 Mirjana Jerkic
Mirjana Jerkic- 1Anesthesia Research, Keenan Research Centre for Biomedical Science, St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada
- 2Molecular Structure and Function Program, Peter Gilgan Centre for Research and Learning, Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
- 3Department of Immunology, University of Toronto, Toronto, ON, Canada
Oxidative stress causes endothelial dysfunction and is implicated in the pathogenesis of cardiovascular diseases. Our studies suggested that reactive oxygen species (ROS) play a crucial role in hereditary hemorrhagic telangiectasia (HHT) disease, a vascular dysplasia affecting 1 in 5,000–8,000 people. Mutations in endoglin (ENG) and activin receptor-like kinase 1 (ACVRL1) genes are responsible for HHT1 and HHT2 and are associated with arteriovenous malformations. ENG and ACVRL1 interact with endothelial nitric oxide synthase (eNOS) and regulate its activation. Mice heterozygous for these genes (Eng+/– and Acvrl1+/–) show reduced ENG or ACVRL1 protein levels in endothelial cells causing eNOS uncoupling, generation of ROS rather than nitric oxide (NO•), leading to impaired NO• mediated vasodilation. ROS production is increased in several organs of Eng+/– and Acvrl1+/– mice, including lungs, liver, and colon, affected in HHT. The major source of increased oxidative stress in these tissues is eNOS-derived ROS and not mitochondrial or NADPH oxidase-dependent ROS. Eng+/– and Acvrl1+/– mice also develop with age signs of pulmonary arterial hypertension attributable to eNOS-derived ROS, which was preventable by antioxidant treatment. To date, only one pilot study has been carried out in HHT patients, and it showed beneficial effects of antioxidant therapy on epistaxis. We suggest that more clinical studies are warranted to investigate whether antioxidants would prevent, delay or attenuate manifestations of disease in individuals with HHT, based on our experimental data in mouse models.
Increased ROS Production and Endothelial Dysfunction in Cardiovascular Diseases
Reactive oxygen species (ROS) are important regulators of many processes including intracellular signaling, response to growth factor stimulation and generation of the inflammatory response (Finkel, 2011). Dysregulated ROS signaling, leading to increased free radical production and/or down regulation of ROS scavengers and antioxidants, may cause or accelerate many pathological conditions, including cardiovascular, neurodegenerative, and malignant diseases. Oxidative stress is associated with endothelial dysfunction in cardiovascular disorders such as systemic and pulmonary hypertension, atherosclerosis, and diabetes (Münzel et al., 2010). Understanding the mechanisms of excessive ROS production and their role in endothelial damage may provide opportunities for pharmacological intervention.
The major enzymatic sources of ROS in endothelial cells are mitochondrial respiratory enzymes, NADPH oxidases and uncoupled endothelial nitric oxide synthase (eNOS). It is important to mention that increased ROS production together with decreased nitric oxide (NO•) bioavailability can further complicate and worsen endothelial injury (Montezano and Touyz, 2012a). It is also recognized that crosstalk between various sources of ROS exists and may represent a feed-forward vicious cycle of ROS production (Dikalov, 2011).
Mitochondria produce ROS during ATP generation, when electrons transferred through the respiratory chain “leak” and create superoxide (•O2–; Widlansky and Gutterman, 2011), which can then be scavenged by the superoxide dismutase 2 (SOD2) present in the mitochondrial matrix. The release of ROS depends on activation of mitochondrial ATP-sensitive potassium channels (mitoK ATP) and opening of the permeability transition pores (PTP). Interestingly, NO• affects mitoK ATP activation (Figure 1) and low levels induce mitochondrial ROS release causing additional cellular oxidative stress (Chang et al., 2010). Excessive production of mitochondrial ROS can be targeted by specific inhibitors of the electron respiratory chain, such as antimycin A (Kudin et al., 2008) or by increased ROS scavenging. Recent animal studies have demonstrated that overexpression of SOD2 reduces fibrosis and pro-apoptotic signaling in the aging mouse heart (Kwak et al., 2014), while MitoTEMPOL, an SOD mimetic drug that concentrates in mitochondria, significantly reduced H2O2- and lipid peroxide-induced endothelial oxidative stress (Dhanasekaran et al., 2005).
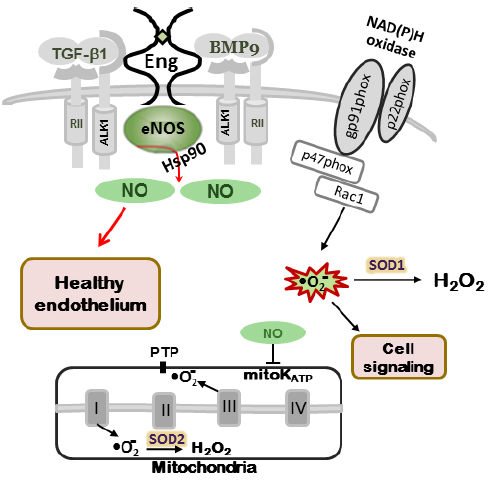
Figure 1. NO production in normal endothelium. In wild type mice (Eng+/+ and Alk1+/+) eNOS is closely associated with caveolae/lipid rafts in endothelial cells. Endoglin and ALK-1 co-localize with eNOS serving as scaffolding molecules for eNOS/Hsp90 association and eNOS activation, leading to NO• production. Normal cellular NO• levels through mitochondrial ATP-sensitive potassium channels (mitoK ATP) and closed permeability transition pores (PTP) prevent mitochondrial ROS “leak” into the cytoplasm. Small amount of ROS, produced by NADPH oxidases, are involved in cell signaling (adapted from Jerkic et al., 2012).
NADPH oxidases are a family of enzyme complexes, which catalyze the transfer of electrons from NADPH to molecular O2 via their “Nox” catalytic subunit, generating •O2– and H2O2 (Dikalov, 2011; Figure 1). The major Nox isoforms present in endothelial cells are Nox4 and Nox5. ROS generated from NADPH oxidases also affect redox-sensitive mitoK ATP activation, since inhibition of NADPH oxidases by apocynin completely prevented mitochondrial dysfunction induced by Angiotensin II (AngII; Doughan et al., 2008). NADPH oxidases also appear to be a major source of ROS in vessels of spontaneously hypertensive rats, as demonstrated by blocking with the novel and more specific NADPH oxidase inhibitor VAS3947 (Wind et al., 2010). Studies on arteries from patients with coronary artery disease and in animals with experimentally induced systemic hypertension, diabetes, or atherosclerosis, suggest that Nox1, Nox2, and Nox5 promote endothelial dysfunction (Drummond and Sobey, 2014, whereas contribution of Nox4 has been proposed in lung vessel remodeling caused by pulmonary hypertension (Barman et al., 2014). The anti-hypertensive effect of angiotensin-converting enzyme (ACE) inhibitors and AT1 receptor antagonists has been attributed in part to their ability to inhibit AngII-induced NADPH oxidase activation and expression (Schramm et al., 2012).
Endothelial nitric oxide synthase is closely associated with caveolae/lipid rafts, and critically involved in NO• production. Multiple factors influence eNOS activation, mediated through Akt-dependent phosphorylation at Ser1177, and inhibited by phosphorylation at Thr459 (Dimmeler et al., 1999). Basal eNOS activity correlates with the amount of tetrahydrobiopterin (BH4) bound to the enzyme. eNOS uncoupling occurs when there is a discrepancy between eNOS levels and NO• production, with a switch in the eNOS enzymatic activity to generate superoxide rather than NO• (Montezano and Touyz, 2012a). eNOS uncoupling is implicated in endothelial dysfunction and has been demonstrated in experimental models and patients with hypertension (essential, renovascular, malignant, and salt-sensitive), diabetes, atherosclerosis, and cardiac failure (Moens and Kass, 2007). Very limited NO• bioavailability combined with overly abundant ROS is one of the key factors causing endothelial damage and vascular dysfunction in these disorders. Moreover, classical antihypertensive agents (β-adrenergic blockers, ACE inhibitors, AT1 receptor blockers and Ca2+-channel blockers) exert some of their beneficial effects by stimulating NO• production and decreasing vascular ROS bioavailability (Weseler and Bast, 2010). Resveratrol, BH4, sepiapterin, and folic acid have also been shown to recouple eNOS and improve endothelial function in animal studies (Li and Forstermann, 2014).
HHT and Oxidative Stress
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular dysplasia with age-related penetrance, affecting 1 in 5,000–8,000 people worldwide. The external visible signs of HHT are recurrent epistaxis (nosebleeds) and small cutaneous telangiectatic lesions. Imaging studies reveal the presence of arteriovenous malformations (AVMs)—defined as abnormal direct connections between arteries and veins—in lungs, liver, and brain. AVMs are life-threatening because of their potentially lethal complications such as stroke, hemorrhage, and brain abscess. HHT is a genetically heterogeneous disorder, HHT type 1 (HHT1) is caused by mutations in the ENG (endoglin) gene located on chromosome 9, region 9q33-q34, whereas HHT type 2 (HHT2) is caused by mutations in the ACVRL1 (activin receptor-like kinase 1 or ALK-1) gene located on chromosome 12, region 12q11-q14. About 15–20% of HHT families remain unresolved after mutation analysis of these two genes (McDonald et al., 2011), and two more genetic loci have been implicated, HHT3 on chromosome 5 (Cole et al., 2005; Govani and Shovlin, 2010) and HHT4 on chromosome 7 (Bayrak-Toydemir et al., 2006), though the causative genes remain unidentified. A subset of patients (2–3%) with a combined syndrome of juvenile polyposis and HHT (defined as JPHT) harbor mutations in the MADH4 (SMAD4) gene (Gallione et al., 2004). More recently, mutations in the BMP9 (GDF2) gene have been associated with an HHT-like syndrome. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with HHT (Wooderchak-Donahue et al., 2013).
The underlying mechanism of HHT1 and HHT2 is haploinsufficiency indicating that reduced amounts of ENG and ACVRL1 lead to endothelial dysfunction and predispose to the clinical signs of disease. The observations that BMP9 is the highest affinity ligand for ACVRL1 and ENG and that Smad4 is a downstream mediator support the hypothesis that BMP9/ACVRL1/ENG/Smad4 mediated pathways are affected in HHT.
Despite the fact that vascular stress and multi-organ vascular dysplasia are hallmarks of HHT (Abdalla and Letarte, 2006), only few studies have been investigating the role of oxidative stress in the pathogenesis of this disease. We will summarize our results in mouse models of HHT, which implicate oxidative stress as an underlying mechanism of endothelial dysfunction in this disease.
Mouse Models of HHT, Endothelial Dysfunction, and eNOS Uncoupling
Endoglin and Acvrl1 heterozygous (Eng+/– and Acvrl1+/–) mice have been used as models for HHT1 and HHT2 respectively, allowing us to dissect the underlying mechanisms of disease (Bourdeau et al., 1999; Srinivasan et al., 2003). Early generations of Eng+/– mice developed multiple signs of HHT in many organs, likely due to modifier genes in the 129/Ola background (Bourdeau et al., 2001). Subsequent backcrossing to the C57BL/6J background gave rise to mice with no apparent symptoms of HHT, but allowed us to study the underlying endothelial dysfunction. Physiological studies revealed that ENG is involved in the control of vascular tone regulating NO-dependent vasodilatation (Jerkic et al., 2004). Resistance arteries from Eng+/– C57BL/6J mice showed enhanced endothelial-dependent dilatation and impaired myogenic response (Toporsian et al., 2005). Furthermore, ENG could be found in caveolae, where it associates with eNOS and likely stabilizes it. eNOS is closely associated with caveolae/lipid rafts in endothelial cells (Pritchard et al., 2001), where ENG and ALK-1 colocalize with eNOS serving as scaffolding molecules for eNOS/Hsp90 association, eNOS activation, and NO• production (Toporsian et al., 2005). ENG-deficient endothelial cells, following Ca2+ activation, showed reduced eNOS/Hsp90 interaction, produced less NO• and generated more eNOS-derived superoxide (•O2–), indicating that ENG is an important regulator in the coupling of eNOS activity (Toporsian et al., 2005). Treatment of Eng+/– resistance arteries with the anti-oxidant Tiron, reversed the vascular abnormalities (Toporsian et al., 2005), confirming that ROS are implicated in the pathobiology of HHT. Acvrl1 was also shown to associate with the eNOS activation complex leading to NO• production (Toporsian et al., 2010). Consequently, in endothelial cells deficient in ENG or Acvrl1, eNOS becomes uncoupled and produces more superoxide than NO•, leading to endothelial damage (Figure 2).
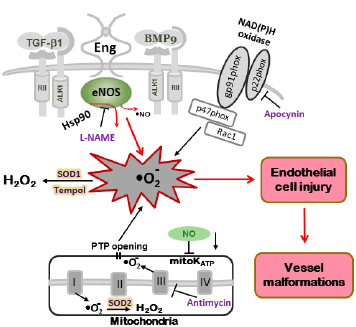
Figure 2. Model of oxidative stress in mutant mice leading to vascular endothelial damage. In Eng+/– and Alk1+/– mice, altered eNOS activation renders the enzyme refractory to regulation by TGF-β/BMP signaling and represents a critical event leading to excessive oxidative stress. Uncoupled eNOS produces low amounts of NO• and high levels of oxygen radicals (•O2–). The NOS inhibitor L-NAME inhibits ROS production in tissues of Eng+/– and Alk1+/– mice. Superoxide dismutase (SOD) and the SOD mimetic compound Tempol, converts •O2– into less harmful hydrogen peroxide (H2O2). A large portion of ROS is produced by mitochondria (Antimycin-inhibitable) and NADPH oxidases (Apocynin-inhibitable), however, that percentage does not differ between mutant and control mice. The low NO• cellular level associated with HHT may also inhibit mitochondrial ATP-sensitive potassium channel (mitoK ATP) opening, trigger permeability transition pores (PTP) opening and further increase the oxidative stress caused by mitochondrial ROS release (adapted from Jerkic et al., 2012).
eNOS-Derived ROS are only Present in Adult Lungs of Eng and Acvrl1 Heterozygous Mice
We addressed the age-dependency of ROS production in lungs of Eng and Acvrl1 heterozygous mice. ROS generation was higher in adult than newborn mice, in both mutant and control mice, and so were the levels of NADPH oxidase and ROS scavenging SOD 1, 2, and 3 isoforms (Belik et al., 2009; Jerkic et al., 2011), indicating maturational changes of these enzymes. There was no difference in ROS levels between newborn mutant mice and their littermate controls, while adult mutant mice showed increased levels of pulmonary oxidative stress than control mice. Protein levels and NADPH activity were normal in adult Eng+/– and Acvrl1+/– lungs as well as the levels of scavenging enzymes and could not account for the increased oxidative stress. However, eNOS-dependent ROS production was increased in adult mutant mice while NO• production was reduced. Thus Eng and Acvrl1 haploinsufficiency results in pulmonary vascular eNOS uncoupling in the adult, but not newborn mice (Belik et al., 2009; Jerkic et al., 2011).
Pulmonary arteries from adult Eng+/– and Acvrl1+/– mice were more dilated compared to the control animals. Moreover, several signs of pulmonary arterial hypertension (PAH), including elevated right ventricular systolic pressure leading to hypertrophy, reduced vascular density, and increased thickness and outward remodeling of pulmonary arterioles, were observed in adult but not newborn Eng+/– and Acvrl1+/– mice relative to wild-type littermate controls. Our observations support the fact that mutations in several genes of the TGF-β superfamily, including BMPR2, ACVRL1, and ENG, predispose to PAH (Girerd et al., 2010). Interestingly, treatment of 3-week-old Eng+/– and Acvrl1+/– mice with the SOD mimetic and anti-oxidant Tempol for 6 weeks prevents the onset of PAH symptoms in these mice, confirming a role for ROS in the pathobiology of the disease. These findings also suggest that an abnormal regulation of ROS might play a role in the formation of pulmonary AVMs in HHT.
Multiple Organs of Eng and Acvrl1 Heterozygous Mice Show Increased ROS
Results of a more recent study (Jerkic et al., 2012) suggest that oxidative stress is indeed a generalized manifestation in organs of Eng and Acvrl1 heterozygous mice. We have found increased ROS production in lungs, liver, and colon of the heterozygous mice, organs most frequently affected in HHT patients. Our results also indicate that increased oxidative stress contributes to endothelial dysfunction in mutant mice. Mitochondrial ROS production was highest in liver while NADPH oxidase was a major source of ROS in the other tissues. However, there was no difference in mitochondrial- or NADPH oxidase-dependent ROS production between mutant and control mice. In tissues of mutant mice ROS overproduction was attributed to NOS, as it was L-NAME inhibitable. As neuronal NOS was not observed and inducible NOS was barely detectable in mouse tissues (Jerkic et al., 2004, 2011), we conclude that the observed increase in ROS production in HHT mice is eNOS-derived and therefore endothelial-dependent.
A recent paper from Ghanian et al. (2014) has explored the mitochondrial redox state in kidneys and eyes of Eng heterozygous mice using optical imaging. They found that mitochondrial oxidative stress was decreased in these organs of Eng+/– mice. We cannot comment on these findings, as we have not explored eye or kidney ROS production and have focused on organs most affected in HHT.
Overall, we have documented in several papers an increased oxidative stress in endothelial cells and tissues of Eng and Acvrl1 heterozygous mice and identified uncoupled eNOS as a source of ROS overproduction (Figure 2). However, increased ROS concentration and decreased NO• bioavailability have further effects on ROS production and/or release from other sources, as discussed previously. Low NO• cellular levels inhibit mitoK ATP and trigger the opening of PTP. This process causes ROS release from mitochondria (Figure 2), worsening oxidative stress and leading eventually to the AVMs characteristic of HHT.
The role of oxidative stress has also been recently suggested in the pathogenesis of endothelial dysfunction in pre-eclampsia, disease associated with increased production from the placenta of a soluble form of ENG (sEng; Venkatesha et al., 2006). Tskitishvili et al. (2010) have demonstrated that cultured placental and amniotic tissues release sEng under oxidative stress, in connection with endothelial dysfunction, systemic inflammation, and pre-eclampsia.
Antioxidants in Cardiovascular Diseases
The excessive generation of ROS and/or reduced antioxidant capacity lead to endothelial dysfunction. One can therefore propose that correction of the abnormally elevated ROS bioavailability might retard, or prevent, endothelial cell injury. Antioxidants could represent a simple way to restore the redox status in the vascular milieu (Münzel et al., 2010). However, experimental and clinical studies that tested preventive or therapeutic use of antioxidants have shown that their use is much more complex and context-dependent.
Tuomilehto et al. (1987) have observed lower cardiovascular mortality in Mediterranean populations compared to those of Northern European countries and attributed the differences to the higher content of antioxidants in the Mediterranean diet. The Zutphen Elderly study confirmed a negative correlation between antioxidant vitamin intake and coronary heart disease and mortality in 805 elderly men without prior history of cardiovascular disease (Hertog et al., 1993). In line with these studies, a meta-analysis of cohort studies including almost 400,000 patients (Ye and Song, 2008) reported that high intake of antioxidant vitamins (C and E) was associated with a lower rate of coronary heart disease. However, none of the above-mentioned clinical trials directly demonstrated that the dose and type of antioxidants used effectively suppressed oxidative stress. In that context, results from the few studies that assessed antioxidant intake based on specific blood assays were more ambiguous. The ATBC study that followed 29,000 male smokers for 19 years showed that higher levels of vitamin E in the serum (but still within the physiological range) were associated with lower mortality for prostate cancer and cardiovascular disease (The Alpha Tocopherol, Beta Carotene Cancer Prevention Study Group, 1994). However, in the same study the group that received beta-carotene showed greater mortality from lung cancer. The NHANES-II study, with 8,000 enrolled subjects, showed that men but not women in the lowest quartile of serum vitamin C had an increased mortality for cancer and cardiovascular disease (Loria et al., 2000). Reasons for these equivocal and sometimes disappointing results relate to the type and dose of antioxidants used, patient cohorts, trial design, and choice of outcome measures. It is also possible that dose and duration of the antioxidant therapy were insufficient. Please refer to Hasnain and Mooradian (2004) and Stephens et al. (2009) for a detailed discussion of these findings.
Another important requirement for the clinical use of antioxidants is to develop and validate markers of oxidative stress, triage the patients with oxidative excess and measure efficacy of the treatment. It is also necessary to know the organs most affected and the major source of ROS in a particular patient, in order to choose the right antioxidant and the most efficient way for its delivery. In controlled preclinical studies and clinical trials of statins and antihypertensive drugs mentioned above, oxidative stress and NO• production were assessed. These drugs were shown to improve vascular function and reduce the incidence of cardiovascular events in patients with cardiovascular disease, at least partly by increasing NO• and reducing ROS production (Chen et al., 2008; Münzel et al., 2010; Weseler and Bast, 2010; Montezano and Touyz, 2012b).
Antioxidants in HHT
We have shown using mouse models that excessive ROS generation may play a pivotal role in the pathobiology of endothelial dysfunction in HHT (Jerkic et al., 2012). Our results suggest that HHT patients may indeed benefit from diets rich in antioxidants, which could prevent or delay the rise in ROS in their blood vessels, due to eNOS uncoupling. Our studies showed that supplementing drinking water with the antioxidant Tempol for 6 weeks prevented rarefaction of the lung vasculature and the onset of PAH in Eng and Acvrl1 heterozygous mice (Toporsian et al., 2010; Jerkic et al., 2011). In HHT patients, the only trial carried out to date was a pilot study in which the antioxidant N-acetylcysteine was given to the patients for 12 weeks (de Gussem et al., 2009). The results of this study showed beneficial effects of this antioxidant therapy with a significant decrease in the frequency and severity of epistaxis in treated HHT patients, improving their quality of life. No preventative dietary study has been carried out, although some patients with HHT have mentioned reduced nosebleeds upon maintenance on diets rich in anti-oxidants.
We propose that antioxidants are promising tools in HHT, both as preventative and therapeutic options. Prevention of endothelial dysfunction in HHT patients may include simple strategies such as an antioxidant-rich diet, abstinence from smoking, a healthier lifestyle, and regular physical activity and exercise. It has been shown that exercise can prevent eNOS uncoupling, reduce NADPH oxidase activity and decrease endothelial oxidative stress and dysfunction in experimental models and patients with coronary artery disease (Adams et al., 2005; Agarwal et al., 2011). The therapeutic use of antioxidants is much more complex and as mentioned above requires considerations of the appropriate compound and proper dosage, the affected organ, individual response to treatment, and monitoring of the therapy. The choice of the right antioxidant is most critical. We have shown that Tempol in particular is an effective antioxidant and it could be a good candidate for clinical trials in HHT patients. Tempol was successfully used in more than 70 animal studies, in diseases associated with oxidative stress, inflammation, and aging (Wilcox, 2010). Two clinical trials are presently ongoing, with the use of Tempol as a topical agent to prevent radiation-induced alopecia and for dermatitis during radiation and chemotherapy for anal cancer (http://www.paidclinicaltrials.org/dir/category/by-medicine/tt/tempol). Our experimental findings suggest that the use of antioxidants could prevent uncoupling of eNOS associated with HHT. Whether it could cause eNOS recoupling in affected organs of HHT patients remains to be tested.
In conclusion, oxidative stress plays a causative role in the pathogenesis of endothelial dysplasia and vascular malformations in HHT. The use of antioxidants in the prevention and treatment of HHT should be of benefit to the patients. More experimental and clinical studies are urgently needed to test the most effective ways to use antioxidants to ameliorate the quality of life for patients with HHT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by grant from the Heart and Stroke Foundation of Canada to Dr. ML (T5598).
References
Abdalla, S. A., and Letarte, M. (2006). Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J. Med. Genet. 43, 97–110. doi: 10.1136/jmg.2005.030833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adams, V., Linke, A., Kränkel, N., Erbs, S., Gielen, S., Möbius-Winkler, S., et al. (2005). Impact of regular physical activity on the NAD(P)H oxidase and angiotensin receptor system in patients with coronary artery disease. Circulation 111, 555–562. doi: 10.1161/01.CIR.0000154560.88933.7E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Agarwal, D., Welsch, M. A., Keller, J. N., and Francis, J. (2011). Chronic exercise modulates RAS components and improves balance between pro- and anti-inflammatory cytokines in the brain of SHR. Basic Res. Cardiol. 106, 1069–1085. doi: 10.1007/s00395-011-0231-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barman, S. A., Chen, F., Su, Y., Dimitropoulou, C., Wang, Y., Catravas, J. D., et al. (2014). NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 34, 1704–1715. doi: 10.1161/ATVBAHA.114.303848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bayrak-Toydemir, P., McDonald, J., Akarsu, N., Toydemir, R. M., Calderon, F., Tuncali, T., et al. (2006). A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am. J. Med. Genet. A. 140A, 2155–2162. doi: 10.1002/ajmg.a.31450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belik, J., Jerkic, M., McIntyre, B. A. S., Pan, J., Leen, J., Yu, L. X., et al. (2009). Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. Am. J. Physiol. Lung. Cell. Mol. Physiol. 297, L1170–L1178. doi: 10.1152/ajplung.00168.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bourdeau, A., Dumont, D. J., and Letarte, M. (1999). A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 104, 1343–1351. doi: 10.1172/JCI8088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bourdeau, A., Faughnan, M. E., McDonald, M. L., Paterson, A. D., Wanless, I. R., and Letarte, M. (2001). Potential role of modifier genes influencing transforming growth factor-beta1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am. J. Pathol. 158, 2011–2020. doi: 10.1016/S0002-9440(10)64673-1
Chang, J. C., Kou, S. J., Lin, W. T., and Liu, C. S. (2010). Regulatory role of mitochondria in oxidative stress and atherosclerosis. World J. Cardiol. 2, 150–159. doi: 10.4330/wjc.v2.i6.150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S., Ge, Y., Si, J., Rifai, A., Dworkin, L. D., and Gong, R. (2008). Candesartan suppresses chronic renal inflammation by a novel antioxidant action independent of AT1R blockade. Kidney Int. 74, 1128–1138. doi: 10.1038/ki.2008.380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, S. G., Begbie, M. E., Wallace, G. M., and Shovlin, C. L. (2005). A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J. Med. Genet. 42, 577–582. doi: 10.1136/jmg.2004.028712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Gussem, E. M., Snijder, R. J., Disch, F. J., Zanen, P., Westermann, C. J., and Mager, J. J. (2009). The effect of N-acetylcysteine on epistaxis and quality of life in patients with HHT: a pilot study. Rhinology 47, 85–88.
Dhanasekaran, A., Kotamraju, S., Karunakaran, C., Kalivendi, S. V., Thomas, S., Joseph, J., et al. (2005). Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic. Biol. Med. 39, 567–583. doi: 10.1016/j.freeradbiomed.2005.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605. doi: 10.1038/21224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496. doi: 10.1161/CIRCRESAHA.107.162800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drummond, G. R., and Sobey, C. G. (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 25, 452–463. doi: 10.1016/j.tem.2014.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gallione, C. J., Repetto, G. M., Legius, E., Rustgi, A. K., Schelley, S. L., Tejpar, S., et al. (2004). A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 363, 852–859. doi: 10.1016/S0140-6736(04)15732-2
Ghanian, Z., Maleki, S., Park, S., Sorenson, C. M., Sheibani, N., and Ranji, M. (2014). Organ specific optical imaging of mitochondrial redox state in a rodent model of hereditary hemorrhagic telangiectasia-1. J. Biophotonics 7, 799–809. doi: 10.1002/jbio.201300033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Girerd, B., Montani, D., Coulet, F., Sztrymf, B., Yaici, A., Jaïs, X., et al. (2010). Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 181, 851–861. doi: 10.1164/rccm.200908-1284OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Govani, F. S., and Shovlin, C. L. (2010). Fine mapping of the hereditary haemorrhagic telangiectasia (HHT)3 locus on chromosome 5 excludes VE-Cadherin-2, Sprouty4 and other interval genes. J. Angiogenes. Res. 2, 15. doi: 10.1186/2040-2384-2-15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hasnain, B. I., and Mooradian, A. D. (2004). Recent trials of antioxidant therapy: what should we be telling our patients? Cleve. Clin. J. Med. 71, 327–334. doi: 10.3949/ccjm.71.4.327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hertog, M. G., Feskens, E. J., Hollman, P. C., Katan, M. B., and Kromhout, D. (1993). Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet 342, 1007–1011. doi: 10.1016/0140-6736(93)92876-U
Jerkic, M., Kabir, M. G., Davies, A., Yu, L. X., McIntyre, B. A. S., Husain, N. W., et al. (2011). Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc. Res. 92, 375–384. doi: 10.1093/cvr/cvr232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jerkic, M., Rivas-Elena, J. V., Prieto, M., Carrón, R., Sanz-Rodríguez, F., Pérez-Barriocanal, A., et al. (2004). Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 18, 609–611. doi: 10.1096/fj.03-0197fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jerkic, M., Sotov, V., and Letarte, M. (2012). Oxidative stress contributes to endothelial dysfunction in mouse models of hereditary hemorrhagic telangiectasia. Oxid. Med. Cell. Longev. 2012:686972. doi: 10.1155/2012/686972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kudin, A. P., Malinska, D., and Kunz, W. S. (2008). Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim. Biophys. Acta 1777, 689–695. doi: 10.1016/j.bbabio.2008.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kwak, H. B., Lee, Y., Kim, J. H., Van Remmen, H., Richardson, A. G., and Lawler, J. M. (2014). MnSOD overexpression reduces fibrosis and pro-apoptotic signaling in the aging mouse heart. J. Gerontol. A. Biol. Sci. Med. Sci. doi: 10.1093/gerona/glu090 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., and Forstermann, U. (2014). Pharmacological prevention of eNOS uncoupling. Curr. Pharm. Des. 20, 3595–3606. doi: 10.2174/13816128113196660749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loria, C. M., Klag, M. J., Caulfield, L. E., and Whelton, P. K. (2000). Vitamin C status and mortality in US adults. Am. J. Clin. Nutr. 72, 139–145.
McDonald, J., Bayrak-Toydemir, P., and Pyeritz, R. E. (2011). Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet. Med. 13, 607–616. doi: 10.1097/GIM.0b013e3182136d32
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moens, A. L., and Kass, D. A. (2007). Therapeutic potential of tetrahydrobiopterin for treating vascular and cardiac disease. J. Cardiovasc. Pharmacol. 50, 238–246. doi: 10.1097/FJC.0b013e318123f854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montezano, A. C., and Touyz, R. M. (2012a). Reactive oxygen species and endothelial function—role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 110, 87–94. doi: 10.1111/j.1742-7843.2011.00785.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montezano, A. C., and Touyz, R. M. (2012b). Molecular mechanisms of hypertension-reactive oxygen species and antioxidants: a basic science update for the clinician. Can. J. Cardiol. 28, 288–295. doi: 10.1016/j.cjca.2012.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Münzel, T., Gori, T., Bruno, R. M., and Taddei, S. (2010). Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 31, 2741–2748. doi: 10.1093/eurheartj/ehq396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pritchard, K. A. Jr, Ackerman, A. W., Gross, E. R., Stepp, D. W., Shi, Y., Fontana, J. T., et al. (2001). Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 276, 17621–17624. doi: 10.1074/jbc.C100084200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schramm, A., Matusik, P., Osmenda, G., and Guzik, T. J. (2012). Targeting NADPH oxidases in vascular pharmacology. Vascul. Pharmacol. 56, 216–231. doi: 10.1016/j.vph.2012.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Srinivasan, S., Hanes, M. A., Dickens, T., Porteous, M. E., Oh, S. P., Hale, L. P., et al. (2003). A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum. Mol. Genet. 12, 473–482. doi: 10.1093/hmg/ddg050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stephens, J. W., Khanolkar, M. P., and Bain, S. C. (2009). The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis 202, 321–329. doi: 10.1016/j.atherosclerosis.2008.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
The Alpha Tocopherol, Beta Carotene Cancer Prevention Study Group. (1994). The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 330, 1029–1035. doi: 10.1056/NEJM199404143301501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toporsian, M., Gros, R., Kabir, M. G., Vera, S., Govindaraju, K., Eidelman, D. H., et al. (2005). A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ. Res. 96, 684–692. doi: 10.1161/01.RES.0000159936.38601.22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toporsian, M., Jerkic, M., Zhou, Y. Q., Kabir, M. G., Yu, L. X., McIntyre, B. A. S., et al. (2010). Spontaneous adult-onset pulmonary arterial hypertension attributable to hemorrhagic telangiectasia increased endothelial oxidative stress in a murine model of hereditary hemorrhagic telangiectasia. Arterioscler. Thromb. Vasc. Biol. 30, 509–517. doi: 10.1161/ATVBAHA.109.200121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tskitishvili, E., Sharentuya, N., Temma-Asano, K., Mimura, K., Kinugasa-Taniguchi, Y., Kanagawa, T., et al. (2010). Oxidative stress-induced S100B protein from placenta and amnion affects soluble Endoglin release from endothelial cells. Mol. Hum. Reprod. 16, 188–199. doi: 10.1093/molehr/gap104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tuomilehto, J., Kuulasmaa, K., and Torppa, J. (1987). WHO MONICA Project: geographic variation in mortality from cardiovascular diseases. Baseline data on selected population characteristics and cardiovascular mortality. World Health Stat. Q. 40, 171–184.
Venkatesha, S., Toporsian, M., Lam, C., Hanai, J., Mammoto, T., Kim, Y. M., et al. (2006). Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 12, 642–649. doi: 10.1038/nm1429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weseler, A. R., and Bast, A. (2010). Oxidative stress and vascular function: implications for pharmacologic treatments. Curr. Hypertens. Rep. 12, 154–161. doi: 10.1007/s11906-010-0103-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Widlansky, M. E., and Gutterman, D. D. (2011). Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid. Redox. Signal. 15, 1517–1530. doi: 10.1089/ars.2010.3642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilcox, C. S. (2010). Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 126, 119–145. doi: 10.1016/j.pharmthera.2010.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wind, S., Beuerlein, K., Eucker, T., Müller, H., Scheurer, P., Armitage, M. E., et al. (2010). Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 161, 885–898. doi: 10.1111/j.1476-5381.2010.00920.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wooderchak-Donahue, W. L., McDonald, J., O’Fallon, B., Upton, P. D., Li, W., Roman, B. L., et al. (2013). Am. J. Hum. Genet. 93, 530–537. doi: 10.1016/j.ajhg.2013.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ye, Z., and Song, H. (2008). Antioxidant vitamins intake and the risk of coronary heart disease: meta-analysis of cohort studies. Eur. J. Cardiovasc. Prev. Rehabil. 15, 26–34. doi: 10.1097/HJR.0b013e3282f11f95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: HHT, reactive oxygen species, nitric oxide, eNOS, antioxidants
Citation: Jerkic M and Letarte M (2015) Contribution of oxidative stress to endothelial dysfunction in hereditary hemorrhagic telangiectasia. Front. Genet. 6:34. doi: 10.3389/fgene.2015.00034
Received: 15 November 2014; Accepted: 25 January 2015;
Published online: 13 February 2015.
Edited by:
Babajan Banganapalli, King Abdulaziz University, Saudi ArabiaReviewed by:
Vita Dolzan, University of Ljubljana, SloveniaXusheng Wang, St. Jude Children’s Research Hospital, USA
Copyright © 2015 Jerkic and Letarte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjana Jerkic, Anesthesia Research, Keenan Research Centre for Biomedical Science, St. Michael’s Hospital, University of Toronto, 209 Victoria Street, Toronto, ON M5B 1T8, Canada E-mail:amVya2ljbUBzbWguY2E=