 Samuel E. Vaughn1,2
Samuel E. Vaughn1,2 Corinne Foley3
Corinne Foley3 Xiaoming Lu2
Xiaoming Lu2 Zubin H. Patel1,2
Zubin H. Patel1,2 Erin E. Zoller2
Erin E. Zoller2 Albert F. Magnusen2Adrienne H. Williams4Julie T. Ziegler4Mary E. Comeau4Miranda C. Marion4Stuart B. Glenn5Adam Adler5Nan Shen1,6
Albert F. Magnusen2Adrienne H. Williams4Julie T. Ziegler4Mary E. Comeau4Miranda C. Marion4Stuart B. Glenn5Adam Adler5Nan Shen1,6 Swapan Nath5
Swapan Nath5 Anne M. Stevens7,8Barry I. Freedman9Betty P. Tsao10Chaim O. Jacob11Diane L. Kamen12
Anne M. Stevens7,8Barry I. Freedman9Betty P. Tsao10Chaim O. Jacob11Diane L. Kamen12 Elizabeth E. Brown13,14Gary S. Gilkeson12Graciela S. Alarcón14John D. Reveille15
Elizabeth E. Brown13,14Gary S. Gilkeson12Graciela S. Alarcón14John D. Reveille15 Juan-Manuel Anaya16
Juan-Manuel Anaya16 Judith A. James5,17Kathy L. Moser5Lindsey A. Criswell18Luis M. Vilá19Marta E. Alarcón-Riquelme5,20Michelle Petri21R. Hal Scofield5,17,22Robert P. Kimberly14Rosalind Ramsey-Goldman23Young Binjoo24Jeongim Choi24Sang-Cheol Bae24Susan A. Boackle25Timothy J. Vyse26Joel M. Guthridge5
Judith A. James5,17Kathy L. Moser5Lindsey A. Criswell18Luis M. Vilá19Marta E. Alarcón-Riquelme5,20Michelle Petri21R. Hal Scofield5,17,22Robert P. Kimberly14Rosalind Ramsey-Goldman23Young Binjoo24Jeongim Choi24Sang-Cheol Bae24Susan A. Boackle25Timothy J. Vyse26Joel M. Guthridge5 Bahram Namjou2Patrick M. Gaffney5Carl D. Langefeld4Kenneth M. Kaufman2,27
Bahram Namjou2Patrick M. Gaffney5Carl D. Langefeld4Kenneth M. Kaufman2,27 Jennifer A. Kelly5
Jennifer A. Kelly5 Isaac T. W. Harley1,2
Isaac T. W. Harley1,2 John B. Harley2,27
John B. Harley2,27 Leah C. Kottyan2,27*
Leah C. Kottyan2,27*- 1Immunology Graduate Program and Medical Scientist Training Program, University of Cincinnati College of Medicine, Cincinnati, OH, USA
- 2Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA
- 3Spelman College, Atlanta, GA, USA
- 4Center for Public Health Genomics and the Department of Biostatistical Sciences, Wake Forest School of Medicine, Winston-Salem, NC, USA
- 5Arthritis and Clinical Immunology Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, USA
- 6Joint Molecular Rheumatology Laboratory of the Institute of Health Sciences and Shanghai Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Institutes for Biological Sciences, and Chinese Academy of Sciences, Shanghai, China
- 7Center for Immunity and Immunotherapies, Seattle Children's Research Institute, Seattle, WA, USA
- 8Division of Rheumatology, Department of Pediatrics, University of Washington, Seattle, WA, USA
- 9Department of Internal Medicine, Section on Nephrology, Wake Forest School of Medicine, Winston-Salem, NC, USA
- 10Division of Rheumatology, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA
- 11Department of Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
- 12Division of Rheumatology, Medical University of South Carolina, Charleston, SC, USA
- 13Department of Epidemiology, University of Alabama at Birmingham, Birmingham, AL, USA
- 14Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA
- 15Rheumatology and Clinical Immunogenetics, University of Texas Health Science Center at Houston, Houston, TX, USA
- 16Center for Autoimmune Disease Research, Universidad del Rosario, Bogota, Colombia
- 17Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
- 18Rosalind Russell/Ephraim P Engleman Rheumatology Research Research Center, Department of Medicine, University of California, San Francisco, San Francisco, CA, USA
- 19Division of Rheumatology, Department of Medicine, University of Puerto Rico Medical Sciences Campus, San Juan, PR, USA
- 20Center for Genomics and Oncological Research, Pfizer-University of Granada-Junta de Andalucia, Granada, Spain
- 21Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 22United States Department of Veterans Affairs Medical Center, Oklahoma City, OK, USA
- 23Division of Rheumatology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
- 24Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul, Korea
- 25Division of Rheumatology, University of Colorado School of Medicine, Aurora, CO, USA
- 26Divisions of Genetics and Molecular Medicine and Immunology, King's College London, London, UK
- 27United States Department of Veterans Affairs Medical Center, Cincinnati, OH, USA
Genome wide association studies have identified variants in PXK that confer risk for humoral autoimmune diseases, including systemic lupus erythematosus (SLE or lupus), rheumatoid arthritis and more recently systemic sclerosis. While PXK is involved in trafficking of epidermal growth factor Receptor (EGFR) in COS-7 cells, mechanisms linking PXK to lupus pathophysiology have remained undefined. In an effort to uncover the mechanism at this locus that increases lupus-risk, we undertook a fine-mapping analysis in a large multi-ancestral study of lupus patients and controls. We define a large (257kb) common haplotype marking a single causal variant that confers lupus risk detected only in European ancestral populations and spans the promoter through the 3′ UTR of PXK. The strongest association was found at rs6445972 with P < 4.62 × 10−10, OR 0.81 (0.75–0.86). Using stepwise logistic regression analysis, we demonstrate that one signal drives the genetic association in the region. Bayesian analysis confirms our results, identifying a 95% credible set consisting of 172 variants spanning 202 kb. Functionally, we found that PXK operates on the B-cell antigen receptor (BCR); we confirmed that PXK influenced the rate of BCR internalization. Furthermore, we demonstrate that individuals carrying the risk haplotype exhibited a decreased rate of BCR internalization, a process known to impact B cell survival and cell fate. Taken together, these data define a new candidate mechanism for the genetic association of variants around PXK with lupus risk and highlight the regulation of intracellular trafficking as a genetically regulated pathway mediating human autoimmunity.
Introduction
Systemic Lupus Erythematosus is the prototypical systemic autoimmune disease. Consequently, considerable effort has been devoted to understanding the genetic component of lupus risk. Now an understanding of causal mechanisms is approaching 10% of the 50 replicated genomic risk-loci that reach genome-wide significance (Vaughn et al., 2012).
Using a genome-wide association study (GWAS) design, we first identified association of a single nucleotide polymorphism (SNP) in the PXK locus with the occurrence of lupus in women of European descent (Harley et al., 2008). The PXK locus association with lupus-risk has since been replicated in several studies (Gateva et al., 2009; Suarez-Gestal et al., 2009). In these studies, the same variant identified in the initial GWAS was assessed in independent cohorts and the lupus-risk association was replicated (Gateva et al., 2009; Suarez-Gestal et al., 2009). These studies did not include any biological or functional genomic follow-up of the replicated association of this variant (Gateva et al., 2009; Suarez-Gestal et al., 2009). A fourth, recent study included a moderately-powered fine mapping analysis of a European cohort and used expression quantitative trait locus analysis of nearby genes to argue for a role of ABHD6 in the increased lupus risk. For the current analysis, we initiated a well-powered fine-mapping study aimed at identifying the likely causal variants and defining the biological mechanisms of lupus risk at this locus.
PXK is part of the sorting nexin (SNX) family of proteins, which are important for receptor internalization, organelle trafficking including endosomal trafficking, and other membrane-centric sorting functions. This is accomplished primarily through the PX-domain mediated binding of PI3P (Xu et al., 2001; Seet and Hong, 2006). PXK was first identified and cloned by two independent groups in 2005 (Mao et al., 2005; Zou et al., 2005). Initial studies established that PXK is detectable in most tissues with a primarily cytoplasmic distribution. A more recent study in COS-7 cells demonstrated that PXK co-localized with the EGFR. Furthermore, PXK facilitated EGFR internalization following ligand binding, which was found to be PX-domain dependent (Takeuchi et al., 2010; Pedersen et al., 2012). PXK is widely expressed in the brain and blood (Zou et al., 2005), especially in B cells (Figure S1).
In this study, we identify a 257 kb region on chromosome 3, including all of PXK and a large region upstream of the gene that contains the lupus association signal. These results are confirmed via Bayesian analysis by which we identify a credible set consisting of 172 variants that explain 95% of the posterior probability in the region (Wellcome Trust Case Control Consortium et al., 2012). Through step-wise logistic regression analysis, we demonstrate that this region contains a single genetic effect.
Many studies in mice and humans highlight a central role of B cells in the etiology of lupus. Using the transcriptome of various B cell subsets, Hu et al. demonstrated that B cells and especially transitional B cells are enriched for transcripts near genetic variants associated with increased lupus risk (Hu et al., 2011). Autoreactive B-cells play a critical role in the development of autoantibodies that lead to immune complex deposition and lupus-associated tissue damage (Grimaldi et al., 2005). In murine studies, mice without B cells are largely protected from lupus-like disease (Shlomchik et al., 1994). Furthermore, murine studies established a clear role for B cells in the dysregulated cytokine production and T cell activation associated with lupus-like autoimmunity (Chan and Shlomchik, 1998; Chan et al., 1999).
B cell signaling through the B cell receptor (BCR) plays a critical role in the development of autoimmunity in lupus (Chaturvedi et al., 2011; Heesters et al., 2014). For example, BCR internalization facilitates Toll like receptor ligand internalization and signaling. Furthermore, type I IFN- a key cytokine known to play a role in lupus- is known to promote rapid BCR internalization (Chaturvedi et al., 2008; Giltiay et al., 2012). Based on its role in regulating EGFR, we hypothesized that PXK participates in BCR internalization and lupus-associated variants in the PXK locus differentially regulate BCR internalization. Indeed, we demonstrate that PXK colocalizes with the BCR and that the risk variants are associated with a decrease in BCR internalization. Knockdown of PXK replicates this phenotype, confirming the direct involvement of PXK in BCR trafficking.
Methods
Subjects and Study Design
We used a large collection of samples from case-control subjects from multiple ethnic groups. These samples were from the collaborative Large Lupus Association Study 2 (LLAS2) (Rasmussen et al., 2011) and were contributed by participating institutions in the United States, Asia, and Europe. According to genetic ancestry, subjects were grouped into ethnic groups including European American (EU), African American (AA), Asian and Asian American (AS), and Hispanic American (HA). Informed consent was obtained from all subjects using Institutional Regulatory Board approved consent documents. All lupus patients met the American College of Rheumatology (ACR) criteria for the classification of lupus (Hochberg, 1997).
Genotyping of Genetic Variants and Sample Quality Control
We genotyped 58 single-nucleotide polymorphisms (SNPs) covering the entire PXK region (58.1–58.5 MB on Chr 3, Build 37), as part of a larger custom genotyping study. The variants were chosen based upon the results of a candidate association study of 720 women of European ancestry and 2337 controls (Harley et al., 2008). Specifically, the variants were chosen to span the association interval identified with the Infinium HumanHap330 array. Genotyping of SNPs was completed with Infinium chemistry on an Illumina iSelect custom array according to the manufacturer's protocol. The following quality-control procedures were implemented to identify SNPs for analysis: well-defined clusters for genotype calling, call rate >90% across all samples genotyped, minor allele frequency (MAF) >1% (except for the rare variant analysis as described below), and p > 0.05 for differential missingness between cases and controls (the total proportion missing was <5%). One marker with evidence of a departure from Hardy-Weinberg proportion expectation (p < 0.0001 in controls) was removed from the initial analysis.
We removed individuals with a call rate <90% or excess heterozygosity. The remaining individuals were examined for excessive allele sharing as estimated by identity-by-descent (IBD). In sample pairs with excessive relatedness (IBD > 0.4), one individual was removed from the analysis on the basis of the following criteria: (1) remove the sample with the lower call rate, (2) remove the control and retain the case, (3) remove the male sample before the female sample, (4) remove the younger control before the older control, and (5) in a situation with two cases, remove the case with the less complete phenotype data available. Discrepancies between self-reported and genetically determined gender were evaluated.
Ascertainment of Population Stratification
Genetic outliers from each ethnic and/or racial group were removed from further analysis as determined by principal component (PC) analysis and admixture estimates (Figure 1 of McKeigue et al., 2000; Price et al., 2006; Lessard et al., 2011). We used 347 ancestral informative markers (AIMs) from the same custom genotyping study that passed quality control in both EIGENSTRAT (Price et al., 2006) and ADMIXMAP (Hoggart et al., 2003, 2004) to distinguish the four continental ancestral populations, allowing identification of the substructure within the sample set (Smith et al., 2004; Halder et al., 2008). The AIMs were selected to distinguish four continental ancestral populations: Africans, Europeans, American Indians, and East Asians. We utilized principal components from EIGENSTRAT outputs to identify outliers of each of the first three PCs for the individual population clusters with visual inspection.
Statistical Analysis—Workflow
The analysis was initiated by assessing the association of genotyped variants in each of the four ancestral cohorts individually as done previously at another locus (Kottyan et al., 2014). Strategically, we analyzed the genotyped, then imputed variants, performed full haplotype analysis, executed linkage disequilibrium analysis, and finally built a statistical model to account for the lupus-associated variability in European ancestry. In building the model of association in European Americans, we comparatively evaluated every variant in the region for its ability to better account for the lupus-associated genetic variation.
Statistical Analysis—Frequentist Approach
We tested each genetic variant for association with lupus using logistic regression models (frequentist approach) that included three admixture proportion estimates as covariates as implemented in PLINK v 1.07 (Marchini et al., 2007; Purcell et al., 2007). The additive genetic model is the primary model of inheritance. Other models were subsequently considered, but only if substantially superior. We performed a Cochran-Armitage trend test, a genotypic test, and tested both the dominant and recessive gene models. Logistic regression using an additive model remained the best model.
Step-wise logistic regression was performed to identify those genetic variants independently associated with the development of lupus in PLINK. For these analyses, the allelic dosage of a specific variant was added to the logistic model as covariates in addition to the admixture estimates. Haplotypic associations were assessed using logistic regression incorporating admixture measurements as covariates.
Linkage disequilibrium (LD) and haplotypes were determined with PLINK and HAPLOVIEW v 4.2 (Barrett et al., 2005; Sankararaman et al., 2008; Barrett, 2009). We calculated haplotype blocks for those haplotypes present at >3% frequency using the 4 gamete rule algorithms with a minimum r2-value of 0.8. Haplotypic associations were performed in PLINK using a 200 kb sliding window approach.
Statistical Analysis—Bayesian Approach
Using SNPTEST, we calculated the Bayes factor (BF) for each genetic variant: the probability of the genotype configuration at that genetic variant in cases and controls under the alternative hypothesis that the variant is associated with disease status divided by the probability of the genotype configuration at that variant in cases and controls under the null hypothesis that disease status is independent of genotype at that variant as previously described (we used the methods developed and introduced in Wellcome Trust Case Control Consortium et al., 2012). We used three admixture estimates as covariates, as we did for the frequentist approach. Large values of the Bayes factor (BF) correlate to robust evidence for association, as small p-values correlate to strong evidence in a frequentist approach. For our well-powered study, the Bayes factors (BFs) of the variants were highly correlated with the p-values (consistent with, Stephens and Balding, 2009). We used the additive model. The linear predictor is log(pi/(1-pi)) = μ + ßGi, and the prior is μ~N(0,12), ß~N(0,0.22) (variables are defined in the supplementary note in Wellcome Trust Case Control Consortium et al., 2012).
To identify the variants most likely to be driving the statistical association we calculated a posterior probability under the assumption that any of the variants within a single genetic effect could be causal and that only one of these variants is causal for each genetic effect. Variants with a low posterior probability are highly unlikely to be causal regardless of the allele frequency or presence of the actual causal variant in the analysis, following the procedure as presented (Wellcome Trust Case Control Consortium et al., 2012).
Re-Sequencing
We re-sequenced the PXK region as described previously (Lessard et al., 2012). DNA from European American subjects included in the current genotyping experiment was sequenced. To assess the accuracy of sequence-based SNP calling, we cross-referenced the sequenced and genotyped allele calls. Briefly, 3–5 micrograms of whole genomic DNA from each sample was sheared and prepared for sequencing with an Illumina Paired-End Genomic DNA Sample Prep Kit. Targeted regions of interest from each sample were then enriched with a SureSelect Target Enrichment System utilizing a custom-designed bait pool (Agilent Technologies). Post-sequence data were processed with Pipeline software v.1.7 (Illumina). All samples were sequenced to minimum average fold coverage of 253. Variant detection and quality control were also performed as previously described (Lessard et al., 2012).
Imputation to Composite 1000 Genomes Reference Panel
To detect associated variants that were not directly genotyped, we imputed the PXK region with IMPUTE2 and using a composite imputation reference panel based on 1000 Genomes Project sequence data freezes from December 2013 (Marchini et al., 2007; Altshuler et al., 2010; Lofgren et al., 2010). Imputed genotypes were required to meet or exceed a probability threshold of 0.5, and information measure of >0.4, and the same quality-control criteria threshold described for the genotyped markers. In the statistical analyses, the probability threshold from each imputed value was incorporated into the statistical analysis using SNPTEST. The overall genotype-imputed variant concordance rate was >93%.
Rare Variant Analysis
Rare variant analysis was performed in SVS Golden Helix SNP and Variation Suite v7.6.10 on the re-sequenced dataset and the imputed dataset. Rare variants were filtered based upon call rate and minor allele frequency <0.01. The one-sided kernel based adaptive cluster algorithm was performed using the hyper geometric kernel type with 1000 permutations in SVS GoldenHelix. For these studies, the initial analysis was performed using sequencing data from 92 lupus cases and 114 controls. Because this initial study was underpowered to identify an increased rare variant burden, we extended this analysis to an imputed dataset of all 4220 European lupus cases and 3803 European lupus controls. The accuracy of rare variant imputation, while limited, has been previously validated (Sung et al., 2012; Chen et al., 2013).
Power Analysis
Power analysis was performed using the Genetic Power Calculator (Purcell et al., 2003).
Cell Culture
Lymphoblastoid cell lines (LCLs) derived from control study participants without lupus were generated as previously described (Rasmussen et al., 2011) and maintained at 37°C in RPMI 1640 media containing 10% heat-inactivated fetal bovine serum (FBS) and antibiotic/antimicotic (Gibco/Life Technologies, Grand Island, NY). Peripheral blood mononucelar cells (PBMCs) were isolated using Ficoll-gradient centrifugation in SepMate tubes (Stemcell Technologies, Vancouver, BC, Canada) from fresh blood collected in acid citrate dextrose (ACD) tubes with informed consent from healthy donors. B cells were isolated using the B-cell isolation kit-II (Miltenyi Biotech, Auburn, CA).
RNA Purification and Expression Analysis
Total RNA was purified from LCLs using the RNeasy kit (Qiagen Valencia, CA). RNA was reverse-transcribed with the High-Capacity RNA-to-cDNA kit (Applied Biosystems, Grand Island, NY). Gene expression was determined by real-time PCR using a 7500 Real-time PCR system or Viia 7 system (Applied Biosystems). Relative quantification was calculated by the comparative CT method (Livak and Schmittgen, 2001). Briefly, expression levels for each target were normalized to levels of 18S ribosomal rRNA in the same well. All samples were then normalized to a selected sample and the data was exponentially transformed.
Protein expression was quantified in LCLs by flow cytometry. Cells were permeabilized and stained using reagents from Beckton, Dickinson and Company (BD, Franklin Lakes, NJ). Following staining, fluorescence was determined on an LSRFortessa analyzer (BD). The geometric median fluorescence intensity (gMFI) was calculated in FlowJo (Tree Star, Ashland, OR) and used as an estimate of protein expression. All experiments were run in triplicate. Statistical analysis and data visualization were done with GraphPad Prism (GraphPad Sftward, La Jolla, CA).
BCR Internalization and Colocalization
BCR internalization was adopted from the method described by Malhotra et al. (2009a,b). Briefly, LCLs or freshly isolated PBMCs from subjects without lupus were incubated with anti-BCR for 30 min at 4°C. Cells were then placed at 37°C for the indicated times. Cross-linked BCR remaining on the surface was then detected with fluorescently labeled secondary antibody with the LSRFortessa Cell Analyzer. Percent internalization was obtained by measuring the gMFI at each time point and then calculating the change as follows: (gMFI no internalization (4° sample) – gMFI internalization (37° sample)) ÷ (gMFI no internalization (4° sample)) × 100. Statistical analysis was performed in R using the packages “nlme” and “lmerTest.” Cells were imaged with a confocal microscope and the ImageStreamX (Amnis, Seattle, WA) for co-localization analysis. All experiments were run in triplicate.
shRNA Transfections
shRNA were obtained from the CCHMC Lenti-shRNA Library Core, utilizing the Sigma Mission system. Viral vector DNA was isolated using Endofree Plasmid Maxi Prep kits (Qiagen). Lentiviral transduction was performed by the CCHMC Viral Vector Core. Viral particles containing the shRNAs were used to transfect LCLs from control subjects. Cells were plated onto retronectin-coated plates, viral particle supernatant added, and spun for 45 min at 1300 × G at 22°C. After resting at 37°C overnight, spinfection was repeated. Cells were transferred to selection media containing 2 ug/ml puromycin after 48 h. After non-transfected cells began to die (approximately 3–5 days) cells were tested for PXK expression and maintained for use in subsequent experiments.
Materials
Anti-BCR (F(ab′)2 antibodies to IgG/IgM and Alexa Fluor 647-conjugated donkey anti-human IgG/IgM) were from Jackson ImmunoResearch (West Grove, PA). Anti-PXK antibody was purchased from Abcam (Cambridge, MA). Anti-LAMP1 antibody was purchased from BioLegend (San Diego, CA). Secondary antibodies were purchased from Life Technologies (Carlsbad, CA). Retronectin was purchased from Clonetech (Mountain View, CA). Puromycin was purchased from Invivogen (San Diego, CA). All other antibiotics and cell culture reagents were purchased from HyClone (Logan, UT) and Gibco (Life Technologies). Quantitative PCR was performed using TaqMan probes from Life Technologies.
Results
We genotyped 57 useful markers from the PXK locus in a transancestral group of 18,286 lupus cases and controls. Our study included 8023 individuals of European descent (EA), 3740 individuals of African American descent (AA), 2481 individuals of Hispanic American descent (HA) and 2652 individuals of Asian descent (AS). To better capture the total variation in the locus, we imputed against a composite reference panel derived from the 1000 Genomes Project (Howie et al., 2011) for a final dataset of 269–835 variant markers, depending on ancestry, all with a minor allele frequency (MAF) greater than 0.01 (Table 1). We performed logistic regression analysis to reveal a large association region of 257 kb extending from just upstream of PXK beginning in ABHD6 and extending through RPP14 in individuals of European descent (Figure 1A). Overall, the most significant SNP was rs6445972 with P < 4.62 × 10−10, OR 0.81 (0.75–0.86). The most significant, directly genotyped SNP was rs4681677 with P < 2.00 × 10−9, OR 0.81 (0.76–0.87). Because rs4681677 was genotyped and not assessed solely through imputation, we used this SNP to classify cell lines as risk or protective for subsequent biological experiments. We found no evidence of the lupus-association at this locus in the other populations studied (Figure 1B). rs6445972 has an allele frequency of 30.0% in the EA cohort, 10.4% in the AA cohort, 22.9% in the HA cohort, and 0.2% in the AS cohort, suggesting that a largely European-derived allele is driving the very modest statistical association in the cohorts with European admixture.
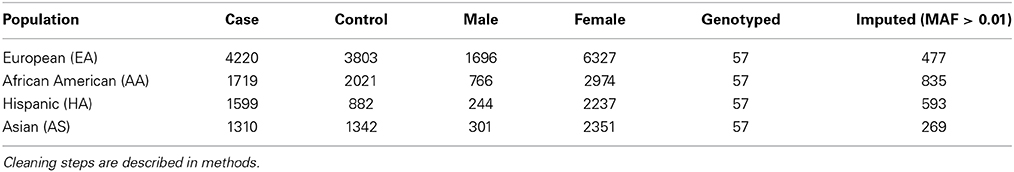
Table 1. Summary statistics for each study population.
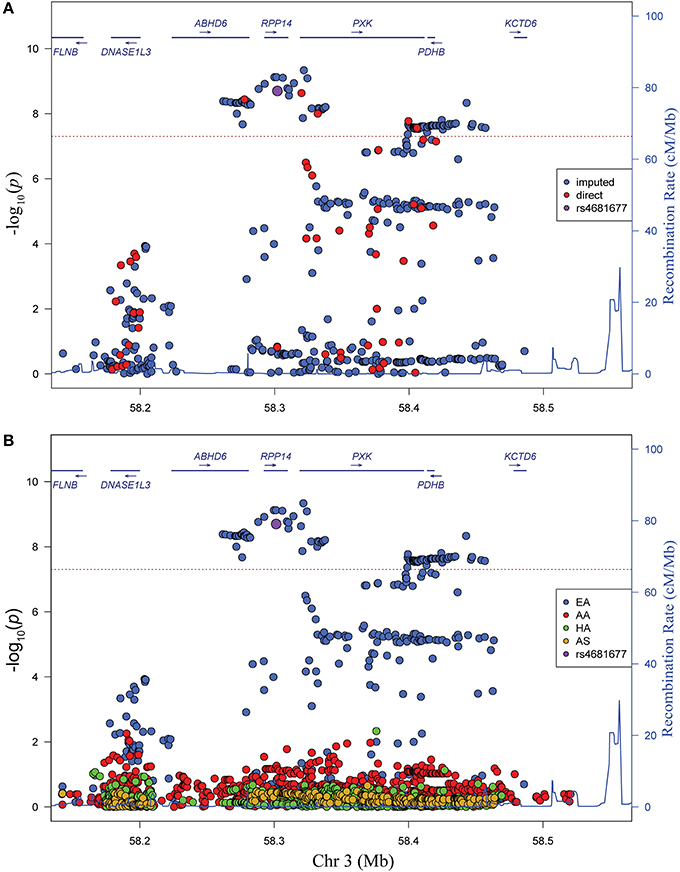
Figure 1. Variants in the PXK locus are associated with lupus. (A) Plot of –log10(p) values for directly genotyped (red) and imputed (blue) SNPs spanning the previously identified region of association with lupus. Gene locations are indicated above with arrows indicating direction of transcription. (B) Plot of –log10(p) values for different ancestral cohorts. EA, European ancestry; AA, African American ancestry; HA, Hispanic and Amerindian ancestry; AS, Asian ancestry. While not the most highly associated SNP, rs4681677 (purple) was used to choose samples for biological studies because it was genotyped; this SNP is in high LD (r2 > 0.9) with the imputed SNPs that are more highly associated.
Analysis of linkage disequilibrium (LD) in the region revealed high LD between the most strongly associated variants (Figure 2A). No single haplotype (using either blocks constructed from continuous groups of variants or the most highly associated variants) outperformed the single variant association model (data not shown), supporting the conclusion that the association in the region is due to a single genetic variant. In a complementary Bayesian analysis, we find a similar pattern of association, consistent with results from our frequentist logistic regression analysis. The 95% credible set (Wellcome Trust Case Control Consortium et al., 2012) contains 172 variants spanning a 202 kb region (chr 3: 58261741-58463411) (Figure 2B).
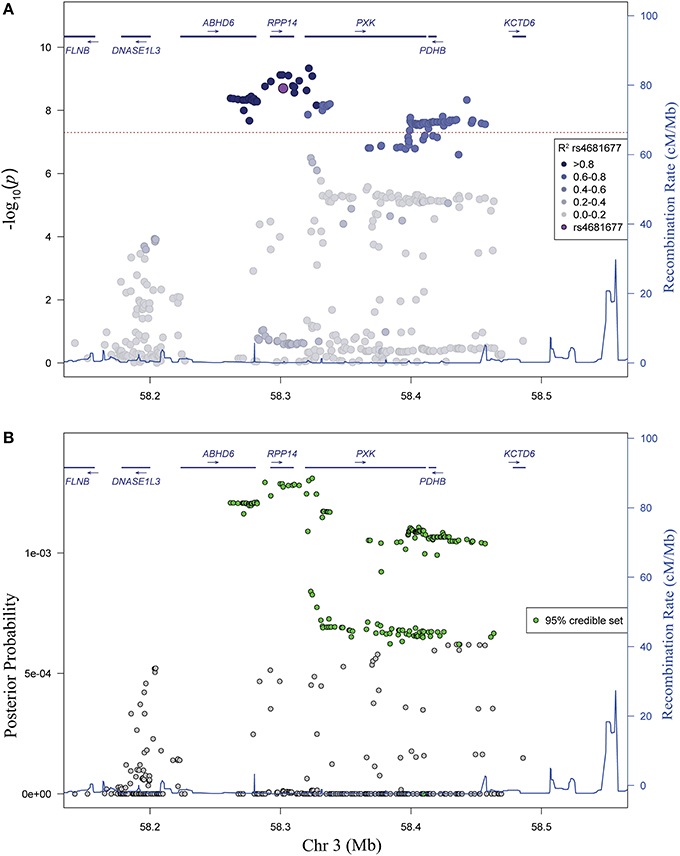
Figure 2. Evaluation of linkage disequilibrium in the PXK locus and Bayesian association analysis. (A) Association plot of genotyped and imputed SNPs shaded to identify the significant LD in the region. Colors correspond to r2-values, with darker shade indicating higher r2-values. (B) Bayesian analysis of variants in the PXK locus. Plot of posterior probability calculated for each variant in the locus. Variants included in the 95% credible set based on posterior probabilities are colored green.
To identify candidate variants in the EA population, we performed stepwise logistic regression to evaluate the ability of variants within the associated haplotype to explain all of the lupus-associated variation. Adjusting for any one of the top variants in the region eliminated the association signal, supporting the model that there is only one association in the region (Figure 3). We sequentially tested each variant in our dataset individually in our conditional analysis in an attempt to isolate groups of variants that may disproportionately carry the association signal in the region. While we did find clear groups, we found those variants that were able to adjust for the largest portion of the lupus-associated variation were in high LD (r2 > 0.8) with the most strongly associated variants. Notably, the SNP identified in our original GWAS study, rs6445975, did not account for lupus risk as completely as the most strongly associated variants in the current study (Figure 3 and Figure S2).
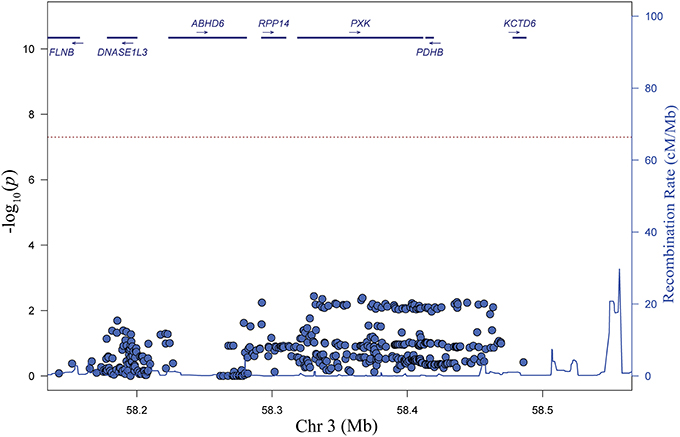
Figure 3. Step-wise logistic regression analysis reveals one independent genetic effect at the PXK locus. Plot of P values (–log10) following adjustment for genotype at rs4681677. Adjustment for other SNPs with r2 > 0.9 with rs4681677 can also account for the genetic association (P < 0.01) in the region.
It remained possible that rare variants were driving the lupus association at the PXK locus. To test this possibility, we performed deep sequencing of the region in 92 cases and 114 controls of European ancestry. We found no statistical association of any of the variants with frequencies less than one percent using a logistic regression analysis with an additive model; furthermore, we found no increased burden of rare variants in any of the genes in the region in the cases compared to the controls (data not shown). Despite the limitations of imputed rare variants, we repeated the rare variant burden test on the complete European population and did not find evidence of increased rare variants in the lupus cases.
In order to assess the hypothesis that lupus-associated variants were affecting the gene expression, we measured expression levels of all 5 genes in transformed B cell lines from control subjects and found no difference between cell lines with homozygous risk and homozygous non-risk genotypes (Figure S3). We did not find any difference in mRNA or total PXK protein expression (Figures 4A,B).
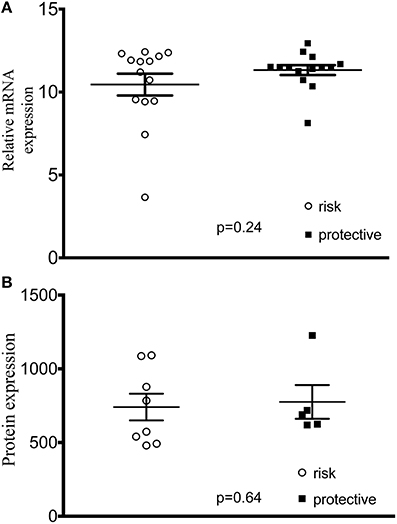
Figure 4. PXK expression in cell lines derived from study participants. (A) Plot of ΔΔCt values of PXK mRNA expression using TaqMan probes. (n = 14 risk/14 protective) (B) PXK expression at the protein level using flow cytometetry. (n = 8 homozygous risk/5 homozygous protective). Data for both A and B are representative of 3 independent experiments using control cell lines. Statistical analysis using a Student's t-test was performed in PRISM.
PXK is highly expressed in the B cell lineage, with the highest expression in mice observed in both transitional and follicular splenic B cells (Hu et al., 2011). The transitional B cell was recently implicated as one of the cell types critical to lupus pathogenesis based on a combined genetic and cell-specific expression analysis (Ramos et al., 2011). Given the expression of PXK in B cells, the fact that PXK has been shown to play a role regulating cell surface receptor expression, and the importance of B cells to the pathology of lupus, we hypothesized that PXK participates in the internalization of the BCR. We first evaluated colocalization between PXK and the BCR in B cells following BCR crosslinking on the cell surface. We found moderate steady-state colocalization between PXK and the BCR at baseline that increased following BCR crosslinking and continued to increase with cellular internalization (Figure 5). We then tested the hypothesis that the lupus-associated risk variants at the PXK locus affected the rate of BCR internalization by measuring the internalization of the BCR in cells derived from individuals with known genotypes. Following BCR crosslinking, we measured receptor internalization with flow cytometry and found that cells carrying the homozygous risk genotype displayed a decrease in the amount of BCR internalization compared to cells homozygous for the protective genotype (Figure 6). This finding was consistent across all the measurements over the time course of the experiment.
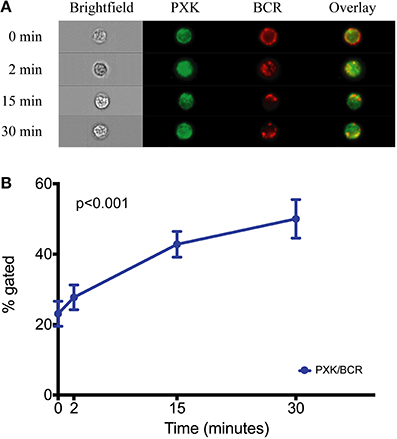
Figure 5. PXK colocalizes with the BCR following receptor crosslinking. (A) Quantification of colocalization between PXK and the BCR following BCR crosslinking in primary B cells from subjects without lupus. (B) Representative images showing colocalization. Statistical analysis using a One-Way ANOVA was performed in PRISM.
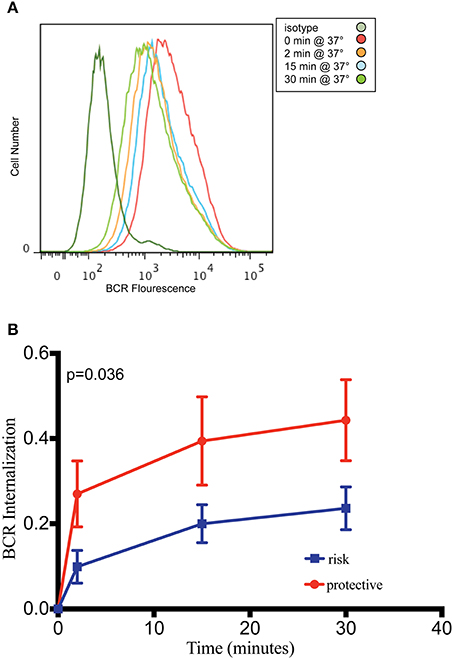
Figure 6. B cells carrying PXK risk allele demonstrate decreased internalization of the BCR. (A) Representative flow cytometry histogram showing decreasing fluorescence following internalization of the BCR from the B cell surface using patient-derived LCLs. (B) Quantification of BCR internalization as measured by the percent internalization (decrease in gMFI) following receptor crosslinking. Results of linear mixed model analysis as performed in R are shown. This experiment was performed using 20 cell lines from subjects without lupus in three independent experiments. The results of these independent experiments were combined and presented in (B).
To assess the role of PXK in the BCR internalization phenotype, we decreased PXK expression using shRNA by transfecting 5 study-derived cell lines from patient without lupus with shRNA targeted against PXK or scrambled controls (Figure 7A). Knockdown of PXK resulted in reduced internalization of the BCR, especially at the early time points (Figure 7B), and this reduction was directly correlated with PXK expression (Figure 7C).
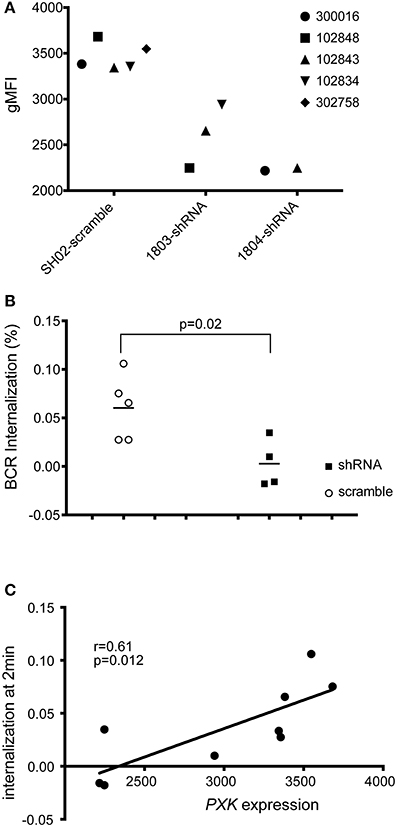
Figure 7. shRNA knockdown of PXK disrupts BCR colocalization. (A) PXK protein expression following shRNA knockdown in 5 LCL lines from non-lupus subjects (expression was assessed in triplicate for each cell line). (B) Summary of BCR internalization at 2 min in LCLs following viral transduction with either an shRNA against PXK or a scrambled control. Results from unpaired Student's t-test shown. (C) Correlation between PXK protein expression and BCR internalization following shRNA knockdown. The significance was assessed using a Pearson correlation. The graphs in (B,C) are representative of the results of three independent experiments.
Discussion
We performed a fine-mapping study to define the genetic variants and the gene most likely to be causal for increased lupus risk. We genotyped 18,286 cases and controls from four ancestral populations, leveraging imputation to increase our coverage of the target locus. We used both logistic regression, and Bayesian methods to verify our findings that the disease risk from this locus is due to highly correlated variants in a large region including PXK. Additionally, we complemented our genetic fine-mapping with biological experiments, identifying a genotypic change in BCR internalization that is associated with the presence of lupus-risk variants.
Analysis of the PXK locus in non-European populations revealed no significant association in the region (Figure 1B). The allele frequency of the most highly associated variants differed amongst the ancestral groups, and given the observed allele frequencies, power analysis indicates that a much larger non-European population will be needed to detect a similar association of this magnitude at this locus (data not shown). Thus, the lack of an association in the other populations may be due entirely to the difference in power.
We found a 50% decrease in BCR internalization kinetics between risk and non-risk genotypes. This change in BCR internalization may have a small effect in the regulation of B cell signaling; however, it could also be meaningful in the context of other additive changes to the B cell signaling pathway (Vaughn et al., 2012). There are several mechanisms through which this functional change could affect the risk of autoimmunity. For example, stimuli that would be sufficient to develop strong B cell activation with subsequent negative selection may now be less likely to result in elimination due to attenuated BCR internalization. Indeed, the persistence of auto-reactive clones in the periphery may play an important role in the context of lupus pathogenesis.
PXK colocalized with the BCR and affected internalization, indicating a potential role for PXK in the regulation of BCR signaling. Importantly, the colocalization increased as the BCR is internalized into the cell, suggesting a role for PXK beyond the cell surface. Furthermore, we find that cells carrying the lupus-risk haplotype have a decreased amount of BCR internalization when compared to cell lines carrying the non-risk haplotype, an allelic functional change that we present as a candidate causal mechanism of increased lupus risk at this locus.
When PXK is specifically knocked down using shRNA, BCR internalization is decreased, confirming a clear role for PXK in regulating BCR internalization. These results support the conclusion that the allele-specific changes in BCR internalization we detect are most likely attributable to PXK. We do not yet understand the genetic mechanism behind this alteration in BCR internalization. We were unable to detect a difference in overall PXK expression. It may be that some other molecular differences are occurring after PXK expression, such as alternative splicing or differential post-translational modification. Future studies will be directed at detecting these changes. While ABHD6 eQTLs were shown to correlate with lupus-association, there were also PXK eQTLs that were associated with lupus (Oparina et al., 2014). The PXK locus remains complicated and future work will be important to continue to unravel the specific genetic variations underlying the lupus association.
We and others refer to this locus as the “PXK locus” (Graham et al., 2008; Harley et al., 2008; Suarez-Gestal et al., 2009; Ramos et al., 2011; Oparina et al., 2014). Although our step-wise logistic regression analysis of common variants (Figure 3) makes it unlikely, it is still possible that there is a contribution of rare variant(s) that we missed. Of the 5 genes in the region, PXK and DNase1l3 are the only genes with appreciable expression in immune cells based on evaluation of public databases (data not shown). DNASE1L3 encodes the protein DNase 1-like 3, and is the only gene that has been investigated functionally in the context of lupus. A loss-of-function frameshift mutation in DNASE1L3 was found to be associated with early-onset lupus and lack of detectable DNASE1L3 transcripts in an autosomal recessive manner in six families of Arab descent (Al-Mayouf et al., 2011). DNASE1L3 was also recently identified as a risk gene in systemic sclerosis (Mayes et al., 2014). The variant identified in that study shows no association with lupus in our dataset (p = 0.3067). DNASE1L3 is removed from the peak association in the frequentist, logistic regression analysis and no members of the 95% credible set in the Bayesian analysis were located in or within 25 kb of DNASE1L3. It remains possible that variants many hundreds of killobases away from the promoter of a gene could affect that genes transcription (Westra et al., 2013); however, while both of our groups found detectable DNASE1L3 transcripts, neither our group nor other groups have identified allelic expression of DNASE1L3 in our cell lines (Oparina et al., 2014). Of the remaining genes, ABHD6 has recently been suggested as the causal gene responsible for the association with lupus at this locus (Oparina et al., 2014). Mutations in ABHD6 have been associated with multiple non-autoimmune phenotypes (Blankman et al., 2007; Alhouayek et al., 2013; Tchantchou and Zhang, 2013; Thomas et al., 2013; Volk et al., 2013), but beyond ABHD6 being in the associated region there is no evidence, at present, for ABHD6 in lupus pathogenesis.
Overall, we identified a limited haplotype of highly associated variants in the promoter and first exon of PXK that account for all of the lupus-association in the region. We performed biochemical analysis to demonstrate that PXK co-localizes with the BCR and affects BCR internalization. Furthermore, we identified an allelic decrease in PXK-BCR co-localization and BCR internalization in subjects expressing the lupus-risk haplotype. Taken together, our work supports a model in which PXK increases lupus risk through the regulation of BCR internalization.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare, Republic of Korea [HI13C2124 to Sang-Cheol Bae], the Alliance for Lupus Research [to Betty P. Tsao], the US Department of Veterans Affairs [IMMA 9 to John B. Harley], the US Department of Defense [PR094002 to John B. Harley] and the National Institutes of Health [R37AI024717 and P01AI083194 to John B. Harley, RO1AR043814 and R21AR065626 to Betty P. Tsao, P01AR049084 to Robert P. Kimberly, AR43727 to Michelle Petri, P60AR062755 and UL1RR029882 to Gary S. Gilkeson and Diane L. Kamen, and K24AI078004, R21AI070304, UL1TR000154 and P30CA046934, T32GM63483 to Samuel E. Vaughn and Zubin H. Patel. We would like to thank Summer Frank and Mai Li Zhu for their assistance in genotyping, quality control analyses, and clinical data management. All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children's Hospital Medical Center, supported in part by NIH AR-47363, NIH DK78392 and NIH DK90971. shRNA were provided by the CCHMC Lenti-shRNA Library core. The lenti-viral vectors were produced by the Cincinnati Children's Hospital Medical Center Viral Vector Core.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2014.00450/abstract
References
Alhouayek, M., Masquelier, J., Cani, P. D., Lambert, D. M., and Muccioli, G. G. (2013). Implication of the anti-inflammatory bioactive lipid prostaglandin D2-glycerol ester in the control of macrophage activation and inflammation by ABHD6. Proc. Natl. Acad. Sci. U.S.A. 110, 17558–17563. doi: 10.1073/pnas.1314017110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Al-Mayouf, S. M., Sunker, A., Abdwani, R., Abrawi, S. A., Almurshedi, F., Alhashmi, N., et al. (2011). Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 43, 1186–1188. doi: 10.1038/ng.975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Altshuler, D. M., Gibbs, R. A., Peltonen, L., Dermitzakis, E., Schaffner, S. F., Yu, F., et al. (2010). Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58. doi: 10.1038/nature09298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrett, J. C. (2009). Haploview: visualization and analysis of SNP genotype data. Cold Spring Harb. Protoc. 2009: pdb. ip71. doi: 10.1101/pdb.ip71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blankman, J. L., Simon, G. M., and Cravatt, B. F. (2007). A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 14, 1347–1356. doi: 10.1016/j.chembiol.2007.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, O., and Shlomchik, M. J. (1998). A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J. Immunol. 160, 51–59.
Chan, O. T., Madaio, M. P., and Shlomchik, M. J. (1999). B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J. Immunol. 163, 3592–3596.
Chaturvedi, A., Dorward, D., and Pierce, S. K. (2008). The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 28, 799–809. doi: 10.1016/j.immuni.2008.03.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chaturvedi, A., Martz, R., Dorward, D., Waisberg, M., and Pierce, S. K. (2011). Endocytosed BCRs sequentially regulate MAPK and Akt signaling pathways from intracellular compartments. Nat. Immunol. 12, 1119–1126. doi: 10.1038/ni.2116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J., Zhang, J. G., Li, J., Pei, Y. F., and Deng, H. W. (2013). On combining reference data to improve imputation accuracy. PLoS ONE 8:e55600. doi: 10.1371/journal.pone.0055600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gateva, V., Sandling, J. K., Hom, G., Taylor, K. E., Chung, S. A., Sun, X., et al. (2009). A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233. doi: 10.1038/ng.468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Giltiay, N. V., Chappell, C. P., and Clark, E. A. (2012). B-cell selection and the development of autoantibodies. Arthritis Res. Ther. 14(Suppl. 4), S1. doi: 10.1186/ar3918
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graham, R. R., Cotsapas, C., Davies, L., Hackett, R., Lessard, C. J., Leon, J. M., et al. (2008). Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 40, 1059–1061. doi: 10.1038/ng.200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grimaldi, C. M., Hicks, R., and Diamond, B. (2005). B cell selection and susceptibility to autoimmunity. J. Immunol. 174, 1775–1781. doi: 10.4049/jimmunol.174.4.1775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Halder, I., Shriver, M., Thomas, M., Fernandez, J. R., and Frudakis, T. (2008). A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: utility and applications. Hum. Mutat. 29, 648–658. doi: 10.1002/humu.20695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harley, J. B., Alarcón-Riquelme, M. E., Criswell, L. A., Jacob, C. O., Kimberly, R. P., Moser, K. L., et al. (2008). Langefeld, Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 40, 204–210. doi: 10.1038/ng.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heesters, B. A., Das, A., Chatterjee, P., and Carroll, M. C. (2014). Do follicular dendritic cells regulate lupus-specific B cells? Mol. Immunol. 62, 283–288. doi: 10.1016/j.molimm.2014.02.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hochberg, M. C. (1997). Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 40, 1725. doi: 10.1002/art.1780400928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoggart, C. J., Parra, E. J., Shriver, M. D., Bonilla, C., Kittles, R. A., Clayton, D. G., et al. (2003). Control of confounding of genetic associations in stratified populations. Am. J. Hum. Genet. 72, 1492–1504. doi: 10.1086/375613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoggart, C. J., Shriver, M. D., Kittles, R. A., Clayton, D. G., and McKeigue, P. M. (2004). Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 74, 965–978. doi: 10.1086/420855
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howie, B., Marchini, J., and Stephens, M. (2011). Genotype imputation with thousands of genomes. G3 (Bethesda) 1, 457–470. doi: 10.1534/g3.111.001198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, X., Kim, H., Stahl, E., Plenge, R., Daly, M., and Raychaudhuri, S. (2011). Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am. J. Hum. Genet. 89, 496–506. doi: 10.1016/j.ajhg.2011.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kottyan, L. C., Zoller, E. E., Bene, J., Lu, X., Kelly, J. A., Rupert, A. M., et al. (2014). The IRF5-TNPO3 association with systemic lupus erythematosus has two components that other autoimmune disorders variably share. Hum. Mol. Genet. doi: 10.1093/hmg/ddu455. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lessard, C. J., Adrianto, I., Ice, J. A., Wiley, G. B., Kelly, J. A., Glenn, S. B., et al. (2012). Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am. J. Hum. Genet. 90, 648–660. doi: 10.1016/j.ajhg.2012.02.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lessard, C. J., Adrianto, I., Kelly, J. A., Kaufman, K. M., Grundahl, K. M., Adler, A., et al. (2011). Identification of a systemic lupus erythematosus susceptibility locus at 11p13 between PDHX and CD44 in a multiethnic study. Am. J. Hum. Genet. 88, 83–91. doi: 10.1016/j.ajhg.2010.11.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lofgren, S. E., Yin, H., Delgado-Vega, A. M., Sanchez, E., Lewen, S., Pons-Estel, B. A., et al. (2010). Promoter insertion/deletion in the IRF5 gene is highly associated with susceptibility to systemic lupus erythematosus in distinct populations, but exerts a modest effect on gene expression in peripheral blood mononuclear cells. J. Rheumatol. 37, 574–578. doi: 10.3899/jrheum.090440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malhotra, S., Kovats, S., Zhang, W., and Coggeshall, K. M. (2009a). Vav and Rac activation in B cell antigen receptor endocytosis involves Vav recruitment to the adapter protein LAB. J. Biol. Chem. 284, 36202–36212. doi: 10.1074/jbc.M109.040089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malhotra, S., Kovats, S., Zhang, W., and Coggeshall, K. M. (2009b). B cell antigen receptor endocytosis and antigen presentation to T cells require Vav and dynamin. J. Biol. Chem. 284, 24088–24097. doi: 10.1074/jbc.M109.014209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mao, H., Ferguson, T. S., Cibulsky, S. M., Holmqvist, M., Ding, C., Fei, H., et al. (2005). MONaKA, a novel modulator of the plasma membrane Na,K-ATPase. J. Neurosci. 25, 7934–7943. doi: 10.1523/JNEUROSCI.0635-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchini, J., Howie, B., Myers, S., McVean, G., and Donnelly, P. (2007). A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913. doi: 10.1038/ng2088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayes, M. D., Bossini-Castillo, L., Gorlova, O., Martin, J.-E., Zhou, X., Chen, W. V., et al. (2014). Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am. J. Hum. Genet. 94, 47–61. doi: 10.1016/j.ajhg.2013.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKeigue, P. M., Carpenter, J. R., Parra, E. J., and Shriver, M. D. (2000). Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann. Hum. Genet. 64, 171–186. doi: 10.1046/j.1469-1809.2000.6420171.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oparina, N. Y., Delgado-Vega, A. M., Martinez-Bueno, M., Magro-Checa, C., Fernández, C., Castro, R. O., et al. (2014). PXK locus in systemic lupus erythematosus: fine mapping and functional analysis reveals novel susceptibility gene ABHD6. Ann. Rheum. Dis. doi: 10.1136/annrheumdis-2013-204909. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pedersen, N. M., Raiborg, C., Brech, A., Skarpen, E., Roxrud, I., Platta, H. W., et al. (2012). The PtdIns3P-binding protein Phafin 2 mediates epidermal growth factor receptor degradation by promoting endosome fusion. Traffic 13, 1547–1563. doi: 10.1111/j.1600-0854.2012.01400.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purcell, S., Cherny, S. S., and Sham, P. C. (2003). Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150. doi: 10.1093/bioinformatics/19.1.149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramos, P. S., Criswell, L. A., Moser, K. L., Comeau, M. E., Williams, A. H., Pajewski, N. M., et al. (2011). A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genet. 7:e1002406. doi: 10.1371/journal.pgen.1002406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rasmussen, A., Sevier, S., Kelly, J. A., Glenn, S. B., Aberle, T., Cooney, C. M., et al. (2011). The lupus family registry and repository. Rheumatology 50, 47–59. doi: 10.1093/rheumatology/keq302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sankararaman, S., Sridhar, S., Kimmel, G., and Halperin, E. (2008). Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82, 290–303. doi: 10.1016/j.ajhg.2007.09.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seet, L.-F., and Hong, W. (2006). The Phox (PX) domain proteins and membrane traffic. Biochim. Biophys. Acta 1761, 878–896. doi: 10.1016/j.bbalip.2006.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shlomchik, M. J., Madaio, M. P., Ni, D., Trounstein, M., and Huszar, D. (1994). The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 180, 1295–1306. doi: 10.1084/jem.180.4.1295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, M. W., Patterson, N., Lautenberger, J. A., Truelove, A. L., McDonald, G. J., Waliszewska, A., et al. (2004). A high-density admixture map for disease gene discovery in african americans. Am. J. Hum. Genet. 74, 1001–1013. doi: 10.1086/420856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stephens, M., and Balding, D. J. (2009). Bayesian statistical methods for genetic association studies. Nature reviews. Genetics 10, 681–690. doi: 10.1038/nrg2615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suarez-Gestal, M., Calaza, M., Endreffy, E., Pullmann, R., Ordi-Ros, J., Sebastiani, G. D., et al. (2009). Replication of recently identified systemic lupus erythematosus genetic associations: a case-control study. Arthritis Res. Ther. 11:R69. doi: 10.1186/ar2698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sung, Y. J., Wang, L., Rankinen, T., Bouchard, C., and Rao, D. C. (2012). Performance of genotype imputations using data from the 1000 Genomes Project. Hum. Hered. 73, 18–25. doi: 10.1159/000334084
Takeuchi, H., Takeuchi, T., Gao, J., Cantley, L. C., and Hirata, M. (2010). Characterization of PXK as a protein involved in epidermal growth factor receptor trafficking. Mol. Cell. Biol. 30, 1689–1702. doi: 10.1128/MCB.01105-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tchantchou, F., and Zhang, Y. (2013). Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury. J. Neurotrauma 30, 565–579. doi: 10.1089/neu.2012.2647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, G., Betters, J. L., Lord, C. C., Brown, A. L., Marshall, S., Ferguson, D., et al. (2013). The serine hydrolase ABHD6 is a critical regulator of the metabolic syndrome. Cell Rep. 5, 508–520. doi: 10.1016/j.celrep.2013.08.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vaughn, S. E., Kottyan, L. C., Munroe, M. E., and Harley, J. B. (2012). Genetic susceptibility to lupus: the biological basis of pathways. J. Leukoc. Biol. 92, 577–591. doi: 10.1189/jlb.0212095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volk, D. W., Siegel, B. I., Verrico, C. D., and Lewis, D. A. (2013). Endocannabinoid metabolism in the prefrontal cortex in schizophrenia. Schizophr. Res. 147, 53–57. doi: 10.1016/j.schres.2013.02.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wellcome Trust Case Control Consortium Maller, J. B., McVean, G., Byrnes, J., Vukcevic, D., Palin, K., et al. (2012). Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 44, 1294–1301. doi: 10.1038/ng.2435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westra, H. J., Peters, M. J., Esko, T., Yaghootkar, H., Schurmann, C., Kettunen, J., et al. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. doi: 10.1038/ng.2756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Seet, L. F., Hanson, B., and Hong, W. (2001). The Phox homology (PX) domain, a new player in phosphoinositide signalling. Biochem. J. 360, 513–530. doi: 10.1042/0264-6021:3600513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zou, X., Qiu, G., Chen, C., Wu, M., Hu, Y., Zheng, H., et al. (2005). Expression pattern and subcellular localization of five splice isoforms of human PXK. Int. J. Mol. Med. 16, 701–707. doi: 10.3892/ijmm.16.4.701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: lupus, PXK, fine-mapping, B cells, BCR
Citation: Vaughn SE, Foley C, Lu X, Patel ZH, Zoller EE, Magnusen AF, Williams AH, Ziegler JT, Comeau ME, Marion MC, Glenn SB, Adler A, Shen N, Nath S, Stevens AM, Freedman BI, Tsao BP, Jacob CO, Kamen DL, Brown EE, Gilkeson GS, Alarcón GS, Reveille JD, Anaya J-M, James JA, Moser KL, Criswell LA, Vilá LM, Alarcón-Riquelme ME, Petri M, Scofield RH, Kimberly RP, Ramsey-Goldman R, Binjoo Y, Choi J, Bae S-C, Boackle SA, Vyse TJ, Guthridge JM, Namjou B, Gaffney PM, Langefeld CD, Kaufman KM, Kelly JA, Harley ITW, Harley JB and Kottyan LC (2015) Lupus risk variants in the PXK locus alter B-cell receptor internalization. Front. Genet. 5:450. doi: 10.3389/fgene.2014.00450
Received: 07 October 2014; Accepted: 09 December 2014;
Published online: 08 January 2015.
Edited by:
Mariza De Andrade, Mayo Clinic, USAReviewed by:
Vikas Bansal, Scripps Translational Science Institute, USAKui Zhang, University of Alabama at Birmingham, USA
Copyright © 2015 Vaughn, Foley, Lu, Patel, Zoller, Magnusen, Williams, Ziegler, Comeau, Marion, Glenn, Adler, Shen, Nath, Stevens, Freedman, Tsao, Jacob, Kamen, Brown, Gilkeson, Alarcón, Reveille, Anaya, James, Moser, Criswell, Vilá, Alarcón-Riquelme, Petri, Scofield, Kimberly, Ramsey-Goldman, Binjoo, Choi, Bae, Boackle, Vyse, Guthridge, Namjou, Gaffney, Langefeld, Kaufman, Kelly, Harley, Harley and Kottyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leah C. Kottyan, Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave., Mail Location 15012, Cincinnati, OH 45229, USA e-mail:bGVhaC5rb3R0eWFuQGNjaG1jLm9yZw==