 Kirk Szafranski
Kirk Szafranski Karan J. Abraham1†
Karan J. Abraham1† Karim Mekhail
Karim Mekhail- 1Department of Laboratory Medicine and Pathobiology, Faculty of Medicine, University of Toronto, Toronto, ON, Canada,
- 2Canada Research Chairs Program, Faculty of Medicine, University of Toronto, Toronto, ON, Canada
Declining brain and neurobiological function is arguably one of the most common features of human aging. The study of conserved aging processes as well as the characterization of various neurodegenerative diseases using different genetic models such as yeast, fly, mouse, and human systems is uncovering links to non-coding RNAs. These links implicate a variety of RNA-regulatory processes, including microRNA function, paraspeckle formation, RNA–DNA hybrid regulation, nucleolar RNAs and toxic RNA clearance, amongst others. Here we highlight these connections and reveal over-arching themes or questions related to recently appreciated roles of non-coding RNA in neural function and dysfunction across lifespan.
Introduction
Neurodegenerative diseases are a group of debilitating neurological disorders typically associated with old age (Eacker et al., 2009). These conditions are characterized by the progressive loss of neurons within one or more regions of the CNS (Eacker et al., 2009). Most neurodegenerative diseases are sporadic in origin, meaning that they arise in patients with no apparent family history or known environmental cause (Robberecht and Philips, 2013). While the biology of sporadic disease remains mysterious, several decades of research on various inherited forms have led to remarkable advances in our understanding of disease genetics. Systematic efforts over the last few decades have led to the identification of a number of causal mutations and key genes (Pulst et al., 1996; Neumann et al., 2006; Kabashi et al., 2008). Building on these findings, many studies have attempted to delineate the precise pathological processes arising from such genetic anomalies. A number of disease mechanisms have been proposed including toxicity induced by misfolded or aberrantly translated proteins, and the mislocalization of important cellular factors (Eacker et al., 2009). Despite encouraging progress, many aspects of these disorders are poorly understood and sadly, many devastating conditions remain untreatable. Furthermore, diseases for which symptom-relieving treatments are available are often difficult to diagnose. As a whole, neurodegenerative disease represents a major source of morbidity and mortality worldwide and is justifiably garnering the attention of both the scientific community and society at large.
Neurodegenerative diseases are associated with a diverse spectrum of clinical presentations, brain pathologies, and health consequences (Eacker et al., 2009). They include relatively well-known conditions like AD, FTLD, PD, and ALS. Additionally, a host of less prevalent disorders including various SCAs and AOA2 also belong to this class of diseases. Conditions, like AD and FTLD are primarily cognitive disorders and represent the two leading causes of dementia worldwide (Mattson, 2004). They are caused by progressive loss of brain regions responsible for reasoning, cognition, and memory. In contrast, ALS and PD are chiefly disorders of motor function, caused by the preferential loss of motor neurons and dopamine-producing neurons in the brain, respectively (Hardiman et al., 2011; Wirdefeldt et al., 2011). Although some conditions can be contained by pharmacotherapy and/or surgery, the majority of neurodegenerative diseases remain untreatable. Furthermore, neurodegenerative diseases represent an increasing fiscal burden in healthcare systems attempting to cope with aging populations. Thus, there is an urgent need to develop approaches that prevent, stall, or cure neurodegenerative disease. Ultimately, this process will hinge on understanding the precise molecular and cellular mechanisms that lead to the premature dysfunction or death of neurons.
Basic research on potential disease mechanisms has benefited greatly from studies using model organisms and/or novel experimental systems. Insights from such studies are providing mounting evidence that ncRNAs and ncRNA-regulatory processes are important players in the pathogenesis of neurodegenerative disease (Eacker et al., 2009; Esteller, 2011; Abe and Bonini, 2013). ncRNAs represent a functionally and structurally diverse class of RNA species that participate in a wide range of basic cellular processes including protein translation, mRNA splicing, chromatin organization, and the regulation of gene expression (Esteller, 2011). Several classes of ncRNAs (e.g., miRNAs, rRNAs, tRNAs, and many lncRNAs) fulfill discrete functions within cells. However, it is becoming clear that a large proportion of the cellular transcript pool is comprised of ncRNAs that lack obvious function (Djebali et al., 2012; Palazzo and Gregory, 2014). These entities are thought to derive primarily from noisy transcription at intergenic sequences and are generally degraded rapidly within the nucleus (Djebali et al., 2012; Palazzo and Gregory, 2014). However, recent studies suggest that under certain conditions, these ncRNAs can trigger processes that are toxic to cells (Figure 1). These processes include the sequestration of crucial RNA-binding proteins as well as the accumulation of genome-destabilizing R-loops, which are structures that can form when nascent RNA stably hybridizes with DNA (Aguilera and Garcia-Muse, 2012; Haeusler et al., 2014; Salvi et al., 2014). Accordingly, cells have evolved a number of mechanisms to constrain both the amount and the ability of transcripts to engage in these detrimental processes (Palazzo and Gregory, 2014; Salvi et al., 2014). In this review, we focus exclusively on ncRNAs and make the broad distinction between ncRNAs that are either functional or lack a discrete role but nevertheless represent important determinants of overall cellular function. Specifically, we discuss the links between neurodegenerative disease and two major classes of functional ncRNAs, namely miRNAs and a group of lncRNAs. Additionally, we explore emerging evidence that deleterious ncRNA-driven processes such as R-loop formation and toxic RNA accumulation represent neurotoxic mechanisms in brain aging and neurodegeneration.
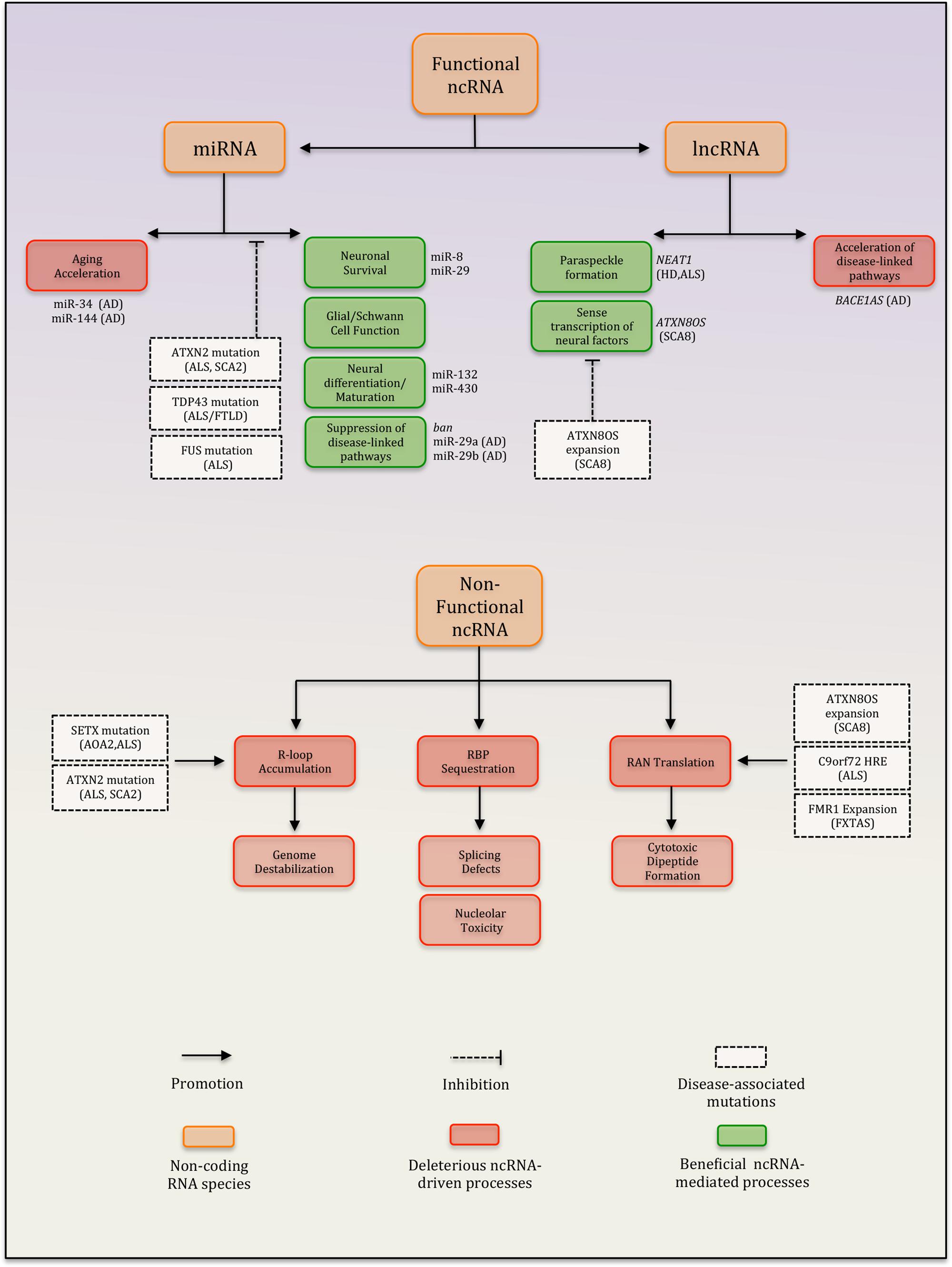
FIGURE 1. Summary of ncRNA-dependent processes implicated in neurodegenerative disease. Diseases highlighted: AD, Alzheimer’s disease; HD, Huntington’s disease; SCA8, spinocerebellar ataxia type 8; SCA2, spinocerebellar ataxia type 2; ALS, amyotrophic lateral sclerosis; AOA2, ataxia with oculomotor apraxia type 2; FXTAS, fragile X-associated tremor/ataxia syndrome; and FTLD, frontotemporal dementia.
Roles of miRNAs, lncRNAs, and other ncRNAs in Neural Function and Aging
Introduction to miRNAs: Biogenesis and Regulatory Mechanisms
microRNAs are an abundant class of short ncRNAs that regulate a variety of cellular processes through the post-transcriptional repression of gene expression (He and Hannon, 2004). Mature miRNAs are generated following a series of well-orchestrated biochemical events that begin in the nucleus and culminate in the cytoplasm (Figure 2; Lee et al., 2002). These steps include the nuclear processing of primary miRNA transcripts (pri-miRNAs) into precursor miRNAs (pre-miRNAs) by the DGCR8/Drosha complex, the cytoplasmic processing of pre-miRNAs into imperfectly paired miRNA duplexes by Dicer, and the preferential incorporation of one strand (the “guide" miRNA strand) onto the RISC (Bartel, 2004). Mature miRNAs complexed to RISC (miRISC) are targeted to mRNAs containing sequences that are complementary to the miRNA “seed" (nucleotides 2–7 of a mature miRNA). Ultimately, the repression of mRNAs occurs via their degradation, destabilization, translational silencing, or combinations thereof. Regardless of the specific mode of repression, the final outcome typically consists of decreased target protein levels and a subsequent biological effect reflecting the cellular function of the targeted transcript.
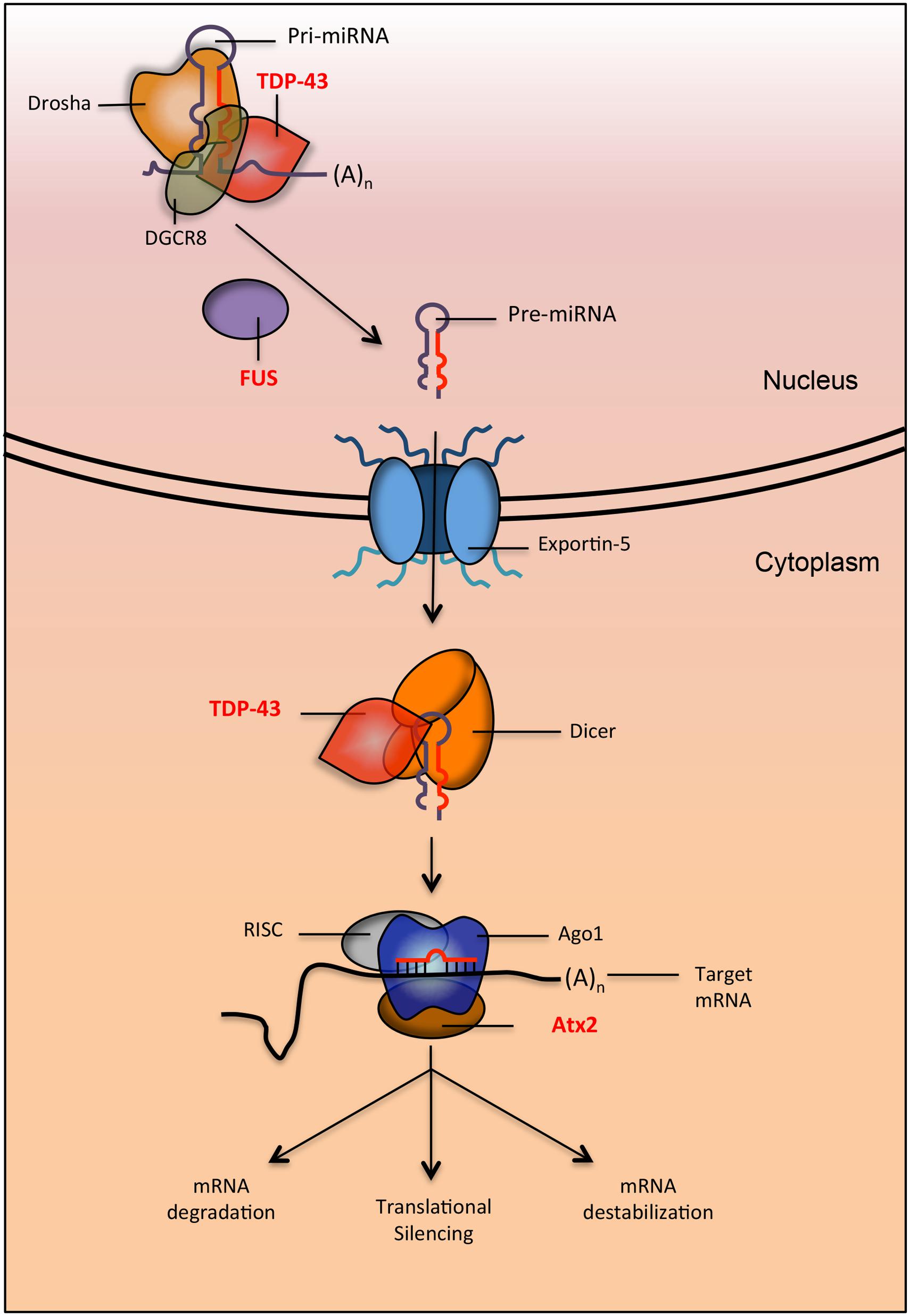
FIGURE 2. Proteins implicated in neurodegenerative diseases are involved in multiple steps of the miRNA biogenesis pathway. TDP-43 and FUS (implicated in ALS and FTLD) interact with Drosha (directly and indirectly respectively) to promote the processing of pri-miRNAs into pre-miRNA. After export, TDP-43 also interacts with dicer to process pre-miRNA into mature miRNA. When miRNA is loaded onto the RISC to be targeted for silencing, it interacts with Ago1. In Drosophila, it was shown that the protein Atx2 (the ortholog of ATXN2, a protein implicated in ALS and SCA2) interacts with Ago1 to promote proper targeting of miRNAs to their mRNA.
microRNAs have been increasingly linked to processes associated with brain aging, declining brain function, and neurodegenerative diseases (Eacker et al., 2009; Abe and Bonini, 2013). Indeed, several studies have reported widespread changes in miRNA expression, while others have identified specific miRNAs as strong correlates of brain aging and/or specific disease states (Li et al., 2011a; Persengiev et al., 2011; Inukai et al., 2012; Sheinerman et al., 2013). Future developments may allow these correlations to be used as powerful diagnostic markers of neurodegenerative diseases. In this section, we discuss the involvement of miRNAs in regulating diverse aspects of neural function based on insights gained from various experimental and genetic model systems. Furthermore, we highlight both direct and indirect evidence for deregulation of miRNA pathways in neurodegenerative disease.
miRNAs as Mediators of Brain Development and Neuronal Differentiation
A crude but effective approach to assess the overall role of miRNAs in the brain is to disable the miRNA biogenesis pathway (Eacker et al., 2009). This can be accomplished by the conditional deletion of Dicer, which results in a failure to generate mature dicer-dependent miRNAs. It is important to note that there have been reports of a relatively small number of dicer-independent miRNAs, which would not be affected by this strategy (Cheloufi et al., 2010; Ha and Kim, 2014). This strategy applied to model organisms has nevertheless helped unearth roles for miRNAs in developmental processes of the CNS including tissue morphogenesis and neuronal differentiation. In zebrafish, the loss of maternal and zygotic Dicer manifests in abnormal organogenesis. A striking component of this phenotype includes severe defects in neurulation and neural differentiation, which leads to failed brain morphogenesis (Giraldez et al., 2005). Several studies in mice have investigated the developmental consequences of disabling miRNA biogenesis in stem or neural precursor populations. One study used a Wnt1-cre promoter to conditionally delete Dicer in neural crest cells and select regions of the brain (Huang et al., 2010). This causes malformed development of the cerebellum and midbrain, and impaired differentiation of neural crest and dopaminergic cells. Similarly, the inactivation of Dicer in olfactory and post-mitotic motor progenitors leads to a failure to generate olfactory and post-mitotic motor neurons, respectively (Choi et al., 2008; Chen and Wichterle, 2012). The importance of miRNAs in neuronal differentiation is also supported by studies employing cell culture models of differentiation. For instance, the inactivation of miRNA biogenesis during the final stages of differentiation of DNs derived from murine embryonic stem cells abolishes terminal differentiation in culture (Kim et al., 2007b). Taken together, these studies identify miRNAs as critical players in fundamental brain development processes such as neuronal differentiation. Thus, disruption of miRNA biogenesis can compromise neurodevelopmental and neural differentiation processes that are known to occur at various stages of life.
Roles for miRNAs in Neuronal Longevity and Survival
Premature death of neurons is a major characteristic of neurodegenerative diseases, and the root cause of many health complications that arise during the course of illness. Mice have been used extensively to address a wide variety of questions relating to neuronal lifespan, including the need for a functional miRNA pathway. To determine the role of miRNAs in the maintenance of neuronal lifespan in vivo, several studies have characterized the neurobiological phenotypes caused by the deletion of Dicer in discrete populations of terminally differentiated cells. In many cases, the absence of a functional miRNA pathway leads to the loss of neurons via both cell autonomous and non-cell autonomous mechanisms. The inactivation of Dicer in post-mitotic murine midbrain neurons for instance, manifests in a drastic reduction in number and axonal projection of DNs residing in the midbrain as evidenced by decreased expression of DN markers on immunohistochemical brain sections (Kim et al., 2007b). This mirrors the pathological changes observed in human PD. Moreover, these degenerative changes are accompanied by marked reductions in locomotion and mobility reminiscent of bradykinesia (i.e., slowing of movement) seen during early stages of PD (Kim et al., 2007b). Region-specific neurodegeneration is observed in cerebellar Purkinje cells (PCs) devoid of Dicer (Schaefer et al., 2007). Interestingly, 10-weeks-old wild-type and Dicer-deficient mice display no differences in morphology or electrophysiological functions of PCs. Between 13 and 17 weeks, Dicer-deficient mice show profound PC apoptosis as revealed by electron microscopy, immunohistochemistry, and TUNEL staining. Furthermore, cerebellar degeneration coincides with onset of ataxia and a mild tremor that ultimately deteriorates to severely impaired coordination and motor function. Similarly, conditional deletion of Dicer in the spinal motor neurons results in deteriorating motor function from week 7 onward and histological changes in muscle that are pathognomonic for denervation-induced muscular atrophy (Schaefer et al., 2007; Damiani et al., 2008; Haramati et al., 2010). Indeed, spinal cord sections from these mice display reduced numbers of lower motor neurons and immunohistochemical evidence of reactive astrocytosis, which is a marker of neurotoxicity (Schaefer et al., 2007; Damiani et al., 2008; Haramati et al., 2010). Thus, the absence of a functional miRNA pathway diminishes survival and recapitulates various clinical and pathological features of human neurodegenerative disease. This highlights the vital role of the miRNA pathway in maintaining normal neuronal lifespan and points to the potential deregulation of this pathway as a driver or contributor to neurodegeneration.
Neurons rely on many supporting cell types (astrocytes, oligodendrocytes, Schwann cells, etc.) that co-exist in the nervous system. Several studies have shown that defective miRNA biogenesis in these cells abrogates critical homeostatic functions and lead to the death of neurons via non-cell autonomous mechanisms. The loss of Dicer in astroglial cells results in impaired neurotransmitter uptake and increased oxidative stress (Tao et al., 2011). Disruption of these critical functions was associated with ataxia at post-natal week 7, followed by prolonged seizures, severe locomotor dysfunction and eventually premature organismal death. Immunohistochemistry in combination with electron microscopy and TUNEL staining in cerebellar sections revealed that these changes were precipitated by apoptosis and axonal degeneration of mature cerebellar granule cells and PCs, respectively. Similarly, the loss of Dicer in Schwann cells and oligodendrocytes triggers inflammatory and oxidative cascades in the brain leading to neuronal injury and degeneration (Shin et al., 2009; Bremer et al., 2010). Thus, in the setting of neurodegenerative disease, loss of key homeostatic functions downstream of miRNA biogenesis can induce non-cell autonomous neuronal degeneration (Figure 1).
It is important to note that miRNAs are required for many aspects of neuronal structure and function, not only their long-term survival. In several studies, disabling miRNA biogenesis manifests in complex phenotypes in which degeneration is preceded by structural and/or functional abnormalities. For instance, Dicer inactivation in retinal cells results in the formation of abnormal rosette structures composed of photoreceptors prior to degeneration (Damiani et al., 2008). Similarly, the deletion of Dicer in cortical and hippocampal neurons is associated with abnormalities in cellular morphology, tissue architecture, and axonal pathfinding followed by profound degeneration within these regions (Davis et al., 2008). Thus, miRNA dysfunction may trigger overt neuronal degeneration as well as more subtle deficits in the setting of neurodegenerative disease.
Regulation of Neurodegenerative Disease-Associated Pathways by miRNAs
While previously discussed studies tested the need for global miRNA function in the brain, many other studies have focused on the relevance of specific miRNAs or miRNA families in regulating either neuroprotective or neurotoxic processes. One family of miRNAs that has emerged as putatively protective is the miR-29 family (Figure 1). Several studies employing mouse models and human cell lines have examined the involvement of miR-29 in neurodegenerative diseases. In agreement with a critical role in the brain, miRNA profiling studies have reported that miR-29a is expressed abundantly in neurons in the adult mouse brain, while miR-29b expression increases in various compartments of the maturing mouse CNS (Kole et al., 2011; Jovicic et al., 2013). Consistent with a role in neuroprotection, diminished levels of miR-29 family members have been reported in both patients or mouse models of AD, HD, and various subtypes of SCA (Lee et al., 2011; Wang et al., 2011b). The notion that these patterns may represent disease-promoting mechanisms rather than just correlational markers stems from the observation that perturbing miR-29 function is associated with compromised neuronal survival. For instance, knocking down miR-29b in the mouse brain using LNA-based antagomirs induces severe cerebellar and hippocampal degeneration, which manifests in ataxia and eventually leads to death (Roshan et al., 2014). This phenotype appears to be mediated in part by the de-repression of VDAC1, an apoptotic effector protein and miR-29b target. Reduced miR-29b levels are also associated with increased susceptibility to neuronal apoptosis in mouse models of acute ischemic stroke (Khanna et al., 2013). In this study, partial phenotypic rescue was achieved by the restoration of miR-29b expression. Lastly, the miR-29 family may also promote neuroprotection via the direct suppression of disease-linked pathways (Figure 1). In the setting of AD, the expression of miR-29a/b-1 is inversely associated with BACE-1 levels (Hebert et al., 2008). BACE-1 is an enzyme that can cleave amyloid precursor protein (APP) into amyloid beta (Aβ) peptides, the chief component of Aβ plaques found in the brains of AD patients (O’Brien and Wong, 2011). In human cell lines modeling Aβ plaque formation, miR-29a/b-1 can directly suppress the expression of BACE-1 via direct seed-3′ UTR interactions and reduce Aβ plaque formation in vitro (Hebert et al., 2008). These results suggest that in disease states like AD, downregulation of miR-29a/b-1 may promote disease susceptibility and/or progression in patients by precipitating Aβ plaque formation. Overall, functional studies focusing on miR-29 align well with its inverse association with neurodegenerative disease and point to a conserved role for this miRNA family in neuroprotection.
Examples of protective miRNAs that can directly target disease-associated proteins also exist in Drosophila melanogaster. For instance, the ban miRNA potently suppresses neurodegeneration induced by polyglutamine Ataxin-3 and tau, pathogenic proteins that are causally linked to SCA3 and AD, respectively (Bilen et al., 2006). Similarly, Drosophila miR-8 functions as a neuroprotective factor by targeting atrophin, a family of proteins linked to the neurodegenerative disease dentatorubral-pallidoluysian atrophy (DRPLA; Figure 1; Ross et al., 1997; Karres et al., 2007). These results raise the possibility that analogous pathways exist in humans, and future studies that explore this further may furnish novel opportunities for miRNA-based therapeutic targeting of disease proteins.
While some miRNAs are clearly protective, others may promote brain aging and/or initiate neurotoxic processes that contribute to specific neurodegenerative diseases (Table 1). For instance, miR-144 is a strong positive correlate of aged brains in humans, chimpanzees, and rhesus macaques (Persengiev et al., 2011). miR-144 is also enriched in post-mortem tissue from SCA1 and AD patients. Importantly, the ATXN1 protein (encoded by the gene mutated in SCA1) is a known target of miR-144. Thus, age-associated miR-144 may contribute to declining brain function in both normal and disease states via the downregulation of longevity/protective factors (Figure 1; Persengiev et al., 2011). Similarly, studies in both D. melanogaster and Caenorhabditis elegans point to the miR-34 family as another important determinant of brain aging. The miR-34 family is a highly conserved set of brain-enriched miRNAs (Bak et al., 2008). Intriguingly, correlative studies in mice and humans seem to point to a potential role for miR-34 in AD. Brain tissue derived from a mouse model of AD and in post-mortem tissue from AD patients are enriched for miR-34a (Wang et al., 2009). Interestingly, the pro-survival protein Bcl2 is a known miR-34a target. Furthermore, the expression of another known target of miR-34a, namely the anti-aging factor SIRT1, correlates inversely with age-associated increases in brain and circulating levels of that miRNA (Li et al., 2011b). SIRT1 knockdown accelerates neurodegeneration due to mutant HTT in mouse models of HD, while overexpression rescues phenotypes (Jiang et al., 2012). Moreover, SIRT1 promotes neuronal survival and suppresses neurodegeneration in a mouse model of AD and ALS (Kim et al., 2007a). Similarly, several reports are consistent with the notion that SIRT1 is neuroprotective (Araki et al., 2004; Chen et al., 2008a; Shindler et al., 2010; Huang et al., 2011; Graff et al., 2013). However, overt neurodegenerative defects have not yet been clearly documented in SIRT1-null mice, which nonetheless exhibit neural migration defects (Di Sante et al., 2015). Thus, SIRT1 may be simply neuroprotective or compensatory processes mediated by other sirtuin proteins may be involved. Consistent with the latter rationale, Sirt6 modulates neural chromatin structure and gene activity (Schwer et al., 2010). Taken together, while the precise links between neurodegeneration and miR-34a remain unclear, repression of protective factors such as Bcl2 and SIRT1 by the latter miRNA may point to two non-mutually exclusive possibilities.
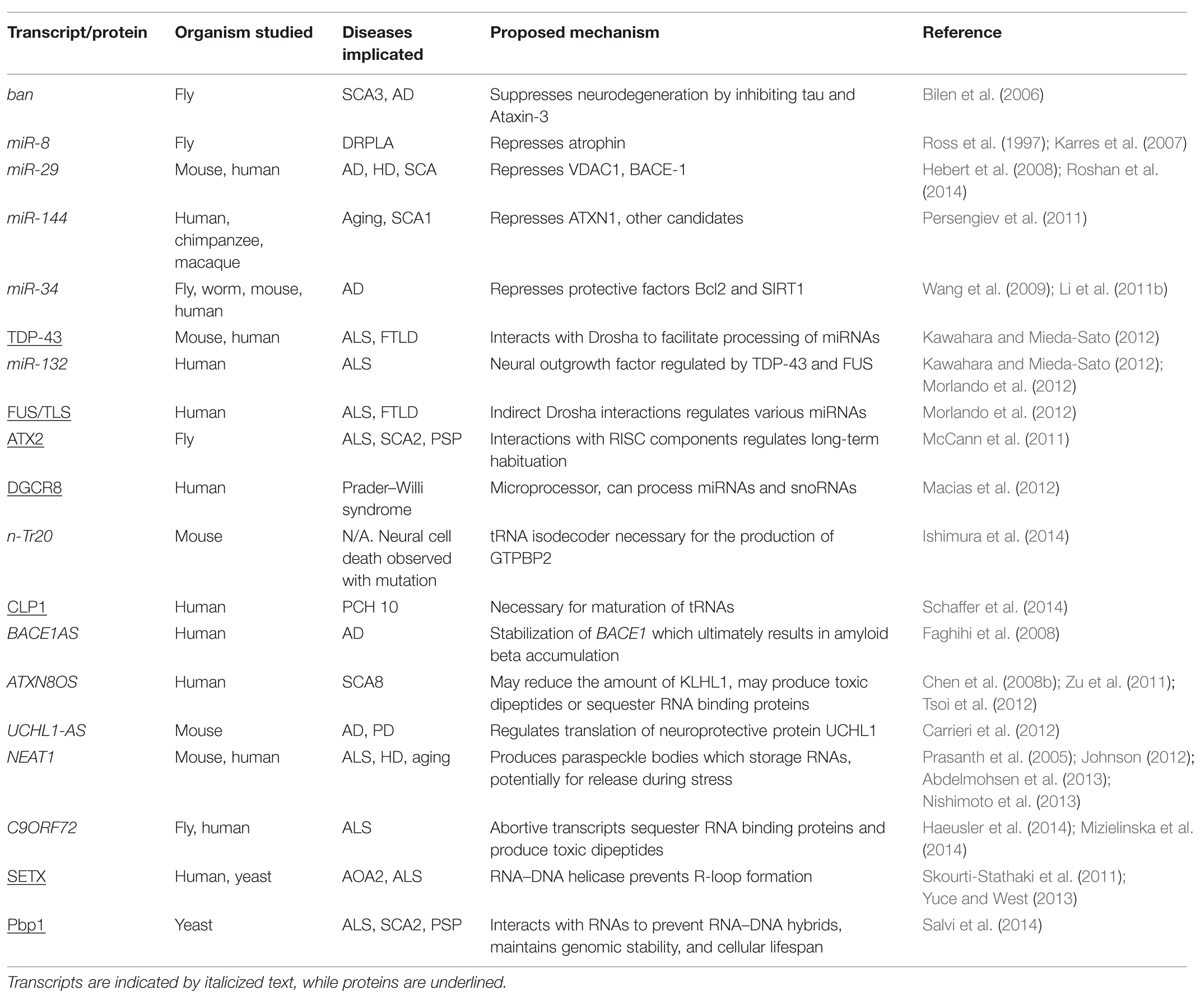
TABLE 1. List of the various transcripts and proteins discussed in this review as well as their associated disease and proposed pathogenic mechanism.
miRNA Dysfunction Downstream of Disease-Associated Proteins
Several neurodegenerative diseases are associated with defects in key RNA-binding proteins including TDP-43, ATXN2, and FUS. TDP-43 and FUS have roles in the regulation of coding RNAs which we do not discuss here and instead refer readers to several excellent reviews about TDP-43 and FUS function (Lagier-Tourenne and Cleveland, 2009; Baloh, 2012). In this section, we explore emerging evidence linking these disease-associated proteins with putative miRNA-regulatory functions and suggest how disruption of these roles may trigger pathological cascades in neurodegenerative diseases.
Tar-DNA binding protein-43 is a ubiquitously expressed RNA-binding protein strongly linked to several neurodegenerative diseases. Mutation of TARDBP (encodes TDP-43) is a relatively rare cause of familial ALS (Kabashi et al., 2008). Moreover, TDP-43 protein is frequently mislocalized to ubiquitinated inclusion bodies in ALS/FTLD, pointing to a role in disease pathogenesis even in the absence of TARDBP mutations (Neumann et al., 2006). In healthy cells, TDP-43 is primarily nuclear and it fulfills diverse roles in the regulation of mRNA transcription, alternative splicing, and ncRNA stability (Robberecht and Philips, 2013). However, it is becoming increasingly apparent that TDP-43 may additionally act as a regulator of miRNA biogenesis and function. Indeed, TDP-43 has been shown to physically interact with Drosha and Dicer in the nucleus and cytoplasm respectively, and these interactions are required for the normal processing of several miRNAs (Kawahara and Mieda-Sato, 2012). Furthermore, TDP-43 can either enhance or diminish the targeting of mRNAs by specific miRNAs (Fan et al., 2014; King et al., 2014). These results identify TDP-43 a component of the miRNA pathway and raise the possibility that mutations and/or mislocalization of TDP-43 may disrupt the biogenesis and/or function of miRNAs (Figure 2). Indeed, knocking down TDP-43 impairs neuronal differentiation of mouse neuroblastoma cells in culture due in part to a failure to properly process pri-miR-132, a miRNA with extensive links to neuronal differentiation (Kawahara and Mieda-Sato, 2012). Additionally, altered expression profiles of multiple TDP-43-regulated miRNAs is observed in serum and lymphoblast cell lines derived from ALS patients (Freischmidt et al., 2013). Taken together, these studies suggest that miRNA dysfunction downstream of disease-linked TDP-43 alterations could represent an important pathogenic mechanism in neurodegenerative disease and warrants further investigation (Figure 1).
Fused in sarcoma is another RNA-binding protein implicated in ALS/FTLD (Kwiatkowski et al., 2009; Vance et al., 2009; Mackenzie et al., 2010). The neuropathology of FUS is remarkably similar to TDP-43 in terms of detection in aberrant cytoplasmic aggregates within affected CNS regions. Unlike TDP-43, FUS shows no direct interaction with Drosha or Dicer (Kawahara and Mieda-Sato, 2012). However, nuclear FUS directly binds pri-mRNA, including the neural outgrowth factor pri-miR-132 (Morlando et al., 2012). Depletion of FUS decreases Drosha levels at miRNA genes, suggesting that FUS facilitates miRNA processing through indirect Drosha interactions (Morlando et al., 2012). FUS also regulates miR-134 and miR-143, both of which display differential neural expression with age (Inukai et al., 2012). Therefore, similarly to TDP-43, FUS may be required in the nucleus to maintain proper miRNA expression in the aging brain (Figure 2).
The ATXN2 protein is yet another disease-associated RNA binding protein with a putative role in miRNA function. Aberrant polyglutamine expansion within the human ATXN2 gene causes spinocerebellar ataxia type 2 (SCA2) and is strongly linked to ALS as well as the atypical parkinsonian disorder called progressive supranuclear palsy (PSP; Pulst et al., 1996; Elden et al., 2010; Ross et al., 2011). Atx2, the D. melanogaster ortholog of ATXN2, plays a critical role in long-term habituation of adult olfactory projection neurons via an Ago1- and Me31B- dependent mechanism (McCann et al., 2011). Both proteins are core components of the miRNA pathway in flies, thus identifying Atx2 as an important component of the D. melanogaster miRNA pathway (Figure 2). Overall, these findings raise the possibility that mutations and/or mislocalization of key RNA binding proteins including TDP-43, FUS, and ATXN2 (Figure 1) may lead to the disruption miRNAs that otherwise fulfill critical functions in the brain.
All in all, the miRNA pathway in general and specific miRNAs in particular are emerging as key players in the nervous system as they are linked to neurogenesis, cell survival/longevity, and a healthy/functional lifespan. However, as we will see in the next section, miRNAs are not the only type of ncRNA with such roles.
Other Small ncRNAs in Neurodegenerative Disease
In addition to miRNAs, a wide variety of small ncRNAs exists. Data linking these latter transcripts with neurodegenerative disorders are relatively limited (Esteller, 2011). However, there is some evidence that neurodegeneration-linked proteins may be responsible for proper regulation of such small ncRNAs. For example, the microprocessor DGCR8 regulates hundreds of small nucleolar RNAs (snoRNAs) independently of Drosha in human cells (Macias et al., 2012). It is important to note that although DGCR8 is also involved in the processing of miRNAs via Drosha interaction, miRNAs are not the most abundant DGCR8 targets (Macias et al., 2012). Furthermore, DGCR8 co-purifies with TDP-43 making it a putative regulator of snoRNA biogenesis (Kawahara and Mieda-Sato, 2012; Macias et al., 2012). Interestingly, defects in snoRNAs are associated with neurodevelopmental disorders such as Prader–Willi syndrome (Sahoo et al., 2008). Furthermore, several snoRNAs are differentially expressed in aged rat brains, so microprocessor action may also be important for the maintenance of proper snoRNA regulation during aging (Wood et al., 2013). Therefore, it is possible that the mislocalization of TDP-43 in ALS, FTLD or AD may impair DGCR8 function. This would in turn inhibit the biogenesis of key neural snoRNAs. Further work is clearly needed to clarify how TDP-43 or other neurodegeneration-related proteins impact snoRNA biogenesis and how this is important for the maintenance of neuronal and brain function.
tRNAs represent another ubiquitous and vital group of small ncRNAs. They recognize RNA codon sequences and allow for the transfer of amino acids, thereby mediating the process of protein synthesis. tRNAs that recognize the same codon, termed isodecoders, are encoded by numerous nuclear genes (Goodenbour and Pan, 2006). These isodecoders are not redundant; mutation of a CNS-specific tRNA isodecoder in mice, n-Tr20, is sufficient to cause the loss of a protein, GTPBP2, and results in widespread neurodegeneration (Ishimura et al., 2014). This highlights the importance of individual tRNAs in maintaining neural health. Additionally, mutations in aminoacyl-tRNA synthetases, which match tRNAs with their respective amino acids, leads to neurodegeneration (Lee et al., 2006). Finally, fidelity of the tRNA splicing pathway is important for maintaining proper brain health. Individuals with a mutation in the kinase CLP1 have very early onset neurodegeneration, termed PCH10. This is likely at least in part due to the role CLP1 plays in the maturation of tRNAs as patient neurons are deficient in mature tRNAs and accumulate unspliced pre-tRNAs. Additionally, transfection of tRNA fragments into patient cells exacerbates neurodegenerative phenotypes (Schaffer et al., 2014). All in all, the proper genesis and function of different types of small ncRNAs is important for maintaining neural cell populations and preventing neurodegenerative disease.
Introduction to lncRNAs
Long non-coding RNAs are a heterogeneously defined group of RNAs, many of which are emerging as regulators of genome expression/stability and modulators of neural function and dysfunction (Figure 1; Wu et al., 2013). LncRNAs can be generally defined as non-coding transcripts that are greater than 200 nucleotides in length (Esteller, 2011). Many lncRNAs are molecularly indistinguishable from mRNAs; they are also produced by Pol II and undergo 5′ capping and polyadenylation (Ip and Nakagawa, 2012). Despite the fact that lncRNAs are not as well-characterized as miRNAs or coding transcripts, they actually make up the majority of the mammalian transcriptome (Esteller, 2011). There are a variety of mechanisms through which lncRNAs can impact gene expression. For example, lncRNAs can inhibit or promote transcription through the recruitment of histone modifying complexes or through the binding of specific transcriptional regulators (Geisler and Coller, 2013). lncRNAs are also capable of altering translation through binding to mRNAs and forming duplexes which can promote stability, alter splicing profiles or mask miRNA binding sites (Geisler and Coller, 2013). Here, we highlight how lncRNAs can impact neurodegeneration.
Antisense lncRNAs Regulate Sense Transcription of Neural Factors
In the context of neurodegenerative diseases, antisense transcripts have emerged as regulators of neural proteins. Antisense transcripts are often lncRNA transcripts that emerge from the opposite strand of a coding RNA region. One such transcript is BACE1AS, which is upregulated in AD (Faghihi et al., 2008). BACE1AS stabilizes its sense transcript BACE1 through the formation of an RNA duplex and this increases production of the BACE1 protein (Faghihi et al., 2008). Excess BACE1 then leads to sequential cleavage of the APP to form Aβ peptides, which constitute the amyloid plaques that are characteristic of AD pathobiology (Faghihi et al., 2008). Indeed, both a mouse and a human cell line model of AD demonstrated delayed plaque formation following BACE1AS knockdown (Modarresi et al., 2011; Liu et al., 2014). Therefore, targeting BACE1AS may be a promising therapeutic avenue for AD.
A less prevalent neurodegenerative disease, spinocerebellar ataxia type 8 (SCA8), is characterized by a mutation within a DNA region yielding a ncRNA. Here, a lncRNA referred to as ATXN8OS has a CTG trinucleotide repeat expansion (Koob et al., 1999; Mutsuddi and Rebay, 2005). This RNA is opposite to the coding strand for a protein referred to as KLHL1 (Nemes et al., 2000). Co-expression analysis of ATXN8OS and KLHL1 RNA in several regions within the human brain suggests that ATXN8OS represses the sense transcription of KLHL1 (Chen et al., 2008b). Intriguingly, siRNA-mediated knockdown of KLHL1 in murine pheochromocytoma PC12 cells markedly decreases neurite outgrowth (Seng et al., 2006). Thus, the CTG repeat expansion in ATXN8OS may lower KLHL1 expression and this may in turn limit neurite outgrowth and contribute to brain dysfunction.
The neural-specific protein ubiquitin-protein hydrolase UCHL1 is also regulated by its antisense transcript (Carrieri et al., 2012). Individuals carrying UCHL1 mutations, which reduce the hydrolase activity of the enzyme, develop early progressive neurodegeneration (Bilguvar et al., 2013). Interestingly, UCHL1 has also been demonstrated to be oxidatively inactivated in the brains of individuals with PD or AD (Choi et al., 2004; Barrachina et al., 2006). Future work should clarify whether perturbations to the UCHL1 antisense transcript alter UCHL1 expression or function in ways that promote neurodegeneration.
lncRNAs form Nuclear Bodies that may Protect Against Neurodegeneration
The formation of disease-linked nuclear bodies can be promoted by some lncRNAs, including NEAT1 (Ip and Nakagawa, 2012). These include the lncRNA NEAT1, whose expression is greatly increased in young proliferating cells, HD neurons, and early stage ALS motor neurons (Johnson, 2012; Abdelmohsen et al., 2013; Nishimoto et al., 2013). NEAT1 is a required component of paraspeckles, which are nuclear ribonucleoprotein-containing bodies (Nakagawa and Hirose, 2012). Interestingly, the formation of paraspeckles has been proposed to represent an important neuronal stress response based on several observations including the fact that NEAT1 is upregulated in early stage ALS and in the brains of heroin users (Michelhaugh et al., 2011; Nakagawa and Hirose, 2012; Nishimoto et al., 2013). Consistent with a putative role in stress responses, paraspeckles store highly edited RNAs that are rapidly released under various stress conditions (Prasanth et al., 2005). Aberrant stress responses are linked to several neurodegenerative diseases and NEAT1/paraspeckle-related defects may thus promote neurodegeneration. Consistent with this notion, FUS localizes to nuclear paraspeckles and is a direct binding partner of NEAT1 (Nishimoto et al., 2013; Shelkovnikova et al., 2014). In many ALS/FTLD cases, FUS is mutated and mislocalized to the cytoplasm and this may compromise nuclear paraspeckles and their role in responding to stress (Shelkovnikova et al., 2014). Indeed, siRNA-mediated knockdown of FUS eliminates paraspeckle formation in a number of human cell lines (Shelkovnikova et al., 2014). Furthermore, both a transgenic mouse model of ALS-FUS and motor neurons from ALS-FUS patients demonstrated that FUS sequestered vital nuclear paraspeckle proteins into cytoplasmic inclusions (Shelkovnikova et al., 2014). Importantly, cytoplasmic aggregations of FUS have also been reported in a number of trinucleotide repeat disorders including HD (Doi et al., 2008, 2010). Thus, failed nuclear paraspeckle formation may contribute to several neurodegenerative diseases.
Taken together, these studies indicate that functional lncRNAs may naturally counteract neurodegeneration through basic roles in the regulation of gene expression and the modulation of cellular stress responses. In contrast, some lncRNAs as well as other RNAs with either unclear or no natural function may contribute to neurodegeneration as we will discuss in the next section.
Deleterious Processes Triggered by Non-Coding RNA in Neurodegenerative Disease
Toxic RNA Sequesters RNA-Binding Proteins
Non-functional RNA can interact with RNA-binding proteins whose functions depend on their specific subcellular distribution. Toxic RNA in patients with neurodegenerative disorders may abnormally sequester these important RNA-binding proteins away from their site of action. For example, the most common genetic cause of both sporadic and familial forms of ALS is a hexanucleotide repeat expansion in an intron of the C9ORF72 gene (DeJesus-Hernandez et al., 2011; Majounie et al., 2012). This mutation confers a toxic gain-of-function onto the C9ORF72 transcript: the repeat expansion itself when transplanted to another portion of the genome is sufficient to cause neurodegeneration in D. melanogaster (Xu et al., 2013). C9ORF72 hexanucleotide repeat expansion causes abortive transcripts rich in G-quadruplexes, which are higher order structures built around hydrogen bonded guanine tetrads. Interestingly, FUS associates with G-quadruplexes pointing to a potential connection between FUS and C9ORF72 (Takahama et al., 2013). These G-quadruplexes bind to and sequester the RNA-binding protein nucleolin (Haeusler et al., 2014). With nucleolin distribution altered, cells show distinct signs of nucleolar stress (Haeusler et al., 2014). While it remains unclear if this nucleolar stress directly causes neurodegeneration, these studies demonstrate a novel mechanism through which toxic RNA may lead to neurodegenerative disease via the aberrant sequestration of various RNA-binding proteins.
Many neurodegenerative diseases are characterized by the presence of a trinucleotide repeat expansion. These include HD (which has a CAG expansion in the HTT protein), several types of SCA, and ATXN2-associated ALS (Elden et al., 2010; Nalavade et al., 2013). Trinucleotide repeats can also trigger neurodegeneration via toxic RNA-mediated sequestration of RNA binding proteins. For example, CAG repeat RNA has been shown to bind to and sequester nucleolin (Tsoi et al., 2012; Tsoi and Chan, 2013). While many diseases with CAG repeats have the repeat within coding regions, studies done in the nematode C. elegans demonstrated that CAG repeats within a 3′ UTR are sufficient to shorten lifespan and inhibit motility in a repeat-length associated manner (Wang et al., 2011a). Studies using flies also demonstrated that the introduction of an untranslated CAG repeat is sufficient to cause neuronal degeneration (Li et al., 2008). Therefore, toxic RNA is capable of being produced from either coding or non-coding regions and it can act as a toxic agent via sequestration of key RNA binding proteins. This sequestration could lead to nucleolar stress or other ribonucleoprotein-related defects yet to be characterized.
Repeat-Associated Non-ATG Translation Generates Toxic Dipeptides
In addition to sequestering RNA binding proteins, repeat-containing transcripts can give rise to toxic proteins through translation initiated from within the repeats (Figure 1). In D. melanogaster, the hexanucleotide repeat present in C9ORF72 produces repeat rich RNA capable of undergoing RAN translation, in which translation occurs on repeats in all three reading frames even in the absence of a start codon (Mizielinska et al., 2014). This produces toxic dipeptide proteins that lead to cellular degeneration and shortened lifespan in flies (Mizielinska et al., 2014). Several other disease-linked mutations including SCA8-associated ATXN8 and ATXN8OS CAG repeat expansions also generate toxic dipeptides through RAN translation even when full length ATXN8 is not expressed at all (Koob et al., 1999; Zu et al., 2011). Therefore, portions of both the coding sense and non-coding antisense transcript ATXN8OS are likely capable of producing toxic dipeptides through RAN translation (Pearson, 2011). Additionally, a CGG repeat in the 5′ UTR of the neurodegenerative disease fragile X-associated tremor/ataxia syndrome (FXTAS)-linked FMR1 also gives rise to RAN translation products (Todd et al., 2013). Therefore, these observations raise the possibility that a number of diseases associated with GC-rich repeats may be driven by pathogenic processes arising from the accumulation of toxic RAN translation products.
R-loops Repress the Expression of Neuroprotective Genes and Threaten Genome Integrity
Nascent ncRNAs transcripts are capable of hybridizing with their parental DNA strands forming RNA–DNA hybrids (Figure 3; Wahba et al., 2011; Aguilera and Garcia-Muse, 2012; Salvi et al., 2014). These RNA–DNA hybrids form structures known as R-loops, which can threaten genome integrity by inducing double stranded breaks and aberrant recombination when collisions occur between R-loops and advancing transcription/replication machineries (Aguilera and Garcia-Muse, 2012). However, R-loops can also regulate the expression of a diverse set of RNAs and are suspected to play a broader role across the genome than it is currently appreciated (Skourti-Stathaki and Proudfoot, 2014).
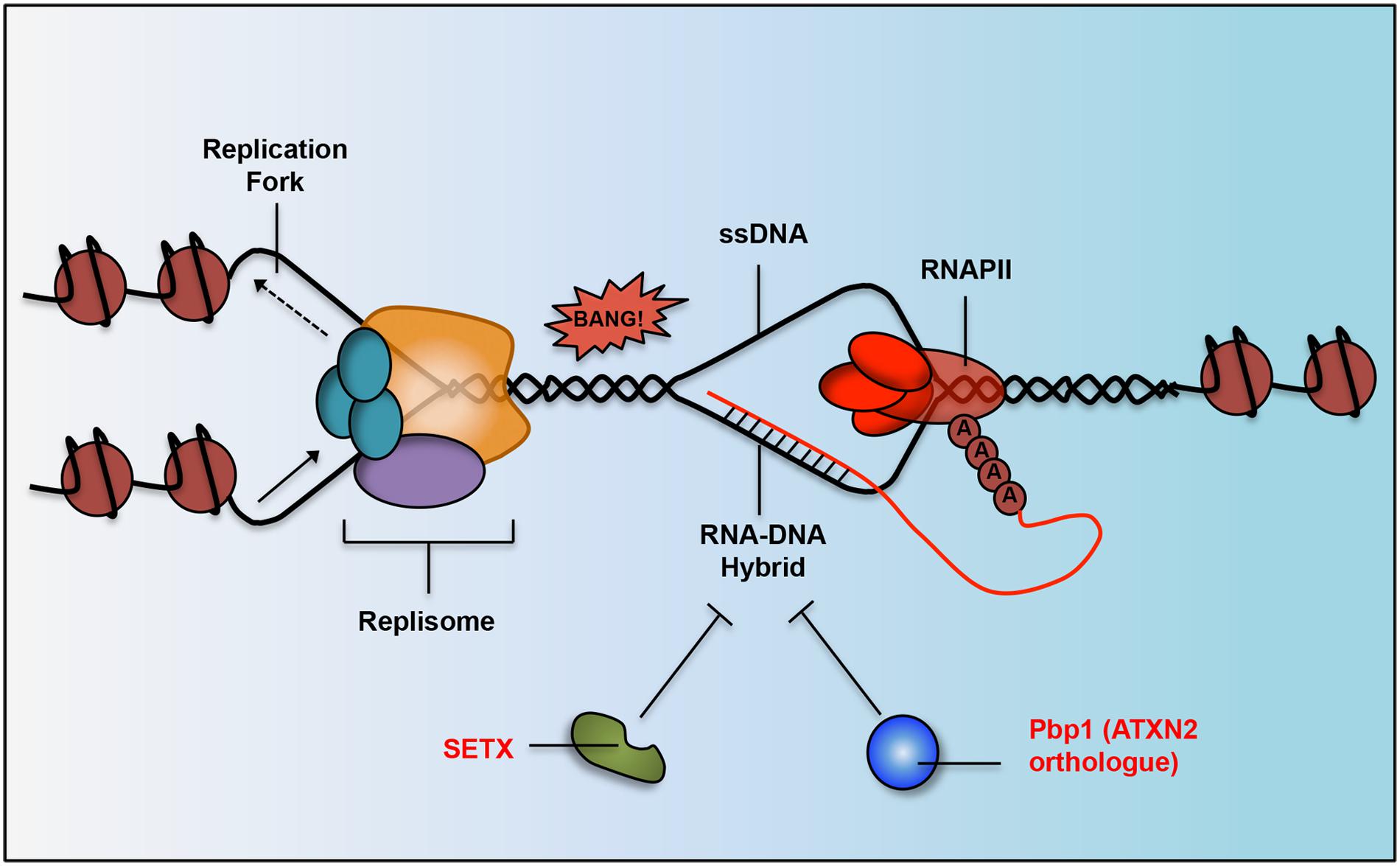
FIGURE 3. Work done in yeast and humans has determined that neurodegeneration-linked proteins and their orthologs are responsible for RNA–DNA hybrid suppression. R-loop structures form when RNAPII is transcribing DNA and the lagging RNA strand (in red) hybridizes with the DNA strand (in black). SETX (implicated in AOA2 and ALS) has helicase activity and can unwind RNA–DNA hybrid structures. Pbp1 (yeast ortholog of human ATXN2 implicated in SCA2 and ALS) has an RNA-binding domain that inhibits hybrids in some other way, possibly by binding to unwound RNA to prevent it from rehybridizing with its DNA strand. R-loops can lead to double stranded breaks and genomic instability when they collide with advancing replication forks.
In several neurodegenerative disorders, R-loops can control the fate of neuroprotective genes (Salvi and Mekhail, 2015). In individuals with fragile X syndrome, a trinucleotide repeat expansion within the promoter of the FXTAS-linked FMR1 triggers the formation of R-loops, which cause the epigenetic silencing of this gene (Colak et al., 2014). Similarly, R-loops formed upon intronic trinucleotide repeat expansion within the Friedrich’s Ataxia gene FXN promote its silencing and are thought to contribute to the disease (Groh et al., 2014). R-loops are also responsible for the abortive transcription observed at the hexanucleotide repeat-expanded C9ORF72 locus (Haeusler et al., 2014). In addition, data from yeast and Arabidopsis point to R-loops as regulators of lncRNA expression but it remains unclear if R-loops regulate neurodegenerative disease-linked lncRNAs such as the AD-linked BACE1AS (Nakama et al., 2012; Sun et al., 2013).
Beyond individual genes, some neurodegenerative disorders may encompass broad defects in R-loop or RNA–DNA hybrid regulation. Early onset or juvenile ALS as well as the neurodegenerative disease AOA2 are linked to mutations within SETX, which has emerged as a conserved RNA–DNA helicase from yeast to human (Skourti-Stathaki et al., 2011; Yuce and West, 2013). Loss of SETX function leads to a drastic increase in the number of R-loops formed within cells (Skourti-Stathaki et al., 2011; Yuce and West, 2013). Interestingly, R-loops were easily detected in dividing germ cells but not in post-mitotic brain cells (Yeo et al., 2014). One possibility may be that the level of R-loops driving neural degeneration is too small for detection using present methods. It is also possible that R-loop formation drives disease by disrupting support cells within the nervous system or certain cell types along the neural lineage including neural stem cells. Future work should clarify the intersection between SETX, R-loops and neurodegeneration.
Similar to SETX-ALS/AOA2 links, another possibly early link between R-loops and ATXN2-associated neurodegeneration has recently emerged from a study conducted in the budding yeast Saccharomyces cerevisiae. It was demonstrated that Pbp1, the yeast ortholog of ATXN2, represses aberrant accumulation of hybrids harboring lncRNA and intergenic DNA within repetitive DNA loci including ribosomal DNA (rDNA) repeats and subtelomeric regions (Salvi et al., 2014). R-loop accumulation in Pbp1-deficient cells triggered aberrant DNA recombination events and shortened replicative lifespan. Whether human ATXN2 functions similarly to suppress deleterious R-loops and if these structures contribute to neuropathology remains to be determined. It is also unclear if the R-loop suppressing functions of ATXN2 and SETX may intersect in ALS and other neurodegenerative diseases.
Therefore, although R-loops can play positive/regulatory roles in the cell, their aberrant regulation may trigger pathological processes leading to neurodegeneration (Figure 3). Aberrant R-loop accumulation can lead to the accumulation of toxic abortive transcripts, suppress the expression of neuroprotective factors, or compromise overall genome integrity. The emerging role of R-loops in various diseases may also point to novel avenues for therapeutic intervention.
Future Questions and Concluding Remarks
While much progress has been made in our understanding of the roles of ncRNAs in neural function, many questions still remain. For example, we know that miRNAs play an important role in neurogenesis, but the role that adult neurogenesis and differentiation plays in many neurodegenerative diseases is unknown. In fact, adult neurogenesis has yet to be detected in most areas of the brain (outside the case of brain damage) including many areas affected in neurodegenerative diseases such as the cerebellum (Ming and Song, 2011) This may be due to detection problems, as the long-lived nature of neurons means that rates of turnover are likely drastically lower when compared to other tissue types. Adult neurogenesis would be an exciting avenue to explore, as it may explain the neural cell-type specificity commonly observed in neurodegenerative diseases. Thus far, it has been demonstrated that premature aging in adult neural stem cells that harbor a LRRK2 mutation characteristic of PD results in a decrease in neurogenesis and improper differentiation (Liu et al., 2012). This hints that Parkinson’s might have defects in adult neurogenesis, although it is also possible that defects in differentiation that occur early in life due to this mutation might predispose neurons to premature death much later in life. Future work examining if neural stem cells are prematurely dying or improperly differentiating in neurodegenerative settings are needed.
More broadly, the reason that neurodegenerative diseases do not manifest until late in life is poorly understood. Why is HD onset typically in the fourth decade when the related HTT mutation is present from birth? One model is that most heritable polymorphisms in neurodegenerative disease-linked factors cause minor molecular defects, as gross genetic failures would exit the gene pool by natural selection. Quality control of protein synthesis decreases with time and there is therefore an increased occurrence/accumulation of misfolded proteins with age (Gaczynska et al., 2001). Such misfolding can contribute to neurodegeneration. Consistently, the unfolded protein response (UPR), an apoptotic defense mechanism against the accumulation of misfolded proteins within the cell, is hyperactivated, perhaps aberrantly, in a number of neurodegenerative diseases including HD, AD, and PD (Halliday and Mallucci, 2014). Another non-exclusive possibility is that several nuclear RNA binding proteins associated with neurodegeneration such as TDP-43 and ATXN2 are recruited to cytoplasmic stress granules and this may prevent their proper RNA regulatory function (Colombrita et al., 2009; Damrath et al., 2012). Stress granule formation under oxidative stress and clearance by autophagy is respectively increased and decreased with age (Colombrita et al., 2009; Buchan et al., 2013; Szafranski and Mekhail, 2014). Thus, the aberrant sequestration of proteins such as TDP-43 and ATXN2 may increase with age and small defects may become amplified over time leading to neurodegeneration. Oxidative stress is also known to cause senescence within astroctyes (Bitto et al., 2011). Senescent astrocytes are enriched in patients with AD relative to age-matched controls (Bhat et al., 2012). This may contribute to disease as these astrocytes have a senescence-associated secretory phenotype wherein they release inflammatory cytokines such as IL-6 (Bhat et al., 2012). Given that astrocytes are the most abundant cell-type in the brain and are more susceptible to oxidative stress than other cell types such as fibroblasts, it is likely that astrocyte senescence is another age-related risk factor for neurodegenerative diseases (Bitto et al., 2011). Future work will undoubtedly clarify the link between aging and neurodegeneration.
Many studies also hinted at links that need further clarification. For example, is miR-34a repressing Bcl2 and SIRT1 in vivo and is this repression leading to neurodegeneration? Is ATXN8OS regulating the transcription of KLHL1? Studies examining orthologs of human transcripts or proteins should also be repeated in human models. For example, miR-8 was found to regulate atrophin levels to prevent neurodegeneration in flies (Karres et al., 2007). Both miR-8 and atrophin are conserved; so is the link to neurodegeneration also conserved? Studies reporting that ATXN2 orthologs impact D. melanogaster RISC and S. cerevisiae R-loop accumulation should serve as foundation for similar studies conducted in mammalian/human model systems. The link between paraspeckles and neuronal stress response also needs to be clarified, as the current literature is mostly correlative.
Overall, commendable gains have been made in understanding the pathobiological processes underlying various neurodegenerative diseases. However, significant research remains to be done in various organisms in order to fully decipher human disease mechanisms. Such mechanistic studies promise to uncover new avenues for targeted therapeutics. For example, understanding the role of neural stem cells in neurodegenerative disease could allow for the development and perfection of stem cell based therapies. Currently, neural stem cell injection therapy for ALS has successfully demonstrated safety and is moving to Phase II trials (Feldman et al., 2014). However, we do not yet know if these stem cells would be resistant to the factors that induce cell death in motor neurons or whether stem cell defects are present in ALS neurons. Other possible therapeutic avenues include ASOs against proteins that are misfolded in neurodegenerative disease. Indeed, studies targeting HTT with ASOs have demonstrated efficacy in primate models (Kordasiewicz et al., 2012). ASO-mediated targeting of C9ORF72 suppressed ALS-linked defects typically observed in patient-derived induced pluripotent stem cells (Donnelly et al., 2013). On another front, it is also possible to target specific miRNAs. For example, repressing miR-34 may alleviate some of the brain degeneration seen in AD (Table 1; Wang et al., 2009; Li et al., 2011b). While no drugs targeting miRNAs in neurodegenerative disease have been developed yet, trials have demonstrated the safety and efficacy of targeted inhibition of a specific miRNA in the treatment of Hepatitis C (Janssen et al., 2013).
As illustrated in this article, ncRNAs, neurodegenerative diseases, and aging are interconnected. miRNAs, lncRNAs, and related factors can impact the nervous system in various positive and negative manners depending on the precise underlying molecular processes. Continued efforts to understand the molecular pathways underlying natural neural function, devastating neurodegenerative diseases and their connection to aging will undoubtedly lead to the development of smarter therapeutics.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Mekhail lab for discussions. KS is supported by an Ontario Graduate Scholarship. KA is supported by a Canadian Institute of Health Research (CIHR) M.D./Ph.D. studentship. KM is funded by CIHR through the Open Operating Grant Program (OOGP) and the Canada Research Chairs (CRC-II) program.
Abbreviations
AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; AOA2, ataxia with oculomotor apraxia type 2; ASO, antisense oligonucleotide;ATXN, Ataxin; BACE-1, beta-secretase 1; BCL, B-cell lymphoma; C9ORF72, Chromosome 9 open reading frame 72; CLP1, cleavage and polyadenylation factor 1 subunit 1; CNS, central nervous system; DN, dopaminergic neuron; FTLD, frontotemporal lobar degeneration; FUS, fused in sarcoma; HD, Huntington’s disease; KLHL1, Kelch-like 1; lncRNA, long non-coding RNA; miRNA, microRNA; ncRNA, non-coding RNA; NEAT1, nuclear enriched abundant transcript 1; PCH, pontocerebellar hypoplasia; PD, Parkinson’s disease; RAN, repeat-associated non-ATG; RISC, RNA-induced silencing complex; SCA, spinocerebellar ataxia; SETX, Senataxin; SIRT1, Sirtuin 1; TDP-43, TAR-DNA binding protein 43.
References
Abdelmohsen, K., Panda, A., Kang, M. J., Xu, J., Selimyan, R., Yoon, J. H.,et al. (2013). Senescence-associated lncRNAs: senescence-associated long noncoding RNAs. Aging Cell 12, 890–900. doi: 10.1111/acel.12115
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Abe, M., and Bonini, N. M. (2013). MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 23, 30–36. doi: 10.1016/j.tcb.2012.08.013
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Aguilera, A., and Garcia-Muse, T. (2012). R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46, 115–124. doi: 10.1016/j.molcel.2012.04.009
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. doi: 10.1126/science.1098014
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Bak, M., Silahtaroglu, A., Moller, M., Christensen, M., Rath, M. F., Skryabin, B.,et al. (2008). MicroRNA expression in the adult mouse central nervous system. RNA 14, 432–444. doi: 10.1261/rna.783108
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Baloh, R. H. (2012). How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr. Opin. Neurol. 25, 701–707. doi: 10.1097/Wco.0b013e32835a269b
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Barrachina, M., Castano, E., Dalfo, E., Maes, T., Buesa, C., and Ferrer, I. (2006). Reduced ubiquitin C-terminal hydrolase-1 expression levels in dementia with Lewy bodies. Neurobiol. Dis. 22, 265–273. doi: 10.1016/j.nbd.2005.11.005
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U.,et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 7:e45069. doi: 10.1371/journal.pone.0045069
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Bilen, J., Liu, N., Burnett, B. G., Pittman, R. N., and Bonini, N. M. (2006). MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol. Cell 24, 157–163. doi: 10.1016/j.molcel.2006.07.030
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Bilguvar, K., Tyagi, N. K., Ozkara, C., Tuysuz, B., Bakircioglu, M., Choi, M.,et al. (2013). Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 3489–3494. doi: 10.1073/pnas.1222732110
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Bitto, A., Sell, C., Crowe, E., Lorenzini, A., Malaguti, M., and Torres, C. (2011). Stress-induced senescence in human and rodent astrocytes. Exp. Gerontol. 46, 213–213. doi: 10.1016/j.exger.2010.11.028
Bremer, J., O’Connor, T., Tiberi, C., Rehrauer, H., Weis, J., and Aguzzi, A. (2010). Ablation of dicer from Murine Schwann Cells increases their proliferation while blocking myelination. PLoS ONE 5:e12450. doi: 10.1371/journal.pone.0012450
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Buchan, J. R., Kolaitis, R. M., Taylor, J. P., and Parker, R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474. doi: 10.1016/j.cell.2013.05.037
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Carrieri, C., Cimatti, L., Biagioli, M., Beugnet, A., Zucchelli, S., Fedele, S.,et al. (2012). Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491, 454–457. doi: 10.1038/Nature11508
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Cheloufi, S., Dos Santos, C. O., Chong, M. M. W., and Hannon, G. J. (2010). A Dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, U584–U576. doi: 10.1038/Nature09092
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Chen, D., Steele, A. D., Hutter, G., Bruno, J., Govindarajan, A., Easlon, E.,et al. (2008a). The role of calorie restriction and SIRT1 in prion-mediated neurodegeneration. Exp. Gerontol. 43, 1086–1093. doi: 10.1016/j.exger.2008.08.050
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Chen, W. L., Lin, J. W., Huang, H. J., Wang, S. M., Su, M. T., Lee-Chen, G. J.,et al. (2008b). SCA8 mRNA expression suggests an antisense regulation of KLHL1 and correlates to SCA8 pathology. Brain Res. 1233, 176–184. doi: 10.1016/j.brainres.2008.07.096
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Chen, J. A., and Wichterle, H. (2012). Apoptosis of limb innervating motor neurons and erosion of motor pool identity upon lineage specific dicer inactivation. Front. Neurosci. 6:69. doi: 10.3389/fnins.2012.00069
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Choi, J., Levey, A. I., Weintraub, S. T., Rees, H. D., Gearing, M., Chin, L. S.,et al. (2004). Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 279, 13256–13264. doi: 10.1074/jbc.M314124200
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Choi, P. S., Zakhary, L., Choi, W. Y., Caron, S., Alvarez-Saavedra, E., Miska, E. A.,et al. (2008). Members of the miRNA-200 family regulate olfactory neurogenesis. Neuron 57, 41–55. doi: 10.1016/j.neuron.2007.11.018
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W. Y., Gerhardt, J.,et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. doi: 10.1126/science.1245831
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E.,et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Damiani, D., Alexander, J. J., O’Rourke, J. R., Mcmanus, M., Jadhav, A. P., Cepko, C. L.,et al. (2008). Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J. Neurosci. 28, 4878–4887. doi: 10.1523/JNEUROSCI.0828-08.2008
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Damrath, E., Heck, M. V., Gispert, S., Azizov, M., Nowock, J., Seifried, C.,et al. (2012). ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old Ataxic Knock-in mice. PLoS Genet. 8:e1002920. doi: 10.1371/journal.pgen.1002920
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Davis, T. H., Cuellar, T. L., Koch, S. M., Barker, A. J., Harfe, B. D., Mcmanus, M. T.,et al. (2008). Conditional loss of dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 28, 4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J.,et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Di Sante, G., Wang, L., Wang, C., Jiao, X., Casimiro, M. C., Chen, K.,et al. (2015). Sirt1-deficient mice have hypogonadotropic hypogonadism due to defective GnRH neuronal migration. Mol. Endocrinol. 29, 200–212. doi: 10.1210/me.2014-1228
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A.,et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Doi, H., Koyano, S., Suzuki, Y., Nukina, N., and Kuroiwa, Y. (2010). The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 66, 131–133. doi: 10.1016/j.neures.2009.10.004
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Doi, H., Okamura, K., Bauer, P. O., Furukawa, Y., Shimizu, H., Kurosawa, M.,et al. (2008). RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J. Biol. Chem. 283, 6489–6500. doi: 10.1074/jbc.M705306200
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Donnelly, C. J., Zhang, P. W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S.,et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Eacker, S. M., Dawson, T. M., and Dawson, V. L. (2009). Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 10, 837–841. doi: 10.1038/nrn2726
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Elden, A. C., Kim, H. J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X.,et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874. doi: 10.1038/Nrg3074
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Faghihi, M. A., Modarresi, F., Khalil, A. M., Wood, D. E., Sahagan, B. G., Morgan, T. E.,et al. (2008). Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 14, 723–730. doi: 10.1038/Nm1784
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Fan, Z., Chen, X., and Chen, R. (2014). Transcriptome-wide analysis of TDP-43 binding small RNAs identifies miR-NID1 (miR-8485), a novel miRNA that represses NRXN1 expression. Genomics 103, 76–82. doi: 10.1016/j.ygeno.2013.06.006
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Feldman, E. L., Boulis, N. M., Hur, J., Johe, K., Rutkove, S. B., Federici, T.,et al. (2014). Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann. Neurol. 75, 363–373. doi: 10.1002/ana.24113
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Freischmidt, A., Muller, K., Ludolph, A. C., and Weishaupt, J. H. (2013). Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 1:42. doi: 10.1186/2051-5960-1-42
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Gaczynska, M., Osmulski, P. A., and Ward, W. F. (2001). Caretaker or undertaker? The role of the proteasome in aging. Mech. Ageing Dev. 122, 235–254. doi: 10.1016/S0047-6374(00)00246-3
Geisler, S., and Coller, J. (2013). RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 14, 699–712. doi: 10.1038/nrm3679
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Giraldez, A. J., Cinalli, R. M., Glasner, M. E., Enright, A. J., Thomson, J. M., Baskerville, S.,et al. (2005). MicroRNAs regulate brain morphogenesis in zebrafish. Science 308, 833–838. doi: 10.1126/science.1109020
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Goodenbour, J. M., and Pan, T. (2006). Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 34, 6137–6146. doi: 10.1093/Nar/Gkl725
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Graff, J., Kahn, M., Samiei, A., Gao, J., Ota, K. T., Rei, D.,et al. (2013). A Dietary regimen of Caloric Restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J. Neurosci. 33, 8951–8960. doi: 10.1523/Jneurosci.5657-12.2013
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Groh, M., Lufino, M. M., Wade-Martins, R., and Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10:e1004318. doi: 10.1371/journal.pgen.1004318
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Ha, M., and Kim, V. N. (2014). Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524. doi: 10.1038/Nrm3838
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A. J., Shaw, P. G., Kim, M. S.,et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/Nature13124
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Halliday, M., and Mallucci, G. R. (2014). Targeting the unfolded protein response in neurodegeneration: a new approach to therapy. Neuropharmacology 76(Pt A), 169–174. doi: 10.1016/j.neuropharm.2013.08.034
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Haramati, S., Chapnik, E., Sztainberg, Y., Eilam, R., Zwang, R., Gershoni, N.,et al. (2010). miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. U.S.A. 107, 13111–13116. doi: 10.1073/pnas.1006151107
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Hardiman, O., van den Berg, L. H., and Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649. doi: 10.1038/nrneurol.2011.153
He, L., and Hannon, G. J. (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. doi: 10.1038/nrg1379
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Hebert, S. S., Horre, K., Nicolai, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N.,et al. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420. doi: 10.1073/pnas.0710263105
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Huang, P. S., Son, J. H., Abbott, L. C., and Winzer-Serhan, U. H. (2011). Regulated expression of neuronal sirt1 and related genes by aging and neuronal beta 2-containing nicotinic cholinergic receptors. Neuroscience 196, 189–202. doi: 10.1016/j.neuroscience.2011.09.007
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Huang, T., Liu, Y., Huang, M., Zhao, X., and Cheng, L. (2010). Wnt1-cre-mediated conditional loss of dicer results in malformation of the midbrain and cerebellum and failure of neural crest and dopaminergic differentiation in mice. J. Mol. Cell Biol. 2, 152–163. doi: 10.1093/jmcb/mjq008
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Inukai, S., De Lencastre, A., Turner, M., and Slack, F. (2012). Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE 7:e40028. doi: 10.1371/journal.pone.0040028
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Ip, J. Y., and Nakagawa, S. (2012). Long non-coding RNAs in nuclear bodies. Dev. Growth Differ. 54, 44–54. doi: 10.1111/j.1440-169X.2011.01303.x
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Ishimura, R., Nagy, G., Dotu, I., Zhou, H. H., Yang, X. L., Schimmel, P.,et al. (2014). Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459. doi: 10.1126/science.1249749
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Janssen, H. L., Reesink, H. W., Lawitz, E. J., Zeuzem, S., Rodriguez-Torres, M., Patel, K.,et al. (2013). Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 368, 1685–1694. doi: 10.1056/NEJMoa1209026
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Jiang, M. L., Wang, J. W., Fu, J. R., Du, L., Jeong, H., West, T.,et al. (2012). Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat. Med. 18, 153–158. doi: 10.1038/Nm.2558
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Johnson, R. (2012). Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol. Dis. 46, 245–254. doi: 10.1016/j.nbd.2011.12.006
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Jovicic, A., Roshan, R., Moisoi, N., Pradervand, S., Moser, R., Pillai, B.,et al. (2013). Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J. Neurosci. 33, 5127–5137. doi: 10.1523/JNEUROSCI.0600-12.2013
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C.,et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Karres, J. S., Hilgers, V., Carrera, I., Treisman, J., and Cohen, S. M. (2007). The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131, 136–145. doi: 10.1016/j.cell.2007.09.020
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kawahara, Y., and Mieda-Sato, A. (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U.S.A. 109, 3347–3352. doi: 10.1073/pnas.1112427109
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Khanna, S., Rink, C., Ghoorkhanian, R., Gnyawali, S., Heigel, M., Wijesinghe, D. S.,et al. (2013). Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J. Cereb. Blood Flow Metab. 33, 1197–1206. doi: 10.1038/jcbfm.2013.68
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kim, D., Nguyen, M. D., Dobbin, M. M., Fischer, A., Sananbenesi, F., Rodgers, J. T.,et al. (2007a). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. doi: 10.1038/sj.emboj.7601758
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kim, J., Inoue, K., Ishii, J., Vanti, W. B., Voronov, S. V., Murchison, E.,et al. (2007b). A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317, 1220–1224. doi: 10.1126/science.1140481
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
King, I. N., Yartseva, V., Salas, D., Kumar, A., Heidersbach, A., Ando, D. M.,et al. (2014). The RNA-binding protein TDP-43 selectively disrupts microRNA-1/206 incorporation into the RNA-induced silencing complex. J. Biol. Chem. 289, 14263–14271. doi: 10.1074/jbc.M114.561902
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kole, A. J., Swahari, V., Hammond, S. M., and Deshmukh, M. (2011). miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. 25, 125–130. doi: 10.1101/gad.1975411
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Koob, M. D., Moseley, M. L., Schut, L. J., Benzow, K. A., Bird, T. D., Day, J. W.,et al. (1999). An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat. Genet. 21, 379–384. doi: 10.1038/7710
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kordasiewicz, H. B., Stanek, L. M., Wancewicz, E. V., Mazur, C., Mcalonis, M. M., Pytel, K. A.,et al. (2012). Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044. doi: 10.1016/j.neuron.2012.05.009
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C.,et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Lagier-Tourenne, C., and Cleveland, D. W. (2009). Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. doi: 10.1016/j.cell.2009.03.006
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Lee, J. W., Beebe, K., Nangle, L. A., Jang, J. S., Longo-Guess, C. M., Cook, S. A.,et al. (2006). Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443, 50–55. doi: 10.1038/Nature05096
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Lee, S. T., Chu, K., Im, W. S., Yoon, H. J., Im, J. Y., Park, J. E.,et al. (2011). Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 227, 172–179. doi: 10.1016/j.expneurol.2010.10.012
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Lee, Y., Jeon, K., Lee, J. T., Kim, S., and Kim, V. N. (2002). MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 21, 4663–4670. doi: 10.1093/emboj/cdf476
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Li, L. B., Yu, Z. M., Teng, X. Y., and Bonini, N. M. (2008). RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453, U1107–U1109. doi: 10.1038/Nature06909
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Li, N., Bates, D. J., An, J., Terry, D. A., and Wang, E. (2011a). Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol. Aging 32, 944–955. doi: 10.1016/j.neurobiolaging.2009.04.020
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Li, X., Khanna, A., Li, N., and Wang, E. (2011b). Circulatory miR34a as an RNAbased, noninvasive biomarker for brain aging. Aging (Albany, NY) 3, 985–1002.
Liu, G. H., Qu, J., Suzuki, K., Nivet, E., Li, M., Montserrat, N.,et al. (2012). Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491, 603–607. doi: 10.1038/nature11557 doi: 10.1038/nature11557
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Liu, T., Huang, Y., Chen, J., Chi, H., Yu, Z., Wang, J.,et al. (2014). Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1AS expression. Mol. Med. Rep. 10, 1275–1281. doi: 10.3892/mmr.2014.2351
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Macias, S., Plass, M., Stajuda, A., Michlewski, G., Eyras, E., and Caceres, J. F. (2012). DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 19, 760–766. doi: 10.1038/Nsmb.2344
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Mackenzie, I. R. A., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S.,et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/Nature02621
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
McCann, C., Holohan, E. E., Das, S., Dervan, A., Larkin, A., Lee, J. A.,et al. (2011). The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc. Natl. Acad. Sci. U.S.A. 108, E655–E662. doi: 10.1073/pnas.1107198108
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Michelhaugh, S. K., Lipovich, L., Blythe, J., Jia, H., Kapatos, G., and Bannon, M. J. (2011). Mining Affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J. Neurochem. 116, 459–466. doi: 10.1111/j.1471-4159.2010.07126.x
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Ming, G. L., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. doi: 10.1016/j.neuron.2011.05.001
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Mizielinska, S., Gronke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A.,et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. doi: 10.1126/science.1256800
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Modarresi, F., Faghihi, M. A., Patel, N. S., Sahagan, B. G., Wahlestedt, C., and Lopez-Toledano, M. A. (2011). Knockdown of BACE1-AS nonprotein-coding transcript modulates beta-amyloid-related hippocampal neurogenesis. Int. J. Alzheimers Dis. 2011, 929042. doi: 10.4061/2011/929042
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Morlando, M., Modigliani, S. D., Torrelli, G., Rosa, A., Di Carlo, V., Caffarelli, E.,et al. (2012). FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 31, 4502–4510. doi: 10.1038/emboj.2012.319
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Mutsuddi, M., and Rebay, I. (2005). Molecular genetics of spinocerebellar ataxia type 8 (SCA8). RNA Biol. 2, 49–52. doi: 10.4161/rna.2.2.1682
Nakagawa, S., and Hirose, T. (2012). Paraspeckle nuclear bodies-useful uselessness? Cell. Mol. Life Sci. 69, 3027–3036. doi: 10.1007/s00018-012-0973-x
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Nakama, M., Kawakami, K., Kajitani, T., Urano, T., and Murakami, Y. (2012). DNA-RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes Cells 17, 218–233. doi: 10.1111/j.1365-2443.2012.01583.x
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Nalavade, R., Griesche, N., Ryan, D. P., Hildebrand, S., and Krauss, S. (2013). Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 4:e752. doi: 10.1038/cddis.2013.276
Nemes, J. P., Benzow, K. A., Moseley, M. L., Ranum, L. P. W., and Koob, M. D. (2000). The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1) (vol 9, pg 1543, 2000). Hum. Mol. Genet. 9, 2777–2777. doi: 10.1093/hmg/9.10.1543
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T.,et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Nishimoto, Y., Nakagawa, S., Hirose, T., Okano, H. J., Takao, M., Shibata, S.,et al. (2013). The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 6:31. doi: 10.1186/1756-6606-6-31
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Palazzo, A. F., and Gregory, T. R. (2014). The case for junk DNA. PLoS Genet. 10:e1004351. doi: 10.1371/journal.pgen.1004351
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Pearson, C. E. (2011). Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 7:e1002018. doi: 10.1371/journal.pgen.1002018
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Persengiev, S., Kondova, I., Otting, N., Koeppen, A. H., and Bontrop, R. E. (2011). Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol. Aging 32, 2316 e2317–2316 e2327. doi: 10.1016/j.neurobiolaging.2010.03.014
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Prasanth, K. V., Prasanth, S. G., Xuan, Z., Hearn, S., Freier, S. M., Bennett, C. F.,et al. (2005). Regulating gene expression through RNA nuclear retention. Cell 123, 249–263. doi: 10.1016/j.cell.2005.08.033
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Pulst, S. M., Nechiporuk, A., Nechiporuk, T., Gispert, S., Chen, X. N., Lopes-Cendes, I.,et al. (1996). Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276. doi: 10.1038/ng1196-269
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Roshan, R., Shridhar, S., Sarangdhar, M. A., Banik, A., Chawla, M., Garg, M.,et al. (2014). Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 20, 1287–1297. doi: 10.1261/rna.044008.113
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Ross, C. A., Becher, M. W., Colomer, V., Engelender, S., Wood, J. D., and Sharp, A. H. (1997). Huntington’s disease and dentatorubral-pallidoluysian atrophy: proteins, pathogenesis and pathology. Brain Pathol. 7, 1003–1016. doi: 10.1111/j.1750-3639.1997.tb00898.x
Ross, O. A., Rutherford, N. J., Baker, M., Soto-Ortolaza, A. I., Carrasquillo, M. M., Dejesus-Hernandez, M.,et al. (2011). Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 20, 3207–3212. doi: 10.1093/Hmg/Ddr227
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Sahoo, T., Del Gaudio, D., German, J. R., Shinawi, M., Peters, S. U., Person, R. E.,et al. (2008). Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 40, 719–721. doi: 10.1038/ng.158
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Salvi, J. S., Chan, J. N. Y., Szafranski, K., Liu, T. T., Wu, J. D., Olsen, J. B.,et al. (2014). Roles for Pbp1 and Caloric Restriction in genome and lifespan maintenance via suppression of RNA-DNA Hybrids. Dev. Cell 30, 177–191. doi: 10.1016/j.devcel.2014.05.013
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Salvi, J. S., and Mekhail, K. (2015). R-loops highlight the nucleus in ALS. Nucleus doi: 10.1080/19491034.2015.1004952 [Epub ahead of print].
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Schaefer, A., O’carroll, D., Tan, C. L., Hillman, D., Sugimori, M., Llinas, R.,et al. (2007). Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 204, 1553–1558. doi: 10.1084/jem.20070823
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Schaffer, A. E., Eggens, V. R., Caglayan, A. O., Reuter, M. S., Scott, E., Coufal, N. G.,et al. (2014). CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157, 651–663. doi: 10.1016/j.cell.2014.03.049
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Schwer, B., Schumacher, B., Lombard, D. B., Xiao, C. Y., Kurtev, M. V., Gao, J.,et al. (2010). Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 21790–21794. doi: 10.1073/pnas.1016306107
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Seng, S., Avraham, H. K., Jiang, S., Venkatesh, S., and Avraham, S. (2006). KLHL1/MRP2 mediates neurite outgrowth in a glycogen synthase kinase 3beta-dependent manner. Mol. Cell. Biol. 26, 8371–8384. doi: 10.1128/MCB.02167-5
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Sheinerman, K. S., Tsivinsky, V. G., Abdullah, L., Crawford, F., and Umansky, S. R. (2013). Plasma microRNA biomarkers for detection of mild cognitive impairment: biomarker validation study. Aging (Albany, NY) 5, 925–938.
Shelkovnikova, T. A., Robinson, H. K., Troakes, C., Ninkina, N., and Buchman, V. L. (2014). Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 23, 2298–2312. doi: 10.1093/hmg/ddt622
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Shin, D., Shin, J. Y., Mcmanus, M. T., Ptacek, L. J., and Fu, Y. H. (2009). Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann. Neurol. 66, 843–857. doi: 10.1002/ana.21927
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Shindler, K. S., Ventura, E., Dutt, M., Elliott, P., Fitzgerald, D. C., and Rostami, A. (2010). Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J. Neuroophthalmol. 30, 328–339. doi: 10.1097/Wno.0b013e3181f7f833
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Skourti-Stathaki, K., and Proudfoot, N. J. (2014). A double-edged sword: r loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 28, 1384–1396. doi: 10.1101/gad.242990.114
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 42, 794–805. doi: 10.1016/j.molcel.2011.04.026
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Sun, Q. W., Csorba, T., Skourti-Stathaki, K., Proudfoot, N. J., and Dean, C. (2013). R-Loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science 340, 619–621. doi: 10.1126/science.1234848
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Szafranski, K., and Mekhail, K. (2014). The fine line between lifespan extension and shortening in response to caloric restriction. Nucleus 5, 56–65. doi: 10.4161/nucl.27929
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Takahama, K., Takada, A., Tada, S., Shimizu, M., Sayama, K., Kurokawa, R.,et al. (2013). Regulation of telomere length by G-quadruplex telomere DNA- and TERRA-binding protein TLS/FUS. Chem. Biol. 20, 341–350. doi: 10.1016/j.chembiol.2013.02.013
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Tao, J., Wu, H., Lin, Q., Wei, W., Lu, X. H., Cantle, J. P.,et al. (2011). Deletion of astroglial dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J. Neurosci. 31, 8306–8319. doi: 10.1523/JNEUROSCI.0567-11.2011
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Todd, P. K., Oh, S. Y., Krans, A., He, F., Sellier, C., Frazer, M.,et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. doi: 10.1016/j.neuron.2013.03.026
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Tsoi, H., and Chan, H. Y. (2013). Expression of expanded CAG transcripts triggers nucleolar stress in Huntington’s disease. Cerebellum 12, 310–312. doi: 10.1007/s12311-012-0447-6
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Tsoi, H., Lau, T. C., Tsang, S. Y., Lau, K. F., and Chan, H. Y. (2012). CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl. Acad. Sci. U.S.A. 109, 13428–13433. doi: 10.1073/pnas.1204089109
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J.,et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wahba, L., Amon, J. D., Koshland, D., and Vuica-Ross, M. (2011). RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell. 44, 978-988. doi: 10.1016/j.molcel.2011.10.017
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wang, L. C., Chen, K. Y., Pan, H., Wu, C. C., Chen, P. H., Liao, Y. T.,et al. (2011a). Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell Mol. Life Sci. 68, 1255–1267. doi: 10.1007/s00018-010-0522-4
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wang, W. X., Huang, Q., Hu, Y., Stromberg, A. J., and Nelson, P. T. (2011b). Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: white matter versus gray matter. Acta Neuropathol. 121, 193–205. doi: 10.1007/s00401-010-0756-0
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wang, X., Liu, P., Zhu, H., Xu, Y., Ma, C., Dai, X.,et al. (2009). miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 80, 268–273. doi: 10.1016/j.brainresbull.2009.08.006
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wirdefeldt, K., Adami, H. O., Cole, P., Trichopoulos, D., and Mandel, J. (2011). Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur. J. Epidemiol. 26(Suppl. 1), S1–S58. doi: 10.1007/s10654-011-9581-6
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wood, S. H., Craig, T., Li, Y., Merry, B., and De Magalhaes, J. P. (2013). Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age 35, 763–776. doi: 10.1007/s11357-012-9410-1
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Wu, P., Zuo, X. L., Deng, H. L., Liu, X. X., Liu, L., and Ji, A. M. (2013). Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res. Bull. 97, 69–80. doi: 10.1016/j.brainresbull.2013.06.001
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Xu, Z. H., Poidevin, M., Li, X. K., Li, Y. J., Shu, L. Q., Nelson, D. L.,et al. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 7778–7783. doi: 10.1073/pnas.1219643110
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Yeo, A. J., Becherel, O. J., Luff, J. E., Cullen, J. K., Wongsurawat, T., Jenjaroenpoon, P.,et al. (2014). R-loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias. PLoS ONE 9:e90219. doi: 10.1371/journal.pone.0090219
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Yuce, O., and West, S. C. (2013). Senataxin, defective in the neurodegenerative disorder Ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 33, 406–417. doi: 10.1128/Mcb.01195-2
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Zu, T., Gibbens, B., Doty, N. S., Gomes-Pereira, M., Huguet, A., Stone, M. D.,et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U.S.A. 108, 260–265. doi: 10.1073/pnas.1013343108
PubMed Abstract | Pubmed Full Text | Full Text | Google Scholar
Keywords: miRNA, lncRNA, R-loops, TDP-43, FUS, ATXN2, SETX, neurodegeneration
Citation: Szafranski K, Abraham KJ and Mekhail K (2015) Non-coding RNA in neural function, disease, and aging. Front. Genet. 6:87. doi: 10.3389/fgene.2015.00087
Received: 01 December 2014; Accepted: 18 February 2015;
Published online: 09 March 2015.
Edited by:
Frank John Slack, Beth Israel Deaconess Medical Center Cancer Center/Harvard Medical School, USAReviewed by:
Douglas Gray, Ottawa Hospital Research Institute, CanadaJohannes Grillari,University of Natural Resources and Life Sciences Vienna, Austria
Fabio Demontis, St. Jude Children’s Research Hospital, USA
Copyright © 2015 Szafranski, Abraham and Mekhail. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Karim Mekhail, Department of Laboratory Medicine and Pathobiology, Faculty of Medicine, University of Toronto, 1 King’s College Circle, Medical Sciences Building, Room 6342, Toronto, ON M5S 1A8, Canadaa2FyaW0ubWVraGFpbEB1dG9yb250by5jYQ==
†These authors have contributed equally to this work.