 Ludmilla B. Zavarez1
Ludmilla B. Zavarez1 Yuri T. Utsunomiya1
Yuri T. Utsunomiya1 Adriana S. Carmo1
Adriana S. Carmo1 Haroldo H. R. Neves2,3
Haroldo H. R. Neves2,3 Roberto Carvalheiro3
Roberto Carvalheiro3 Maja Ferenčaković4
Maja Ferenčaković4 Ana M. Pérez O'Brien5
Ana M. Pérez O'Brien5 Ino Curik4
Ino Curik4 John B. Cole6
John B. Cole6 Curtis P. Van Tassell6
Curtis P. Van Tassell6 Marcos V. G. B. da Silva7
Marcos V. G. B. da Silva7 Tad S. Sonstegard6
Tad S. Sonstegard6 Johann Sölkner5
Johann Sölkner5 José F. Garcia1,8*
José F. Garcia1,8*- 1Departamento de Medicina Veterinária Preventiva e Reprodução Animal, Faculdade de Ciências Agrárias e Veterinárias, UNESP – Univ Estadual Paulista, Jaboticabal, São Paulo, Brazil
- 2GenSys Consultores Associados, Porto Alegre, Rio Grande do Sul, Brazil
- 3Departamento de Zootecnia, Faculdade de Ciências Agrárias e Veterinárias, UNESP – Univ Estadual Paulista, Jaboticabal, São Paulo, Brazil
- 4Department of Animal Science, Faculty of Agriculture, University of Zagreb, Zagreb, Croatia
- 5Division of Livestock Sciences, Department of Sustainable Agricultural Systems, BOKU - University of Natural Resources and Life Sciences, Vienna, Austria
- 6Animal Genomics and Improvement Laboratory, United States Department of Agriculture, Agricultural Research Service, Beltsville, MD, USA
- 7Bioinformatics and Animal Genomics Laboratory, Embrapa Dairy Cattle, Juiz de Fora, Minas Gerais, Brazil
- 8Laboratório de Bioquímica e Biologia Molecular Animal, Departamento de Apoio, Produção e Saúde Animal, Faculdade de Medicina Veterinária de Araçatuba, UNESP – Univ Estadual Paulista, Araçatuba, São Paulo, Brazil
The use of relatively low numbers of sires in cattle breeding programs, particularly on those for carcass and weight traits in Nellore beef cattle (Bos indicus) in Brazil, has always raised concerns about inbreeding, which affects conservation of genetic resources and sustainability of this breed. Here, we investigated the distribution of autozygosity levels based on runs of homozygosity (ROH) in a sample of 1,278 Nellore cows, genotyped for over 777,000 SNPs. We found ROH segments larger than 10 Mb in over 70% of the samples, representing signatures most likely related to the recent massive use of few sires. However, the average genome coverage by ROH (>1 Mb) was lower than previously reported for other cattle breeds (4.58%). In spite of 99.98% of the SNPs being included within a ROH in at least one individual, only 19.37% of the markers were encompassed by common ROH, suggesting that the ongoing selection for weight, carcass and reproductive traits in this population is too recent to have produced selection signatures in the form of ROH. Three short-range highly prevalent ROH autosomal hotspots (occurring in over 50% of the samples) were observed, indicating candidate regions most likely under selection since before the foundation of Brazilian Nellore cattle. The putative signatures of selection on chromosomes 4, 7, and 12 may be involved in resistance to infectious diseases and fertility, and should be subject of future investigation.
Introduction
Autozygosity is the homozygote state of identical-by-descent alleles, which can result from several different phenomena such as genetic drift, population bottleneck, mating of close relatives, and natural and artificial selection (Falconer and Mackay, 1996; Keller et al., 2011; Curik et al., 2014). In the past 20 years, the heavy use of relatively low number of sires in Brazilian Nellore breeding programs (Bos indicus) is deemed to have mimicked all these triggers of autozygosity, especially considering the increasing use of artificial insemination over the decades. As inbreeding has been incriminated in reduced fitness and reproductive performance in other cattle populations under artificial selection (Bjelland et al., 2013; Leroy, 2014), avoidance of mating of close relatives is a typical practice of many Nellore breeders. Therefore, there is a growing interest in characterizing and monitoring autozygosity in this breed to preserve genetic diversity and allow the long-term sustainability of breeding programs in Brazil.
Evidence from whole-genome sequencing studies in humans indicate that highly deleterious variants are common across healthy individuals (MacArthur et al., 2012; Xue et al., 2012), and although no such systematical survey has been conducted in cattle to the present date, it is highly expected that unfavorable alleles also segregate in cattle populations. Therefore, the use of ever-smaller numbers of animals as founders is expected to inadvertently increase autozygosity of such unfavorable alleles (Szpiech et al., 2013), potentially causing economic losses.
Recently, the use of high-density single nucleotide polymorphism (SNP) genotypes to scan individual genomes for contiguous homozygous chromosomal fragments has been proposed as a proxy for the identification of identical-by-descent haplotypes (Gibson et al., 2006; Lencz et al., 2007). As the length of autozygous chromosomal segments is proportional to the number of generations since the common ancestor (Howrigan et al., 2011), the identification of runs of homozygosity (ROH) can reveal recent and remote events of inbreeding, providing invaluable information about the genetic relationships and demographic history of domesticated cattle (Purfield et al., 2012; Ferenčaković et al., 2013a; Kim et al., 2013). Also, given the stochastic nature of recombination, the occurrence of ROH is highly heterogeneous across the genome, and hotspots of ROH across a large number of samples (hereafter referred as common ROH) may be indicative of selective pressure. Moreover, the fraction of an individual's genome covered by ROH can be used as an estimate of its genomic autozygosity or inbreeding coefficient (McQuillan et al., 2008; Curik et al., 2014).
Here, we investigated the occurrence of ROH in high-density SNP genotypes in order to characterize autozygosity in the genomes of a sample of 1,278 Nellore cows under artificial selection for weight, carcass and reproductive traits. We aimed at characterizing the distribution of ROH length and genome-wide levels of autozygosity, as well as detecting common ROH that may be implicated in past events of selection.
Materials and Methods
Ethical Statement
The present study was exempt of the local ethical committee evaluation as genomic DNA was extracted from stored hair samples of animals from commercial herds.
Genotyping and Data Filtering
A total of 1,278 cows were genotyped with the Illumina® BovineHD Genotyping BeadChip assay (HD), according to the manufacturer's protocol (http://support.illumina.com/array/array_kits/bovinehd_dna_analysis_kit.html). These animals comprised part of the genomic selection reference population from a commercial breeding program. These dams were born between 1993 and 2008, being under routine genetic evaluation for weight, carcass and reproductive traits by the DeltaGen program, an alliance of Nellore cattle breeders from Brazil. Data filtering was performed using PLINK v1.07 (Purcell et al., 2007), and markers were removed from the dataset if GenTrain score lower than 70% or a call rate lower than 98% was observed. All genotyped samples exhibited call rates greater than 90%, thus no animals were filtered from further analyses. Minor allele frequency (MAF) was not used as an exclusion criterion in this analysis, so that the detection of homozygous segments was not compromised. Both autosomal and X-linked markers were included.
Estimates of Genomic Individual Autozygosity
Genomic autozygosity was measured based on the percentage of an individual's genome that is covered by ROH. Stretches of consecutive homozygous genotypes were identified for each animal using SNP & Variation Suite v7.6.8 (Golden Helix, Bozeman, MT, USA http://www.goldenhelix.com), and chromosomal segments were declared ROH under the following criteria: 30 or more consecutive homozygous SNPs, a density of at least 1 SNP every 100 kb, gaps of no more than 500 kb between SNPs, and no more than 5 missing genotypes across all individuals. In order to account for genotyping error and avoid underestimation of long ROH (Ferenčaković et al., 2013b), heterozygous genotype calls were allowed under conditions where there were 2 heterozygous genotypes for ROH ≥ 4 Mb, or no heterozygous genotypes for ROH < 4 Mb. Autozygosity was estimated according to McQuillan et al. (2008):
Where LROHj is the length of ROH j, and Ltotal is the total size of the genome covered by markers, calculated from the sum of inter-marker distances in the UMD v3.1 assembly. In order to facilitate comparisons with other studies, FROH was calculated using both the genome size based on autosomal and autosomal + X chromosomes. For each animal, FROH was calculated based on ROH of different minimum lengths: 0.5, 1, 2, 4, 8 or 16 Mb, representing autozygosity events that occurred approximately 100, 50, 25, 13, 6, and 3 generations in the past, respectively (Howrigan et al., 2011; Ferenčaković et al., 2013b). Additionally, chromosome-wise FROH was also computed.
An alternative measure of autozygosity was obtained by computing the diagonal elements of a modified realized genomic relationship matrix (VanRaden, 2008; VanRaden et al., 2011), calculated as:
Where Z is a centered genotype matrix and pl is the reference allele frequency at locus l. Matrix Z is obtained by subtracting from the genotype matrix M (with genotype scores coded as 0, 1 or 2 for alternative allele homozygote, heterozygote, and reference allele homozygote, respectively) the matrix P, whose elements of column l are equal to 2pl. The diagonal elements of G (Gi, i) represent the relationship of an animal with itself, and thus encapsulate autozygosity information. Following VanRaden et al. (2011), Gi, i can provide a more suitable proxy for the pedigree-based inbreeding coefficient when assuming pl = 0.5, rather than using base population allele frequencies estimates (which could be difficult to estimate especially in absence of complete pedigree data). Thus, matrix G was computed using allele frequencies fixed at 0.5.
Detection of Common Runs of Homozygosity
Chromosomal segments presenting ROH hotspots were defined as ROH islands or common ROH. In order to identify such genomic regions, we used two different strategies. First, we used the clustering algorithm implemented in SNP & Variation Suite v7.6.8, which identifies clusters of contiguous set of SNPs with size > smin, where every SNP has at least nmin samples presenting a run. Clusters were identified based on a fixed minimum cluster size of smin = 0.5 Mb for varying minimum number of samples: 127 (10%), 255 (20%), 319 (25%), and 639 (50%). In order to assess the sensitivity of the algorithm to parameter settings in ROH detection, we repeated the analysis using minimum numbers of 30 or 150 SNPs in a run, maximum gap sizes of 100 kb or 500 kb, and 0 or 2 heterozygous genotypes as variable parameters.
Alternatively, we calculated locus autozygosity (FL) following Kim et al. (2013). Briefly, for each SNP, animals were scored as autozygous (1) or non-autozygous (0) based on the presence of a ROH encompassing the SNP. Then, the locus autozygosity was simply computed as:
where Si is the autozygosity score of individual i, and n is the number of individuals. In essence, FL represents the proportion of animals with scores equal to 1 (i.e., that present a ROH enclosing the marker), thus it summarizes the level of local autozygosity in the sample.
Results and Discussion
Distribution of ROH Length
After filtering, 668,589 SNP marker genotypes across 1,278 animals were retained for analyses. The average, median, minimum and maximum ROH length detected across all chromosomes were 1.26, 0.70, 0.50, and 70.91 Mb, respectively, suggesting this specific Nellore cattle population experienced both recent and remote autozygosity events. Segments as large as 10 Mb are traceable to inbreeding that occurred within the last five generations (Howrigan et al., 2011), and a total of 942 samples (73.7%) presented at least one homozygous fragment larger than 10 Mb. Therefore, it is likely that these long ROH are signatures of the extended use of recent popular sires.
Distribution of Genome-Wide Autozygosity
The distributions of Gi, i and FROH based on autosomal ROH of different minimum lengths (>0.5, >1, >2, >4, >8 or >16 Mb) are shown in Figure 1. Although the inclusion of the X chromosome did not cause substantial differences in the calculation of genome-wide FROH (Supplementary Figure S1), we focused on the estimates using only autosomes for the ease of comparison with other studies. The skewness of the autosomal FROH distribution increased as the minimum fragment length increased, ranging from 1.56 for FROH > 0.5 Mb to 3.98 for FROH > 16 Mb. The number of animals with FROH = 0 also increased as the minimum ROH length increased, starting at 12 (0.94%) for FROH > 2 Mb and increasing to 827 (64.71%) for FROH > 16 Mb. Under the assumption of the relationship between ROH length and age of autozygosity, these findings show that varying the minimum ROH length in the calculation of FROH can be useful to discriminate animals with recent and remote autozygosity.
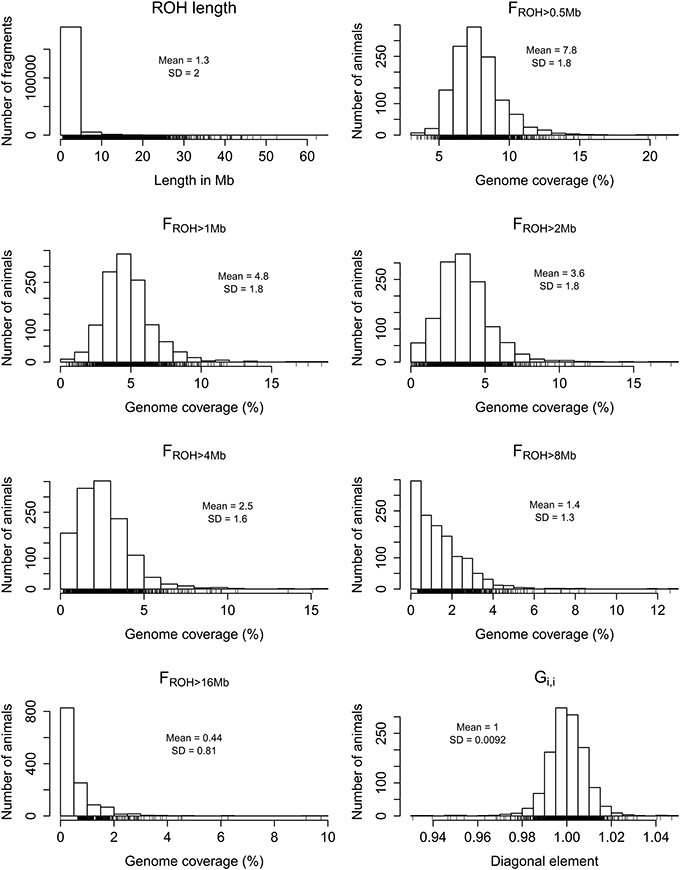
Figure 1. Frequency distributions of all detected runs of homozygosity (ROH) across samples, percentage of autosomal genome coverage by ROH (FROH) of different minimum lengths (>0.5, >1, >2, >4, >8, and >16 Mb), and diagonal elements of the realized genomic relationship matrix (Gi, i).
As shown in Figure 2, the correlation between autosomal FROH > 1 Mb and Gi, i (r = 0.69) was close to the ones reported by Ferenčaković et al. (2013b) for the comparison between FROH > 1 Mb derived from the HD panel and pedigree estimates in Brown Swiss (r = 0.61), Pinzgauer (r = 0.62), and Tyrol Gray (r = 0.75). Similar correlations were observed when the X chromosome was included in the analysis (Supplementary Figure S2). McQuillan et al. (2008) also reported correlations between FROH and pedigree estimates in human European populations ranging from 0.74 to 0.82. Considering that VanRaden (2008) proposed G as a proxy for a numerator relationship matrix obtained from highly reliable and recursive pedigree data, we expect that the correlations found for Gi, i are fair approximations to the ones we would have found if complete pedigree data was available.
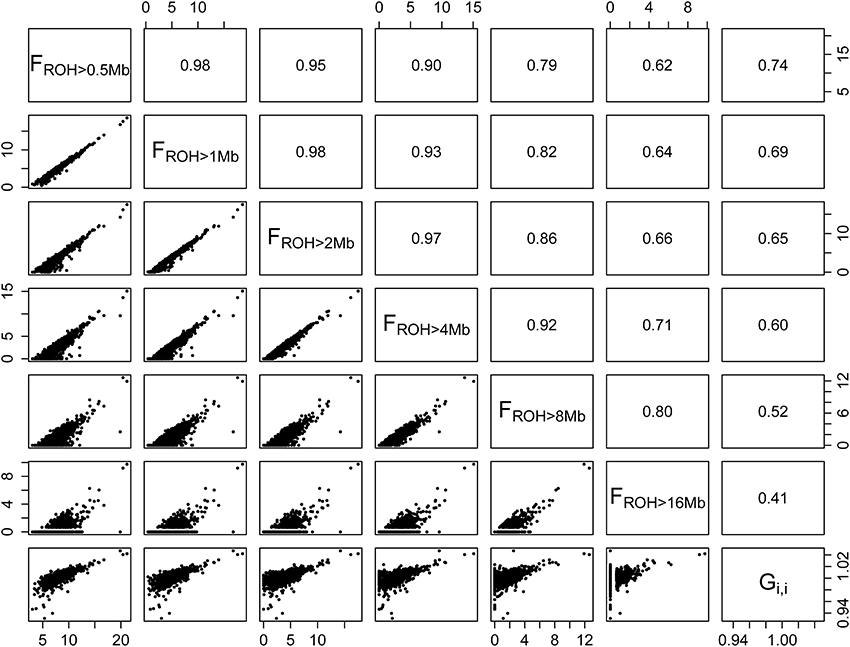
Figure 2. Scatterplots (lower panel) and correlations (upper panel) of percentage of autosomal genome coverage by runs of homozygosity (FROH) of different minimum lengths (>0.5, >1, >2, >4, >8, and >16 Mb) and diagonal elements of the realized genomic relationship matrix (Gi, i). The last column of panels on the right indicates that the correlation between FROH and Gi, i decreases as a function of minimum fragment size.
In the present study, correlations between FROH and Gi, i decreased as a function of different ROH length (Figure 2). This may be due to the properties of the G matrix, which is based on individual loci, whereas FROH is based on chromosomal segments. Ferenčaković et al. (2013b) showed that medium density SNP panels, such as the Illumina® BovineSNP50, systematically overestimate FROH when segments shorter than 4 Mb are included in the calculations, while the Illumina® BovineHD panel is robust for the detection of shorter segments. Hence, although the inclusion of short length ROH in the calculation of FROH may be desirable for autozygosity estimates accounting for remote inbreeding, there is a compromise between SNP density, minimum ROH length and false discovery of ROH. Since the HD panel allows for the detection of short ROH, in this section we focused on the results obtained with FROH > 1 Mb as it presented the second highest correlation with Gi, i and is comparable with previous studies.
The minimum, average, median, and maximum autosomal FROH > 1 Mb across all animals were 0.43, 4.79, 4.58, and 18.55%, respectively. The animal presenting the highest autozygosity value (18.55%) exhibited 69 ROH > 1 Mb encompassing 465.66 Mb of the total autosomal genome extension covered by markers (2.51 Gb), with a mean ROH length of 6.75 ± 9.20 Mb, and a maximum segment length of 43.79 Mb. The least inbred animal presented 8 ROH > 1 Mb, summing up only 10.72 Mb, with an average length of 1.34 ± 0.46 Mb and a maximum of 2.43 Mb.
The coefficient of variation (here denoted as the ratio of the standard deviation to the mean) of the FROH > 1 Mb distribution was 37.5%, indicating moderate variability in autozygosity levels in this sample. In spite of the average genome coverage by ROH of 4.58% may seem to indicate moderate inbreeding levels for classical standards, it has to be considered that incomplete pedigree data usually fails to capture remote inbreeding, so that traditional inbreeding estimates based on pedigree are only comparable with FROH calculated over large ROH lengths, which in the present study were close to 0%.
Compared to other cattle populations, this sample of Nellore cows presented a lower average autozygosity. For instance, Ferenčaković et al. (2013b) reported average autosomal FROH > 1 Mb of 15.1%, 6.2%, and 6.6% for samples of the Bos taurus breeds Brown Swiss, Pinzgauer, and Tyrol Gray, respectively. Also, the effective population size estimated for this Nellore sample was approximately 362 animals (Supplementary Material), which is consistent with the low genome average LD reported by other studies (McKay et al., 2007; Espigolan et al., 2013; Pérez O'Brien et al., 2014) and indicative of a non-inbred population.
Distribution of Chromosome-Wise Autozygosity
The averages of the chromosome-wise FROH > 0.5 Mb across samples are shown in Figure 3. Chromosome X exhibited a substantially higher average autozygosity when compared to the autosomes. Importantly, we found no evidence for a smaller effective population size for the X chromosome in comparison to the autosomal genome (Supplementary Material). This may be due to the mode of inheritance of the X chromosome, which is hemizygous in the male lineage and therefore more susceptible to bottlenecks and drift even under assumptions of balanced numbers of males and females (Gottipati et al., 2011).
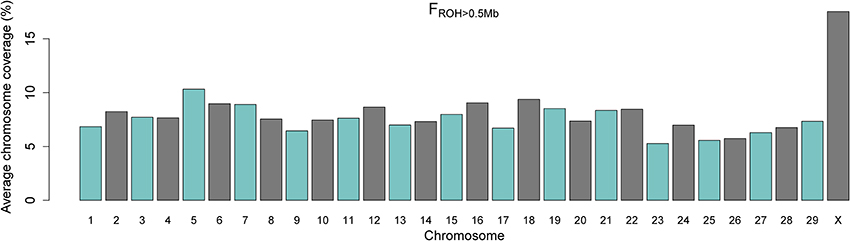
Figure 3. Barplot of average percentage of chromosome coverage by runs of homozygosity (FROH) of minimum length of 0.5 Mb.
An alternative explanation is that the gene content and the sex-specific copy number of the X chromosome is under stronger selective pressure in comparison to autosomal DNA (Hammer et al., 2010; Deng et al., 2014). In both hypotheses, this higher autozygosity may reflect historical and demographical events. In the early 20th century, when more frequent importation of Nellore cattle to Brazil was initiated, the indigenous herds mainly consisted of descendants from taurine (Bos taurus) cattle imported since the late 15th century after the discovery of America (Ajmone-Marsan et al., 2010). In spite of the use of taurine dams for breeding during the early establishment of Nellore cattle in Brazil, the decades that followed were marked by intense backcrossing to Nellore bulls, causing most of the taurine contribution to be swept out from the Nellore autosomal genome (Utsunomiya et al., 2014). However, it is well-established that taurine mitochondrial DNA is prevalent in Nellore cattle, as it is a strict maternal contribution (Meirelles et al., 1999). Therefore, the X chromosome may have experienced a greater drift than the autosomal genome due to limited number of founders. The levels of taurine introgression still segregating in the X chromosome in this herd remain unclear.
Identification of Common ROH
Table 1 presents the results obtained from the ROH clustering analysis. The algorithm was robust in respect to gap size between SNPs, but substantial differences were observed when the number of consecutive SNPs and the number of heterozygous genotypes were modified. Few common ROH were identified even when the minimum number of samples in the cluster was 10%, indicating that ROH distribution is not uniform across the genome. In fact, despite of the occurrence of 99.98% of the SNPs within a ROH of at least one individual, only 19.37% markers were encompassed by ROH observed in 10% or more of the samples. This finding is similar to that reported by Ferenčaković et al. (2013b), and is consistent with the stochastic nature of meiotic recombination. This suggests that the ongoing selection for weight, carcass and reproductive traits in this population has not yet created detectable ROH-based selection signatures related to production.
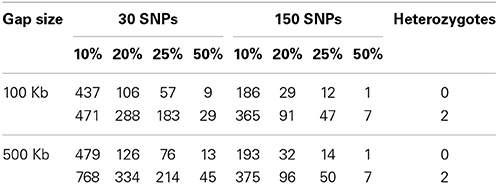
Table 1. Detection of common runs of homozygosity according to different number of consecutive SNPs, percentage of animals, gap size, and number of heterozygous genotypes.
The calculations of locus autozygosity were consistent with the cluster analysis using 150 SNPs and 2 heterozygous genotypes, regardless of permitted gap size (Figure 4). Seven distinct genomic regions, four of them on chromosome X, presented strong hotspots of autozygosity, where over half of the samples (n = 639) contained a ROH. The common ROH on the X chromosome are difficult to be discussed as they span several millions of bases, encompassing hundreds of genes and making functional explorations unfeasible. Besides, the assembly status of X chromosome is poorer than the autosomal ones. Hence, we focused on the three autosomal regions on chromosomes 4, 7, and 12. The three regions were relatively short, ranging from 0.73 to 1.43 Mb. For this range of ROH length, the expected number of generations since the common ancestor is estimated between 35 and 69 (Howrigan et al., 2011). Assuming a cattle generation interval of 5 years, these inbreeding events may have occurred between 175 and 345 years ago. Although this estimate does not account for birth date and overlapping generations, these remote autozygosity events are likely to predate the foundation of the Nellore breeding programs, and therefore expected to be related to natural selection, random drift or population bottlenecks.
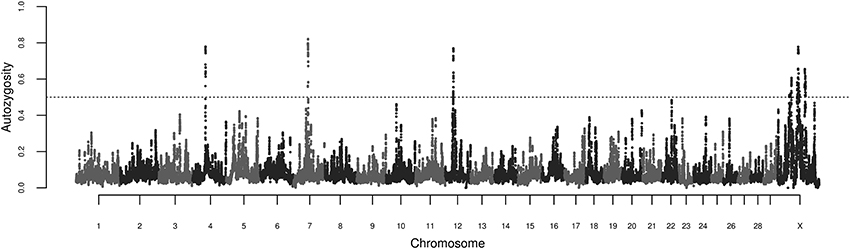
Figure 4. Manhattan plot of genome-wide locus autozygosity in Nellore cows. The dashed line represents the 50% threshold.
The most autozygous locus was found at chromosome 7:51605639-53035752. This region was previously reported in genome-wide scans for signatures of selection in cattle through the comparison of Bos taurus and indicus breeds via FST analysis (Bovine HapMap Consortium, 2009; Porto-Neto et al., 2013) and was detected as a ROH hotspot in an analysis of three taurine and indicine breeds each (Sölkner et al., 2014). This region has been implicated in the control of parasitemia in cattle infected by Trypanosoma congolense (Hanotte et al., 2003), and is orthologous to the human chromosome segment 5q31-q33, known as the Th2 cytokine gene cluster, which has been shown to be implicated in the control of allergy and resilience against infectious diseases such as malaria (Garcia et al., 1998; Rihet et al., 1998; Flori et al., 2003; Hernandez-Valladares et al., 2004) and leishmaniasis (Jeronimo et al., 2007). The region also flanks SPOCK1, a candidate gene for puberty both in humans (Liu et al., 2009) and cattle (Fortes et al., 2010). Although fertility and resistance to infectious diseases are candidate biological drivers of this ROH hotspot, the gene and the phenotype underlying this putative selection signature are unknown.
The common ROH at 12:28433881-29743057 identified in the present study also overlaps a common ROH hotspot (Sölkner et al., 2014) and a region of divergent selection between Bos taurus and Bos indicus cattle (Gautier et al., 2009; Porto-Neto et al., 2013), and the segment encompasses the human ortholog BRCA2, involved in Fanconi anemia in humans (Howlett et al., 2002). A signature of selection nearby the 4:46384250-47113352 region detected here has also been reported by Gautier and Naves (2011), but the genes involved and the selective pressure remain uncharacterized.
Conclusions
We used high-density SNP genotypes to successfully characterize autozygosity in Nellore cows under artificial selection for reproductive, carcass and weight traits. We have shown that, although the massive use of relatively few sires and artificial insemination has generated long stretches of homozygous haplotypes in the genomes of over 70% of these animals, inbreeding levels were considerably low in this population. We also found few genomic regions with high homozygosity across individuals, suggesting that the ongoing selection for reproductive, weight and carcass traits in this population is not very intensive or too recent to have left selection signatures in the form of ROH. Furthermore, the current common breeding practices of avoiding inbreeding in the mating schemes are antagonistic to additive trait selection, making it hard to maintain ROH signatures in the herds. The three candidate regions under selection identified herein were likely to be contributions from remote ancestors, predating the foundation of the Nellore breeding programs. The selective pressure effects and the genes involved in these regions should be subject of future investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Guilherme Penteado Coelho Filho and Daniel Biluca for technical assistance in sample acquisition. We also thank to Fernando Sebastian Baldi Rey for the manuscript revision and pertinent suggestions. This research was supported by: National Counsel of Technological and Scientific Development (CNPq - http://www.cnpq.br/) (process 560922/2010-8 and 483590/2010-0); and São Paulo Research Foundation (FAPESP - http://www.fapesp.br/) (process 2013/15869-2 and 2014/01095-8). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Mention of trade name proprietary product or specified equipment in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the authors or their respective institutions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2015.00005/abstract
References
Ajmone-Marsan, P., Garcia, J. F., Lenstra, J. A., and The Globaldiv Consortium (2010). On the origin of cattle: how aurochs became cattle and colonized the world. Evol. Anthropol. 19, 148–157. doi: 10.1002/evan.20267
Bjelland, D. W., Weigel, K. A., Vukasinovic, N., and Nkrumah, J. D. (2013). Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 96, 4697–4706. doi: 10.3168/jds.2012-6435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bovine HapMap Consortium (2009). Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–532. doi: 10.1126/science.1167936
Curik, I., Ferenčaković, M., and Sölkner, J. (2014). Inbreeding and runs of homozygosity: a possible solution to an old problem. Livest. Sci. 166, 26–34. doi: 10.1016/j.livsci.2014.05.034
Deng, X., Berletch, J. B., Nguyen, D. K., and Disteche, C. M. (2014). X chromosome regulation: diverse patterns in development, tissues and disease. Nat. Rev. Genet. 15, 367–378. doi: 10.1038/nrg3687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Espigolan, R., Baldi, F., Boligon, A. A., Souza, F. R., Gordo, D. G., Tonussi, R. L., et al. (2013). Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genomics. 14:305. doi: 10.1186/1471-2164-14-305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falconer, D. S., and Mackay, T. F. C. (1996). Introduction to Quantitative Genetics, 4 Edn. (Harlow, UK: Addison-Wesley Longman), 464.
Ferenčaković, M., Hamziæ, E., Gredler, B., Solberg, T. R., Klemetsdal, G., Curik, I., et al. (2013a). Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 130, 286–293. doi: 10.1111/jbg.12012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferenčaković, M., Sölkner, J., and Curik, I. (2013b). Estimating autozygosity from high-throughput information: effects of SNP density and genotyping errors. Genet. Sel. Evol. 45:42. doi: 10.1186/1297-9686-45-42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flori, L., Kumulungui, B., Aucan, C., Esnault, C., Traore, A. S., Fumoux, F., et al. (2003). Linkage and association between Plasmodium falciparum blood infection levels and chromosome 5q31-q33. Genes Immun. 4, 265–268. doi: 10.1038/sj.gene.6363960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fortes, M. R. S., Reverter, A., Zhang, Y., Collis, E., Nagaraj, S. H., Jonsson, N. N., et al. (2010). Association weight matrix for the genetic dissection of puberty in beef cattle. Proc. Natl. Acad. Sci. U.S.A. 107, 13642–136427. doi: 10.1073/pnas.1002044107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia, A., Marquet, S., Bucheton, B., Hillaire, D., Cot, M., Fievet, N., et al. (1998). Linkage analysis of blood Plasmodium falciparum levels: interest of the 5q31-q33 chromosome region. Am. J. Trop. Med. Hyg. 58, 705–709.
Gautier, M., Flori, L., Riebler, A., Jaffrézic, F., Laloé, D., Gut, I., et al. (2009). A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics. 10:550. doi: 10.1186/1471-2164-10-550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gautier, M., and Naves, M. (2011). Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 20, 3128–3143. doi: 10.1111/j.1365-294X.2011.05163.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibson, J., Morton, N. E., and Collins, A. (2006). Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 15, 789–795. doi: 10.1093/hmg/ddi493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gottipati, S., Arbiza, L., Siepel, A., Clark, A. G., and Keinan, A. (2011). Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nat. Genet. 43, 741–743. doi: 10.1038/ng.877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hammer, M. F., Woerner, A. E., Mendez, F. L., Watkins, J. C., Cox, M. P., and Wall, J. D. (2010). The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nat. Genet. 42, 830–831. doi: 10.1038/ng.651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanotte, O., Ronin, Y., Agaba, M., Nilsson, P., Gelhaus, A., Horstmann, R., et al. (2003). Mapping of quantitative trait loci controlling trypanotolerance in a cross of tolerant West African N'Dama and susceptible East African Boran cattle. Proc. Natl. Acad. Sci. U.S.A. 100, 7443–7448. doi: 10.1073/pnas.1232392100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernandez-Valladares, M., Rihet, P., ole-MoiYoi, O. K., and Iraqi, F. A. (2004). Mapping of a new quantitative trait locus for resistance to malaria in mice by a comparative mapping approach with human chromosome 5q31-q33. Immunogenetics 56, 115–117. doi: 10.1007/s00251-004-0667-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howlett, N. G., Taniguchi, T., Olson, S., Cox, B., Waisfisz, Q., De Die-Smulders, C., et al. (2002). Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297, 606–609. doi: 10.1126/science.1073834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howrigan, D. P., Simonson, M. A., and Keller, M. C. (2011). Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics 12:460. doi: 10.1186/1471-2164-12-460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeronimo, S. M., Holst, A. K., Jamieson, S. E., Francis, R., Martins, D. R., Bezerra, F. L., et al. (2007). Genes at human chromosome 5q31.1 regulate delayed-type hypersensitivity responses associated with Leishmania chagasi infection. Genes Immun. 8, 539–551. doi: 10.1038/sj.gene.6364422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keller, M. C., Visscher, P. M., and Goddard, M. E. (2011). Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189, 237–249. doi: 10.1534/genetics.111.130922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, E. S., Cole, J. B., Huson, H., Wiggans, G. R., Van Tassell, C. P., Crooker, B. A., et al. (2013). Effect of artificial selection on runs of homozygosity in u.s. Holstein cattle. PLoS ONE 8:e80813. doi: 10.1371/journal.pone.0080813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lencz, T., Lambert, C., DeRosse, P., Burdick, K. E., Morgan, V., Kane, J. M., et al. (2007). Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 104, 19942–19947. doi: 10.1073/pnas.0710021104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leroy, G. (2014). Inbreeding depression in livestock species: review and meta-analysis. Anim. Genet. 45, 618–628. doi: 10.1111/age.12178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y. Z., Guo, Y. F., Wang, L., Tan, L. J., Liu, X. G., Pei, Y. F., et al. (2009). Genome-wide association analyses identify SPOCK as a key novel gene underlying age at menarche. PLoS Genet. 5:e1000420. doi: 10.1371/journal.pgen.1000420
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacArthur, D. G., Balasubramanian, S., Frankish, A., Huang, N., Morris, J., Walter, K., et al. (2012). A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823. doi: 10.1126/science.1215040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKay, S. D., Schnabel, R. D., Murdoch, B. M., Matukumalli, L. K., Aerts, J., Coppieters, W., et al. (2007). Whole genome linkage disequilibrium maps in cattle. BMC Genet. 8:74. doi: 10.1186/1471-2156-8-74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McQuillan, R., Leutenegger, A. L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372. doi: 10.1016/j.ajhg.2008.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meirelles, F. V., Rosa, A. J. M., Lobo, R. B., Garcia, J. M., Smith, L. C., and Duarte, F. A. M. (1999). Is the American zebu really Bos indicus? Genet. Mol. Biol. 22, 543–546. doi: 10.1590/S1415-47571999000400013
Pérez O'Brien, A. M., Mészáros, G., Utsunomiya, Y. T., Sonstegard, T. S., Garcia, F. J., Van Tassell, C. P., et al. (2014). Linkage disequilibrium levels in Bos indicus and Bos taurus cattle using medium and high density SNP chip data and different minor allele frequency distributions. Livest. Sci. 166, 121–132. doi: 10.1016/j.livsci.2014.05.007
Porto-Neto, L. R., Sonstegard, T. S., Liu, G. E., Bickhart, D. M., Da Silva, M. V., Machado, M. A., et al. (2013). Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics 14:876. doi: 10.1186/1471-2164-14-876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purfield, D. C., Berry, D. P., McParland, S., and Bradley, D. G. (2012). Runs of homozygosity and population history in cattle. BMC Genet. 13:70. doi: 10.1186/1471-2156-13-70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rihet, P., Traoreì, Y., Abel, L., Aucan, C., Traoreì-Leroux, T., and Fumoux, F. (1998). Malaria in humans: Plasmodium falciparum blood infection levels are linked to chromosome 5q31-q33. Am. J. Hum. Genet. 63, 498–505. doi: 10.1086/301967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sölkner, J., Ferenčaković, M., Karimi, Z., Perez O'Brien, A. M., Mészáros, G., Eaglen, S., et al. (2014). “Extremely non-uniform: patterns of runs of homozygosity in bovine populations,” in Proceedings of the 10th World Congress of Genetics Applied to Livestock Production (Vancouver, BC).
Szpiech, Z. A., Xu, J., Pemberton, T. J., Peng, W., Zollner, S., Rosenberg, N. A., et al. (2013). Long runs of homozygosity are enriched for deleterious variation. Am. J. Hum. Genet. 93, 90–102. doi: 10.1016/j.ajhg.2013.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Utsunomiya, Y. T., Bomba, L., Lucente, G., Colli, L., Negrini, R., Lenstra, J. A., et al. (2014). Revisiting AFLP fingerprinting for an unbiased assessment of genetic structure and differentiation of taurine and zebu cattle. BMC Genet. 15:47. doi: 10.1186/1471-2156-15-47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
VanRaden, P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423. doi: 10.3168/jds.2007-0980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
VanRaden, P. M., Olson, K. M., Wiggans, G. R., Cole, J. B., and Tooker, M. E. (2011). Genomic inbreeding and relationships among Holsteins, Jerseys, and Brown Swiss. J. Dairy Sci. 94, 5673–5682. doi: 10.3168/jds.2011-4500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xue, Y., Chen, Y., Ayub, Q., Huang, N., Ball, E. V., Mort, M., et al. (2012). Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 91, 1022–1032. doi: 10.1016/j.ajhg.2012.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Bos indicus, runs of homozygosity, selection, cattle, fertility, disease resistance
Citation: Zavarez LB, Utsunomiya YT, Carmo AS, Neves HHR, Carvalheiro R, Ferenčaković M, Pérez O'Brien AM, Curik I, Cole JB, Van Tassell CP, da Silva MVGB, Sonstegard TS, Sölkner J and Garcia JF (2015) Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 6:5. doi: 10.3389/fgene.2015.00005
Received: 31 October 2014; Paper pending published: 03 December 2014;
Accepted: 07 January 2015; Published online: 29 January 2015.
Edited by:
Paolo Ajmone Marsan, Università Cattolica del S. Cuore, ItalyReviewed by:
Yniv Palti, United States Department of Agriculture, USANicolo Pietro Paolo Macciotta, University of Sassari, Italy
Copyright © 2015 Zavarez, Utsunomiya, Carmo, Neves, Carvalheiro, Ferenčaković, Pérez O'Brien, Curik, Cole, Van Tassell, da Silva, Sonstegard, Sölkner and Garcia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José F. Garcia, Laboratório de Bioquímica e Biologia Molecular Animal, Departamento de Apoio, Produção e Saúde Animal, Faculdade de Ciências Agrárias e Veterinárias, UNESP – Universidade Estadual Paulista, Rua Clóvis Pestana 793, Araçatuba, São Paulo, 16050-680, Brazil e-mail:amZnYXJjaWFAZm12YS51bmVzcC5icg==