 Michael E. Scharf
Michael E. Scharf- Department of Entomology, Purdue University, West Lafayette, IN, USA
Many recent breakthroughs in our understanding of termite biology have been facilitated by “omics” research. Omic science seeks to collectively catalog, quantify, and characterize pools of biological molecules that translate into structure, function, and life processes of an organism. Biological molecules in this context include genomic DNA, messenger RNA, proteins, and other biochemicals. Other permutations of omics that apply to termites include sociogenomics, which seeks to define social life in molecular terms (e.g., behavior, sociality, physiology, symbiosis, etc.) and digestomics, which seeks to define the collective pool of host and symbiont genes that collaborate to achieve high-efficiency lignocellulose digestion in the termite gut. This review covers a wide spectrum of termite omic studies from the past 15 years. Topics covered include a summary of terminology, the various kinds of omic efforts that have been undertaken, what has been revealed, and to a degree, what the results mean. Although recent omic efforts have contributed to a better understanding of many facets of termite and symbiont biology, and have created important new resources for many species, significant knowledge gaps still remain. Crossing these gaps can best be done by applying new omic resources within multi-dimensional (i.e., functional, translational, and applied) research programs.
Introduction
Overview and Terminology
In a broad sense, the underlying goals of omic1 science are to catalog, quantify, and characterize pools of biological molecules that translate into structure, function, and life processes of an organism or environment. The types of biological molecules receiving focus in omics2 include genomic DNA, messenger RNA (mRNA), protein, and metabolites (Figure 1). DNA, mRNA, and protein are respectively the foci of genomics, transcriptomics, methylomics, and proteomics. Genomics, methylomics, and transcriptomics rely on nucleic acid sequencing, whereas proteomics utilizes peptide sequencing procedures. By contrast, metabolomics is rooted more in analytical chemistry and focuses on biochemicals, metabolites, or pathways. Another relevant omic approach is the cataloging of bacterial and protist symbionts using high-throughput 16S and 18S rRNA sequencing.
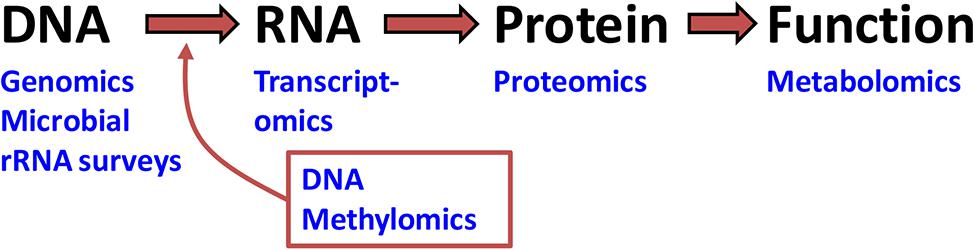
FIGURE 1. The continuum of biological organization and function addressed by omic research. The three bio-molecules listed (DNA, RNA, and protein) constitute the Central Dogma of Biology. Omic approaches that target these molecules can at best infer function. Proving function requires metabolomics and other functional or translational approaches not covered in this review (Scharf, 2015).
Termite omic research has focused on the host termite, individual gut microbial symbionts or entire populations of gut microbes. In the latter case, these “meta” analyses focusing broadly on collective microbiota occurring in the gut microenvironment have been popular, particularly with microbiologists specializing in termite intestinal microbiology. Although it presents significant bioinformatic challenges, a more inclusive approach that considers host and symbionts together as a single functional unit is the best approach for appreciating the full functional capacity of termites. A fundamental advantage of omic research over more traditional organismal research is that it enables direct mechanistic insights into termite and symbiont physiology and biochemistry. The use of omic technologies has led to new insights into behavior, social structure, digestion, and host-symbiont/symbiont–symbiont interactions, and many other aspects of termite biology. However, also as addressed throughout this review, omic science has limits for being able to define biological function.
Termite Symbiosis and the Holobiont Concept
Termites are perhaps best known for their symbiotic associations with gut microbes (König et al., 2013; Brune, 2014) that are often linked to digestive processes, although lignocellulose digestion is not mediated entirely by gut microbes (Watanabe and Tokuda, 2010; Figure 2A). The more ancestral lower termites have tri-partite symbioses that include host, bacteria and protozoa; whereas in higher termites, symbiosis has been reduced to a two-way association between host and bacteria (but some higher termites also maintain ecto-symbiotic associations with fungi; Brune, 2014). The host component of termite symbiotic systems adds substantially to the digestive process both in terms of contributing enzymes and maintaining a favorable gut microenvironment for symbiosis and digestion to occur (Watanabe et al., 1998; Tartar et al., 2009; Scharf et al., 2011; Sethi et al., 2013a; Tokuda et al., 2014). Because of the high degree of interplay that occurs between the termite host and gut symbionts, a key idea moving forward will be to consider termites from the perspective of the “holobiont” (a single functional unit in which host and symbionts are physiologically tightly connected). Omic research has enabled a multifaceted systemic understanding of gut digestomes that is central to understanding the termite holobiome from an applied perspective (Scharf, 2015).
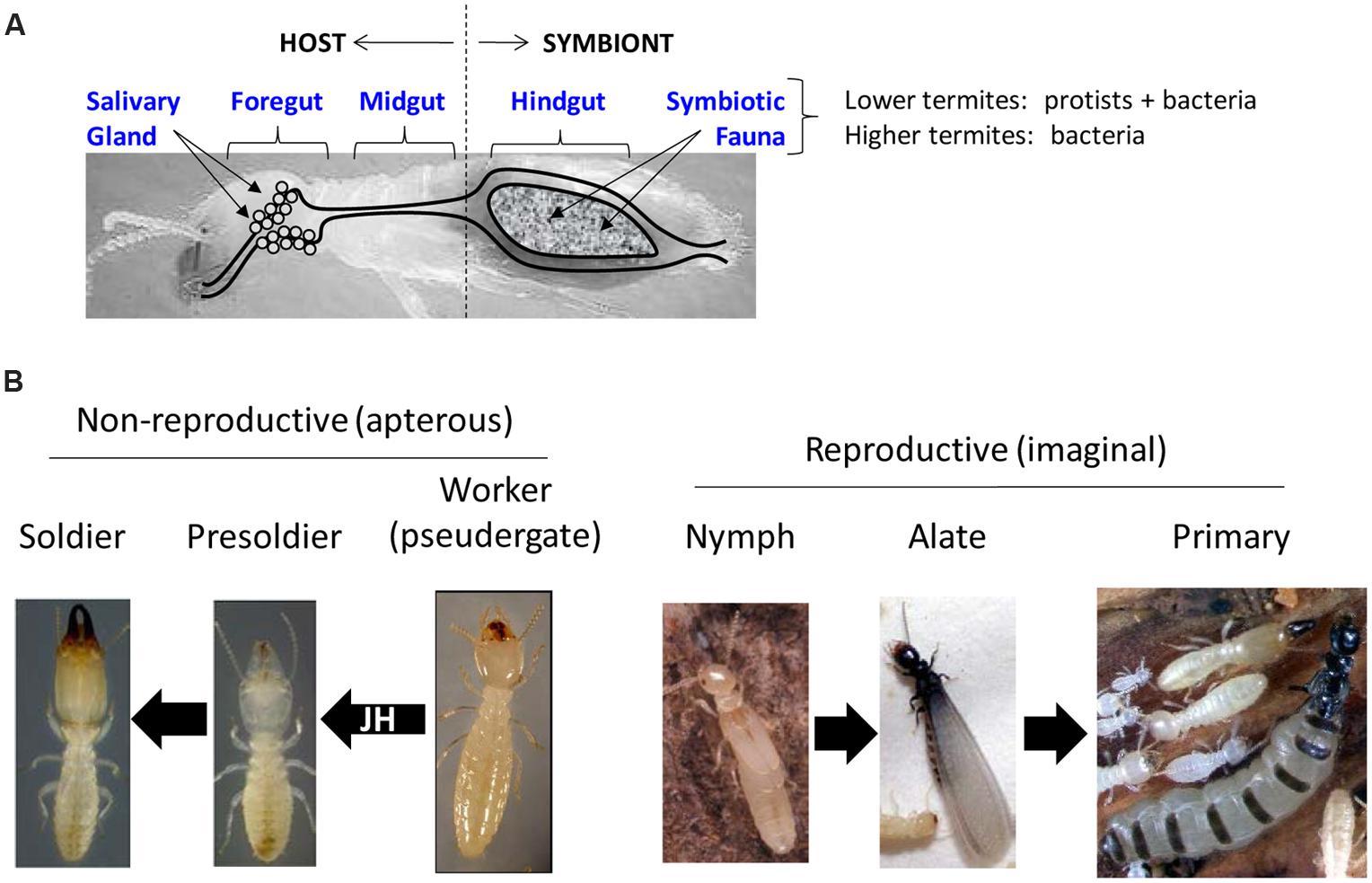
FIGURE 2. Fundamental ideas behind digestomic and sociogenomic research in termites. (A) Key components associated with termite digestomes and digestomic research. Different gut regions have been studied in an attempt to dissect host and symbiont contributions to digestion. An important distinction between lower and higher termites is the presence of protist and bacterial symbiota in lower termites, and only bacteria in higher termites. (B) Caste and phenotype-associated transitions addressed through sociogenomic research. Left: non-reproductive or “apterous” (wingless) phenotypes of lower termites. Presoldiers and soldiers differentiate from workers in response to elevated juvenile hormone (JH) titers. Right: nymphs give rise to alates that become primary reproductives; a process akin to typical hemimetabolous insect development.
Sociogenomics and Digestomics
The term sociogenomics was coined to describe the use of omic approaches for defining social life in molecular terms, which began with studies on the honey bee, Apis mellifera (Robinson et al., 2005). A parallel idea cited as rationale for many omic studies in social insects, including termites, is that solitary genes and traits were likely co-opted for new functions as solitary ancestors transitioned to social lifestyles (West-Eberhard, 2003; Nelson et al., 2007). Understanding such traits is essential for understanding termite social evolution (Miura and Scharf, 2011; Figure 2B). Another term used specifically in relation to digestive research is digestomics, which was coined to describe the collective pool of host and symbiont genes that collaborate to achieve high-efficiency lignocellulose digestion in the termite gut (Scharf and Tartar, 2008; Tartar et al., 2009; Figure 2A). Such terminology is useful because of the large number of symbionts that occupy termite guts and collaborate with the host in lignocellulose digestion. A related term is termitosphere, which is the full complement of gut and ectosymbiotic (nest) microbes present in termites, termite colonies, and their surrounding nest structures (Roose-Amsaleg et al., 2004; Bastien et al., 2013). Whether in relation to social, solitary or symbiont genes, proteins or other biomolecules, sociogenomic and digestomic research in termites has created an explosion of new sequence data.
Omic Studies in Termites: What has been Done?
Based on a recent literature survey (Table 1), at the time of writing this article around 70 papers had been published describing omic efforts in termite systems. These studies include all the themes introduced above, as well as microbial 16S and 18S surveys.
Taxonomic Distribution
In total, 82 termite species have been investigated using various omic approaches, with greater representation by lower than higher termites (72 vs. 28%). Among lower termites the top genera studied are important pest groups (Reticulitermes and Coptotermes), followed by non-pests from Hodotermopsis, Mastotermes, and Cryptotermes. Among higher termite genera, Nasutitermes dominate, followed by Odontotermes, Trinervitermes, and several other minor groups. Two termite genome sequences have now been published from the lower termite Zootermopsis angusticollis and the higher termite Macrotermes natalensis (see below).
Host vs. Symbiont Investigation
Of the various omic studies to date considering symbiosis and symbiotic partnerships in termite systems, the majority have taken an exclusive symbiont-oriented approach (>60%), whereas a minority have considered the host termite separately (<20%). The remainder have considered host and symbiont together (∼20%). In the latter category of host and symbiont combined, some studies have been a case of “accidental metatranscriptomics” (because protist symbionts have polyadenylated transcripts that are represented in cDNA libraries along with host transcripts; e.g., Scharf et al., 2003, 2005; Steller et al., 2010), but others have been deliberate metatranscriptomic studies (e.g., Tartar et al., 2009; Raychoudhury et al., 2013; Sen et al., 2013). The greater emphasis on gut symbiota compared to the host termite is likely because of the stereotypically well-recognized presence of gut microbes in termites.
Experimental Approaches and Types of Sequencing
In terms of experimental approaches taken, there has been an approximately equal split between descriptive and hypothesis-driven studies. Regarding the types of sequencing performed, transcriptomics and metatranscriptomics have been the dominant approaches (25 and 21% of studies), followed by microbial surveys for cataloging purposes (23%). The transcriptomic approaches used can be further divided into different methodologies such as cDNA library sequencing (Sanger, pyrosequencing or Illumina RNA-seq) and microarrays. Other efforts have targeted symbiont metagenomes (15%), symbiont or termite genomes (9%), proteomes (3%), and DNA methylomes (3%).
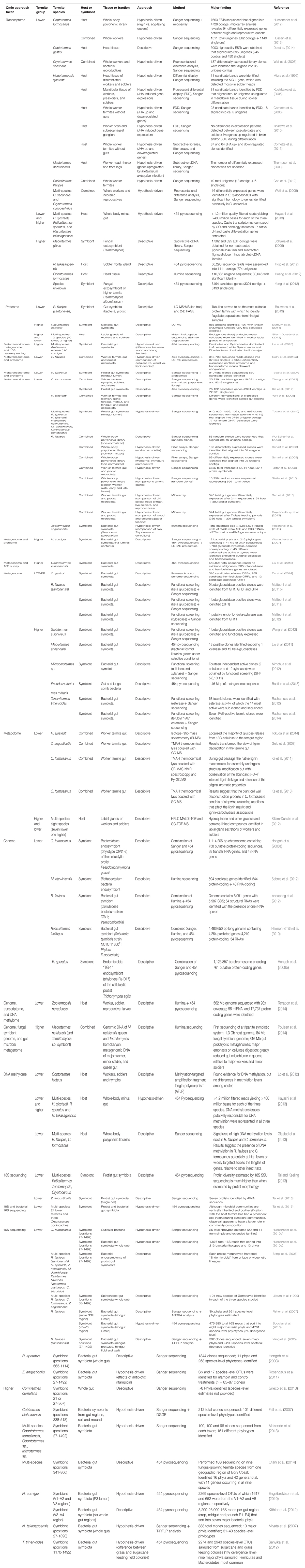
TABLE 1. A comprehensive literature summary of termite omic research, organized by approaches taken.
Omic Studies in Termites: What has been Revealed?
Genomics
Host Termite Genomes
At present only two termite genome sequences are available (Table 1); one from the lower termite Zootermopsis nevadensis (Terrapon et al., 2014) and one from the higher termite M. natalensis (Poulsen et al., 2014). Z. nevadensis was selected for sequencing based on its small genome size of 562 Mb relative to other termites, most of which are over 1000 Mb (Koshikawa et al., 2008). The Z. nevadensis sequencing approach involved shotgun genome sequencing of genomic DNA from symbiont-free soldier heads (n = 50 and 150 heads for 2 and 20 kb libraries, respectively). The transcriptomes of castes and various phenotypes were also sequenced for both gene prediction and comparative transcriptomic purposes. Transcriptome data were also used to search for DNA methylation machinery and methylation/epigenetic differences among castes and developmental stages.
The Z. nevadensis genome provided the first hints into how termites differ at the genome level from their eusocial counterparts in the order Hymenoptera, which evolved sociality independently. For making socio-evolutionary comparisons, emphasis was placed on gene family expansions, male fertility, chemoreception, immunity, polyphenism/division of labor, and potential epigenetic caste regulation. An expansion of genes related to male fertility and upregulated gene expression in male reproductives are consistent with differences in mating biology between termites and Hymenoptera. Regarding chemoreception, divergent numbers of genes and gene families relative to Hymenoptera were identified, as were variations in chemoreception gene expression among castes. Regarding caste polyphenism and division of labor, caste-associated gene expression profiles were readily identifiable. Key caste-regulatory and reproduction-associated genes identified through preceding work (e.g., hexamerins, vitellogenins, and CYP genes) were further defined and verified as gene families at the genomic level. Interestingly, there are 76 cytochrome P450 genes in the Z. nevadensis genome; which is nearly 2x as many as encoded by the honey bee genome (Honey Bee Genome Sequencing Consortium, 2006). Lastly, DNA methylation signatures and patterns of alternative splicing provided some evidence to suggest epigenetic caste regulation (see later).
The M. natalensis sequencing considered not only the host genome, but also the entire tri-partite system of this higher fungus-growing termite. This included the 1.3 Gb host genome, the 84 Mb genome of the Termitomyces sp. fungal symbiont and 816 Mb of prokaryotic gut metagenome from major workers, minor soldiers, and queens. Emphasis was placed mostly on cellulose digestion, which revealed a rich complement of glycosyl hydrolases from host, fungi, and gut microbes that likely collaborate in lignocellulose digestion. Another major finding was that gut microbiota composition is reduced by over 50% in queens relative to workers and soldiers, suggesting that queen gut microbiota undergo substantial compositional changes during colony founding, which points toward the local environment or other external factors as sources of microbiota as incipient colonies grow and age. Moving forward, the Z. nevadensis and M. natalensis genomes will be important resources for termitologists, and will also provide important scaffolds for assembly of additional termite genomes that will facilitate study of genes related to many evolutionary and biological processes.
Individual Symbiont Genomes
Five individual symbiont genomes have been sequenced (Table 1), with several others published or in progress since the writing of this article. No protist genomes have yet been sequenced. Two bacterial endosymbionts of hindgut protists from Coptotermes formosanus and Reticulitermes speratus (phylum Elusimicrobia or “TG1”) were the first symbiont genomes sequenced; they were obtained from isolated individual cells after whole-genome amplification (Hongoh et al., 2008a,b). No lignocellulase genes were identified; however, both genomes encoded capabilities to fix nitrogen, recycle host nitrogen wastes for amino acid and cofactor biosynthesis, and import glucose and xylose as energy and carbon sources. The next symbiont genomes were from gut bacteria in the phyla Verrucomicrobia and Fusobacteria, from the termites Reticulitermes flavipes and R. lucifugus (Harmon-Smith et al., 2010; Isanapong et al., 2012). These genomes were from culturable isolates and were found to encode genes related to cellulose degradation and nitrogen fixation. Another example is the genome of an obligate fat body endosymbiont Blattabacterium from the basal termite Mastotermes darwiniensis (Sabree et al., 2012). This bacterium displays a reduction in genome size and loss of genes required for amino acid production relative to free-living gut bacteria, which is consistent with its ability to recycle nitrogenous wastes and its role as a co-evolved endosymbiotic partner of the host termite.
Symbiont Metagenomes
At the time of writing this article, at least 12 prokaryotic metagenomes had been partially sequenced (Table 1). Most metagenome publications have reported on lignocellulase identification from genome sequences of gut bacteria that selectively grew on lignocellulose media (Liu et al., 2011; Mattéotti et al., 2011a,b, 2012; Nimchua et al., 2012; Rashamuse et al., 2012, 2014; Wang et al., 2012). Another study used targeted xylanase screening from gut and ectosymbiotic fungi-associated bacteria of the higher termite Pseudacanthotermes militaris (Bastien et al., 2013). Other studies took broader approaches to sequence from gut bacterial communities of higher termites. By combining metagenome sequencing with 16S surveys and metatranscriptomics, these studies revealed new information on bacterial cellulase diversity from termites with different symbiosis strategies (i.e., with and without fungal ectosymbionts; Warnecke et al., 2007; Liu et al., 2013) and from different feeding guilds (dung vs. wood; He et al., 2013). While these studies provided a wealth of new high-impact information on bacterial symbionts, they did not consider how symbionts from the gut and/or nest termitosphere collaborate with or complement the host termite.
Transcriptomics
Host Transcriptome
Around 15 transcriptomic studies to date have focused on physiological processes or tissues in the host termite (Table 1). Early studies looked for caste-biased gene expression, but the approaches employed had low resolving power and typically revealed only small numbers of differentially expressed genes. These studies mainly used subtractive hybridizations or cDNA “macro” arrays (reviewed by Miura and Scharf, 2011). Also, these early studies in lower termites often fell into the category of “accidental metatranscriptomics” as described earlier. The majority of focus in termite transcriptomic work has been on differences among castes or during caste differentiation (reviewed by Miura and Scharf, 2011). Mainly, newer studies are considered here.
Because of the importance of juvenile hormone (JH) to soldier caste differentiation and the reliability of JH treatment for inducing soldier caste differentiation, continuing focus has been placed on this transition in hypothesis-driven studies that combine JH assays with transcriptomics (e.g., Cornette et al., 2013; Sen et al., 2013). Caste-regulatory primer pheromones and the social environment have also been studied in the same context (Tarver et al., 2010; Sen et al., 2013). Other studies have included tissue-directed subtractive hybridizations, random/de novo cDNA library sequencing and/or cDNA oligonucleotide microarrays to reveal caste-biased gene expression (Weil et al., 2009; Ishikawa et al., 2010; Leonardo et al., 2011; Hojo et al., 2012; Huang et al., 2012; Husseneder et al., 2012; Terrapon et al., 2014). The over-arching themes emerging from this work include caste and morphogenesis-associated gene expression, endocrine signaling, vitellogenesis, reproduction-related processes, and regulatory mechanisms that maintain juvenile worker states in lower termites.
The immune response is another aspect of host termite physiology investigated through transcriptomics. Four studies have revealed responses to immune challenges by both stereotypical and unprecedented immune-responsive genes (Thompson et al., 2003; Yuki et al., 2008; Gao et al., 2012; Hussain et al., 2013). Finally, an emerging theme has been to investigate pathogen-xenobiotic interactions at the transcriptome level (Husseneder and Simms, 2014; Sen et al., 2015).
Symbiont-Host Metatranscriptomes
In addition to host-targeted studies noted above, other studies have considered symbiont or host-symbiont metatranscriptome composition (Table 1). Early examples in this category showed worker-biased expression of protist cellulases (Scharf et al., 2003) and differential expression of symbiont cellulases between dispersing and non-dispersing adult reproductives (Scharf et al., 2005). Subsequent studies focused on metatranscriptome composition of bacteria, protist and/or fungal symbionts, mostly for the purpose of identifying digestive cellulases (reviewed by Scharf and Tartar, 2008). Recent work has probed deeper into gut metatranscriptomes by taking advantage of both traditional and next-generation sequencing technology (Todaka et al., 2010; Rosenthal et al., 2011; Xie et al., 2012; Zhang et al., 2012; He et al., 2013). Other work has sought to partition host and symbiont digestive contributions and identify candidate enzymes expressed specifically in response to wood (i.e., complex lignocellulose), cellulose and lignin feeding (Tartar et al., 2009; Raychoudhury et al., 2013; Sethi et al., 2013a).
One microarray study investigated gut metatranscriptome changes in responses to JH, primer pheromones and socio-environmental conditions, suggesting interesting linkages between gut symbiota and caste differentiation (Sen et al., 2013). Another microarray study investigated host and symbiont gene expression in response to pathogen and nicotinoid-insecticide challenges, providing new insights into immunological roles played by bacterial and protist gut symbionts in defending against invading fungal and bacterial pathogens (Sen et al., 2015), building on the ideas of extended disease resistance as conferred by fecal nest bacteria (Chouvenc et al., 2013) and gut microbiota (Rosengaus et al., 2014).
Proteomics
Proteomics (Table 1) is important to validate transcriptome studies, particularly for determining if a gene’s presence and/or its transcription and translation are proportional. For example, proteomic studies in a higher termite were unable to identify most of the bacterial cellulase proteins predicted by metagenome sequencing (Warnecke et al., 2007; Burnum et al., 2011). Alternatively, proteomic studies in lower termites were able to identify both protist cellulases and other host lignocellulases initially identified via metatranscriptome sequencing (Todaka et al., 2007; Sethi et al., 2013a). Another study investigated proteins present in labial gland secretions of 12 lower and higher termite species, identifying endogenous GHF9 cellulases as dominant components of worker labial gland secretions in most species investigated (Sillam-Dussès et al., 2012). Another study used proteomics to catalog gut microbial communities, but with limited resolution (Bauwens et al., 2013). Clearly, more proteomic efforts are needed to resolve issues related to: (1) congruency between nucleic acid and protein sequencing approaches, and (2) to verify open reading frames predicted by metagenome and transcriptome sequencing.
DNA Methylomes
Four studies to date have looked at methylation signatures across termite castes with somewhat differing results. A seminal study used a methylation-targeted amplification fragment length polymorphism (AFLP) approach in Coptotermes lacteus to look for methylation signature differences among castes (Lo et al., 2012). Evidence of methylation was found, but no significant caste-associated methylation patterns were identified.
A subsequent study was done in silico using database sequences from R. flavipes and C. formosanus (Glastad et al., 2013). In this study and the two described below, transcriptome data were mined to determine the specific distribution of CpG dinucleotides (i.e., 5′–3′ cytosine followed by guanine), in order to predict DNA methylation levels in silico. Evidence of DNA methylation machinery and methylation signatures was found at high levels among expressed genes. Results also suggested that DNA methylation in R. flavipes is targeted to genes with ubiquitous (rather than differential) expression among castes and morphs. A third study examined host transcriptomes of three termite species that included two lower (Hodotermopsis sjostedti, R. speratus) and one higher termite (Nasutitermes takasagoensis; Hayashi et al., 2013). Pyrosequencing was done in combination with 69 caste and phenotypic libraries from the three termite species. Sequence analysis revealed that DNA methyltransferases potentially responsible for DNA methylation were present in each species, and verified the presence of methylation signatures. However, only limited evidence of caste-associated methylation profiles was detectable across the three species.
Finally, DNA methylation was assessed in Z. nevadensis as part of genome and transcriptome sequencing efforts (Terrapon et al., 2014). Transcriptome data were used to determine the specific distribution of CpG dinucleotides, in order to make in silico predictions of DNA methylation levels and explore for epigenetic differences among castes. In addition to verifying the presence of genes that encode for DNA methylation machinery (i.e., DNA methyltransferases 1 and 3), results showed greater methylation of genes rather than intergenic DNA, and a greater presence in introns than exons. This evidence, along with findings that alternatively spliced genes have greater degrees of methylation, suggests intronic methylation may impact alternative splicing.
While it is clear that DNA methylation exists in termites, so-far inconclusive results have been obtained to suggest epigenetic caste regulation. As concluded previously in relation to genetic caste determination (Vargo and Husseneder, 2009), the field of epigenetic caste regulation is in its infancy and epigenetic phenomena may or may not be relevant in natural colonies. More importantly, in silico methylation studies can only suggest that methylation may exist and which genes might be differentially methylated. Functional/translational research will be required to verify whether or not such genes truly are methylated, as well as the functions of those genes.
Metabolomics
Metabolomic studies are useful for assessing in situ processes, both as an exploratory approach and for functional/translational studies to verify nucleotide sequences. Soldier defensive secretions previously received much attention in this respect (Prestwich, 1984; Nelson et al., 2001). A more recent study investigated chemical components of labial gland secretions in soldier and worker termites from 7 lower and 1 higher termite (Sillam-Dussès et al., 2012). This study confirmed hydroquinone and other glucose and benzene-linked compounds as common labial gland secretions among most species.
Other metabolomic studies have focused on lignocellulose digestion. One main question addressed has been: does lignin digestion or modification occur during passage through the termite gut? Several studies over the past 25 years have addressed this question (reviewed by Ni and Tokuda, 2013) but recent metabolomic studies have been particularly informative (Geib et al., 2008; Ke et al., 2011, 2013). In general, findings are consistent regarding modification of lignin during passage through the gut, but evidence of actual lignin depolymerization has been more elusive. One possible reason for this could relate to insufficient detection procedures. Another possibility is that lignin-ether bonds, broken during depolymerization, only remain in this state for a short time and thus appear as intact lignin in frass. The induction of numerous antioxidant and detoxification enzymes by lignin feeding, as well as increased saccharification in the presence of lignin-associated phenoloxidases, supports the latter possibility (Sethi et al., 2013a). Despite convincing evidence of lignin modification during passage through the termite gut, and related omic studies revealing lignin-associated changes in host oxidative enzymatic machinery, the topic of lignin digestion/modification in termite guts remains contentious (Brune, 2014).
Another aspect of termite metabolomic research considers cellulose digestion and relative contributions of host and symbiont to this process. A recent metabolomic study investigated in situ digestion of 13C-labeled crystalline cellulose by H. sjostedti (Tokuda et al., 2014). Novel insights obtained related to both cellulose digestion and nitrogen metabolism. The results not only confirmed preceding work showing that endogenous cellulose digestion by the host is substantial, but also suggested other novel possibilities; for example (i) a significant digestive contribution by hindgut bacteria is phosphorolysis of cello-oligosaccharides to glucose-1-phosphate, and (ii) essential amino acid acquisition occurs via lysis of hindgut microbes obtained through proctodeal trophallaxis. The rapid buildup of glucose observed in the foregut agrees well with prior studies showing that host foregut cellulases can produce high levels of glucose directly from wood lignocellulose (Scharf et al., 2011; Sethi et al., 2013a,b). Additionally, higher glucose levels observed in the hindgut than other regions agrees with estimates that glucose release from lignocellulose is about 1/3 host and 2/3 symbiont (Scharf et al., 2011). However, since this study only focused on metabolite identification in gut tissue, it could not account for nutrients/metabolites transported out of the foregut and catabolized in other areas of the body.
Symbiont 16S and 18S Surveys
Bacterial 16S rRNA sequence surveys have been used extensively for cataloging bacteria and archaea (Wang and Qian, 2009), whereas 18S small subunit (SSU) rRNA surveys are just beginning to gain attention for cataloging protist symbionts (Tai and Keeling, 2013). Over 20 bacterial 16S surveys have been published to date using both cloning-dependent and -independent, high- and low-throughput approaches (Table 1). Highly variable species-level compositions have been obtained across the different termite species investigated, but, in general, six major bacterial phyla are represented across higher and lower termites: Bacteroidetes, Firmicutes, Spirochaetes, Proteobacteria, Fibrobacteres, and Elusimicrobia (Brune, 2014). Surveys conducted in parallel with higher-termite metagenome studies have been very informative for matching functional and taxonomic diversity (Warnecke et al., 2007; He et al., 2013); however, a study comparing multiple colonies through pyrosequencing of 16S amplicons found that bacterial compositions were different among colonies and likely influenced by local environment (Boucias et al., 2013). Additionally, 16S surveys revealed that lignocellulosic diet shifts have no short-term impacts on termite and cockroach microbiota composition (Sanyika et al., 2012; Boucias et al., 2013; Schauer et al., 2014). Another 16S survey of fungus-growing termites suggested a core microbiota of 42 genera that was shared among all nine termite species tested (Otani et al., 2014). This core microbiota was very different from other higher and lower termites, leading the authors to conclude the 42 common genera represent a core microbiota of fungus-growing termites. Conversely, since the termites were sampled from a limited geographic area it is possible that the core genera represent common microbes acquired from the local environment.
In comparison to prokaryotic 16S surveys, comparatively few protist 18S SSU surveys have been conducted (Table 1). These studies, conducted using a combination of cloning-dependent and independent approaches, have been transformative. Two studies provided new evidence to suggest greater protist symbiont diversity than originally indicated by traditional morphological identification (James et al., 2013; Tai et al., 2013). Two other studies used high-throughput 16S and 18S SSU sequencing to compare 24 lower termites with three wood-feeding cockroaches (Tai and Keeling, 2013; Tai et al., 2015). Like their predecessors, these studies found protist diversity to be higher than when estimated by morphology, and also that protist symbiont taxa tend to be highly endemic to a host genus, which is different than relationships between termite hosts and bacterial symbiota. These findings illustrate the significant opportunities that exist for development of high-throughput techniques for assessing protist symbiont communities and studying protist-bacterial symbiont relationships.
Needs and Opportunities
Termite omic research in the last 10–15 years has led to a new era of understanding for termite and symbiont biology. Omics has also enabled the development of new unparalleled resources (i.e., transcriptome, genome, proteome, metabolome, symbiont meta-omic, and symbiont rDNA) useful for moving ahead with targeted functional work. The stage is now set for making significant headway in many aspects of termite research, including, but not limited to digestion, symbiosis, caste differentiation, and social evolution. However, key needs and opportunities remain in specific areas that seem particularly relevant for filling in knowledge gaps and potentially leading to transformative, paradigm-shifting outcomes.
Having the Z. nevadensis and M. natalensis genomes available not only facilitates further study of genes related to a range of evolutionary and biological processes, but these resources also provide important scaffolds for assembly of additional lower and higher termite genomes. Once multiple termite genomes are available, this would certainly better inform our view of termite social evolution. On the topic of host-symbiont “hologenomes,” sequencing more host genomes and symbiont metagenomes from the same termites concurrently (as recently done for M. natalensis), would provide unprecedented insights into the scope of interactions and synergies occurring in termite holobiomes. Such efforts could further reveal important differences between clades of higher and lower termites, leading to new evolutionary insights. Such datasets would also provide unmatched resources for advancing integrative sociogenomic, digestomic, termitosphere, and other research topics.
On the topic of proteomics, more studies are needed in species that have had genomes, transcriptomes, metagenomes, or metatranscriptomes sequenced. Combining proteomics with nucleic acid sequencing will better resolve gene prediction models and better test for congruency between transcription and translation profiles. On the topic of metabolomics, termite digestion remains an area much in need of metabolomic research focusing on how complex lignocellulose is broken down in termite guts and converted to energy. Also, tracking metabolites as they leave the gut and are utilized in the termite body would be very informative for testing hypotheses on the relative importance of nutrient flow into symbiont metabolic pathways.
On the topic of DNA methylomics, while it is now clear that DNA methylation happens in termites, so-far inconclusive results have been obtained regarding the role of DNA methylation in caste regulation. In silico methylation studies as performed can only suggest that methylation may exist and which genes are potentially differentially methylated. Functional and translational research is needed to understand the roles of such genes.
Substantial opportunities and needs still remain for 16S and 18S rRNA-based symbiont cataloging. Protist 18S SSU cataloging capabilities in particular have recently been developed, and can continue to improve provided that several conditions are met, such as: (1) appropriate primers can be developed, (2) statistically sound sampling regimes can be developed at biologically relevant scales, (3) single-cell microbiology and other data sources can be integrated, and (4) appropriate analytical tools developed (Tai and Keeling, 2013). This line of research has already begun to transform the view of protist diversity and co-evolution with host termites but more studies are needed in different termite species with established omic resources.
Finally, regarding prokaryotic 16S surveys, much has already been done, but an important gap in knowledge is the extent to which environment influences bacterial microbiota composition. This is important information for understanding differences in behavior and physiology across the geographic range for a termite species, as well as potentially for limiting the extent to which generalizations can be made about the relative importance of individual microbes or core microbiota in gut communities.
Conclusion
This review has covered many aspects related to outcomes, findings and trends resulting from termite omic research. To date, omic research in diverse termite species has provided key insights into caste differentiation, digestion, pathogen defense and microbiomes, and most recently has provided two termite genome sequences. Termite omics has also created important tools and resources for conducting targeted, functional, translational, and applied research. However, these resources have only received limited attention to date for asking hypothesis-driven questions to elucidate the functional and evolutionary significance for pools of identified genes, proteins, and microbes. In recent years sequencing has rapidly moved into the realm of super high-throughput, with accompanying assembly and analyses requiring proportional super-computing power and bioinformatics expertise, but only limited resolution of biology or function. Transitioning from research that produces lists of genes, proteins and microbes, to research that determines their functional significance, is where the most important challenges lie for the next phases of termite science.
Funding
Work conducted in the author’s laboratory was supported by the following funding sources: USDA-CSREES-NRI grant no. 2007-35607-17777, USDA-NIFA-AFRI grant nos. 2009-05245 and 2010-65106-30727, Consortium for Plant Biotechnology Research-DOE grant no. DE-FG36-02GO12026, DOE-SBIR grant nos. DE-FG02-08ER85063 and DE-85538 S08-II, NSF grant no. 1233484CBET, and the O.W. Rollins/Orkin Endowment at Purdue University. M.E.S. is an inventor on the following patents: US Patent No. 7,968,525, US Patent No. 8,445,240, US Provisional Patent No. 61/602,149, and US Provisional Patent No. 61/902,472.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Apologies are extended to investigators whose research could not be cited because of space limitations. The author thanks Priya Rajarapu, Brittany Peterson, and Andres Sandoval for manuscript review, Vera Tai for sharing prepublication data, as well as his collaborators and all members of his laboratory, past and present, for their contributions and input.
Footnotes
- ^The singular term “omic" is used as an adjective in this review.
- ^The plural term “omics" is used as a noun.
References
Bastien, G., Arnal, G., Bozonnet, S., Laguerre, S., Ferreira, F., Fauré, R.,et al. (2013). Mining for hemicellulases in the fungus-growing termite Pseudacanthotermes militaris using functional metagenomics. Biotechnol. Biofuels. 6:78. doi: 10.1186/1754-6834-6-78
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bauwens, J., Millet, C., Tarayre, C., Brasseur, C., Destain, J., Vandenbol, M.,et al. (2013). Symbiont diversity in Reticulitermes santonensis: investigation strategy through proteomics. Environ. Entomol. 42, 882–887. doi: 10.1603/EN13112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boucias, D. G., Cai, Y., Sun, Y., Lietze, V. U., Sen, R., Raychoudhury, R.,et al. (2013). The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol. Ecol. 22, 1836–1853. doi: 10.1111/mec.12230
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burnum, K. E., Callister, S. J., Nicora, C. D., Purvine, S. O., Hugenholtz, P., Warnecke, F.,et al. (2011). Proteome insights into the symbiotic relationship between a captive colony of Nasutitermes corniger and its hindgut microbiome. ISME J. 5, 161–164. doi: 10.1038/ismej.2010.97
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chouvenc, T., Efstathion, C. A., Elliott, M. L., and Su, N. Y. (2013). Extended disease resistance emerging from the faecal nest of a subterranean termite. Proc. Biol. Sci. 280:20131885. doi: 10.1098/rspb.2013.1885
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cornette, R., Koshikawa, S., Hojo, M., Matsumoto, T., and Miura T. (2006). Caste-specific cytochrome P450 in the damp-wood termite Hodotermopsis sjostedti. Insect Mol. Biol. 15, 235–244. doi: 10.1111/j.1365-2583.2006.00632.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cornette, R., Hayashi, Y., Koshikawa, S., and Miura, T. (2013). Differential gene expression in response to juvenile hormone analog treatment in the damp-wood termite Hodotermopsis sjostedti. J. Insect Physiol. 59, 509–518. doi: 10.1016/j.jinsphys.2013.02.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Do, T. H., Nguyen, T. T., Nguyen, T. N., Le, Q. G., Nguyen, C., Kimura, K.,et al. (2014). Mining biomass-degrading genes through Illumina-based de novo sequencing and metagenomic analysis of free-living bacteria in the gut of the lower termite Coptotermes gestroi harvested in Vietnam. J. Biosci. Bioeng. 6, 665–671. doi: 10.1016/j.jbiosc.2014.05.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Engelbrektson, A., Kunin, V., Wrighton, K. C., Zvenigorodsky, N., Chen, F., Ochman, H., et al. (2010). Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 4, 642–647. doi: 10.1038/ismej.2009.153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fall, S., Hamelin, J., Ndiaye, F., Assigbetse, K., Aragno, M., Chotte, J. L.,et al. (2007). Differences between bacterial communities in the gut of a soil-feeding termite (Cubitermes niokoloensis) and its mounds. Appl. Environ. Microbiol. 73, 5199–5208. doi: 10.1128/AEM.02616-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fisher, M., Miller, D., Brewster, C., Husseneder, C., and Dickerman, A. (2007). Diversity of gut bacteria of Reticulitermes flavipes as examined by 16S rRNA gene sequencing and amplified rDNA restriction analysis. Curr. Microbiol. 55, 254–259. doi: 10.1007/s00284-007-0136-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, Q., Tancredi, S. E., and Thompson, G. J. (2012). Identification of mycosis-related genes in the eastern subterranean termite by suppression subtractive hybridization. Arch. Insect Biochem. Physiol. 80, 63–76. doi: 10.1002/arch.21026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Geib, S. M., Filley, T. R., Hatcher, P. G., Hoover, K., Carlson, J. E., Jimenez-Gasco Mdel, M.,et al. (2008). Lignin degradation in wood-feeding insects. Proc. Natl. Acad. Sci. U.S.A. 105, 12932–12937. doi: 10.1073/pnas.0805257105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. D. (2013). Evidence of a conserved functional role for DNA methylation in termites. Insect Mol. Biol. 22, 143–154. doi: 10.1111/imb.12010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grieco, M. A., Cavalcante, J. J., Cardoso, A. M., Vieira, R. P., Machado, E. A., Clementino, M. M.,et al. (2013). Microbial community diversity in the gut of the South American termite Cornitermes cumulans. Microb. Ecol. 65, 197–204. doi: 10.1007/s00248-012-0119-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harmon-Smith, M., Celia, L., Chertkov, O., Lapidus, A., Copeland, A., Glavina Del Rio, T.,et al. (2010). Complete genome sequence of Sebaldella termitidis type strain (NCTC 11300). Stand. Genomic Sci. 2, 220–227. doi: 10.4056/sigs.811799
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayashi, Y., Shigenobu, S., Watanabe, D., Toga, K., Saiki, R., Shimada, K.,et al. (2013). Construction and characterization of normalized cDNA libraries by 454 pyrosequencing and estimation of DNA methylation levels in three distantly related termite species. PLoS ONE 8:e76678. doi: 10.1371/journal.pone.0076678
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
He, S., Ivanova, N., Kirton, E., Allgaier, M., Bergin, C., Scheffrahn, R. H.,et al. (2013). Comparative metagenomic and metatranscriptomic analysis of hindgut paunch microbiota in wood- and dung-feeding higher termites. PLoS ONE 8:e61126. doi: 10.1371/journal.pone.0061126
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hojo, M., Maekawa, K., Saitoh, S., Shigenobu, S., Miura, T., Hayashi, Y.,et al. (2012). Exploration and characterization of genes involved in the synthesis of diterpene defence secretion in nasute termite soldiers. Insect Mol. Biol. 21, 545–557. doi: 10.1111/j.1365-2583.2012.01162.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honey Bee Genome Sequencing Consortium. (2006). Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949. doi: 10.1038/nature05260
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hongoh, Y., Ohkuma, M., and Kudo, T. (2003). Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus. FEMS Microbiol. Ecol. 44, 231–242. doi: 10.1016/S0168-6496(03)00026-6
Hongoh, Y., Sharma, V. K., Prakash, T., Noda, S., Toh, H., Taylor, T. D.,et al. (2008a). Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 322, 1108–1109. doi: 10.1126/science.1165578
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hongoh, Y., Sharma, V. K., Prakash, T., Noda, S., Taylor, T. D., Kudo, T.,et al. (2008b). Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc. Natl. Acad. Sci. U.S.A. 105, 5555–5560. doi: 10.1073/pnas.0801389105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, Q., Sun, P., Zhou, X., and Lei, C. (2012). Characterization of head transcriptome and analysis of gene expression involved in caste differentiation and aggression in Odontotermes formosanus. PLoS ONE 7:e50383. doi: 10.1371/journal.pone.0050383
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hussain, A., Li, Y. F., Cheng, Y., Liu, Y., Chen, C. C., and Wen, S. Y. (2013). Immune-related transcriptome of Coptotermes formosanus Shiraki workers: the defense mechanism. PLoS ONE 8:e69543. doi: 10.1371/journal.pone.0069543
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Husseneder, C., Ho, H. Y., and Blackwell, M. (2010a). Comparison of the bacterial symbiont composition of the Formosan subterranean termite from its native and introduced range. Open Microbiol. J. 4, 53–66. doi: 10.2174/1874285801004010053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Husseneder, C., Simms, D. M., Aluko, G. K., and Delatte, J. (2010b). Colony breeding system influences cuticular bacterial load of Formosan subterranean termite workers. Environ. Entomol. 39, 1715–1723. doi: 10.1603/EN09238
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Husseneder, C., McGregor, C., Lang, R. P., Collier, R., and Delatte, J. (2012). Transcriptome profiling of female alates and egg-laying queens of the Formosan subterranean termite. Comp. Biochem. Physiol. D. 7, 14–27.
Husseneder, C., and Simms, D. M. (2014). Effects of caste on the expression of genes associated with septic injury and xenobiotic exposure in the Formosan subterranean termite. PLoS ONE 9:e105582. doi: 10.1371/journal.pone.0105582
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Isanapong, J., Goodwin, L., Bruce, D., Chen, A., Detter, C., Han, J.,et al. (2012). High-quality draft genome sequence of the Opitutaceae bacterium strain TAV1, a symbiont of the wood-feeding termite Reticulitermes flavipes. J. Bacteriol. 194, 2744–2745. doi: 10.1128/JB.00264-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishikawa, Y., Okada, Y., Ishikawa, A., Miyakawa, H., Koshikawa, S., and Miura, T. (2010). Gene expression changes during caste-specific neuronal development in the damp-wood termite Hodotermopsis sjostedti. BMC Genomics 11:314. doi: 10.1186/1471-2164-11-314
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
James, E. R., Tai, V., Scheffrahn, R. H., and Keeling, P. J. (2013). Trichonympha burlesquei n. sp. from Reticulitermes virginicus and evidence against a cosmopolitan distribution of Trichonympha agilis in many termite hosts. Int. J. Syst. Evol. Microbiol. 63, 3873–3876. doi: 10.1099/ijs.0.054874-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johjima, T., Taprab, Y., Noparatnaraporn, N., Kudo, T., and Ohkuma, M. (2006). Large-scale identification of transcripts expressed in a symbiotic fungus (Termitomyces) during plant biomass degradation. Appl. Microbiol. Biotechnol. 73, 195–203. doi: 10.1007/s00253-006-0570-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, J., Laskar, D. D., and Chen, S. (2013). Tetramethylammonium hydroxide (TMAH) thermochemolysis for probing in situ softwood lignin modification in each gut segment of the termite. J. Agric. Food Chem. 61, 1299–1308. doi: 10.1021/jf3048548
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, J., Laskar, D. D., Singh, D., and Chen, S. (2011). In situ lignocellulosic unlocking mechanism for carbohydrate hydrolysis in termites: crucial lignin modification. Biotechnol. Biofuels 4:17. doi: 10.1186/1754-6834-4-17
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Köhler, T., Dietrich, C., Scheffrahn, R. H., and Brune, A. (2012). High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 78, 4691–4701. doi: 10.1128/AEM.00683-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
König, H., Li, L., and Fröhlich, J. (2013). The cellulolytic system of the termite gut. Appl. Microbiol. Biotechnol. 97, 7943–7962. doi: 10.1007/s00253-013-5119-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koshikawa, S., Cornette, R., Hojo, M., Maekawa, K., Matsumoto, T., and Miura, T. (2005). Screening of genes expressed in developing mandibles during soldier differentiation in the termite Hodotermopsis sjostedti. FEBS Lett. 579, 1365–1370. doi: 10.1016/j.febslet.2005.01.031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koshikawa, S., Miyazaki, S., Cornette, R., Matsumoto, T., and Miura, T. (2008). Genome size of termites (Insecta, Dictyoptera, Isoptera) and wood roaches (Insecta, Dictyoptera, Cryptocercidae). Naturwissenschaften 95, 859–867. doi: 10.1007/s00114-008-0395-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leonardo, F. C., da Cunha, A. F., da Silva, M. J., Carazzolle, M. F., Costa-Leonardo, A. M., Costa, F. F.,et al. (2011). Analysis of the workers head transcriptome of the Asian subterranean termite, Coptotermes gestroi. Bull. Entomol. Res. 101, 383–391. doi: 10.1017/S0007485310000556
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lilburn, T. G., Schmidt, T. M., and Breznak, J. A. (1999). Phylogenetic diversity of termite gut spirochaetes. Environ. Microbiol. 1, 331–345. doi: 10.1046/j.1462-2920.1999.00043.x
Liu, N., Yan, X., Zhang, M., Xie, L., Wang, Q., Huang, Y.,et al. (2011). Microbiome of fungus-growing termites: a new reservoir for lignocellulase genes. Appl. Environ. Microbiol. 77, 48–56. doi: 10.1128/AEM.01521-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, N., Zhang, L., Zhou, H., Zhang, M., Yan, X., Wang, Q.,et al. (2013). Metagenomic insights into metabolic capacities of the gut microbiota in a fungus-cultivating termite (Odontotermes yunnanensis). PLoS ONE 8:e69184. doi: 10.1371/journal.pone.0069184
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lo, N., Li, B., and Ujvari, B. (2012). DNA methylation in the termite Coptotermes lacteus. Insect Soc. 59, 257–261. doi: 10.1007/s00040-011-0213-7
Makonde, H. M., Boga, H. I., Osiemo, Z., Mwirichia, R., Mackenzie, L. M., Göker, M.,et al. (2013). 16S-rRNA-based analysis of bacterial diversity in the gut of fungus-cultivating termites (Microtermes and Odontotermes species). Antonie Van Leeuwenhoek 104, 869–883. doi: 10.1007/s10482-013-0001-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mattéotti, C., Bauwens, J., Brasseur, C., Tarayre, C., Thonart, P., Destain, J.,et al. (2012). Identification and characterization of a new xylanase from Gram-positive bacteria isolated from termite gut (Reticulitermes santonensis). Protein Expr. Purif. 83, 117–127. doi: 10.1016/j.pep.2012.03.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mattéotti, C., Haubruge, E., Thonart, P., Francis, F., De Pauw, E., Portetelle, D.,et al. (2011a). Characterization of a new β-glucosidase/β-xylosidase from the gut microbiota of the termite (Reticulitermes santonensis). FEMS Microbiol. Lett. 314, 147–157. doi: 10.1111/j.1574-6968.2010.02161.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mattéotti, C., Thonart, P., Francis, F., Haubruge, E., Destain, J., Brasseur, C.,et al. (2011b). New glucosidase activities identified by functional screening of a genomic DNA library from the gut microbiota of the termite Reticulitermes santonensis. Microbiol. Res. 166, 629–642. doi: 10.1016/j.micres.2011.01.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miura, T., Kamikouchi, A., Sawata, M., Takeuchi, H., Natori, S., Kubo, T.,et al. (1999). Soldier caste-specific gene expression in the mandibular glands of Hodotermopsis japonica. Proc. Natl. Acad. Sci. U.S.A. 96, 13874–13879. doi: 10.1073/pnas.96.24.13874
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miura, T., and Scharf, M. E. (2011). “Molecular mechanisms underlying caste differentiation in termites,” in Biology of Termites: A Modern Synthesis, eds D. E. Bigness, Y. Roisin, and N. Lo (Dordrecht: Springer), 211–253.
Miyata, R., Noda, N., Tamaki, H., Kinjyo, K., Aoyagi, H., Uchiyama, H.,et al. (2007). Influence of feed components on symbiotic bacterial community structure in the gut of the wood-feeding higher termite Nasutitermes takasagoensis. Biosci. Biotechnol. Biochem. 71, 1244–1251. doi: 10.1271/bbb.60672
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nelson, C. M., Ihle, K. E., Fondrk, M. K., Page, R. E., and Amdam, G. V. (2007). The gene vitellogenin has multiple coordinating effects on social organization. PLoS Biol. 5:e62. doi: 10.1371/journal.pbio.0050062
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nelson, L. J., Cool, L. G., Forschler, B. T., and Haverty, M. I. (2001). Correspondence of soldier defense secretion mixtures with cuticular hydrocarbon phenotypes for chemotaxonomy of the termite genus Reticulitermes in North America. J. Chem. Ecol. 27, 1449–1479. doi: 10.1023/A:1010325511844
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ni, J., and Tokuda, G. (2013). Lignocellulose-degrading enzymes from termites and their symbiotic microbiota. Biotechnol. Adv. 31, 838–850. doi: 10.1016/j.biotechadv.2013.04.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nimchua, T., Thongaram, T., Uengwetwanit, T., Pongpattanakitshote, S., and Eurwilaichitr, L. (2012). Metagenomic analysis of novel lignocellulose-degrading enzymes from higher termite guts inhabiting microbes. J. Microbiol. Biotechnol. 22, 462–469. doi: 10.4014/jmb.1108.08037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Koné, N. A., Sørensen, S. J.,et al. (2014). Identifying the core microbial community in the gut of fungus-growing termites. Mol. Ecol. 23, 4631–4644. doi: 10.1111/mec.12874
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poulsen, M., Hu, H., Li, C., Chen, Z., Xu, L., Otani, S.,et al. (2014). Complementary symbiont contributions to plant decomposition in a fungus-farming termite. Proc. Natl. Acad. Sci. U.S.A. 111, 14500–14505. doi: 10.1073/pnas.1319718111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Prestwich, G. D. (1984). Defense mechanisms of termites. Ann. Rev. Entomol. 29, 201–232. doi: 10.1146/annurev.en.29.010184.001221
Rashamuse, K., Mabizela-Mokoena, N., Sanyika, T. W., Mabvakure, B., and Brady D. (2012). Accessing carboxylesterase diversity from termite hindgut symbionts through metagenomics. J. Mol. Microbiol. Biotechnol. 22, 277–286. doi: 10.1159/000342447
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rashamuse, K., Ronneburg, T., Sanyika, W., Mathiba, K., Mmutlane, E., and Brady, D. (2014). Metagenomic mining of feruloyl esterases from termite enteric flora. Appl. Microbiol. Biotechnol. 98, 727–737. doi: 10.1007/s00253-013-4909-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Raychoudhury, R., Sen, R., Cai, Y., Sun, Y., Lietze, V. U., Boucias, D. G.,et al. (2013). Comparative metatranscriptomic signatures of wood and paper feeding in the gut of the termite Reticulitermes flavipes. Insect Mol. Biol. 22, 155–171. doi: 10.1111/imb.12011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Robinson, G. E., Grozinger, C. M., and Whitfield, C. W. (2005). Sociogenomics: social life in molecular terms. Nat. Rev. Genet. 6, 257–270. doi: 10.1038/nrg1575
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roose-Amsaleg, C., Brygoo, Y., and Harry, M. (2004). Ascomycete diversity in soil-feeding termite nests and soils from a tropical rainforest. Environ. Microbiol. 6, 462–469. doi: 10.1111/j.1462-2920.2004.00579.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosengaus, R. B., Schultheis, K. F., Yalonetskaya, A., Bulmer, M. S., DuComb, W. S., Benson, R. W.,et al. (2014). Symbiont-derived β-1,3-glucanases in a social insect: mutualism beyond nutrition. Front. Microbiol. 5:607. doi: 10.3389/fmicb.2014.00607
Rosengaus, R. B., Zecher, C. N., Schultheis, K. F., Brucker, R. M., and Bordenstein, S. R. (2011). Disruption of the termite gut microbiota and its prolonged consequences for fitness. Appl. Environ. Microbiol. 77, 4303–4312. doi: 10.1128/AEM.01886-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosenthal, A. Z., Matson, E. G., Eldar, A., and Leadbetter, J. R. (2011). RNA-seq reveals cooperative metabolic interactions between two termite-gut spirochete species in co-culture. ISME J. 5, 1133–1142. doi: 10.1038/ismej.2011.3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sabree, Z. L., Huang, C. Y., Arakawa, G., Tokuda, G., Lo, N., Watanabe, H.,et al. (2012). Genome shrinkage and loss of nutrient-providing potential in the obligate symbiont of the primitive termite Mastotermes darwiniensis. Appl. Environ. Microbiol. 78, 204–210. doi: 10.1128/AEM.06540-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanyika, T. W., Rashamuse, K. J., Hennesy, F., and Brady, D. (2012). Luminal hindgut bacterial diversities of the grass and sugarcane feeding termite Trinervitermes trinervoides. African J. Microbiol. Res. 6, 2639–2648.
Scharf, M. E. (2015). Termites as targets and models for biotechnology. Ann. Rev. Entomol. 60, 77–102. doi: 10.1146/annurev-ento-010814-020902
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scharf, M. E., Karl, Z. J., Sethi, A., and Boucias, D. G. (2011). Multiple levels of synergistic collaboration in termite lignocellulose digestion. PLoS ONE 6:e21709. doi: 10.1371/journal.pone.0021709
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scharf, M. E., Scharf, W. D., Pittendrigh, B. R., and Bennett, G. W. (2003). Caste- and development-associated gene expression in a lower termite. Genome Biol. 4:R62. doi: 10.1186/gb-2003-4-10-r62
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scharf, M. E., and Tartar, A. (2008). Termite digestomes as sources for novel lignocellulases. Biofuels Bioprod. Bioref. 2, 540–552. doi: 10.1002/bbb.107
Scharf, M. E., Wu-Scharf, D., Zhou, X., Pittendrigh, B. R., and Bennett, G. W. (2005). Gene expression profiles among immature and adult reproductive castes of the termite Reticulitermes flavipes. Insect Mol. Biol. 14, 31–44. doi: 10.1111/j.1365-2583.2004.00527.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schauer, C., Thompson, C., and Brune, A. (2014). Pyrotag sequencing of the gut microbiota of the cockroach Shelfordella lateralis reveals a highly dynamic core but only limited effects of diet on community structure. PLoS ONE 9:e85861. doi: 10.1371/journal.pone.0085861
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sen, R., Raychoudhury, R., Cai, Y., Sun, Y., Lietze, V. U., Boucias, D. G.,et al. (2013). Differential impacts of juvenile hormone, soldier head extract and alternate caste phenotypes on host and symbiont transcriptome composition in the gut of the termite Reticulitermes flavipes. BMC Genomics 14:491. doi: 10.1186/1471-2164-14-491
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sen, R., Raychoudhury, R., Cai, Y., Sun, Y., Lietze, V. U., Boucias, D. G.,et al. (2015). Metatranscriptomic signatures of nicotinoid-pathogen synergy in the termite gut. PLoS ONE (in press).
Sethi, A., Slack, J. M., Kovaleva, E. S., Buchman, G. W., and Scharf, M. E. (2013a). Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect Biochem. Mol. Biol. 43, 91–101. doi: 10.1016/j.ibmb.2012.10.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sethi, A., Kovaleva, E. S., Slack, J. M., Brown, S., Buchman, G. W., and Scharf, M. E. (2013b). A GHF7 cellulase from the protist symbiont community of Reticulitermes flavipes enables more efficient lignocellulose processing by host enzymes. Arch. Insect Biochem. Physiol. 84, 175–193. doi: 10.1002/arch.21135
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sillam-Dussès, D., Krasulová, J., Vrkoslav, V., Pytelková, J., Cvačka, J., Kutalová, K.,et al. (2012). Comparative study of the labial gland secretion in termites. PLoS ONE 7:e46431. doi: 10.1371/journal.pone.0046431
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steller, M. M., Kambhampati, S., and Caragea, D. (2010). Comparative analysis of expressed sequence tags from three castes and two life stages of the termite Reticulitermes flavipes. BMC Genomics 11:463. doi: 10.1186/1471-2164-11-463
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stingl, U., Radek, R., Yang, H., and Brune, A. (2005). “Endomicrobia”: cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Appl. Environ. Microbiol. 71, 1473–1479. doi: 10.1128/AEM.71.3.1473-1479.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tai, V., James, E. R., Nalepa, C. A., Scheffrahn, R. H., Perlman, S. J., and Keeling, P. J. (2015). The role of host phylogeny varies in shaping microbial diversity in the hindguts of lower termites. Appl. Environ. Microbiol. 81, 1059–1070. doi: 10.1128/AEM.02945-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tai, V., James, E. R., Perlman, S. J., and Keeling, P. J. (2013). Single-Cell DNA barcoding using sequences from the small subunit rRNA and internal transcribed spacer region identifies new species of Trichonympha and Trichomitopsis from the hindgut of the termite Zootermopsis angusticollis. PLoS ONE 8:e58728. doi: 10.1371/journal.pone.0058728
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tai, V., and Keeling, P. J. (2013). Termite hindguts and the ecology of microbial communities in the sequencing age. J. Eukaryot. Microbiol. 60, 421–428. doi: 10.1111/jeu.12048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tartar, A., Wheeler, M. M., Zhou, X., Coy, M. R., Boucias, D. G., and Scharf, M. E. (2009). Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite R. flavipes. Biotechnol. Biofuels 2:25. doi: 10.1186/1754-6834-2-25
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tarver, M. R., Zhou, X., and Scharf, M. E. (2010). Socio-environmental and endocrine influences on developmental and caste-regulatory gene expression in the eusocial termite Reticulitermes flavipes. BMC Mol. Biol. 11:28. doi: 10.1186/1471-2199-11-28
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Terrapon, N., Li, C., Robertson, H. M., Ji, L., Meng, X., Booth, W.,et al. (2014). Molecular traces of alternative social organization in a termite genome. Nat. Commun. 5:3636. doi: 10.1038/ncomms4636
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thompson, G. J., Crozier, Y. C., and Crozier, R. H. (2003). Isolation and characterization of a termite transferrin gene up-regulated on infection. Insect Mol. Biol. 12, 1–7. doi: 10.1046/j.1365-2583.2003.00381.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Todaka, N., Inoue, T., Saita, K., Ohkuma, M., Nalepa, C. A., Lenz, M.,et al. (2010). Phylogenetic analysis of cellulolytic enzyme genes from representative lineages of termites and a related cockroach. PLoS ONE 5:e8636. doi: 10.1371/journal.pone.0008636
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Todaka, N., Moriya, S., Saita, K., Hondo, T., Kiuchi, I., Takasu, H.,et al. (2007). Environmental cDNA analysis of the genes involved in lignocellulose digestion in the symbiotic protist community of Reticulitermes speratus. FEMS Microbiol. Ecol. 59, 592–599. doi: 10.1111/j.1574-6941.2006.00237.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tokuda, G., Tsuboi, Y., Kihara, K., Saitou, S., Moriya, S., Lo, N.,et al. (2014). Metabolomic profiling of 13C-labelled cellulose digestion in a lower termite: insights into gut symbiont function. Proc. Biol. Sci. 281, 1789. doi: 10.1098/rspb.2014.0990
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vargo, E. L., and Husseneder, C. (2009). Biology of subterranean termites: insights from molecular studies of Reticulitermes, and Coptotermes. Annu. Rev. Entomol. 54, 379–403. doi: 10.1146/annurev.ento.54.110807.090443
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Q., Qian, C., Zhang, X. Z., Liu, N., Yan, X., and Zhou, Z. (2012). Characterization of a novel thermostable β-glucosidase from a metagenomic library of termite gut. Enzyme Microb. Technol. 51, 319–324. doi: 10.1016/j.enzmictec.2012.07.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., and Qian, P. Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 4:e7401. doi: 10.1371/journal.pone.0007401
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Warnecke, F., Luginbühl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T.,et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watanabe, H., Noda, H., Tokuda, G., and Lo, N. (1998). A cellulase gene of termite origin. Nature 394, 330–331. doi: 10.1038/28527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watanabe, H., and Tokuda, G. (2010). Cellulolytic systems in insects. Annu. Rev. Entomol. 55, 609–632. doi: 10.1146/annurev-ento-112408-085319
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weil, T., Korb, J., and Rehli, M. (2009). Comparison of queen-specific gene expression in related lower termite species. Mol. Biol. Evol. 26, 1841–1850. doi: 10.1093/molbev/msp095
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weil, T., Rehli, M., and Korb, J. (2007). Molecular basis for the reproductive division of labour in a lower termite. BMC Genomics 8:198. doi: 10.1186/1471-2164-8-198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
West-Eberhard, M. J. (2003). Developmental Plasticity and Evolution. Oxford: Oxford University Press.
Wu-Scharf, D., Scharf, M. E., Pittendrigh, B. R., and Bennett, G. W. (2003). Expressed sequence tags from a polyphenic Reticulitermes flavipes cDNA library. Sociobiology 41, 479–490.
Xie, L., Zhang, L., Zhong, Y., Liu, N., Long, Y., Wang, S.,et al. (2012). Profiling the metatranscriptome of the protistan community in Coptotermes formosanus with emphasis on the lignocellulolytic system. Genomics 99, 246–255. doi: 10.1016/j.ygeno.2012.01.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, H., Schmitt-Wagner, D., Stingl, U., and Brune, A. (2005). Niche heterogeneity determines bacterial community structure in the termite gut (Reticulitermes santonensis). Environ. Microbiol. 7, 916–932. doi: 10.1111/j.1462-2920.2005.00760.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, F., Xu, B., Li, J., and Huang, Z. (2012). Transcriptome analysis of Termitomyces albuminosus reveals the biodegradation of lignocellulose. Wei Sheng Wu Xue Bao 52, 466–477.
Yuki, M., Moriya, S., Inoue, T., and Kudo, T. (2008). Transcriptome analysis of the digestive organs of Hodotermopsis sjostedti, a lower termite that hosts mutualistic microorganisms in its hindgut. Zoolog. Sci. 25, 401–406. doi: 10.2108/zsj.25.401
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, D., Lax, A. R., Henrissat, B., Coutinho, P., Katiya, N., Nierman, W. C.,et al. (2012). Carbohydrate-active enzymes revealed in Coptotermes formosanus transcriptome. Insect Mol. Biol. 21, 235–245. doi: 10.1111/j.1365-2583.2011.01130.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: holobiome, digestome, sociogenomics, symbiosis, metabolomics, DNA methylation, sociobiology, socioevolution
Citation: Scharf ME (2015) Omic research in termites: an overview and a roadmap Front. Genet. 6:76 doi: 10.3389/fgene.2015.00076
Received: 02 September 2014; Accepted: 13 February 2015;
Published online: 13 March 2015.
Edited by:
Juergen Rudolf Gadau, Arizona State University, USAReviewed by:
Judith Korb, University of Freiburg, GermanyEdward L. Vargo, North Carolina State University, USA
Copyright © 2015 Scharf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael E. Scharf, Department of Entomology, Purdue University, 901 West State Street, West Lafayette, IN 47907-2089, USAbXNjaGFyZkBwdXJkdWUuZWR1