 Moulay A. Alaoui-Jamali
Moulay A. Alaoui-Jamali Grégoire B. Morand
Grégoire B. Morand Sabrina Daniela da Silva
Sabrina Daniela da Silva- 1Departments of Medicine and Oncology, Segal Cancer Centre and Lady Davis Institute for Medical Research, Sir Mortimer B. Davis-Jewish General Hospital, McGill University, Montreal, QC, Canada
- 2Department of Otolaryngology-Head and Neck Surgery, Sir Mortimer B. Davis-Jewish General Hospital, McGill University, Montreal, QC, Canada
Advances in high-throughput genomic-scanning have expanded the repertory of genetic variations in DNA sequences encoding ErbB tyrosine kinase receptors in humans, including single nucleotide polymorphisms (SNPs), polymorphic repetitive elements, microsatellite variations, small-scale insertions and deletions. The ErbB family members: EGFR, ErbB2, ErbB3, and ErbB4 receptors are established as drivers of many aspects of tumor initiation and progression to metastasis. This knowledge has provided rationales for the development of an arsenal of anti-ErbB therapeutics, ranging from small molecule kinase inhibitors to monoclonal antibodies. Anti-ErbB agents are becoming the cornerstone therapeutics for the management of cancers that overexpress hyperactive variants of ErbB receptors, in particular ErbB2-positive breast cancer and non-small cell lung carcinomas. However, their clinical benefit has been limited to a subset of patients due to a wide heterogeneity in drug response despite the expression of the ErbB targets, attributed to intrinsic (primary) and to acquired (secondary) resistance. Somatic mutations in ErbB tyrosine kinase domains have been extensively investigated in preclinical and clinical setting as determinants for either high sensitivity or resistance to anti-ErbB therapeutics. In contrast, only scant information is available on the impact of SNPs, which are widespread in genes encoding ErbB receptors, on receptor structure and activity, and their predictive values for drug susceptibility. This review aims to briefly update polymorphic variations in genes encoding ErbB receptors based on recent advances in deep sequencing technologies, and to address challenging issues for a better understanding of the functional impact of single versus combined SNPs in ErbB genes to receptor topology, receptor-drug interaction, and drug susceptibility. The potential of exploiting SNPs in the era of stratified targeted therapeutics is discussed.
An Overview of ErbB Receptor Signaling
Since the pioneering discovery of epidermal growth factor receptor (EGFR), the founding member of the ErbB protein tyrosine kinase family by Stanley Cohen, a contribution recognized as 1986 Nobel Prize of Medicine, a remarkable progress has been made in the characterization of this receptor family, which in addition to EGFR/ErbB1/HER1, includes ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4 (Chinkers and Cohen, 1981). These homologous receptors are type 1 transmembrane proteins that share a common structure with a large glycosylated ligand binding extracellular region composed of domains I-IV, a single hydrophobic transmembrane region, a cytoplasmic domain with tyrosine kinase activity, and a C-terminal tail containing multiple phosphorylation sites required for propagation of downstream signaling (Figure 1). The strongest homology among ErbB receptors is seen in the kinase domain (59–82%) followed by the extracellular domain (36–48%) and the C-terminal tail (24–33%) (Gschwind et al., 2004). There are unique features characterizing two members of this family: the first is the lack of kinase activity in ErbB3 receptor, a deficiency proposed to be due to substitution of a conserved aspartate (Asp/D) residue to an asparagine (Asn/N) in the kinase domain. Therefore, ErbB3 activation relies on heterodimerization with the other members of the family (Citri et al., 2003). The second is the orphan ErbB2, which lacks a specific ligand. Structural studies have revealed that the ligand-binding site of ErbB2 is constitutionally in activate conformation, and thus prone for heterodimerization and transphosphorylation through interactions with the other ligand-activated ErbB receptors (Garrett et al., 2003).
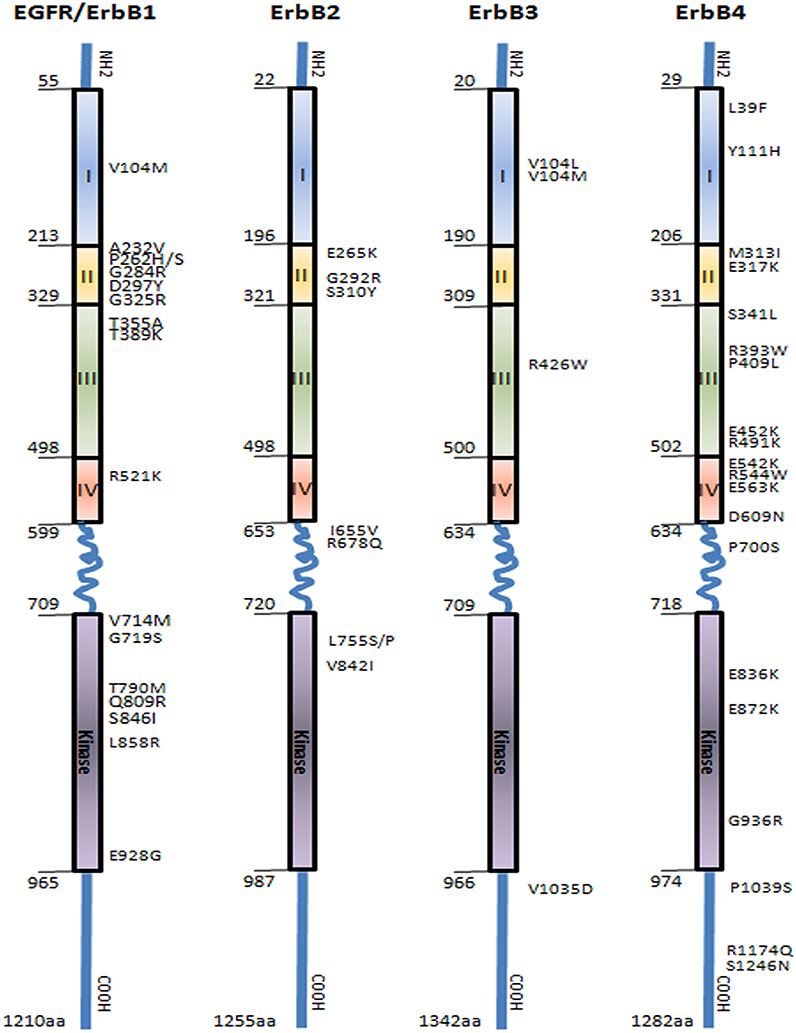
Figure 1. Representation of epidermal growth factor receptors family (EGFR/ErbB1, ErbB2, ErbB3, and ErbB4) showing the distribution of the main SNPs mutations in the extracellular domain, transmembrane region, and intracellular domain comprising the tyrosine kinase and autophosphorylation sites. Boxes represent functional domains: I, extracellular domain sub-region I; II, extracellular domain sub-region II; III, extracellular domain sub-region III; IV, extracellular domain sub-region IV. (Prickett et al., 2009; Yarden and Pines, 2012).
ErbB receptors have a propensity to be activated by a wide range of ligands, which include epidermal growth factor (EGF), transforming growth factor α (TGFα), heparin-binding epidermal growth factor, amphiregulin, betacellulin, epiregulin, and neuregulins. These ligands induce a broad array of ErbB receptor homo- and heterodimerization culminating into a diverse cell transduction signaling of utmost importance to both physiological and pathological contexts, including embryonic development (Gassmann et al., 1995; O-Charoenrat et al., 2002; Stein and Staros, 2006; Birchmeier, 2009). The general mode of activation of ErbB receptors involves changes in allosteric conformations due to intermolecular mechanisms triggered by ligand activation and/or activating mutations (Monsey et al., 2010; Lemmon et al., 2014). Both homo- and heterodimerization with partners involve primarily the kinase domain and C-lobe surface structures of ErbB receptors (Burgess et al., 2003; Zhang et al., 2006; Monsey et al., 2010). For example, the ligand binds between domains I and III (extracellular region) of the EGFR and ErbB3 receptors to induce conformational change and subsequent dimerization and kinase activation. A widely accepted model of activation stipulates that dimerization occurs via an asymmetric mode where a C-lobe of an acceptor monomer and the N-lobe of a donor monomer in the intracellular juxtamembrane region interacts. Mutations in the asymmetric dimer interface can abolish kinase activation (Zhang et al., 2006, 2007; Red Brewer et al., 2009). In this mode of activation, C-lobe surface amino acids, the extracellular domain, the transmembrane domain, and the intracellular juxtamembrane region all can influence receptor dimerization and the intensity of activation (Burgess et al., 2003; Thiel and Carpenter, 2007; Lemmon, 2009). This suggests that subtle molecular changes in the receptor structure due to single nucleotide polymorphisms (SNPs) can impact on ErbB kinase activity as well as ErbB receptor interaction with ligands, downstream partners, or with inhibitors including anti-ErbB monoclonal antibodies (Shigematsu et al., 2005; Fan et al., 2008).
ErbB Receptors: A Success Story in Discovery of Therapeutic Targets and Limitations
In general, ErbB receptors are expressed at low levels on the surface of normal adult epithelial cells (O-Charoenrat et al., 2002). Extensive preclinical studies in cell lines and transgenic mouse models established the contribution of dysfunctional ErbB signaling, due to overexpression and hyperactivation, to carcinogenesis (Bargmann et al., 1986; Bouchard et al., 1989). In cancer cells, aberrant activation due to gene overexpression and/or amplification and (less commonly) mutation scan promote enhanced cell proliferation, resistance to apoptosis, and higher cell invasiveness. In several cancer types, in particular breast, ovarian, gastric, and non-small cell carcinoma of the lung, amplification and/or overexpression of specific members of the ErbB receptor family have been associated with poor clinical prognosis, including high incidence of metastasis and recurrence. In breast cancer, molecular profiling studies have enabled identification of ErbB2-positivebreast cancer molecular subtype, which represents up to 30% of breast cancers. In this subtype, the amplified and/or overexpressed ErbB2 induces enrichment of several downstream ErbB2-regulated genes (Slamon et al., 1989; Cancer Genome Atlas Network, 2012).
The remarkable progress in the understanding the biology of ErbB signaling in cancer has led to the discovery of several targeted agents against ErbB members, including small molecule tyrosine kinase inhibitors (TKIs) and monoclonal antibodies (MAbs). At present, small molecule inhibitors under clinical trials or approved by the US Food and Drug Administration (FDA) are either reversible or irreversible inhibitors that bind to the ATP-binding site in the kinase domain of ErbB receptors (Table 1). In addition to TKIs, several MAbs that target either EGFR (e.g., cetuximab), ErbB2 (trastuzumab), or ErbB2 dimerization (pertuzumab), are becoming major therapeutics, in particular for the ErbB2-positive breast cancer subtype where combination of anti-ErbB2, e.g., trastuzumab with conventional chemotherapy, has drastically changed the patients' prognosis and outcome (Piccart-Gebhart et al., 2005; Romond et al., 2005). As well, combination of MAbs with TKIs, e.g., trastuzumab plus anti-EGFR/anti-ErbB2 such as lapatinib or pertuzumab, revealed of clinical benefit for recurrent cancers following trastuzumab treatment for instance (Geyer et al., 2006; Joensuu et al., 2006). More recently, a double-blind placebo-controlled international multicentre clinical trial (CLEOPATRA)designed to evaluate the efficacy and safety of pertuzumab+trastuzumab+docetaxel (pertuzumab arm) versus placebo+trastuzumab+docetaxel (control arm) showed a survival improvement in the pertuzumab arm and also demonstrated that ErbB2 marker is suited for patient selection for the pertuzumab-based regimen in ErbB2-positive metastatic breast cancer or locally recurrent unresectable tumor (Baselga et al., 2014; Fleeman et al., 2015).
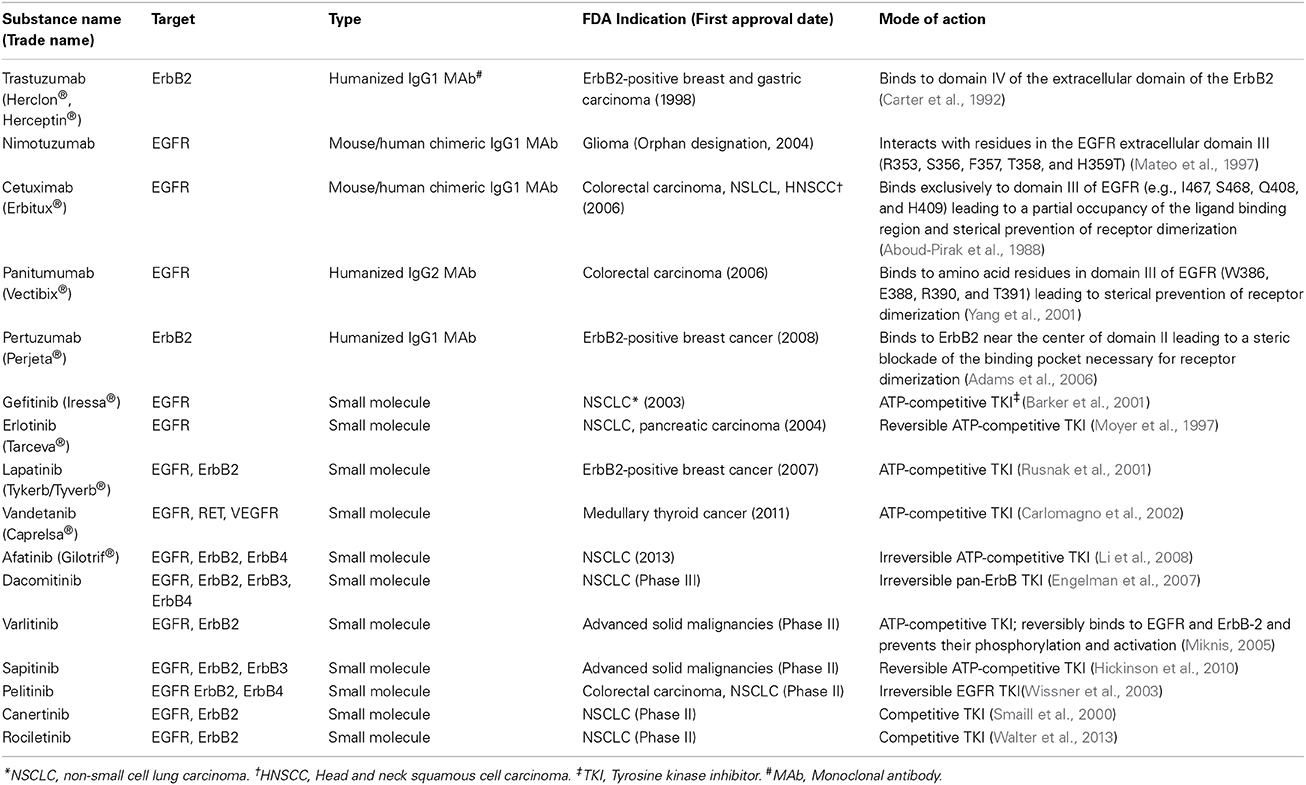
Table 1. Representative FDA approved and experimental anti-ErbB therapeutic agents.
Despite of these successes, there remain major obstacles in achieving sustained response or cure with anti-ErbB inhibitors. The first obstacle refers to de novo or intrinsic resistance seen in patients expressing the ErbB targets yet failing to respond to anti-ErbB. This form of resistance is estimated to occur in up to ~20 and ~70% of ErbB2-positive patients with early and metastatic breast cancer treated with trastuzumab monotherapy, respectively (Harris et al., 2007; Wolff et al., 2007). The second type of resistance is the acquired form attributed to drug selection and can be seen in over 50% of patients who initially respond to anti-ErbB therapeutics but later become refractory to these drugs (Harris et al., 2007; Wolff et al., 2007).
Studies in preclinical models revealed intrinsic and acquired resistance to anti-ErbB therapeutics to involve multifactorial mechanisms both tumor- and host-related (Rexer and Arteaga, 2012). Briefly, mechanisms of primary drug resistance include emergence of pre-existing tumor cell subpopulations with (i) specific mutations in ErbB genes affecting the drug-target interaction; (ii) alternate splicing of ErbB gene leading to truncated isoforms of the receptors not recognized by the inhibitor, e.g., trastuzumab resistance in breast cancer has been associated with the expression of a truncated p95-ErbB2 receptor isoform that lacks trastuzumab antibody binding site; (iii) decreased MAb-induced cell-mediated cytotoxicity in ErbB2-positive cells such as due to an alteration in the binding of immune cells to Fc region of the MAb; and (iv) failure of MAb such as trastuzumab to induce ErbB2 receptor shedding, internalization, and/or degradation by ubiquitination (Rexer and Arteaga, 2012).
In contrast to intrinsic resistance, a broader range of mechanisms induced by drug pressure can mediate acquired resistance. These include secondary mutations that affect drug-ErbB target interaction (the most common are mutations in the TK domain), activation of compensatory signaling pathways able to bypass signaling blockade by the ErbB inhibitors, inefficient cellular transport/uptake of the drug, enhanced drug inactivation such as by phase II enzymes, up-regulation of survival signals, and altered drug pharmacokinetics and pharmacodistribution in the host. Targeting some of these mechanisms has provided alternative approaches to overcome resistance to anti-ErbB, e.g., combination of MAb such as trastuzumab with lapatinib or pertuzumab, the use of ado-trastuzumabemtansine (T-DM1), or combinations of trastuzumab with heat shock protein-90 (HSP90) inhibitors, PI3K inhibitors, and immune checkpoint modulators in combination with trastuzumab (Amiri-Kordestani et al., 2014).
In contrast to cancer-associated somatic mutations, single nucleotide polymorphisms (SNPs) are widespread in ErbB genes. In general, SNPs represent the most common genetic variations that occur in over 1.5% of healthy human population. In contrast somatic mutations are acquired genetic changes present in only a subset of cells. While most SNPs are silent with no apparent impact on physiological functions, some SNPs may impact on individual's susceptibility to anti-ErbB target therapeutics. The following chapters review the biological impact and predictive value of SNPs in ErbB genes.
Potential Impact of Single Nucleotide Polymorphisms in ErbB Genes on Receptor Activity, Drug-Receptor Interaction and Drug Efficacy
SNPs are genetic variations that occur at a single position in a DNA sequence with fairly high frequency in the general population. SNPs are widespread in genes encoding all the 4 members of ErbB receptor family. Polymorphic point mutations could lead to variations in the amino acid sequence, however, SNPs can also occur in noncoding regions of DNA. In addition, mounting evidence support that genetic polymorphisms can also occur in microRNA-encoding sequences and this can contribute to phenotypic differences seen in earlier studies examining SNPs in relation to cancer via distinct regulatory mechanisms (Jin and Lee, 2013). Clearly, the contribution of SNPs in miRNA to pharmacogenetic in general requires in-depth investigations.
Genetic association studies have supported specific SNPs in ErbB genes to impact on ErbB biological activity and hence may represent potential markers for understanding individual susceptibility to anti-ErbB therapeutics. Below we are discussing the status of SNPs challenges to exploit these for optimization of the efficacy of anti-ErbB targeted therapeutics.
EGFR
Several polymorphic variations in the promoter region of EGFR have been reported to impact on gene expression and function (Figure 1, Supplemental Table 1). The CA-simple sequence repeat 1 (CA-SSR1) is a highly polymorphic locus (14–21 CA dinucleotide repeats) situated in the first intron of EGFR. A lower CA-CSSR1 repeat number was found to modulate EGFR transcription in vivo and in vitro and correlated with increased transcription and protein expression (Gebhardt et al., 1999). The allele size distribution of CA-SSR1 demonstrates ethnic differences with East Asian having longer allele than individuals of European descent or African-Americans (Liu et al., 2011). Conversely, low CA-SSR1 number was associated with increased breast cancer risk in young women (Brandt et al., 2004), while a high CA-SSR1 number was correlated with better prognosis in luminal A breast cancer (Leite et al., 2014). Studies analyzing Asian population showed that the low CA repeat polymorphism is associated with better drug response and prolonged survival (Ichihara et al., 2007; Han et al., 2014). A second key polymorphism within EGFR pathways (R521K, previously assigned as R497K, rs2227983) is a single nucleotide change (G–A) in the codon 497 of the EGFR (Figure 1) leading to an arginine-lysine substitution in the extracellular domain of the subdomain IV. This variant is thought to decrease the ligand binding affinity of the receptor, thus dampening downstream impact of activation of downstream target such a CMYC and VEGF (Wang et al., 2007; Hsieh et al., 2012). R521K polymorphism has been associated with good prognostic features in breast cancer especially in patients with luminal A subtypes, as well as non-small cell lung, colorectal, and head and neck cancers (Wang et al., 2007; Sasaki et al., 2009; Hsieh et al., 2012; Leite et al., 2014). Other studies have reported that R521K polymorphism is associated with favorable outcome in cetuximab-based and 5-FU-based chemotherapy but shows negative correlation in gefitinib-based chemotherapy (Wang et al., 2007; Lurje et al., 2008; Sasaki et al., 2009; Dahan et al., 2011; Hsieh et al., 2012; Leite et al., 2014). This indicate that SNP in individual gene, whether it would affect cancer cell proliferation or not, may influence the chromosome instability (CIN) and even that cancer progression is not altered remarkably it can result different profiles of clinical response to chemotherapy; however, further investigation still is necessary and the field is very promising.
Recent genome-wide studies (GWAS) have identified an EGFR polymorphisms (rs2252586 and rs11979158) to be associated with increased glioma risk (Sanson et al., 2011). Two further polymorphisms in the promoter region of EGFR, SNPs -216 (rs712829) and -191 (rs712830), were associated with increased protein synthesis; these SNPs are rare in East Asian populations compared to other ethnicities (Liu et al., 2011). SNPs -216 variant is located -216 upstream from the initiator ATG and the change of nucleoside is guanine to thymine (G/T or T/T) in a important binding site for the transcription factor SP1 that is necessary for activation of EGFR promoter activity (Liu et al., 2005). This variant is frequent in individuals of European descent and African-American than Asians (Liu et al., 2005) and has been associated with increased intrinsic gene expression (Liu et al., 2011). Furthermore, patients harboring SNP-216 and CA-SSR1 germline polymorphisms were more likely to present somatic mutations of EGFR, in particular microdeletions of exon 19. These deletions are common in lung cancer and were reported to strongly predict response to TKI (Liu et al., 2011).
Besides polymorphic variations, somatic mutations identified in ErbB genes, including EGFR (Figures 1, 2) greatly influence in the receptor activity and interaction with TKIs, it is of fundamental importance to investigate the cooperation between SNPs and cancer-associated somatic mutations in ErbB genes to better understand the utility of ErbB polymorphisms to predict individual susceptibility to anti-ErbB therapeutics.
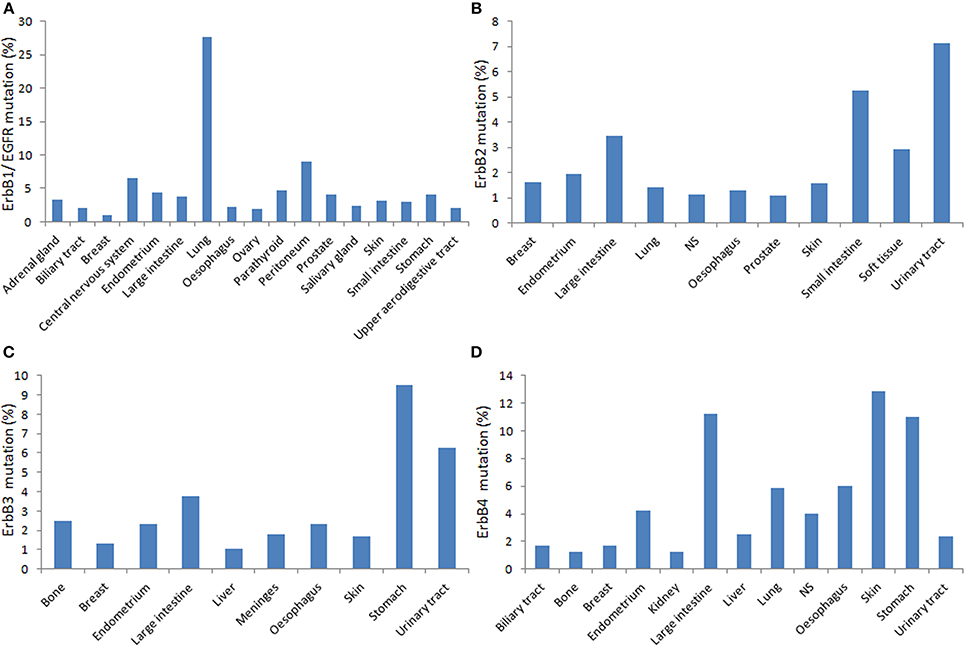
Figure 2. Distribution of the relative percentage of somatic mutations among different cancers types according to the cataloge of somatic mutations in cancer (COSMIC) database in human primary tumors. Each panel shows mutations rates for corresponding ErbB (A. EGFR; B. ErbB2; C. ErbB3; D. ErbbB4) per tumor tissue affected (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).
ERBB2
The most investigated ERBB2 polymorphism related to cancer risk is I655V (rs1136201) located at the codon 655 (ATC/isoleucine to GTC/valine) in the transmembrane domain of the receptor (Figure 1, Supplemental Table 2) (Kuraoka et al., 2003; Puputti et al., 2006; Han et al., 2014). The amino acid change could result in increased protein tyrosine kinase activity. Tumors harboring the I655V polymorphism showed higher tumorigenic potential in preclinical models and breast cancers expressing this variant also manifest an aggressive phenotype (Han et al., 2014). I655Vpolymorphism confers a higher risk of trastuzumab-associated cardiotoxicity but does not directly affect patients' survival, and hence it may be useful for the prevention of cardiovascular events in patient receiving after therapy (Beauclair et al., 2007). Further, a systematic review and meta-analysis studying ErbB2 polymorphism in over 45,000 breast cancer patients showed that there is a lack of experiments replication for association studies between the polymorphic variants and cancer risk, as well as lack of quality control of genotyping assays or publication bias influencing the results (Dahabreh and Murray, 2011).
ERBB3
Compared to the other members of the ErbB family, ErbB3 receptor has been under-investigated in part because it was initially identified as an inactive kinase receptor whose function depends on interaction with ErbB partners, primarily through heterodimerization. Therefore, functional polymorphisms in ERBB3 were long neglected for this pseudo-kinase (Shi et al., 2010). Recent studies revealed at least 1091 polymorphic forms in human ERBB3 gene (dbSNP database; http://www.ncbi.nlm.nih.gov/snp) (Figure 1, Supplemental Table 3). However, only one study reported clinical relevance of the polymorphism 276C/T (rs2271188) in the promoter region of the ERBB3. This variant affected gene expression contributing to genetic susceptibility and was associated with higher risk of lung cancer in never-smokers (Sung et al., 2012). Noticeable, one research group reported the association of ErbB3 overexpression with resistance to tamoxifen as well as to small molecule TKI in breast cancer; however, a possible contribution of SNPs was not addressed (Liu et al., 2007).
ERBB4
Multiple SNPs in the ERBB4 promoter region were associated with adverse clinical features (Figure 1, Supplemental Table 4). The frequency and prognostic significance of two ERBB4 promoter region variants, -782G/T (rs62626348) and -815A/T (rs62626347), were investigated in a large cohort of breast cancer patients. 782G/T conferred highest cancer risk (Rokavec et al., 2007) but another study observed an association of this variant with well-differentiated breast tumor (Kurppa et al., 2014). The variant815A/T was correlated with poor survival and proposed as a prognostic marker in high-risk early breast cancer (Kurppa et al., 2014). A genome-wide association study (GWAS) identified a breast cancer risk variant in ERBB4 at 2q34 (rs13393577) to occur in European and Chinese populations. However, the impact of ErbB4 polymorphisms on response to anti-ErbB therapeutics remains to be established.
Conclusion and Perspectives
Anti-ErbB drugs are becoming cornerstone therapeutics for many cancers and in particular for lung and breast cancers subtype where combination of anti-ErbB with conventional chemotherapy has drastically changed the patient's outcome. Yet a dilemma for the oncologist is how to predict with certainty patients likely will respond to primary treatment from those that may be intrinsically resistant despite the presence of the ErbB target. With the advent of the genome-wide scanning technologies, remarkable advances have been made in the identification of single polymorphic variants in ErbB genes. However, the significance of most SNPs leading to amino acid substitutions to ErbB receptor signaling and cell susceptibility to anti-ErbB therapeutics remains to be established. With the availability of technologies to manipulate gene expression in intact cells, such as the clustered regularly interspaced short palindromic repeat (CRISPER) system, allow to develop cell models expressing wild-type versus polymorphic variants for functional studies, which are essential for surveying the implications of ErbB SNPs landscape in greater details, in particular in relation to receptor activity, receptor topology, and response to anti-ErbB therapeutics. This represents the backbone to draw testable hypotheses on SNP-impacts on receptor structure activity and interaction with the inhibitor. This knowledge can be later exploited for the design of more meaningful large-scale SNP association studies. In this respect, among challenging questions is that relevant SNPs may likely produce borderline resistance or sensitivity to anti-ErbB, which is difficult to investigate in the clinical setting. Alternatives for standard clinical trials such as clinical trial simulations may be required to fill this gap and ultimately design alternative molecules that target relevant SNP genetic variants. In summary, identification of ErbB genetic variations that can predict drug response or resistance is a first step toward exploiting the utility of pharmacogenetics to tailor individual ErbB-based therapy regimens, a prerequisite to optimize care cancer.
Author Contributions
Original idea and study design by Moulay A. Alaoui-Jamali. Figures by Sabrina Daniela da Silva, Moulay A. Alaoui-Jamali, Grégoire B. Morand, and Sabrina Daniela da Silva all have actively participated to manuscript drafting, elaboration of tables and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Moulay A. Alaoui-Jamali is supported by the Quebec Breast Cancer Foundation and the Canadian Institutes for Health Research. Grégoire B. Morand is supported by the Swiss Cancer League (BIL KFS-3002-08-2012).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2015.00017/abstract
References
Aboud-Pirak, E., Hurwitz, E., Pirak, M. E., Bellot, F., Schlessinger, J., and Sela, M. (1988). Efficacy of antibodies to epidermal growth factor receptor against KB carcinoma in vitro and in nude mice. J. Natl. Cancer Inst. 80, 1605–1611. doi: 10.1093/jnci/80.20.1605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adams, C. W., Allison, D. E., Flagella, K., Presta, L., Clarke, J., Dybdal, N., et al. (2006). Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunol. Immunother. 55, 717–727. doi: 10.1007/s00262-005-0058-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amiri-Kordestani, L., Wedam, S., Zhang, L., Tang, S., Tilley, A., Ibrahim, A., et al. (2014). First FDA Approval of neoadjuvant therapy for breast cancer: pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin. Cancer Res. 20, 5359–5364. doi: 10.1158/1078-0432.CCR-14-1268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cancer Genome Atlas Network, (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. doi: 10.1038/nature11412
Bargmann, C. I., Hung, M. C., and Weinberg, R. A. (1986). Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45, 649–657. doi: 10.1016/0092-8674(86)90779-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barker, A. J., Gibson, K. H., Grundy, W., Godfrey, A. A., Barlow, J. J., Healy, M. P., et al. (2001). Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 11, 1911–1914. doi: 10.1016/S0960-894X(01)00344-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baselga, J., Cortes, J., Im, S. A., Clark, E., Ross, G., Kiermaier, A., et al. (2014). Biomarker analyses in CLEOPATRA: a Phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J. Clin. Oncol. 32, 3753–3761. doi: 10.1200/JCO.2013.54.5384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beauclair, S., Formento, P., Fischel, J., Lescaut, W., Largillier, R., Chamorey, E., et al. (2007). Role of the HER2 [Ile655Val] genetic polymorphism in tumorogenesis and in the risk of trastuzumab-related cardiotoxicity. Ann. Oncol. 18, 1335–1341. doi: 10.1093/annonc/mdm181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Birchmeier, C. (2009). ErbB receptors and the development of the nervous system. Exp. Cell Res. 315, 611–618. doi: 10.1016/j.yexcr.2008.10.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouchard, L., Lamarre, L., Tremblay, P. J., and Jolicoeur, P. (1989). Stochastic appearance of mammary tumors in transgenic mice carrying the MMTV/c-neu oncogene. Cell 57, 931–936. doi: 10.1016/0092-8674(89)90331-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandt, B., Hermann, S., Straif, K., Tidow, N., Buerger, H., and Chang-Claude, J. (2004). Modification of breast cancer risk in young women by a polymorphic sequence in the egfr gene. Cancer Res. 64, 7–12. doi: 10.1158/0008-5472.CAN-03-2623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burgess, A. W., Cho, H. S., Eigenbrot, C., Ferguson, K. M., Garrett, T. P., Leahy, D. J., et al. (2003). An open-and-shut case? recent insights into the activation of EGF/ErbB receptors. Mol. Cell 12, 541–552. doi: 10.1016/S1097-2765(03)00350-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carlomagno, F., Vitagliano, D., Guida, T., Ciardiello, F., Tortora, G., Vecchio, G., et al. (2002). ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 62, 7284–7290.
Carter, P., Presta, L., Gorman, C. M., Ridgway, J. B., Henner, D., Wong, W. L., et al. (1992). Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 89, 4285–4289. doi: 10.1073/pnas.89.10.4285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chinkers, M., and Cohen, S. (1981). Purified EGF receptor-kinase interacts specifically with antibodies to Rous sarcoma virus transforming protein. Nature 290, 516–519. doi: 10.1038/290516a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Citri, A., Skaria, K. B., and Yarden, Y. (2003). The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp. Cell Res. 284, 54–65. doi: 10.1016/S0014-4827(02)00101-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dahabreh, I. J., and Murray, S. (2011). Lack of replication for the association between HER2 I655V polymorphism and breast cancer risk: a systematic review and meta-analysis. Cancer Epidemiol 35, 503–509. doi: 10.1016/j.canep.2011.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dahan, L., Norguet, E., Etienne-Grimaldi, M. C., Formento, J. L., Gasmi, M., Nanni, I., et al. (2011). Pharmacogenetic profiling and cetuximab outcome in patients with advanced colorectal cancer. BMC Cancer 11, 496. doi: 10.1186/1471-2407-11-496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Engelman, J. A., Zejnullahu, K., Gale, C. M., Lifshits, E., Gonzales, A. J., Shimamura, T., et al. (2007). PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 67, 11924–11932. doi: 10.1158/0008-5472.CAN-07-1885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fan, Y. X., Wong, L., Ding, J., Spiridonov, N. A., Johnson, R. C., and Johnson, G. R. (2008). Mutational activation of ErbB2 reveals a new protein kinase autoinhibition mechanism. J. Biol. Chem. 283, 1588–1596. doi: 10.1074/jbc.M708116200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fleeman, N., Bagust, A., Beale, S., Dwan, K., Dickson, R., Proudlove, C., et al. (2015). Pertuzumab in combination with trastuzumab and docetaxel for the treatment of her2-positive metastatic or locally recurrent unresectable breast cancer. Pharmacoeconomics 33, 13–23. doi: 10.1007/s40273-014-0206-2
Garrett, T. P., Mckern, N. M., Lou, M., Elleman, T. C., Adams, T. E., Lovrecz, G. O., et al. (2003). The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 11, 495–505. doi: 10.1016/S1097-2765(03)00048-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., et al. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390–394. doi: 10.1038/378390a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gebhardt, F., Zanker, K. S., and Brandt, B. (1999). Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J. Biol. Chem. 274, 13176–13180. doi: 10.1074/jbc.274.19.13176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geyer, C. E., Forster, J., Lindquist, D., Chan, S., Romieu, C. G., Pienkowski, T., et al. (2006). Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743. doi: 10.1056/NEJMoa064320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gschwind, A., Fischer, O. M., and Ullrich, A. (2004). The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 4, 361–370. doi: 10.1038/nrc1360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, X., Diao, L., Xu, Y., Xue, W., Ouyang, T., Li, J., et al. (2014). Association between the HER2 Ile655Val polymorphism and response to trastuzumab in women with operable primary breast cancer. Ann. Oncol. 25, 1158–1164. doi: 10.1093/annonc/mdu111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harris, L. N., You, F., Schnitt, S. J., Witkiewicz, A., Lu, X., Sgroi, D., et al. (2007). Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin. Cancer Res. 13, 1198–1207. doi: 10.1158/1078-0432.CCR-06-1304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickinson, D. M., Klinowska, T., Speake, G., Vincent, J., Trigwell, C., Anderton, J., et al. (2010). AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin. Cancer Res. 16, 1159–1169. doi: 10.1158/1078-0432.CCR-09-2353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsieh, Y. Y., Tzeng, C. H., Chen, M. H., Chen, P. M., and Wang, W. S. (2012). Epidermal growth factor receptor R521K polymorphism shows favorable outcomes in KRAS wild-type colorectal cancer patients treated with cetuximab-based chemotherapy. Cancer Sci. 103, 791–796. doi: 10.1111/j.1349-7006.2012.02225.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ichihara, S., Toyooka, S., Fujiwara, Y., Hotta, K., Shigematsu, H., Tokumo, M., et al. (2007). The impact of epidermal growth factor receptor gene status on gefitinib-treated Japanese patients with non-small-cell lung cancer. Int. J. Cancer 120, 1239–1247. doi: 10.1002/ijc.22513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, Y., and Lee, C. G. (2013). Single nucleotide polymorphisms associated with microRNA regulation. Biomolecules 3, 287–302. doi: 10.3390/biom3020287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joensuu, H., Kellokumpu-Lehtinen, P. L., Bono, P., Alanko, T., Kataja, V., Asola, R., et al. (2006). Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N. Engl. J. Med. 354, 809–820. doi: 10.1056/NEJMoa053028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuraoka, K., Matsumura, S., Hamai, Y., Nakachi, K., Imai, K., Matsusaki, K., et al. (2003). A single nucleotide polymorphism in the transmembrane domain coding region of HER-2 is associated with development and malignant phenotype of gastric cancer. Int. J. Cancer 107, 593–596. doi: 10.1002/ijc.11450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurppa, K. J., Rokavec, M., Sundvall, M., Kellokumpu-Lehtinen, P. L., Joensuu, H., Brauch, H., et al. (2014). ERBB4 promoter polymorphism is associated with poor distant disease-free survival in high-risk early breast cancer. PLoS ONE 9:e102388. doi: 10.1371/journal.pone.0102388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leite, M., Giacomin, L., Piranda, D., Festa-Vasconcellos, J., Indio-Do-Brasil, V., Koifman, S., et al. (2014). Epidermal growth factor receptor gene polymorphisms are associated with prognostic features of breast cancer. BMC Cancer 14:190. doi: 10.1186/1471-2407-14-190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemmon, M. A. (2009). Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 315, 638–648. doi: 10.1016/j.yexcr.2008.10.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemmon, M. A., Schlessinger, J., and Ferguson, K. M. (2014). The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 6:a020768. doi: 10.1101/cshperspect.a020768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, D., Ambrogio, L., Shimamura, T., Kubo, S., Takahashi, M., Chirieac, L. R., et al. (2008). BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27, 4702–4711. doi: 10.1038/onc.2008.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M., Zhang, Z., Li, X., Ye, J., Wu, X., Tan, Z., et al. (2014). Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 46, 872–876. doi: 10.1038/ng.3030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, B., Ordonez-Ercan, D., Fan, Z., Edgerton, S. M., Yang, X., and Thor, A. D. (2007). Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int. J. Cancer 120, 1874–1882. doi: 10.1002/ijc.22423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, W., He, L., Ramirez, J., Krishnaswamy, S., Kanteti, R., Wang, Y. C., et al. (2011). Functional EGFR germline polymorphisms may confer risk for EGFR somatic mutations in non-small cell lung cancer, with a predominant effect on exon 19 microdeletions. Cancer Res. 71, 2423–2427. doi: 10.1158/0008-5472.CAN-10-2689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, W., Innocenti, F., Wu, M. H., Desai, A. A., Dolan, M. E., Cook, E. H. Jr., et al. (2005). A functional common polymorphism in a Sp1 recognition site of the epidermal growth factor receptor gene promoter. Cancer Res. 65, 46–53. doi: 10.1158/0008-5472.CAN-03-2868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lurje, G., Nagashima, F., Zhang, W., Yang, D., Chang, H. M., Gordon, M. A., et al. (2008). Polymorphisms in cyclooxygenase-2 and epidermal growth factor receptor are associated with progression-free survival independent of K-ras in metastatic colorectal cancer patients treated with single-agent cetuximab. Clin. Cancer Res. 14, 7884–7895. doi: 10.1158/1078-0432.CCR-07-5165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mateo, C., Moreno, E., Amour, K., Lombardero, J., Harris, W., and Perez, R. (1997). Humanization of a mouse monoclonal antibody that blocks the epidermal growth factor receptor: recovery of antagonistic activity. Immunotechnology 3, 71–81. doi: 10.1016/S1380-2933(97)00065-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miknis, G. (2005). “ARRY-334543, a potent, orally active small molecule inhibitor of EGFR and ErbB-2,” in AACR Meeting Abstracts (Anaheim, CA).
Monsey, J., Shen, W., Schlesinger, P., and Bose, R. (2010). Her4 and Her2/neu tyrosine kinase domains dimerize and activate in a reconstituted in vitro system. J. Biol. Chem. 285, 7035–7044. doi: 10.1074/jbc.M109.096032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moyer, J. D., Barbacci, E. G., Iwata, K. K., Arnold, L., Boman, B., Cunningham, A., et al. (1997). Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 57, 4838–4848.
O-Charoenrat, P., Rhys-Evans, P. H., Modjtahedi, H., and Eccles, S. A. (2002). The role of c-erbB receptors and ligands in head and neck squamous cell carcinoma. Oral. Oncol. 38, 627–640. doi: 10.1016/S1368-8375(02)00029-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piccart-Gebhart, M. J., Procter, M., Leyland-Jones, B., Goldhirsch, A., Untch, M., Smith, I., et al. (2005). Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672. doi: 10.1056/NEJMoa052306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prickett, T. D., Agrawal, N. S., Wei, X., Yates, K. E., Lin, J. C., Wunderlich, J. R., et al. (2009). Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 41, 1127–1132. doi: 10.1038/ng.438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Puputti, M., Sihto, H., Isola, J., Butzow, R., Joensuu, H., and Nupponen, N. N. (2006). Allelic imbalance of HER2 variant in sporadic breast and ovarian cancer. Cancer Genet. Cytogenet. 167, 32–38. doi: 10.1016/j.cancergencyto.2004.09.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Red Brewer, M., Choi, S. H., Alvarado, D., Moravcevic, K., Pozzi, A., Lemmon, M. A., et al. (2009). The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 34, 641–651. doi: 10.1016/j.molcel.2009.04.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rexer, B. N., and Arteaga, C. L. (2012). Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit. Rev. Oncog. 17, 1–16. doi: 10.1615/CritRevOncog.v17.i1.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rokavec, M., Justenhoven, C., Schroth, W., Istrate, M. A., Haas, S., Fischer, H. P., et al. (2007). A novel polymorphism in the promoter region of ERBB4 is associated with breast and colorectal cancer risk. Clin. Cancer Res. 13, 7506–7514. doi: 10.1158/1078-0432.CCR-07-0457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Romond, E. H., Perez, E. A., Bryant, J., Suman, V. J., Geyer, C. E. Jr., Davidson, N. E., et al. (2005). Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684. doi: 10.1056/NEJMoa052122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rusnak, D. W., Affleck, K., Cockerill, S. G., Stubberfield, C., Harris, R., Page, M., et al. (2001). The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: potential therapy for cancer. Cancer Res. 61, 7196–7203.
Sanson, M., Hosking, F. J., Shete, S., Zelenika, D., Dobbins, S. E., Ma, Y., et al. (2011). Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum. Mol. Genet. 20, 2897–2904. doi: 10.1093/hmg/ddr192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sasaki, H., Okuda, K., Shimizu, S., Takada, M., Kawahara, M., Kitahara, N., et al. (2009). EGFR R497K polymorphism is a favorable prognostic factor for advanced lung cancer. J. Cancer Res. Clin. Oncol. 135, 313–318. doi: 10.1007/s00432-008-0464-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, F., Telesco, S. E., Liu, Y., Radhakrishnan, R., and Lemmon, M. A. (2010). ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 7692–7697. doi: 10.1073/pnas.1002753107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shigematsu, H., Takahashi, T., Nomura, M., Majmudar, K., Suzuki, M., Lee, H., et al. (2005). Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 65, 1642–1646. doi: 10.1158/0008-5472.CAN-04-4235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J. A., Wong, S. G., Keith, D. E., et al. (1989). Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, 707–712. doi: 10.1126/science.2470152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smaill, J. B., Rewcastle, G. W., Loo, J. A., Greis, K. D., Chan, O. H., Reyner, E. L., et al. (2000). Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J. Med. Chem. 43, 1380–1397. doi: 10.1021/jm990482t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stein, R. A., and Staros, J. V. (2006). Insights into the evolution of the ErbB receptor family and their ligands from sequence analysis. BMC Evol. Biol. 6:79. doi: 10.1186/1471-2148-6-79
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sung, J. S., Jin, L., Jo, U., Park, K. H., and Kim, Y. H. (2012). Association between -276 C/T polymorphism of the ERBB3 gene and lung cancer risk in a Korean population. Anticancer Res. 32, 4433–4437.
Thiel, K. W., and Carpenter, G. (2007). Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc. Natl. Acad. Sci. U.S.A. 104, 19238–19243. doi: 10.1073/pnas.0703854104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walter, A. O., Sjin, R. T., Haringsma, H. J., Ohashi, K., Sun, J., Lee, K., et al. (2013). Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 3, 1404–1415. doi: 10.1158/2159-8290.CD-13-0314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, W. S., Chen, P. M., Chiou, T. J., Liu, J. H., Lin, J. K., Lin, T. C., et al. (2007). Epidermal growth factor receptor R497K polymorphism is a favorable prognostic factor for patients with colorectal carcinoma. Clin. Cancer Res. 13, 3597–3604. doi: 10.1158/1078-0432.CCR-06-2601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wissner, A., Overbeek, E., Reich, M. F., Floyd, M. B., Johnson, B. D., Mamuya, N., et al. (2003). Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J. Med. Chem. 46, 49–63. doi: 10.1021/jm020241c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolff, A. C., Hammond, M. E., Schwartz, J. N., Hagerty, K. L., Allred, D. C., Cote, R. J., et al. (2007). American society of clinical oncology/college of American pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 131, 18–43. doi: 10.1043/1543-2165(2007)131[18:ASOCCO]2.0.CO;2
Yang, X. D., Jia, X. C., Corvalan, J. R., Wang, P., and Davis, C. G. (2001). Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit. Rev. Oncol. Hematol. 38, 17–23. doi: 10.1016/S1040-8428(00)00134-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yarden, Y., and Pines, G. (2012). The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 12, 553–563. doi: 10.1038/nrc3309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149. doi: 10.1016/j.cell.2006.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Pickin, K. A., Bose, R., Jura, N., Cole, P. A., and Kuriyan, J. (2007). Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 450, 741–744. doi: 10.1038/nature05998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ErbB receptors, cancer, SNPs, anti-ErbB therapeutics, drug response, resistance
Citation: Alaoui-Jamali MA, Morand GB and da Silva SD (2015) ErbB polymorphisms: insights and implications for response to targeted cancer therapeutics. Front. Genet. 6:17. doi: 10.3389/fgene.2015.00017
Received: 28 October 2014; Accepted: 14 January 2015;
Published online: 04 February 2015.
Edited by:
Marcelo Rizzatti Luizon, University of California, San Francisco, USAReviewed by:
Juergen Reichardt, James Cook Univerrsity, AustraliaWanqing Liu, Purdue University, USA
Copyright © 2015 Alaoui-Jamali, Morand and da Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moulay A. Alaoui-Jamali and Sabrina Daniela da Silva, Segal Cancer Centre and Lady Davis Institute for Medical Research, Sir Mortimer B. Davis-Jewish General Hospital, McGill University, 3755 Côte Ste-Catherine Road, Montreal, QC H3T 1E2, Canada e-mail:bW91bGF5LmFsYW91aS1qYW1hbGlAbWNnaWxsLmNh;c2FicmluYWRhbmllbGFAaG90bWFpbC5jb20=