) in the cancer cell line HCT116, compared to the normal IMR90 (Botcheva and McCorkle, 2014). Epigenetic dysregulation at repeats is a major cancer landmark, and hypomethylated LINE repeats (normally methylated) are associated with bad colon cancer prognosis (Ogino et al., 2008); thus, p53 enrichment at LINE repeats in the colorectal cell line HCT116, might be due to cancer-associated LINE hypomethylation (Botcheva and McCorkle, 2014). The distinct p53 binding patterns in HCT116 and IMR90 likely reflect differences in the epigenetic landscapes in these cell lines, due to cancer-associated changes (accumulated in HCT116), overlaid on tissue-specific differences (HCT116 has epithelial, while IMR90 has mesenchymal origin; Botcheva and McCorkle, 2014). In the future, it would be very interesting to investigate the involvement of the p53 transcriptional network in the process of cancer-associated EMT (epithelial mesenchymal transition).
Sensing the Epigenome
The epigenetic impact on p53 genome-wide binding (Botcheva et al., 2011), and p53-dependent transcription regulation (Leonova et al., 2013) has far reaching implications. The p53 genomic targets selection is important for tumor suppression, yet the mechanisms are unclear. These studies suggest p53 genomic binding dependent on DNA methylation (Figure 2). While it is known that transcription factor binding can be affected by the methylation status of the CpGs in their binding sites (Chen et al., 2011), the probable mechanisms remain to be defined. Interestingly, although the OCT4 motif does not contain CpGs, in human embryonic stem cells this protein is excluded from target sites residing in DNA methylated locations (You et al., 2011; Jones, 2012).
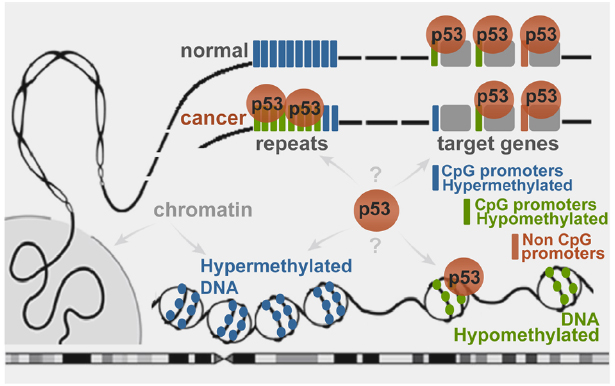
Figure 2. Schematic representation of wild type p53 binding to genomic targets in a DNA methylation-dependent manner. In normal cells the stress-induced p53 binds to sites residing in hypomethylated regions. Cancer-associated epigenetic changes may lead to local p53 depletion from hypermethylated target promoters and enrichment at globally hypomethylated regions, such as repeats (Botcheva et al., 2011; Botcheva and McCorkle, 2014).
p53 has been long known as a keeper of the genomic integrity, and sensing the epigenome might be important part of it. While searching for binding sites, p53 may be “screening” the genome for DNA hypomethylation at regions “normally” hypermethylated (residing in inaccessible chromatin context), which would become available for p53 binding upon hypomethylation. Global DNA hypomethylation on a large scale could expose numerous sites, causing global redistribution of the p53 binding and readjusting its transcriptional program to maintain genomic stability.
While “guarding the genome” does not involve direct recognition of genetic lesions by p53, sensing the epigenome may imply direct recognition of the regions of epigenetic changes. Moreover, the extensive network of p53 binding sites at repeats (Wang et al., 2007; Harris et al., 2009; Zemojtel et al., 2009; Cui et al., 2011; Botcheva and McCorkle, 2014) could serve as a system of sensors for detection of epigenetic perturbations, which could destabilize the repeats and the whole genome. Therefore, the reason for the existence of p53 binding sites at repeats may be monitoring for epigenetic aberrations. Upon comparing p53 binding in cancer cell lines under different stress, common global trends may emerge (besides the “default program”) that are dependent on the epigenetic changes, shared at least to some extent by these cell lines. Considering the magnitude of such epigenetic changes, the common trends could dominate the p53 binding landscape, obscuring the detection of more subtle stress-specific patterns and making the binding look “non-selective.”
p53 has been regularly reported bound to some of its genomic targets in absence of stress, often interpreted as allowing for fast transcriptional response upon stress induction. Considering the p53 binding at hypomethylated DNA, it is possible that p53 occupies constitutively some of its key targets to protect them from hypermethylation, which may make them inaccessible for p53 binding, abrogating its tumor suppression program.
Mammalian cells acquire epigenetic hallmarks of human cancer during immortalization (Tommasi et al., 2013). If p53 indeed is sensing the epigenome at the level of genomic binding, one may expect p53 binding patterns in immortalized cells to be more similar to those in cancer cells, rather than to those in normal cells. In support of that, ChIP-chip study examining p53 binding in different cell contexts demonstrated that immortalized fibroblasts behaved as cancer cell lines, unlike the normal cells (Shaked et al., 2008). Studying the changing p53 binding patterns during immortalization could reveal information about the epigenetic modulation of the p53 network functions early in tumorogenesis.
Do these findings have clinical implications? In certain contexts, wild type p53 may serve “oncogenic” functions and may not need further activation. Interestingly, there are colorectal cancer subgroups with CGI methylator phenotype (CIMP-high, CIMP-low) less likely to accumulate p53 mutations (Hinoue et al., 2012). It may be because they need wild type p53 to protect them from the deleterious effects of aberrant DNA methylation. In such cases, use of demethylating agents could be explored. One such drug, Decitabine, has been approved by FDA since 2006 for treating myelodysplastic syndromes, and has shown promising results on epithelial tumor cells as well (Tsai et al., 2012).
Crowd Control Navigation
The high-confidence p53 binding sites annotated by a given genome-wide study represent only a small subset of the total sites detected; the vast majority of the sites are not associated with particular function. Considering the degenerate p53 consensus binding motif, computational models estimate hundreds of thousands sites in the human genome (Hoh et al., 2002; Veprintsev and Fersht, 2008). Why so many, if only few are functional? Degenerate binding motifs are often found for sequence specific DNA binding proteins in higher eukaryotes, unlike in prokaryotes (Kadonaga, 2004). Interestingly, despite the smaller genome, prokaryotic transcription factors have more specific binding motifs; despite the larger genomes, eukaryotic transcription factors have degenerate, lower specificity DNA binding motifs. Applying information theory to the genomic sites recognition by DNA binding proteins, minimum information content is required to specify unique position in the genome. Calculations of the average information content of prokaryotic and eukaryotic motifs (based on the motif length and the base frequency at particular positions) demonstrated that in prokaryotes single binding site is sufficient to address unique location in the genome, but not in eukaryotes (Wunderlich and Mirny, 2009). The average information content of a prokaryotic motif I ≈ 23 bits is slightly above the required Imin ≈ 22 bits to specify unique position in a prokaryotic genome (106–107 bp); the average information content of a multicellular eukaryotic motif I ≈ 12.1 bits is far below the minimum information required Imin ≈ 30 bits to specify unique position in eukaryotic genome of ∼109 bp (Wunderlich and Mirny, 2009).
One may expect that eukaryotic DNA binding proteins, locating their sites in larger genomes, would have motifs with higher information content than prokaryotes. Instead it is the opposite. Because of motifs degeneracy, numerous DNA binding sites, besides the “functional” ones, are present for a given DNA binding protein (p53 including). Thus, “functionality” is specified by additional means, such as clustering of binding sites, combinatorial and cooperative binding (Kadonaga, 2004), and three-dimensional organization of the chromatin (Lieberman-Aiden et al., 2009; Edelman and Fraser, 2012; Amouyal, 2014). The point is, why on the first place, eukaryotic DNA binding proteins evolved to have degenerate motifs, meaning promiscuous binding to the genome, and requiring additional means to specify functionality? Why not evolving higher content information motifs which would specify unambiguously unique positions in the genome? It may be because the unique positions have to be not only specified, but found in the large genome.
The mode employed by eukaryotic DNA binding proteins to find their genomic sites may make sense from navigation point of view. According to current models for DNA binding sites search (Halford and Marko, 2004), and by p53 (McKinney et al., 2004; Tafvizi et al., 2011), a protein spends time “on” and “off” DNA, and “moving along DNA” is important (here, terms as “sliding” and “diffusing” are omitted since the point is not about how a protein propels on DNA, but how it navigates it). There are many DNA binding proteins presumably searching for their “functional” sites on DNA (Zabet and Adryan, 2013). It may look contra intuitive but the many degenerate binding sites, by transiently binding their cognate proteins, may actually serve the role to navigate and ensure “smooth traffic” on DNA, so that the “functional” sites could be reached efficiently (in everyday life, traffic lights and stop signs are placed in the most crowded traffic spots). Interestingly, “crowd control” theories have been long applied to animal kingdom and group decision making has been hypothesized to be beneficial in complex environments (Berdahl et al., 2013). It is possible that the DNA binding proteins in the large eukaryotic genomes rely on “crowd control navigation” provided by the multiple binding sites, in order to reach their “functional” binding sites and regulate transcription.
Conclusion
The p53 network is perfectly fit to be interrogated by genome-wide approaches. That would allow otherwise hidden global patterns to be revealed and studied, to help interpreting the p53-dependent tumor suppression program in the context of dynamic chromatin and to dissect the interplay between genetic and epigenetic changes associated with cancer. The discoveries from the past 35 years expanded greatly the p53 knowledge and underlined the importance of addressing the p53 network complexity for the pathway’s full potential to be unlocked and applied in the clinic, because cancer progression and tumor suppression are two faces of the same coin and the p53 mark is undeniably stamped on both.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Krassimira Botcheva thanks Mina Bissell (Lawrence Berkeley National Laboratory) for the support, and Sean McCorkle (Brookhaven National Laboratory) and Ben Brown (Lawrence Berkeley National Laboratory) for the discussions.
References
Akdemir, K. C., Jain, A. K., Allton, K., Aronow, B., Xu, X., Cooney, A. J., et al. (2014). Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 42, 205–223. doi: 10.1093/nar/gkt866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amouyal, M. (2014). From adjacent activation in Escherichia coli and DNA cyclization to eukaryotic enhancers: the elements of a puzzle. Front. Genet. 5:371. doi: 10.3389/fgene.2014.00371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baylin, S. B., and Jones, P. A. (2011). A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer 11, 726–734. doi: 10.1038/nrc3130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beckerman, R., and Prives, C. (2010). Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2:a000935. doi: 10.1101/cshperspect.a000935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belyi, V. A., Ak, P., Markert, E., Wang, H., Hu, W., Puzio-Kuter, A., et al. (2010). The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2:a001198. doi: 10.1101/cshperspect.a001198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berdahl, A., Torney, C. J., Ioannou, C. C., Faria, J. J., and Couzin, I. D. (2013). Emergent sensing of complex environments by mobile animal groups. Science 339, 574–576. doi: 10.1126/science.1225883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Botcheva, K., and McCorkle, S. R. (2014). Cell context dependent p53 genome-wide binding patterns and enrichment at repeats. PLoS ONE 9:e113492. doi: 10.1371/journal.pone.0113492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Botcheva, K., McCorkle, S. R., McCombie, W. R., Dunn, J. J., and Anderson, C. W. (2011). Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle 10, 4237–4249. doi: 10.4161/cc.10.24.18383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bourdon, J. C., Fernandes, K., Murray-Zmijewski, F., Liu, G., Diot, A., Xirodimas, D. P., et al. (2005). p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137. doi: 10.1101/gad.1339905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braithwaite, A. W., Del Sal, G., and Lu, X. (2006). Some p53-binding proteins that can function as arbiters of life and death. Cell Death Differ. 13, 984–993. doi: 10.1038/sj.cdd.4401924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandt, T., Townsley, F. M., Teufel, D. P., Freund, S. M., and Veprintsev, D. B. (2012). Molecular basis for modulation of the p53 target selectivity by KLF4. PLoS ONE 7:e48252. doi: 10.1371/journal.pone.0048252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cawley, S., Bekiranov, S., Ng, H. H., Kapranov, P., Sekinger, E. A., Kampa, D., et al. (2004). Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116, 499–509. doi: 10.1016/S0092-8674(04)00127-8
Chang, G. S., Chen, X. A., Park, B., Rhee, H. S., Li, P., Han, K. H., et al. (2014). A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep. 8, 514–527. doi: 10.1016/j.celrep.2014.06.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J., and Sadowski, I. (2005). Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc. Natl. Acad. Sci. U.S.A. 102, 4813–4818. doi: 10.1073/pnas.0407069102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, P. Y., Feng, S., Joo, J. W., Jacobsen, S. E., and Pellegrini, M. (2011). A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 12:R62. doi: 10.1186/gb-2011-12-7-r62
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cui, F., Sirotin, M. V., and Zhurkin, V. B. (2011). Impact of Alu repeats on the evolution of human p53 binding sites. Biol. Direct 6:2. doi: 10.1186/1745-6150-6-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deaton, A. M., and Bird, A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. doi: 10.1101/gad.2037511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edelman, L. B., and Fraser, P. (2012). Transcription factories: genetic programming in three dimensions. Curr. Opin. Genet. Dev. 22, 110–114. doi: 10.1016/j.gde.2012.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
El-Deiry, W. S., Kern, S. E., Pietenpol, J. A., Kinzler, K. W., and Vogelstein, B. (1992). Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49. doi: 10.1038/ng0492-45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Espinosa, J. M. (2008). Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene 27, 4013–4023. doi: 10.1038/onc.2008.37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freed-Pastor, W. A., and Prives, C. (2011). Dissimilar DNA binding by p53 in normal and tumor-derived cells. Cell Cycle 10:4207. doi: 10.4161/cc.10.24.18723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funk, W. D., Pak, D. T., Karas, R. H., Wright, W. E., and Shay, J. W. (1992). A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 12, 2866–2871.
Guccione, E., Martinato, F., Finocchiaro, G., Luzi, L., Tizzoni, L., Dall’ Olio, V., et al. (2006). Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 8, 764–770. doi: 10.1038/ncb1434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guertin, M. J., and Lis, J. T. (2013). Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr. Opin. Genet. Dev. 23, 116–123. doi: 10.1016/j.gde.2012.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Halford, S. E., and Marko, J. F. (2004). How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 32, 3040–3052. doi: 10.1093/nar/gkh624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harris, C. R., Dewan, A., Zupnick, A., Normart, R., Gabriel, A., Prives, C., et al. (2009). p53 responsive elements in human retrotransposons. Oncogene 28, 3857–3865. doi: 10.1038/onc.2009.246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hearnes, J. M., Mays, D. J., Schavolt, K. L., Tang, L., Jiang, X., and Pietenpol, J. A. (2005). Chromatin immunoprecipitation-based screen to identify functional genomic binding sites for sequence-specific transactivators. Mol. Cell. Biol. 25, 10148–10158. doi: 10.1128/MCB.25.22.10148-10158.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hinoue, T., Weisenberger, D. J., Lange, C. P., Shen, H., Byun, H. M., Van Den Berg, D., et al. (2012). Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 22, 271–282. doi: 10.1101/gr.117523.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoh, J., Jin, S., Parrado, T., Edington, J., Levine, A. J., and Ott, J. (2002). The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl. Acad. Sci. U.S.A. 99, 8467–8472. doi: 10.1073/pnas.132268899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hollstein, M., Sidransky, D., Vogelstein, B., and Harris, C. C. (1991). p53 mutations in human cancers. Science 253, 49–53. doi: 10.1126/science.1905840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horvath, M. M., Wang, X., Resnick, M. A., and Bell, D. A. (2007). Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS Genet. 3:e127. doi: 10.1371/journal.pgen.0030127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joerger, A. C., and Fersht, A. R. (2010). The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2:a000919. doi: 10.1101/cshperspect.a000919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi: 10.1038/nrg3230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kadonaga, J. T. (2004). Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 116, 247–257. doi: 10.1016/S0092-8674(03)01078-X
Kaneshiro, K., Tsutsumi, S., Tsuji, S., Shirahige, K., and Aburatani, H. (2007). An integrated map of p53-binding sites and histone modification in the human ENCODE regions. Genomics 89, 178–188. doi: 10.1016/j.ygeno.2006.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kenzelmann Broz, D., Spano Mello, S., Bieging, K. T., Jiang, D., Dusek, R. L., Brady, C. A., et al. (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031. doi: 10.1101/gad.212282.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koeppel, M., van Heeringen, S. J., Kramer, D., Smeenk, L., Janssen-Megens, E., Hartmann, M., et al. (2011). Crosstalk between c-Jun and TAp73alpha/beta contributes to the apoptosis-survival balance. Nucleic Acids Res. 39, 6069–6085. doi: 10.1093/nar/gkr028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koonin, E. V. (2013). Elaborate security TRAINing to fight against expression of genomic junk. Cell Cycle 12, 553–554. doi: 10.4161/cc.23717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kouwenhoven, E. N., van Heeringen, S. J., Tena, J. J., Oti, M., Dutilh, B. E., Alonso, M. E., et al. (2010). Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet. 6:e1001065. doi: 10.1371/journal.pgen.1001065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landt, S. G., Marinov, G. K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831. doi: 10.1101/gr.136184.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leonova, K. I., Brodsky, L., Lipchick, B., Pal, M., Novototskaya, L., Chenchik, A. A., et al. (2013). p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc. Natl. Acad. Sci. U.S.A. 110, E89–E98. doi: 10.1073/pnas.1216922110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leroy, B., Anderson, M., and Soussi, T. (2014). TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum. Mutat. 35, 672–688. doi: 10.1002/humu.22552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levine, A. J., and Greenbaum, B. (2012). The maintenance of epigenetic states by p53: the guardian of the epigenome. Oncotarget 3, 1503–1504.
Lewis, S. L. (2006). Tropical forests and the changing earth system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 195–210. doi: 10.1098/rstb.2005.1711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, B., Carey, M., and Workman, J. L. (2007). The role of chromatin during transcription. Cell 128, 707–719. doi: 10.1016/j.cell.2007.01.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M., He, Y., Dubois, W., Wu, X., Shi, J., and Huang, J. (2012). Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell 46, 30–42. doi: 10.1016/j.molcel.2012.01.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lidor Nili, E., Field, Y., Lubling, Y., Widom, J., Oren, M., and Segal, E. (2010). p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res. 20, 1361–1368. doi: 10.1101/gr.103945.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. doi: 10.1126/science.1181369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcel, V., Fernandes, K., Terrier, O., Lane, D. P., and Bourdon, J. C. (2014). Modulation of p53beta and p53gamma expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 21, 1377–1387. doi: 10.1038/cdd.2014.73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martynova, E., Pozzi, S., Basile, V., Dolfini, D., Zambelli, F., Imbriano, C., et al. (2012). Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 3, 132–143.
McDade, S. S., Patel, D., Moran, M., Campbell, J., Fenwick, K., Kozarewa, I., et al. (2014). Genome-wide characterization reveals complex interplay between TP53 and TP63 in response to genotoxic stress. Nucleic Acids Res. 42, 6270–6285. doi: 10.1093/nar/gku299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKinney, K., Mattia, M., Gottifredi, V., and Prives, C. (2004). p53 linear diffusion along DNA requires its C terminus. Mol. Cell 16, 413–424. doi: 10.1016/j.molcel.2004.09.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meek, D. W., and Anderson, C. W. (2009). Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1:a000950. doi: 10.1101/cshperspect.a000950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Melo, C. A., Drost, J., Wijchers, P. J., van de Werken, H., de Wit, E., Oude Vrielink, J. A., et al. (2013). eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 49, 524–535. doi: 10.1016/j.molcel.2012.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Menendez, D., Nguyen, T. A., Freudenberg, J. M., Mathew, V. J., Anderson, C. W., Jothi, R., et al. (2013). Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 41, 7286–7301. doi: 10.1093/nar/gkt504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. doi: 10.1038/nature06008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Millau, J. F., Bandele, O. J., Perron, J., Bastien, N., Bouchard, E. F., Gaudreau, L., et al. (2011). Formation of stress-specific p53 binding patterns is influenced by chromatin but not by modulation of p53 binding affinity to response elements. Nucleic Acids Res. 39, 3053–3063. doi: 10.1093/nar/gkq1209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Millau, J. F., Mai, S., Bastien, N., and Drouin, R. (2010). p53 functions and cell lines: have we learned the lessons from the past? Bioessays 32, 392–400. doi: 10.1002/bies.200900160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Natoli, G. (2009). Control of NF-kappaB-dependent transcriptional responses by chromatin organization. Cold Spring Harb. Perspect. Biol. 1:a000224. doi: 10.1101/cshperspect.a000224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ng, P., Tan, J. J., Ooi, H. S., Lee, Y. L., Chiu, K. P., Fullwood, M. J., et al. (2006). Multiplex sequencing of paired-end ditags (MS-PET): a strategy for the ultra-high-throughput analysis of transcriptomes and genomes. Nucleic Acids Res. 34:e84. doi: 10.1093/nar/gkl444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikulenkov, F., Spinnler, C., Li, H., Tonelli, C., Shi, Y., Turunen, M., et al. (2012). Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 19, 1992–2002. doi: 10.1038/cdd.2012.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogino, S., Kawasaki, T., Nosho, K., Ohnishi, M., Suemoto, Y., Kirkner, G. J., et al. (2008). LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int. J. Cancer 122, 2767–2773. doi: 10.1002/ijc.23470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680. doi: 10.1038/nrg2641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petrovich, M., and Veprintsev, D. B. (2009). Effects of CpG methylation on recognition of DNA by the tumour suppressor p53. J. Mol. Biol. 386, 72–80. doi: 10.1016/j.jmb.2008.11.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rashi-Elkeles, S., Warnatz, H. J., Elkon, R., Kupershtein, A., Chobod, Y., Paz, A., et al. (2014). Parallel profiling of the transcriptome, cistrome, and epigenome in the cellular response to ionizing radiation. Sci. Signal. 7:rs3. doi: 10.1126/scisignal.2005032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Riley, T., Sontag, E., Chen, P., and Levine, A. (2008). Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412. doi: 10.1038/nrm2395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rinn, J. L., and Huarte, M. (2011). To repress or not to repress: this is the guardian’s question. Trends Cell Biol. 21, 344–353. doi: 10.1016/j.tcb.2011.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlereth, K., Heyl, C., Krampitz, A. M., Mernberger, M., Finkernagel, F., Scharfe, M., et al. (2013). Characterization of the p53 cistrome–DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 9:e1003726. doi: 10.1371/journal.pgen.1003726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shaked, H., Shiff, I., Kott-Gutkowski, M., Siegfried, Z., Haupt, Y., and Simon, I. (2008). Chromatin immunoprecipitation-on-chip reveals stress-dependent p53 occupancy in primary normal cells but not in established cell lines. Cancer Res. 68, 9671–9677. doi: 10.1158/0008-5472.CAN-08-0865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smeenk, L., van Heeringen, S. J., Koeppel, M., Gilbert, B., Janssen-Megens, E., Stunnenberg, H. G., et al. (2011). Role of p53 serine 46 in p53 target gene regulation. PLoS ONE 6:e17574. doi: 10.1371/journal.pone.0017574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smeenk, L., van Heeringen, S. J., Koeppel, M., van Driel, M. A., Bartels, S. J., Akkers, R. C., et al. (2008). Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 36, 3639–3654. doi: 10.1093/nar/gkn232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taberlay, P. C., Statham, A. L., Kelly, T. K., Clark, S. J., and Jones, P. A. (2014). Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res. 24, 1421–1432. doi: 10.1101/gr.163485.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tafvizi, A., Huang, F., Fersht, A. R., Mirny, L. A., and van Oijen, A. M. (2011). A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. U.S.A. 108, 563–568. doi: 10.1073/pnas.1016020107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tavana, O., and Gu, W. (2013). p53 and DNA methylation suppress the TRAIN to cell death. Cell Cycle 12, 9–10. doi: 10.4161/cc.23324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tokino, T., Thiagalingam, S., el-Deiry, W. S., Waldman, T., Kinzler, K. W., and Vogelstein, B. (1994). p53 tagged sites from human genomic DNA. Hum. Mol. Genet. 3, 1537–1542. doi: 10.1093/hmg/3.9.1537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tommasi, S., Zheng, A., Weninger, A., Bates, S. E., Li, X. A., Wu, X., et al. (2013). Mammalian cells acquire epigenetic hallmarks of human cancer during immortalization. Nucleic Acids Res. 41, 182–195. doi: 10.1093/nar/gks1051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, H. C., Li, H., Van Neste, L., Cai, Y., Robert, C., Rassool, F. V., et al. (2012). Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 21, 430–446. doi: 10.1016/j.ccr.2011.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veprintsev, D. B., and Fersht, A. R. (2008). Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 36, 1589–1598. doi: 10.1093/nar/gkm1040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vogelstein, B., Lane, D., and Levine, A. J. (2000). Surfing the p53 network. Nature 408, 307–310. doi: 10.1038/35042675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vousden, K. H., and Lu, X. (2002). Live or let die: the cell’s response to p53. Nat. Rev. Cancer 2, 594–604. doi: 10.1038/nrc864
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, B., Niu, D., Lam, T. H., Xiao, Z., and Ren, E. C. (2014). Mapping the p53 transcriptome universe using p53 natural polymorphs. Cell Death Differ. 21, 521–532. doi: 10.1038/cdd.2013.132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, B., Xiao, Z., and Ren, E. C. (2009). Redefining the p53 response element. Proc. Natl. Acad. Sci. U.S.A. 106, 14373–14378. doi: 10.1073/pnas.0903284106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T., Zeng, J., Lowe, C. B., Sellers, R. G., Salama, S. R., Yang, M., et al. (2007). Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. U.S.A. 104, 18613–18618. doi: 10.1073/pnas.0703637104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, C.-L., Wu, Q., Vega, V. B., Chiu, K. P., Ng, P., Zhang, T., et al. (2006). A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219. doi: 10.1016/j.cell.2005.10.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wunderlich, Z., and Mirny, L. A. (2009). Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 25, 434–440. doi: 10.1016/j.tig.2009.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
You, J. S., Kelly, T. K., De Carvalho, D. D., Taberlay, P. C., Liang, G., and Jones, P. A. (2011). OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc. Natl. Acad. Sci. U.S.A. 108, 14497–14502. doi: 10.1073/pnas.1111309108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zabet, N. R., and Adryan, B. (2013). The effects of transcription factor competition on gene regulation. Front. Genet. 4:197. doi: 10.3389/fgene.2013.00197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zemojtel, T., Kielbasa, S. M., Arndt, P. F., Chung, H. R., and Vingron, M. (2009). Methylation and deamination of CpGs generate p53-binding sites on a genomic scale. Trends Genet. 25, 63–66. doi: 10.1016/j.tig.2008.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: p53 tumor suppressor network, genome-wide binding, epigenetic regulation, chromatin, CpG islands, repeats, navigation
Citation: Botcheva K (2014) p53 binding to human genome: crowd control navigation in chromatin context. Front. Genet. 5:447. doi: 10.3389/fgene.2014.00447
Received: 28 October 2014; Accepted: 05 December 2014;
Published online: 22 December 2014.
Edited by:
Michèle Amouyal, Centre National de la Recherche Scientifique, FranceReviewed by:
Dmitry Veprintsev, Paul Scherrer Institute, SwitzerlandJean-Christophe Bourdon, University of Dundee, UK
Copyright © 2014 Botcheva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krassimira Botcheva, Cell and Molecular Biology Department, Life Sciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, Mail Stop 977, Berkeley, CA 94720, USA e-mail:a2FsZXhpZXZAbGJsLmdvdg==