 Alessandra Maresca
Alessandra Maresca Mirko Zaffagnini
Mirko Zaffagnini Leonardo Caporali
Leonardo Caporali Valerio Carelli
Valerio Carelli Claudia Zanna
Claudia Zanna- 1Unit of Neurology, Department of Biomedical and NeuroMotor Sciences, University of Bologna, Bologna, Italy,
- 2Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy,
- 3IRCCS Institute of Neurological Sciences of Bologna, Bellaria Hospital, Bologna, Italy
Autosomal dominant cerebellar ataxia-deafness and narcolepsy (ADCA-DN) and Hereditary sensory neuropathy with dementia and hearing loss (HSN1E) are two rare, overlapping neurodegenerative syndromes that have been recently linked to allelic dominant pathogenic mutations in the DNMT1 gene, coding for DNA (cytosine-5)-methyltransferase 1 (DNMT1). DNMT1 is the enzyme responsible for maintaining the nuclear genome methylation patterns during the DNA replication and repair, thus regulating gene expression. The mutations responsible for ADCA-DN and HSN1E affect the replication foci targeting sequence domain, which regulates DNMT1 binding to chromatin. DNMT1 dysfunction is anticipated to lead to a global alteration of the DNA methylation pattern with predictable downstream consequences on gene expression. Interestingly, ADCA-DN and HSN1E phenotypes share some clinical features typical of mitochondrial diseases, such as optic atrophy, peripheral neuropathy, and deafness, and some biochemical evidence of mitochondrial dysfunction. The recent discovery of a mitochondrial isoform of DNMT1 and its proposed role in methylating mitochondrial DNA (mtDNA) suggests that DNMT1 mutations may directly affect mtDNA and mitochondrial physiology. On the basis of this latter finding the link between DNMT1 abnormal activity and mitochondrial dysfunction in ADCA-DN and HSN1E appears intuitive, however, mtDNA methylation remains highly debated. In the last years several groups demonstrated the presence of 5-methylcytosine in mtDNA by different approaches, but, on the other end, the opposite evidence that mtDNA is not methylated has also been published. Since over 1500 mitochondrial proteins are encoded by the nuclear genome, the altered methylation of these genes may well have a critical role in leading to the mitochondrial impairment observed in ADCA-DN and HSN1E. Thus, many open questions still remain unanswered, such as why mtDNA should be methylated, and how this process is regulated and executed?
DNA Methylation
Several epigenetic signals participate in cell specific gene expression, including DNA methylation and demethylation, post-translational modifications of histone proteins (i.e., acetylation, methylation, phosphorylation, and ubiquitination), incorporation of histone variants and gene regulation by non-coding RNAs (Rivera and Ren, 2013; Kanherkar et al., 2014a). DNA methylation, which occurs in all prokaryotic and eukaryotic organisms, with rare exception for yeast, roundworm, and fruit fly, is a key epigenetic process involved in the regulation of gene expression (Lande-Diner et al., 2007; Lee et al., 2010) and parental imprinting (Sha, 2008; Ishida and Moore, 2013), in chromosome X inactivation (Straub and Becker, 2007) as well as in the development of the immune system (Cedar and Bergman, 2011; Kondilis-Mangum and Wade, 2013; Li et al., 2013) and in cellular reprogramming (Reik, 2007; Krishnakumar and Blelloch, 2013; Papp and Plath, 2013; Kanherkar et al., 2014b). Furthermore, it is engaged in the maintenance of the genome integrity through protection against endogenous retroviruses and transposons (Howard et al., 2008). In humans aberrant DNA methylation patterns are associated with several diseases, including various cancers (Jones and Baylin, 2007; Esteller, 2008; Iacobuzio-Donahue, 2009), immune system disorders (Feinberg, 2007) and neurodegeneration (Jakovcevski and Akbarian, 2012; Qureshi and Mehler, 2013).
Classical Model of DNA Methylation
In prokaryotes, DNA methylation occurs on both cytosine and adenine bases and is one of the host restriction systems to distinguish self and non-self DNA (Jeltsch, 2006). In mammals, following the classical model of DNA methylation, it takes place in the cytosine residues at their C5 positions, primarily in the CG dinucleotides (CpG), acting mainly as a repressive tag to silence chromatin and inhibit transcription. In the human genome there are 56 million CG sites, about 60–80% of which are methylated, corresponding to 4–6% of all cytosines (Laurent et al., 2010). Methylation levels and patterns are cell and tissue specific. In mammalian genomes, the CpG are poorly represented compared to other dinucleotides, because of the higher mutagenic property of the 5-methyl-cytosine base compared to the unmethylated one (Pfeifer et al., 2000). The irregular CpG distribution is reflected by their depletion in intergenic and intragenic sequences, whereas their presence is less suppressed in repetitive DNA (such as transposons and retroviruses) and CpG islands, regions of at least 550 bp, with a ratio of observed CpG/expected CpG higher than 0.65 (Takai and Jones, 2004). Around 70% of human genes promoter regions present the CpG islands and an inverse correlation between CpG islands density and the promoter methylation status exists. Moreover, active genes usually show hypomethylation at the transcriptional start site (TSS) and high levels of methylation in the gene body, which is supposed to block aberrant transcription initiation inside the gene, avoiding the production of truncated mRNAs and proteins. The splicing sites are regions characterized by a change in DNA methylation, since exons have higher methylation than introns, suggesting a strong implication of DNA methylation in the splicing process (Laurent et al., 2010; Chatterjee and Vinson, 2012).
DNA Methyltransferase Enzymes
DNA methylation is catalyzed by a group of enzymes called DNA (cytosine-5)-methyltransferases (DNMTs) that transfer a methyl group from a cofactor molecule S-adenosyl-l-methionine (AdoMet or SAM) to the C5 position of the cytosine residues to generate 5-methylcytosine (5 mC) and S-adenosyl-l-homocysteine (AdoHcy, SAH; Figure 1A). The mammalian DNMT family includes four members: DNMT1, DNMT3A, DNMT3B, and DNMT3L. These enzymes comprise two parts: a C-terminal catalytic portion and a large N-terminal region of variable size containing regulatory domains involved in the interaction with DNA, chromatin, and other proteins (Figure 2). Furthermore, the N-terminal region contains 621 amino acids required for discriminating between hemi-methylated and unmethylated DNA. The C-terminal catalytic domain, being highly conserved between eukaryotes and prokaryotes, is composed by 500 amino acids and harbors the active center of the enzyme, which contains amino acids motifs characteristic of the cytosine-C5 methyltransferases, called the “AdoMet-dependent MTase fold" (Jurkowska et al., 2011a). Motifs I and X of this domain are involved in cofactor binding whereas motifs IV, VI, and VIII have a catalytic function. The non-conserved region between motifs VIII and IX is the target recognition domain (TRD), crucial for DNA recognition and specificity (Cheng, 1995; Jeltsch, 2002; Cheng and Blumenthal, 2008). Considering the high variability of the N-terminus of DNTMs, a detailed description of this region for each DNMT isoforms will be provided in the following paragraphs.
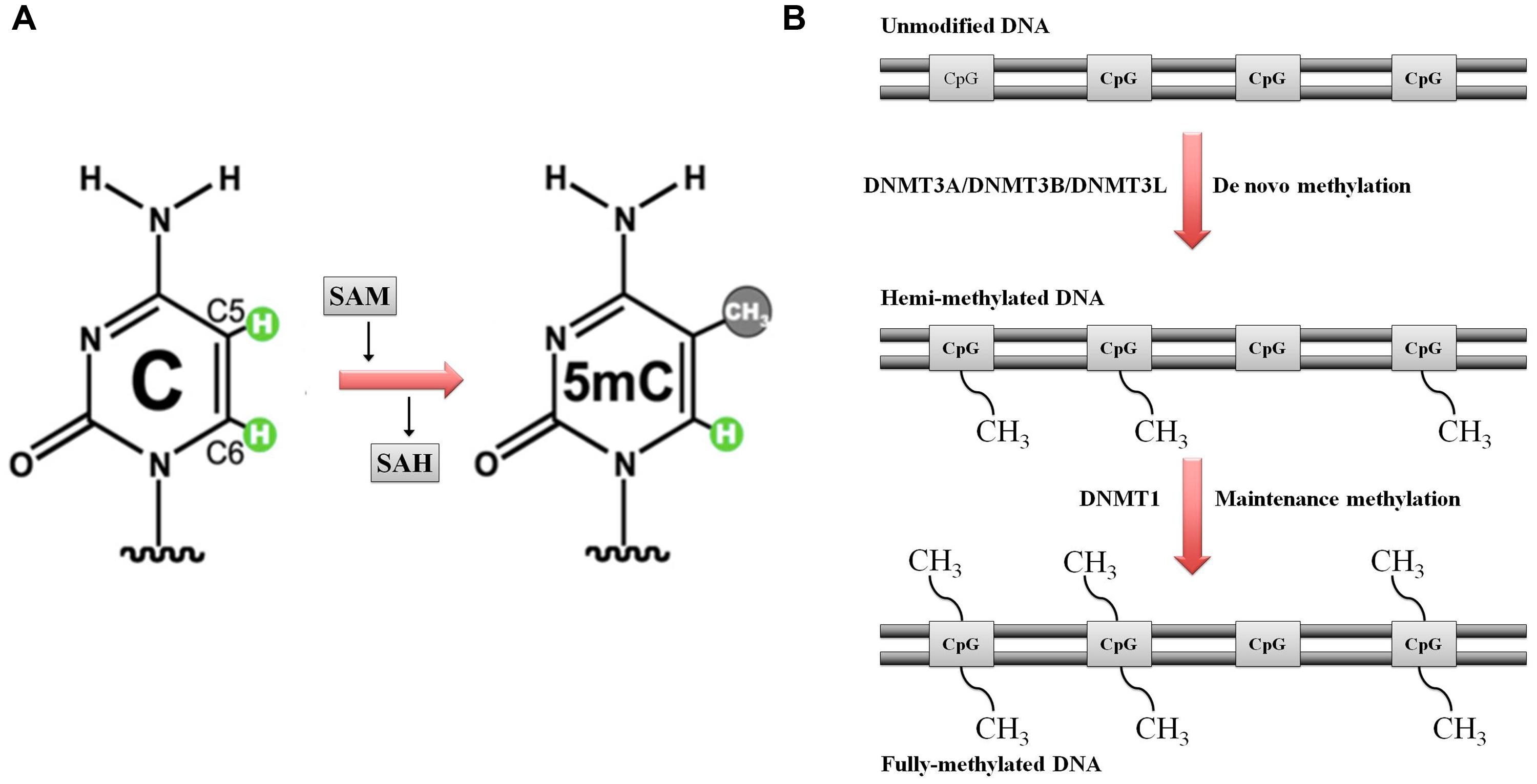
FIGURE 1. Schematic representation of DNA methylation. (A) DNA (cytosine-5)-methyltransferases (DNMTs) transfer the methyl group at the DNA cytosine ring carbon C5 by using S-adenosyl methionine (SAM) as methyl donor. S-adenosyl homocysteine (SAH) is the cofactor product. (B) DNA methylation: classical model. Maintenance versus de novo methylation. Figure modified by Cheng and Blumenthal (2008)
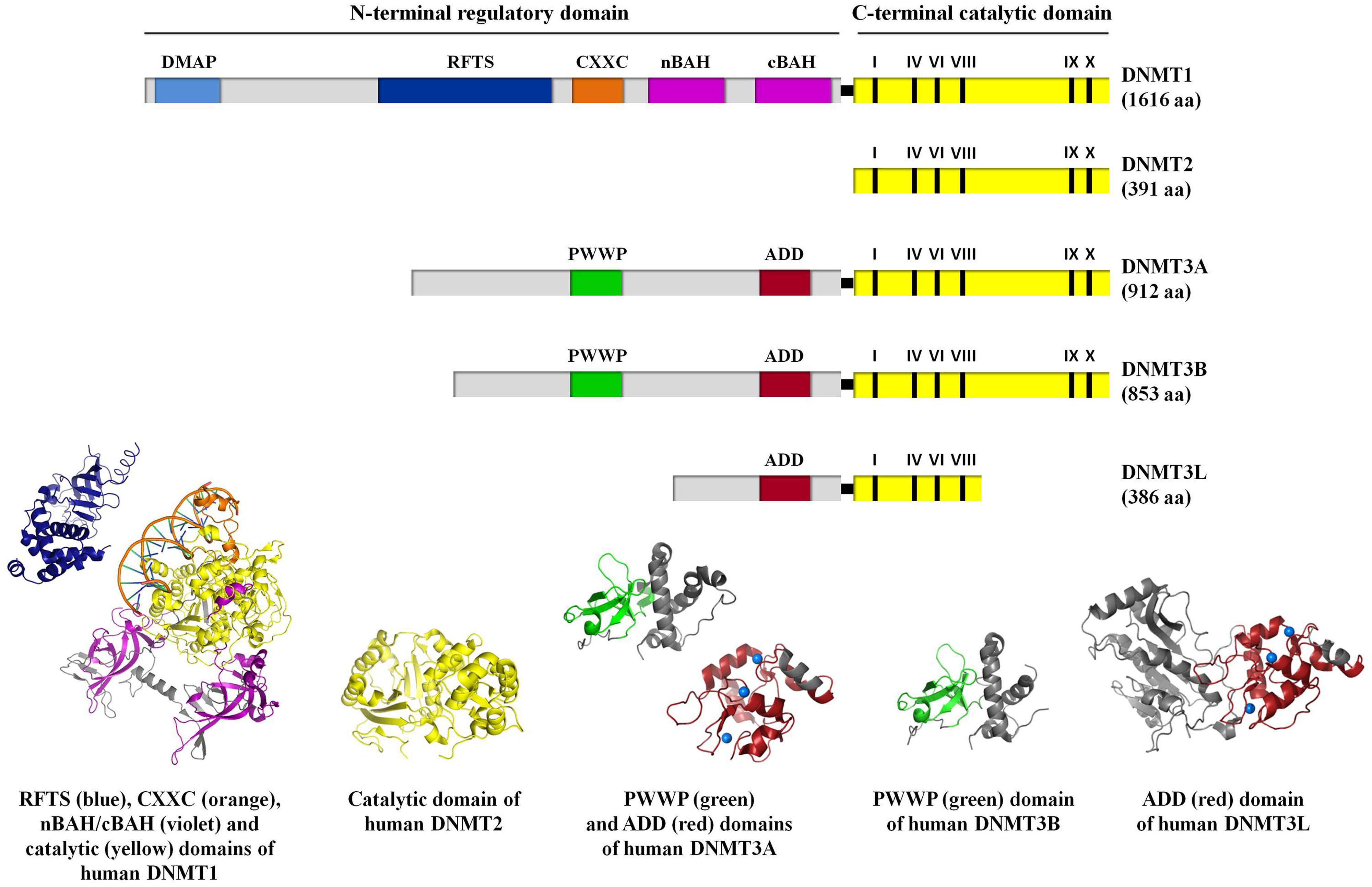
FIGURE 2. A schematic representation of the domain structure of human DNMT isoforms. For human DNMT1, DNA methyltransferase associated protein (DMAP), replication foci targeting sequence (RFTS), CXXC, bromo-adjacent homology (nBAH/cBAH), and catalytic domains are represented by cyan, blue, orange, violet, and yellow bars, respectively. For human DNMT2, catalytic domain is represented by yellow bar. For human DNMT3A, DNMT3B, and DNMT3L, PWWP, ADD, and catalytic domains are represented by green, red, and yellow bars, respectively. In the catalytic domain (yellow bar), the conserved C5 DNA MTase motifs in the C-terminal part of each DNMT isoforms are labeled. The interdomain linker regions are shown as light gray bars. Numbers in parentheses indicate the length of each protein. The crystal structures of the human DNMT1 RFTS domain (residues 351–597, PDB ID: 3EPZ; Syeda et al., 2011), human DNMT1 CXXC, nBAH/cBAH, and catalytic domains bound to DNA-containing unmethylated CpG sites (residues 646–1600, PDB ID: 3PTA; Song et al., 2011), human DNMT2 catalytic domain (residues 1–188 and 248–391, PDB ID: 1G55; Dong et al., 2001), human DNMT3A PWWP (residues 275–427, PDB ID: 3LLR), and ADD domains (residues 476–614, PDB ID: 3A1A; Otani et al., 2009; Wu et al., 2011, respectively), human DNMT3B PWWP domain (residues 293–442; PDB ID: 3QKJ; Wu et al., 2011), human DNMT3L ADD domain (residues 34–277 and 279–380, PDB ID: 2PV0; Ooi et al., 2007), are shown. For further details see “Text."
The initial methylation pattern is established by de novo DNMTs (DNMT3 family in mammals). This pattern is perpetuated for the rest of the life (with small tissue-specific changes) by a mechanism first proposed by Holliday and Pugh (1975) and Riggs (1975).
Each round of DNA replication produces hemimethylated DNA with the methylation marks in the parental stand and the new synthesized daughter unmethylated strand (Figure 1B). The maintenance DNA methyltransferase (DNMT1 in mammals), showing a preference for hemimethylated sites, copies the existing methylation pattern (Jones and Liang, 2009; Denis et al., 2011).
DNMT3 Family
During the embryogenesis the DNMT3 family establishes the initial CpG methylation pattern (Chen and Li, 2006; Jurkowska et al., 2011a). The DNMT3 family includes two active de novo DNMTs, DNMT3A, and DNMT3B, without significant preference between hemimethylated and unmethylated DNA, and one catalytically inactive regulatory factor, the DNMT3-Like protein (DNMT3L; Bestor, 2000; Jurkowska et al., 2011a).
Knockout of DNMT3A or DNMT3B is lethal, being both essential for embryonic development in mice. Mouse DNMT3B knockout embryos die in utero, whereas the DNMT3A knockout animals die shortly after birth (Okano et al., 1999). DNMT3L knockout mice are viable, but male are sterile, failing to produce mature sperm (Hata et al., 2002). Mutations in the human DNMT3B gene are associated with a rare autosomal disease, called ICF (Immunodeficiency, Centromere instability, Facial abnormalities) syndrome, which is accompanied by hypomethylation of classical satellites of the pericentromeric regions of chromosomes 1, 9, and 16, probably due to reduction of enzyme catalytic efficiency or alteration of its localization (Xu et al., 1999; Weemaes et al., 2013). De novo mutations in DNMT3A have also been identified in patients with overgrowth disorders (Tatton-Brown et al., 2014). Despite high sequence homology and similar biochemical properties, DNMT3A and 3B show partially non-overlapping biological functions with DNMT3A being involved in the setting of parental imprints and DNMT3B in the methylation of pericentromeric repeats (Okano et al., 1999; Kaneda et al., 2004).
From the structural point of view, the DNMT3 enzymes contain a N-terminal variable portion (around 280 and 220 amino acids for DNMT3A and DNMT3B, respectively), followed by two conserved regions: the regulatory PWWP and ADD domains and C-terminal catalytic portion. DNMT3L lacks the PWWP domain as well as the DNMTs motifs IX and X and all the important catalytic residues in its C-terminal portion (Figure 2). The PWWP is a variable module of 100–150 amino acids characterized by the presence of a strictly conserved proline-tryptophan-tryptophan-proline motif, also found in chromatin associated eukaryotic protein from yeast to mammals. It comprises a globular domain with a five-stranded β-barrel followed by a five-helix bundle (Figure 2; Qiu et al., 2002; Chen et al., 2004). In vitro experiments showed that the PWWP domain of DNMT3B mediate DNA binding, but it is not required for CpG methylating activity (Qiu et al., 2002). Subsequently, it has been demonstrated that it exhibits a dual function: binding to both DNA and methylated lysine on histone proteins (Chen et al., 2004; Dhayalan et al., 2010; Wu et al., 2011). The ADD domain (ATRX-DNMT3-DNMT3L), also known as PHD domain (plant homodomain), is present in all the proteins belonging to DNMT3 family and in the ATRX (alpha thalassemia/mental retardation syndrome X-linked) protein. This domain is a cysteine-rich module that binds zinc ions (comprising six CXXC motifs; Figure 2) and constitutes a platform for various protein–protein interactions. It has been reported that DNMT3L, which only owns this characteristic motif, recognizes specifically the unmethylated lysine 4 of histone H3 through its ADD domain and induces de novo DNA methylation by recruitment or activation of DNMT3A, thanks to an interaction between its C-terminal domain and the catalytic portion of DNMT3A. These data support the idea that DNMT3L has the dual function of binding the unmethylated histone tail and activating DNA methyltransferase (Jia et al., 2007; Ooi et al., 2007). Nevertheless, the ADD domains of DNMT3A and 3B share considerable homology with DNMT3L and it has been reported a direct binding of the DNMT3A and DNMT3B ADD domains to H3 tails unmodified at lysine 4 (Otani et al., 2009; Zhang et al., 2010). Since the ADD domains of DNMT3A/B and DNMT3L clearly show the same binding specificity, the recruitment of DNMT3A to chromatin with unmethylated H3 tail is probably not the primary function of DNMT3L (Zhang et al., 2010).
Jurkowska et al. (2011b) reported that DNMT3A forms oligomers predicted to bind several DNA molecules oriented in parallel and required to tightly tie to heterochromatin by the ADD and PWWP domains, which recognize H3 tails unmodified at lysine 4 and H3 trimethylated at lysine 36, respectively (Dhayalan et al., 2010; Zhang et al., 2010). In addition, the association of the DNMT3A oligomers to DNMT3L changes its subnuclear localization, from heterochromatin to euchromatin, thereby increasing its availability and DNA methylation activity for the generation of DNA methylation imprints (Jurkowska et al., 2011b).
DNMT1
Whereas DNMT3 family works as de novo methylases, DNMT1 maintains the existing pattern of methylation during chromosomes replication (Chen and Li, 2006; Jurkowska et al., 2011a) and repair (Mortusewicz et al., 2005; Loughery et al., 2011). Human DNMT1, a large enzyme of 1616 amino acids, is the most abundant DNMT involved in preserving and propagating the existing methylation patterns during cell division (Jurkowska et al., 2011a; Kar et al., 2012). It shows a preference for hemimethylated DNA (Goyal et al., 2006) and it is localized at the DNA replication foci during the S-phase of cell cycle (Leonhardt et al., 1992). In addition, DNMT1, being a highly processive enzyme, is able to methylate long stretches of DNA without dissociation (Hermann et al., 2004; Vilkaitis et al., 2005). These two features make it suitable for its role in the maintenance of methylation patterns. The expression of DNMT1 is ubiquitous and high in proliferating cells and varies in a cell-cycle-dependent manner, being maximal during the S-phase and extremely low in non-dividing cells (Robertson et al., 1999; Kimura et al., 2003). Concerning the subcellular localization of DNMT1, it is diffusely distributed in the nucleus during the interphase and moves to the replication foci for early and mid S-phase creating a characteristic punctuate pattern (O’Keefe et al., 1992; Easwaran et al., 2008). DNMT1 mouse knockout shows extensive demethylation of the genome and embryonic lethality shortly after gastrulation, underlying the crucial role of DNMT1 in early development. Embryonic stem cells lacking DNMT1 are viable, despite the low level of DNA methylation, but die after differentiation induction (Li et al., 1992; Chen et al., 1998). Complete inactivation of DNMT1 in human colorectal carcinoma cells leads to severe mitotic defects and progressive cell death (Chen et al., 2007).
Mutations affecting the DNMT1 gene have been associated with two distinct autosomal dominant neurodegenerative diseases: hereditary sensory and autonomic neuropathy with dementia and hearing loss type 1E (HSN1E) and autosomal dominant cerebellar ataxia-deafness and narcolepsy (ADCA-DN; Klein et al., 2011; Winkelmann et al., 2012). All these mutations are localized in the DNA replication foci targeting sequence (RFTS) domain, essential in mediating the association of DNMT1 to heterochromatin. Mutations in exon 20 of DNMT1 have been associated with HSN1E, whereas mutations in exon 21 have been found in ADCA-DN (Klein et al., 2013).
From the structural point of view, DNMT1 is a multi-domain enzyme composed by a N-terminal regulatory region and a C-terminal catalytic domain joined by a series of KG (lysine-glycine) repeats (Figure 2; Dhe-Paganon et al., 2011; Jurkowska et al., 2011a; Kar et al., 2012). The N-terminal portion contains several motifs:
- DMAP1 (DNA methyltransferase associated protein 1) charged rich domain is involved in the interaction of DNMT1 with the transcriptional repressor DMAP1 as well as in the stability of the enzyme and in its binding to DNA CpG sites (Rountree et al., 2000; Ding and Chaillet, 2002).
- PBD (PCNA-proliferating cell nuclear antigen-binding) domain address DNMT1 to the replication foci through the interaction between DNMT1 and PCNA.
- NLS (nuclear localization sequence) are at least three.
- RFTS (replication foci targeting sequence) is crucial for the localization of DNMT1 at the centromeric chromatin and replication foci as well as for its dimerization (Easwaran et al., 2008; Fellinger et al., 2009).
- CXXC zinc domain, similar to the cysteine-rich motif present in other chromatin associated proteins. It contains eight conserved cysteine residues clustered in two CXXCXXC repeats that bind to two zinc ions and is necessary for the recognition of unmethylated CpG, thus influencing the catalytic activity of the enzyme (Pradhan et al., 2008).
- PBHD (polybromo homology domain) domain is composed by the BAH1 and BAH2 (Bromo-adjacent homology 1 and 2) motifs, typical of proteins involved in transcriptional regulation. It has been proposed to act as a protein–protein interaction module specialized in gene silencing even if its functional role in DNMT1 remain unknown.
Thus, the N-terminal regulatory region is designed to recognize the methylation target site (DMAP1, PBD, and RFTS domains), to bind nucleic acids trough an allosteric site (CXXC domain) and to allow interaction with other proteins (PBHD domains), making DNMT1 ideally suited for its role as the maintenance methyltransferase.
The KG linker between the N- and C-terminal regions of the enzyme is composed by a series of lysine and glycine residues, which might contribute to address DNMT1 to the region next to the replication fork.
Finally, the C-terminal region encloses the catalytic center of the enzyme bearing the conserved motifs I–X, and is folded in large and small domains, separated by a huge cleft. The large domain (motifs I-VIII and part of motif X) participates in SAM cofactor binding substrate (cytosine) targeting and other essential catalytic events. The small domain includes a variable region between motif VIII and IX, named TRD (target recognition domain), the conserved motif IX and part of motif X. The catalytic domain allows the binding of the target DNA in the active site and various other regulatory molecules in the allosteric sites, which support a multiple levels of enzyme regulation (Dhe-Paganon et al., 2011; Jurkowska et al., 2011a; Kar et al., 2012).
In order to avoid runaway methylation, DNMT1 undergoes to several auto-inhibitory mechanisms. In particular, to keep DNMT1 inactive the RFTS domain is positioned and stabilized deep inside the catalytic domain in a task where the hemimethylated DNA is expected to fit, thus, masking the catalytic core of the enzyme (Qin et al., 2011; Syeda et al., 2011; Bashtrykov et al., 2014). The auto-inhibitory function of the CXXC domain is more controversial. Song et al. (2011). reported that CXXC specifically binds to unmethylated CpG, activating the CXXC-BH21 linker, which then occupies the catalytic pocket and interferes with its function. By contrast, Bashtrykov et al. (2012) refused this auto-inhibitory mechanism, proposing instead that the recognition of the hemimethylated state of target sites resides within the catalytic domain. While the unmethylated DNA emerges from the replication fork, DNMT1 is tethered and kept auto-inhibited to avoid unauthorized de novo methylation of the DNA. As soon as the hemimethylated sites are released from the replication complex, the TRD region of DNMT1 in combination with UHRF1 (ubiquitin-like with PHD and ring finger domains 1) recognizes and marks them as prime targets for methylation to guarantee the correct methylation patterns across successive generations (Kar et al., 2012).
DNMT2
DNA (cytosine-5)-methyltransferases 2 is the smallest mammalian DNMT, now named TRDMT1, which has the potential to methylate RNA instead of DNA (Goll et al., 2006; Schaefer and Lyko, 2010). It completely lacks the regulatory N-terminal region and the C-terminal domain contains all 10 sequence motifs that are conserved among DNMTs, including the consensus S-adenosyl-l-methionine-binding motifs (Okano et al., 1998; Dong et al., 2001; Figure 2). Despite the high sequence and structural similarity with other DNMTs and its capability to bind the DNA, DNMT2 has failed to show a significant transmethylase activity (Hermann et al., 2003). Nonetheless, it methylates cytosine 38 in the anticodon loop of tRNAAsp (Goll et al., 2006), by using a DNA methyl transferase-like catalytic mechanism (Jurkowski et al., 2008). Thus, DNMT2 seems to have intermediate properties between a DNA methyl transferase and a RNA methyl transferase, with which it shares the structural/catalytic features and the nuclear-cytoplasmic localization, respectively (Schaefer et al., 2008).
Stochastic Model of DNA Methylation
For long time the classical model of DNA methylation at CpG sites in mammals, with the involvement of both DNMT3 and DNMT1 (i.e., de novo and maintenance DNA methyltransferases, respectively), has been considered a paradigm for epigenetic information transfer (Holliday and Pugh, 1975; Riggs, 1975). Over the years, several experimental observations highlighted that the site-specific maintenance methylation model had to be revised. Thus, Jeltsch and Jurkowska (2014) proposed a modified stochastic DNA methylation model which includes a new description of both de novo and maintenance methylation process at CpG and non-CpG sites.
An efficient methylation of both DNA strands, starting from unmethylated DNA, can be achieved by the tight cooperation of DNMT3 enzymes with DNMT1. Thus, de novo and maintenance methylation could not be regarded as two distinct events. DNMT3 enzymes, and in particular DNMT3A, can bind adjacent DNA molecules, but only one of the two strands is preferentially methylated. By contrast, the hemi-methylated DNA is the favorite substrate for DNMT1, which copies the methylation on the second strand (Fatemi et al., 2002). In addition, DNMT1 shows considerable de novo methylation activity on unmethylated DNA both in vitro (Goyal et al., 2006) and in vivo, as observed in DNMT3A/3B double knockout embryos (Okano et al., 1999). Arand et al. (2012). have confirmed these results by reporting DNMT1-dependent de novo methylation in cells with single or combined DNMTs knockout. On the other side, deletion of DNMT3A/3B or DNMT3B alone led to a reduction of maintenance DNA methylation at repetitive elements, despite the presence of functional DNMT1 (Chen et al., 2003; Dodge et al., 2005; Arand et al., 2012).
Thus, neither de novo methylation nor maintenance methylation can be exclusively assigned to DNMT3A/3B or DNMT1, respectively, but a synchronized cooperation of all the DNMTs seems crucial.
The classical model of maintenance methylation assumes that the methylation patterns of single CpG sites are stably inherited and that exists for this purpose a perfect enzyme, characterized by a working efficiency of 100%.
In reality, average methylation densities of DNA regions are maintained, instead of exact CpG site-specific methylation patterns (Zhang et al., 2010), and changes in methylation levels occur through stochastic processes (Landan et al., 2012). In fact, despite the 10–40-fold preference of DNMT1 for hemi-methylated DNA, this feature is not enough to copy accurately the site-specific methylation status of around 56 million CpG sites in the human genome over several rounds of DNA replication (Jeltsch and Jurkowska, 2014).
In addition, patterns of non-CpG DNA methylation, typical of plants, have been reported also in the human genome, taking place in CpA sites, and specifically introduced by DNMT3A. This clearly indicates a permanent de novo methylation activity of this enzyme, which is not in agreement with the classical methylation model (Arand et al., 2012; Guo et al., 2014).
Finally, also the DNA demethylation has to be considered in the establishment and maintenance of DNA methylation pattern. This process could be passive, during the replication, or active, performed by the family of ten eleven translocation (TET) dioxygenases. These enzymes oxidize the methyl groups of methylcytosine and are expressed both during early development and also in later stages, suggesting a permanent DNA demethylation. Hydroxymethylation of DNA by TET enzymes has been proposed to keep CpG islands in unmethylated state by counteracting stochastic DNA methylation (Kohli and Zhang, 2013; Wu and Zhang, 2014).
In conclusion, the DNA methylation is influenced by all the events above described, that are combined in the stochastic model of Jeltsch and Jurkowska (2014):
- DNA methylation at each site is determined by the local rates of methylation and demethylation
- The local rates of methylation depend on targeting/regulation of DNMTs/demethylases and on chromatine remodeling/DNA accessibility
- DNA methylation occurs on both CpG and non-CpG sites de novo and maintenance methylation are combined in a unified mechanism
- The average DNA methylation level of DNA regions is inherited rather than the methylation state of individual CpG sites.
Mitochondrial DNA Methylation
Unlike nuclear DNA, the methylation of mtDNA is a highly controversial topic that after 40 years is still matter of debate among researchers. In fact, although recent studies have demonstrated the presence of 5 mC in mtDNA, skepticism regarding the sensitivity and reproducibility of the methods used and the putative biological function of methylation in mitochondria still remains. Indeed, the observed methylation levels are very low (1–5%) and, furthermore, mtDNA has a different organization compared to nuclear DNA (i.e., no histones, lack of introns, multicopy genomes), which should imply a different regulatory mechanism of gene expression through DNA methylation. In fact, mammalian cells typically contain 1,000–10,000 copies of mtDNA, which are organized into nucleoprotein complexes termed nucleoids, being TFAM (mitochondrial transcriptor factor A) the main protein component. The TFAM/mtDNA ratio finely regulates the fraction of active mtDNA molecules available for mitochondrial replication/transcription (Bogenhagen, 2012; Campbell et al., 2012; Farge et al., 2014).
The first approaches to investigate mtDNA methylation date back to 1970s, when Vanyushin et al. (1971) found a DNMT activity in mitochondria isolated from loach embryos, demonstrated the presence of 5 mC in mtDNA extracted from beef heart (Vanyushin and Kirnos, 1974) and evidenced a different specificity for DNA methylases isolated from nucleus and mitochondria, being the mitochondrial specific for mono-pyrimidines and the nuclear for di- and tri-pyrimidines (Vanyushin and Kirnos, 1977). However, in the same years the absence of 5 mC in mtDNA from mouse, hamster, frog, and HeLa cells was also reported (Nass, 1973; Dawid, 1974). Shmookler and Goldstein (1983) and Pollack et al. (1984), studying both human fibroblasts and mouse fibroblastoid cells, clarified that methylation occurred in mtDNA with a frequency of 1.5–5% and only in CpG di-nucleotides, which, however, are underrepresented in mtDNA (Pollack et al., 1984; Cardon et al., 1994). After many years, in 2004, the presence of methylated cytosines in mtDNA, from gastric and colorectal cancers, was once again denied using bisulfite-PCR-single-stranded DNA conformation polymorphism on three selected regions of mtDNA containing 37 CpG sites (Maekawa et al., 2004).
Rebelo et al. (2009) failed to observe mtDNA methylation in human osteosarcoma 143B, HEK293, and HeLa cells, under standard culture conditions, but the same authors also reasoned that percentages of methylation <5% have not been detected by the approach employed (site-specific methylation restriction enzymes). However, in this study, some methylation of mtDNA was observed after the induced-expression of two different mitochondria-targeted bacterial methyltransferases and, additionally, the levels of this methylation increased during mtDNA replication, when it is assumed that nucleoids are remodeled and mtDNA could be less protected by proteins (i.e., TFAM) and more accessible to DNMTs (Rebelo et al., 2009).
The issue of mtDNA methylation came back again in 2011, when two different groups demonstrated the presence of 5 mC and DNMTs in mitochondria through novel and more sensitive approaches (Chestnut et al., 2011; Shock et al., 2011). Surprisingly, a mitochondria targeted DNMT1 isoform (mtDNMT1), conserved in different species, has been found by Shock et al. (2011); the mtDNMT1 seems to be translated from an unconventional ATG site, located immediately upstream of the canonical one, and localized to mitochondria where it bounds to mtDNA. Moreover, the mtDNMT1 gene expression was induced by NRF1 and PGC1α, two master regulators of mitochondrial biogenesis, in HCT116 cells (Shock et al., 2011). Interestingly, gene expression of mtDNA-encoded genes was altered after mtDNMT1 over-expression, with reduced levels of MT-ND6 in the L-strand and increased levels of MT-ND1 in the H-strand, suggesting an opposite role for mtDNMT1, and cytosine methylation in the two strands; however, the expression levels of MT-CO1 and MT-ATP6, both in the H-strand, were unaltered (Shock et al., 2011). The authors speculate that mtDNMT1 interferes with MTERF-dependent transcription termination, inducing an increased transcription of MT-ND1 through the HSP1, with no effects on the polycistronic mRNA produced by HSP2. Lastly, in this study the presence of 5 mC in mtDNA was demonstrated by a methylated DNA-immunoprecipitation (MeDIP) approach, followed by real time-PCR (Shock et al., 2011).
Concomitantly, Chestnut et al. (2011) demonstrated the presence of DNMT3A in mitochondria isolated from mouse brain and from human motor cortex, whereas DNMT1 was faintly detectable in mitochondria from these tissues. Furthermore, co-localization of 5 mC and mitochondria was shown by immunofluorescence experiments using antibodies recognizing 5 mC and a specific mitochondrial marker (superoxide dismutase isoform 2, SOD2; Chestnut et al., 2011).
The mtDNA methylation was also investigated by a liquid chromatography-electrospray ionization tandem-mass spectrometry (LC-ESI-MS) method, proving the existence of 5 mC in the mitochondrial genome (Infantino et al., 2011), and soon after also 5 hmC have been identified in mtDNA by an ELISA approach (Dzitoyeva et al., 2012).
In a study focused on the intragenic methylation of PolgA in different mouse cells/tissues as possibly correlated with PolgA expression and mtDNA content regulation, the authors found in the eluted fraction of MeDIP analysis some mtDNA encoded genes (mt-cytb, mt-co1) and the D-loop region, indicating that these genes have 5 mC and 5 hmC, although at very low levels (Kelly et al., 2012).
Pirola et al. (2013) further evidence of methylated mtDNA has been published. The methylation of MT-ND6, MT-CO1 and of the D-loop of mtDNA was assessed by quantitative methylation specific-PCR in the context of non-alcoholic fatty liver disease. The authors found a significant association between the condition of non-alcoholic steatohepatitis (NASH) and the methylation of MT-ND6 gene, which inversely correlates with MT-ND6 transcription and protein expression in the liver of subject affected by NASH (Pirola et al., 2013).
The methylation of the D-loop region was also confirmed in mammals by bisulfite-sequencing (30 clones for each sample sequenced) and by MeDIP (Bellizzi et al., 2013). Methylation was limited to the L-strand of the D-loop and the majority of methylated cytosines were located outside the CpG nucleotides; different tissues were analyzed (blood cells, fibroblasts, HeLa cells) and tissue-specific patterns of methylation were identified, HeLa cells showing the highest percentage of methylated cytosines (Bellizzi et al., 2013). In the same study, DNMT1 and DNMT3B expression in mitochondria from HeLa and mouse 3T3-L1 cells was evidenced (Bellizzi et al., 2013).
On the contrary, Wong et al. (2013) proved that in mouse skeletal muscle DNMT1 was not present within the organelle, but probably bound to the outer membrane (detectable in the crude mitochondrial fraction), whereas DNMT3A was present in the pure mitochondrial fraction isolated from both mouse skeletal muscle and spinal cord; DNMT3B was not detectable in any of the mitochondrial preparations. DNMT3A was also present in mitochondria from human cerebral cortex, but not in mitochondria from HEK293 cells, showing a preferential expression in mitochondria from excitable tissues in humans and mouse (Wong et al., 2013). In addition, methylated cytosines in the mouse D-loop and mt-rnr2 (coding for 16S RNA) were identified by bisulfite treatment and pyrosequencing, and different levels of methylation in brain (highest percentage of 5 mC), liver and testes were shown (Wong et al., 2013). Moreover, as previously observed by Chestnut, the co-localization of 5 mC, and mitochondria by immunofluorescence was confirmed and interestingly, it was observed a co-localization of 5 mC with autophagosome (LC3A positive staining), indicating that mtDNA methylation may be involved in mitophagy (Wong et al., 2013).
Also Byun et al. (2013) carried out bisulfite-pyrosequencing to investigate methylation of three mtDNA regions, MT-TF (tRNA phenylalanine gene, two CpG sites), MT-RNR1 (12S RNA gene, two CpG sites), and the D-loop (three CpG sites), in association with effect of airborne pollutants. The authors were able to detect around 5–6% of 5 mC in the selected regions.
Hong et al. (2013) critically revised all methods employed to quantify methylated mtDNA in the last years. Contextually, these authors established the absence of CpG methylation in human mtDNA performing bisulfite-sequencing and analyzing data of bisulfite-next generation sequencing (NGS) previously published by Akalin et al. (2012) and Hong et al. (2013). Bisulfite-sequencing failed to reveal 5 mC in four selected mtDNA regions (MT-RNR1, MT-RNR2, MT-CO2, MT-ATP6) of HEK293 cells, and identified very low frequencies of 5 mC in the same regions of HCT116 and blood cells mtDNA (<0.5%), considered not relevant because comparable to the non-convertion rate of bisulfite; cross-contamination of nuclear DNA sequences of mitochondrial origins (NUMTs; Lascaro et al., 2008) has been also excluded based on the specificity of the analyzed regions for mtDNA sequence (Hong et al., 2013). Comparable levels of 5 mC (<0.5%), both in CpG or not-CpG sites, came from the re-analysis of bisulfite-NGS data on HCT116 cells previously published (Akalin et al., 2012), but the mean coverage obtained of 94x may be not sufficient, considering that HCT116 cells have a mtDNA content of about 4000 copies, as mentioned by the authors (Hong et al., 2013).
Lastly, a comparative analysis of mitochondrial methylomes in 39 different cell lines was carried out extracting public data from the NIH Roadmap Epigenomics project (Ghosh et al., 2014). The re-analyzed data, deriving from MeDIP-sequencing included human brain, breast, blood, penis, and two cell lines, H1 and neurosphere cultured cells; moreover, some tissues were analyzed at different developmental time points. The authors identified tissue- and development-specific pattern of 5 mC methylation in mtDNA, with MT-ND6 and MT-ATP6 showing progressive reduction in methylation correlated with brain development. However, also in this case, methylated cytosines resulted underrepresented (<0.5%) and an extremely low minimum coverage of 5x was considered (Ghosh et al., 2014).
In conclusion, since 2011 several studies claimed the occurrence of mtDNA methylation, but a few others also asserted the absence of 5 mC in mtDNA (Figure 3). Many of the published approaches had some limitations, such as the reproducibility of the methods based on the use of antibodies (ELISA, MeDIP), or the enrichment by amplification before sequencing that may create a bias, or the very low coverage considered for the analysis of NGS data that may influence the quantification of 5 mC referred to total mtDNA copies. In fact, the analysis of mtDNA methylation should take into account that the mitochondrial genome is multicopy, differing for copy number depending on cell types and tissues (Campbell et al., 2012). None of the sequencing studies cited above have considered the percentage of methylated sites respect to total mtDNA molecules present in the cells/tissues analyzed (“methylation heteroplasmy"), but only the percentage of 5 mC respect to total cytosines, as is usually done for the nuclear genome.
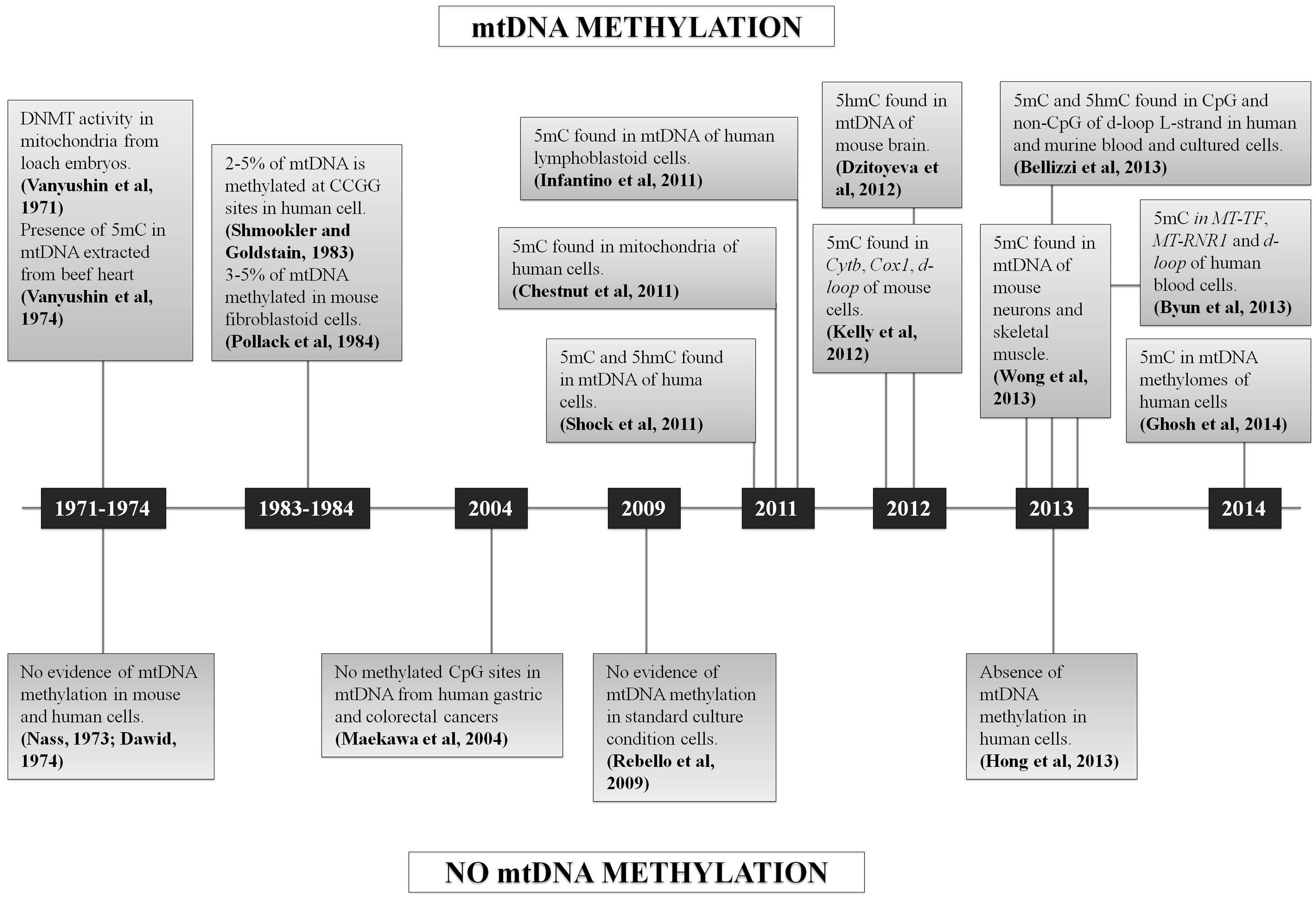
FIGURE 3. Chronological sequence of mtDNA methylation studies. Schematic representation of all the studies which support or denied the mtDNA methylation.
The scenario becomes even blurrier when considering the the controversial results on DNMTs localization (Tables 1 and 2), since DNMT1 presence within mitochondria was demonstrated (Shock et al., 2011; Bellizzi et al., 2013), and then denied by Wong et al. (2013), who in addition observed DNMT3A within mitochondria, but not DNMT3B, which, however, has been detected in these organelles by others (Bellizzi et al., 2013). Therefore, it remains truly unclear which DNMTs might be implicated in mtDNA methylation. Overall, further experiments, focused on both global/site-specific methylation and the functional effects of this modification are necessary to unequivocally demonstrate methylation of the mitochondrial genome, its biological function, and the possible links to pathological conditions.
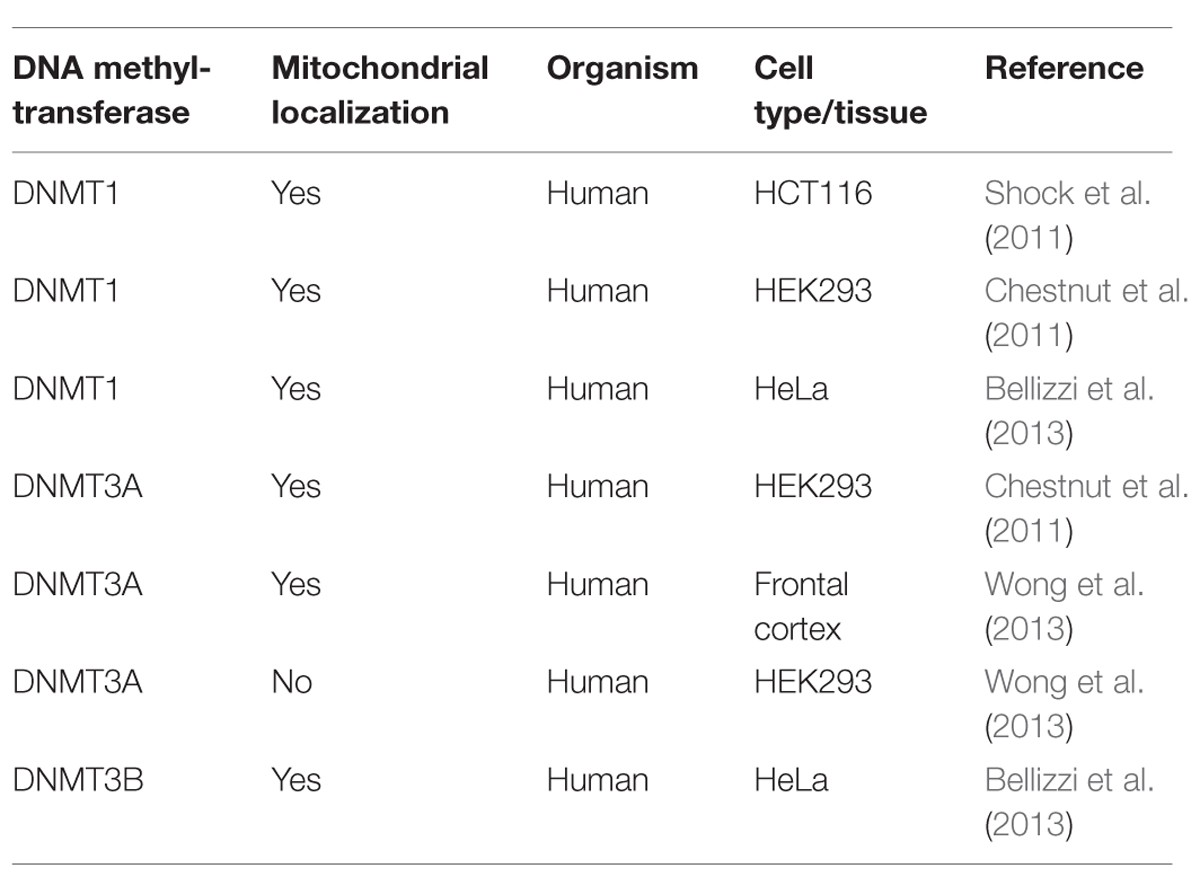
TABLE 1. Mitochondrial localization of DNA (cytosine-5)-methyltransferases – DNMT1, DNMT3A, and DNMT3B, as evaluated by Western blot, in different human cell types and tissues.
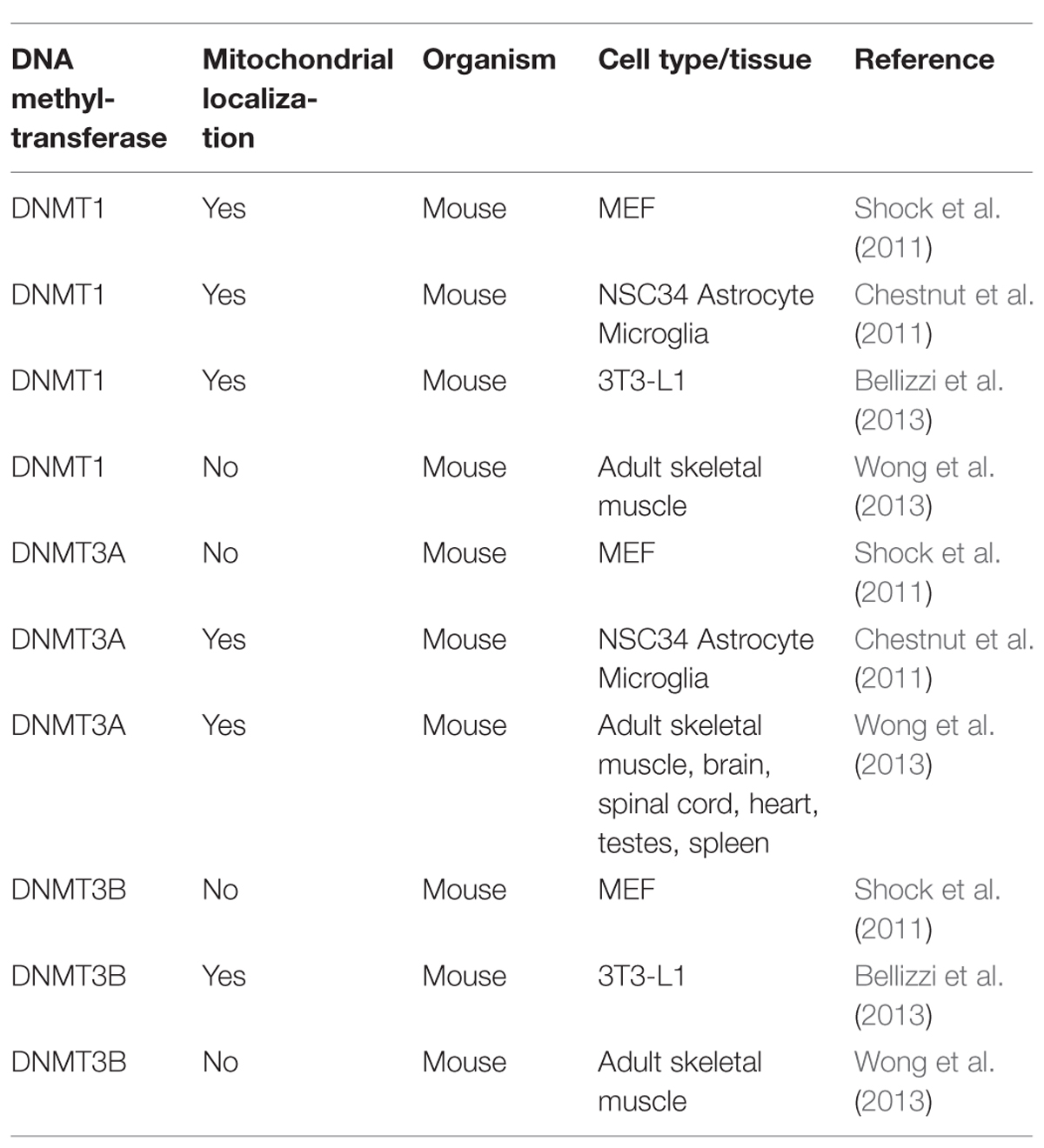
TABLE 2. Mitochondrial localization of DNMT1, DNMT3A, and DNMT3B, as evaluated by Western blot, in different mouse cell types and tissues.
Defective DNA Methylation and Neurological Diseases
Altered DNA methylation is a common hallmark of cancer, and 200, 590, and 320 somatic mutations in DNMT1, DNMT3A, and DNMT3B, respectively, have been reported in the catalog of somatic mutations in cancers (COSMIC). Up and down-regulation of DNMTs has also been observed in different types of cancer (Forbes et al., 2008; Subramaniam et al., 2014; Wu et al., 2014).
In addition to cancer, alterations of the DNA methylation machinery also cause a few neurodegenerative and neurodevelopmental diseases, some of these recently described thanks to whole-exome sequencing (Klein et al., 2011; Winkelmann et al., 2012; Tatton-Brown et al., 2014). Genetic defects may produce an epigenetic deregulation at different levels, affecting both the enzymes responsible for de novo/maintenance methylation (DNMT3/DNMT1), or proteins with a role in the recognition and binding of CpG methylated sites (MBDs, MeCP2; Weissman et al., 2014). As mentioned previously, in Klein et al. (2011) and Winkelmann et al. (2012), two independent exome-sequencing studies revealed mutations in the RFTS domain of DNMT1 gene causing two neurodegenerative disorders with overlapping features: hereditary sensory HSN1E and ADCA-DN. More recently, de novo mutations in DNMT3A gene affecting functional conserved domains of the protein, have been identified by exome-sequencing in patients with overgrowth disorders (Tatton-Brown et al., 2014), resembling clinical features of histone defects and imprinting disorders (Weissman et al., 2014).
Besides primary epigenetic defects, deregulation of DNA methylation may also influence the pathogenesis of other neurodegenerative disorders, like amyotrophic lateral sclerosis (ALS) or Alzheimer disease (AD), Parkinson disease (PD), or neurodevelopmental diseases, such as Down syndrome (DS; Lu et al., 2013). Global DNA hypomethylation in AD has been evidenced in a post-mortem study of monozygotic twins discordant for AD (Mastroeni et al., 2009) and later confirmed by others (Sung et al., 2011; Chouliaras et al., 2013), whereas global hypermethylation has been described in other cases (Bakulski et al., 2012; Rao et al., 2012; Coppieters et al., 2014). In contradiction, Lashley et al. (2014) excluded alterations in global DNA methylation and hydroxymethylation in AD. Hypomethylation of specific genes related to AD (i.e., PSEN1, NEP, BIN1) have also been observed (Lu et al., 2013; Yu et al., 2014). An additional link between DNA methylation and AD came from the association of the chromosome location of DNMT1 (19p13.2) with familial late-onset AD (FLOAD; Wijsman et al., 2004), although sequencing of exon 20 and 21 of DNMT1 in 364 FLOAD cases failed to identify pathogenic mutations in these regions, but only known polymorphisms with expected minor allele frequencies based on European HapMap data (Klein et al., 2013).
Hypomethylation of intron 1 of the α-synuclein (SNCA) gene might be relevant for PD pathogenesis, inducing an increased expression of α-synuclein in the substantia nigra of PD patients and possibly contributing to the Lewy Body formation (Jowaed et al., 2010; Matsumoto et al., 2010; Lu et al., 2013). Moreover, low levels of nuclear DNMT1 in post-mortem brain of PD patients have been observed, indicating a possible link between SNCA hypomethylation and the methylase (Desplats et al., 2011).
DNA methylation seems to have a role also in the pathogenesis of ALS, as proposed by Chestnut et al. (2011), by demonstrating that the levels of DNMT3A, DNMT1, and 5 mC are increased in motor neurons of ALS affected patients. Furthermore, abnormal mtDNA methylation has been found in ALS (Wong et al., 2013) and also in patients affected by Down’s syndrome (Infantino et al., 2011), and for both diseases a mitochondrial dysfunction has been documented (Valenti et al., 2011; Cozzolino et al., 2013).
Based on the mounting evidence of mitochondrial dysfunction in AD and PD, including organelle bioenergetics, dynamics, and quality control (Schon and Przedborski, 2011; Burté et al., 2015), DNA methylation may influence the pathogenesis of these neurodegenerative diseases, acting both on nuclear and mitochondrial DNAs. Moreover, mitochondria participate in the production of the universal methyl donor SAM, through synthesis of ATP and folate, whose deficiency, together with high levels of homocysteine, have been associated with dementia, and neurodegenerative diseases, including AD and PD (Iacobazzi et al., 2013; Ansari et al., 2014). To the best of our knowledge, mtDNA methylation has never been investigated in AD and PD, and neither methylation of nuclear genes encoding for mitochondrial proteins.
ADCA-DN and HSN1E: Defective Methylation Diseases with Mitochondrial Involvement
Hereditary sensory neuropathy with dementia and hearing loss (OMIM 614116), an adult-onset neurodegenerative disorder, is the first Mendelian inherited “methylopathy" identified due to mutations in the DNMT1 gene affecting the RFTS domain (Klein et al., 2011). Shortly after, another adult-onset neurodegenerative disease, ADCA-DN (OMIM 604121), has been associated to mutations in DNMT1 located in the same functional domain (Winkelmann et al., 2012). HSN1E and ADCA-DN have been initially considered as two distinct clinical entities, but more recently the evidence of overlapping clinical features, often subclinical, has emerged, strongly suggesting that they can be better considered as phenotypes belonging to the same neurodegenerative spectrum (Moghadam et al., 2014).
Hereditary sensory neuropathy with dementia and hearing loss is a severe disorder characterized by both central and peripheral nervous system involvement, with peripheral neuropathy leading to extremity injuries and infections, frequently needing amputations, severe early onset hearing loss, and middle-age dementia (Hojo et al., 1999; Klein et al., 2011). By a whole-exome sequencing analysis, two heterozygous mutations in DNMT1 have been identified as the genetic cause of HSN1E in four unrelated families from different geographical area (Klein et al., 2011). A point mutation leading to the p.Tyr511Cys (NP_001124295.1) aminoacid substitution was found in three families, whereas three nucleotide changes causing the substitution of two contiguous aminoacids, p.Asp506Glu-Pro507Arg (NP_001124295.1) were found in the fourth pedigree of this study; both mutations were located in exon 20 and affected the RFTS domain of DNMT1 (Klein et al., 2011). Through an accurate functional investigation, Klein et al. (2011) demonstrated that these mutations cause a premature degradation of the protein, reduced methyltransferase activity, and impaired binding to heterochromatin in G2 phase, leading to global DNA hypomethylation and site-specific hypermethylation.
The abnormal status of DNA methylationinduced by the p.Tyr511Cys mutation has been thoroughly analyzed by whole-genome bisulfite sequencing in three pairs of HSN1E patients compared to gender and age-matched siblings, providing an evaluation of methylation at single base-resolution (Sun et al., 2014). The results showed prevalent hypomethylation in intergenic regions and around the transcription start sites, with the highest reduction of methylation in chromosomes X and 18 (Sun et al., 2014). Furthermore, using the Ingenuity Pathway Analysis, the differentially methylated regions identified were linked to “neurological disease," including progressive neuropathy, PD, AD, ALS, narcolepsy (NC), “psychological disorders," “skeletal and muscular disorders" and “cancer," whereas the most compromised pathway resulted the NAD+/NADH metabolism, which is also implicated in neurodegeneration (Sun et al., 2014).
In 2013 two additional cases of HSN1E with psychiatric manifestations and seizures were identified, again caused by mutations in the exon 20 of DNMT1, one of them previously identified (p.Tyr511Cys), and one previously unreported mutation p.Tyr511His (NP_001124295.1), affecting both the same amino acid residue in the RFTS domain (Klein et al., 2013). Two other HSN1E families has been described in 2014, presenting mutations in exon 20 of DNMT1: one point mutation (p.Pro506Arg, NP_001124295.1) hitting the same amino acid position of another previously reported case (Klein et al., 2011), and a novel trinucleotide deletion (p.Lys521del, NP_001124295.1; Moghadam et al., 2014). The extensive characterization of the HSN1E patients in this latter study showed that subclinical symptoms usually characterizing ADCA-DN, such as NC (without cataplexy and with normal hypocretin-1 level in cerebrospinal fluid) and optic atrophy, may be present in patients affected by HSN1E, highlighting aphenotypic overlap between these two diseases (Moghadam et al., 2014).
Moreover, the strict association between exon 20 and HSN1E has been broken by Yuan et al. (2013) a unique case of HSN1E caused by a novel mutation (p.His569Arg, NP_001124295.1) in exon 21, that is usually associated with ADCA-DN), but still affecting the RFTS domain and further remarking the idea of a continuum.
Autosomal dominant cerebellar ataxia-deafness and narcolepsy was first described by Melberg et al. (1995), but the genetic cause has been identified only recently in Winkelmann et al. (2012). ADCA-DN is initially characterized by late-onset NC with or without cataplexy, complicated in later stages of the disease by sensorineural deafness, cerebellar ataxia, and dementia appear (Melberg et al., 1995). Mild polyneuropathy, optic atrophy, epilepsy, psychosis, diabetes mellitus, cardiomyopathy, and progressive cerebral, cerebellar, and brainstem atrophy may also be present (Melberg et al., 1999; Winkelmann et al., 2012; Moghadam et al., 2014). DNMT1 mutations in exon 21 have been found to cause ADCA-DN (Winkelmann et al., 2012), thus qualifying DNMT1 as the third mutant leading to genetically determined NC (Peyron et al., 2000; Hor et al., 2011). A previous link between DNMT1 and NC emerged from a genome-wide study associating NC with the SNP rs4804122, located in a region of high linkage disequilibrium spanning several genes including DNMT1, and a weak correlation between this NC-associated allele and lower DNMT1 mRNA expression was also documented in peripheral blood mononuclear cells (Kornum et al., 2011).
Initially, three missense mutations have been identified in four different families, all affecting the RFTS domain: p.Ala570Val, p.Val606Phe, p.Gly605Ala (NP_001124295.1). The authors speculated that these mutations might affect the interaction with other proteins, i.e., HDAC2, or the DNA- binding, being close to three phenylalanines critical for the anchoring of the RFTS domain to the DNA-binding pocket (Takeshita et al., 2011; Winkelmann et al., 2012). Concerning the role of mutant DNMT1 in the pathogenesis of NC in ADCA-DN, hypocretin cells, whose loss cause NC, may be particularly susceptible to altered methylation induced by the DNMT1 mutations (Winkelmann et al., 2012). Alternatively, the mutations may impair the regulation and differentiation of immune cells possibly promoting an autoimmune reaction (Winkelmann et al., 2012), thus supporting autoimmunity as the main pathogenic mechanism of NC (Fontana et al., 2010). This hypothesis is reinforced by the critical role played by DNMT1for T-cells development, function and survival (Lee et al., 2001; Wang et al., 2013). However, the HLA-DQB1∗06:02, which represents the major genetic risk factor for NC (Tafti et al., 2014), was negative in all except for two of the ADCA-DN patients with DNMT1 mutations (Winkelmann et al., 2012; Pedroso et al., 2013; Moghadam et al., 2014), weakening the autoimmune hypothesis for this disease. An additional case of ADCA-DN due to a novel missense mutation in exon 21 of DNMT1, p.Cys596Arg (NP_001124295.1), has been reported; this patient displayed the typical, previously described features of the disease (Pedroso et al., 2013).
Interestingly, HSN1E and ADCA-DN present common features resembling mitochondrial encephalomyopathies, such as sensorineural deafness, optic atrophy, cerebellar involvement, and peripheral neuropathy (Moghadam et al., 2014). Furthermore, a mitochondrial dysfunction at the biochemical level was already documented in skeletal muscle of an ADCA-DN patient in Melberg et al. (1995). In addition, the alteration of NAD+/NADH-related pathways that emerged from the methylome study on HSN1E (Sun et al., 2014) also reinforces the possibility of mitochondrial dysfunction in the complex pathogenesis of this disorder.
Furthermore, hypomethylation of nuclear genes with mitochondrial function has been observed in HSN1E patients compared to sex and age-matched controls, although a statistical significance was not reached (Sun et al., 2014).
In conclusion, 10 different mutations in exons 20 and 21 of the DNMT1 gene have been identified to date, six causing HSN1E and four causing ADCA-DN, two distinct diseases now considered as clinical phenotypes of the same disease spectrum (Moghadam et al., 2014). In fact, all the mutations affect the RFTS domain, which has a regulatory function for the DNMT1 activity. In particular, mounting evidence demonstrate that this domain has an auto-inhibitory role (Bashtrykov et al., 2014) and it has been suggested that its deletion activates DNMT1 for euchromatic DNA-binding, but at the same time decreases heterochromatin binding probably through a missing protein interaction, thus producing passive DNA demethylation (Wu et al., 2014). Moreover, the RFTS deletion seems to produce a methylation status typical of cancer, with global hypomethylation and promoter hypermethylation, and additional 26 mutations in this domain have been found in different tumors (Forbes et al., 2008; Wu et al., 2014).
Open questions still have to be addressed. For example, it is unclear whether mutations in exon 20 and 21 of DNMT1 act through the same pathogenic mechanism, and the methylation status of ADCA-DN patients has not been investigated yet. Furthermore, although the altered methylation pattern observed in HSN1E patients resembles that of tumorigenesis (Sun et al., 2014) and loss of function of RFTS domain enhances tumorigenicity (Wu et al., 2014), none of the DNMT1 mutant patients seem to develop cancer. Lastly, the possible role of mitochondrial dysfunction deserves to be unraveled by functional studies, to shed light on the potential role of mito-epigenetics (from both nuclear and mitochondrial genomes) in the pathogenic mechanisms of DNMT1 mutations. Tackling all these open questions will be crucial to understand the pathogenesis of DNMT1 related neurodegeneration, including the narcoleptic features, but ultimately will also help to resolve the question of mtDNA methylation.
Final Remarks
The fascinating field of epigenetic regulation of gene expression is fast evolving, as well as the implications in human pathology. One point highlighted by reviewing the state of art of this field, and prompted by the recent identification of neurodegenerative disorders due to pathogenic mutations in the DNMT1 gene, is the role played by the mitochondrial methylome as a whole, includes obviously the tissue and cell-specific methylation pattern of the nuclear mitochondrial proteome (about 1500–2000 nuclear genes), and the still very controversial existence of mtDNA methylation. This latter longstanding question needs to be resolved, and the possible biological function played by mtDNA methylation in cell physiology may add to the complex inter-genomic dialog between nuclear and mitochondrial DNAs. The rapidly evolving NGS approaches should allow the resolution of the controversies, in conjunction with functional studies, paying attention to the peculiar features of the mitochondrial genome, which include gene organization, transcription, and replication, and most importantly its multicopy nature. The nuclear counterpart represented by the growing list of nuclear genes implicated in mitochondrial biology is equally crucial for understanding the complexities of cancer and neurodegeneration, now fuelled by the existence of an ideal model to investigate, the DNMT1-related human diseases.
References
Akalin, A., Garrett-Bakelman, F. E., Kormaksson, M., Busuttil, J., Zhang, L., Khrebtukova, I.,et al. (2012). Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8:e1002781. doi: 10.1371/journal.pgen.1002781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ansari, R., Mahta, A., Mallack, E., and Luo, J. J. (2014). Hyperhomocysteinemia and neurologic disorders: a review. J. Clin. Neurol. 10, 281–288. doi: 10.3988/jcn.2014.10.4.281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arand, J., Spieler, D., Karius, T., Branco, M. R., Meilinger, D., Meissner, A.,et al. (2012). In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 8:e1002750. doi: 10.1371/journal.pgen.1002750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bakulski, K. M., Dolinoy, D. C., Sartor, M. A., Paulson, H. L., Konen, J. R., Lieberman, A. P.,et al. (2012). Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimers Dis. 29, 571–588. doi: 10.3233/JAD-2012- 111223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bashtrykov, P., Jankevicius, G., Smarandache, A., Jurkowska, R. Z., Ragozin, S., and Jeltsch, A. (2012). Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 19, 572–578. doi: 10.1016/j.chembiol.2012.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bashtrykov, P., Rajavelu, A., Hackner, B., Ragozin, S., Carell, T., and Jeltsch, A. (2014). Targeted mutagenesis results in an activation of DNA methyltransferase 1 and confirms an autoinhibitory role of its RFTS domain. Chembiochem 15, 743–748. doi: 10.1002/cbic.201300740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bellizzi, D., D’Aquila, P., Scafone, T., Giordano, M., Riso, V., Riccio, A.,et al. (2013). The control region of mitochondrial DNA Shows an unusual CpG and non-CpG methylation pattern. DNA Res. 20, 537–547. doi: 10.1093/dnares/dst029
Bestor, T. H. (2000). The DNA methyltransferases of mammals. Hum. Mol. Genet. 9, 2395–2402. doi: 10.1093/hmg/9.16.2395
Bogenhagen, D. F. (2012). Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 1819, 914–920. doi: 10.1016/j.bbagrm.2011.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burté, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Byun, H. M., Panni, T., Motta, V., Hou, L., Nordio, F., Apostoli, P.,et al. (2013). Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 10, 18. doi: 10.1186/1743-8977-10-18
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campbell, C. T., Kolesar, J. E., and Kaufman, B. A. (2012). Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 1819, 921–929. doi: 10.1016/j.bbagrm.2012.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cardon, L. R., Burge, C., Clayton, D. A., and Karlin, S. (1994). Pervasive CpG suppression in animal mitochondrial genomes. Proc. Natl. Acad. Sci. U.S.A. 91, 3799–3803. doi: 10.1073/pnas.91.9.3799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cedar, H., and Bergman, Y. (2011). Epigenetics of haematopoietic cell development. Nat. Rev. Immunol. 11, 478–488. doi: 10.1038/nri2991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chatterjee, R., and Vinson, C. (2012). CpG methylation recruits sequence specific transcription factors essential for tissue specific gene expression. Biochim. Biophys. Acta 1819, 763–770. doi: 10.1016/j.bbagrm.2012.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, R. Z., Pettersson, U., Beard, C., Jackson-Grusby, L., and Jaenisch, R. (1998). DNA hypomethylation leads to elevated mutation rates. Nature 395, 89–93. doi: 10.1038/25779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, T., Hevi, S., Gay, F., Tsujimoto, N., He, T., Zhang, B.,et al. (2007). Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 39, 391–396. doi: 10.1038/ng1982
Chen, T., and Li, E. (2006). Establishment and maintenance of DNA methylation patterns in mammals. Curr. Top Microbiol. Immunol. 301, 179–201. doi: 10.1007/3-540-31390-7_6
Chen, T., Tsujimoto, N., and Li, E. (2004). The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 24, 9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, T., Ueda, Y., Dodge, J. E., Wang, Z., and Li, E. (2003). Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheng, X. (1995). Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct. 24, 293–318. doi: 10.1146/annurev.bb.24.060195.001453
Cheng, X., and Blumenthal, R. M. (2008). Mammalian DNA methyltransferases: a structural perspective. Structure 16, 341–50. doi: 10.1016/j.str.2008.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chestnut, B. A., Chang, Q., Price, A., Lesuisse, C., Wong, M., and Martin, L. J. (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 31, 16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R.,et al. (2013). Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 34, 2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coppieters, N., Dieriks, B. V., Lill, C., Faull, R. L., Curtis, M. A., and Dragunow, M. (2014). Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 35, 1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cozzolino, M., Ferri, A., Valle, C., and Carrì, M. T. (2013). Mitochondria and ALS: implications from novel genes and pathways. Mol. Cell. Neurosci. 55, 44–49. doi: 10.1016/j.mcn.2012.06.001
Dawid, I. B. (1974). 5-methylcytidylic acid: absence from mitochondrial DNA of frogs and HeLa cells. Science 184, 80–81. doi: 10.1126/science.184.4132.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denis, H., Ndlovu, M. N., and Fuks, F. (2011). Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 12, 647–656. doi: 10.1038/embor.2011.11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C.,et al. (2011). Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 286, 9031–9037. doi: 10.1074/jbc.C110.212589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dhayalan, A., Rajavelu, A., Rathert, P., Tamas, R., Jurkowska, R. Z., Ragozin, S.,et al. (2010). The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 285, 26114–26120. doi: 10.1074/jbc.M109.089433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dhe-Paganon, S., Syeda, F., and Park, L. (2011). DNA methyl transferase 1: regulatory mechanisms and implications in health and disease. Int. J. Biochem. Mol. Biol. 2, 58–66.
Ding, F., and Chaillet, J. R. (2002). In vivo stabilization of the Dnmt1 (cytosine-5)-methyltransferase protein. Proc. Natl. Acad. Sci. U.S.A. 99, 14861–14866. doi: 10.1073/pnas.232565599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dodge, J. E., Okano, M., Dick, F., Tsujimoto, N., Chen, T., Wang, S.,et al. (2005). Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 280, 17986–17991. doi: 10.1074/jbc.M413246200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, A., Yoder, J. A., Zhang, X., Zhou, L., Bestor, T. H., and Cheng, X. (2001). Structure of human DNMT2, an enigmatic DNA methyltransferase homolog that displays denaturant-resistant binding to DNA. Nucleic Acids Res. 29, 439–448. doi: 10.1093/nar/29.2.439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dzitoyeva, S., Chen, H., and Manev, H. (2012). Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 33, 2881–2891. doi: 10.1016/j.neurobiolaging.2012.02.006
Easwaran, H. P., Schermelleh, L., Leonhardt, H., and Cardoso, M. C. (2008). Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep. 5, 1181–1186. doi: 10.1038/sj.embor.7400295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Esteller, M. (2008). Epigenetics in cancer. N. Engl. J. Med. 358, 1148–1159. doi: 10.1056/NEJMra072067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farge, G., Mehmedovic, M., Baclayon, M., van den Wildenberg, S. M., Roos, W. H., Gustafsson, C. M.,et al. (2014). In vitro-reconstituted nucleoids can block mitochondrial DNA replication and transcription. Cell Rep. 8, 66–74. doi: 10.1016/j.celrep.2014.05.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fatemi, M., Hermann, A., Gowher, H., and Jeltsch, A. (2002). Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 269, 4981–4984. doi: 10.1046/j.1432-1033.2002.03198.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feinberg, A. P. (2007). Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440. doi: 10.1038/nature05919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fellinger, K., Rothbauer, U., Felle, M., Längst, G., and Leonhardt, H. (2009). Dimerization of DNA methyltransferase 1 is mediated by its regulatory domain. J. Cell. Biochem. 106, 521–528. doi: 10.1002/jcb.22071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fontana, A., Gast, H., Reith, W., Recher, M., Birchler, T., and Bassetti, C. L. (2010). Narcolepsy: autoimmunity, effector T cell activation due to infection, or T cell independent, major histocompatibility complex class II induced neuronal loss? Brain 133, 1300–1311. doi: 10.1093/brain/awq086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forbes, S. A., Bhamra, G., Bamford, S., Dawson, E., Kok, C., Clements, J.,et al. (2008). The catalogue of somatic mutations in cancer (COSMIC). Curr. Protoc. Hum. Genet. 10, 11. doi: 10.1002/0471142905.hg1011s57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghosh, S., Sengupta, S., and Scaria, V. (2014). Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 18, 58–62. doi: 10.1016/j.mito.2014.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder, J. A., Hsieh, C. L., Zhang, X.,et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. doi: 10.1126/science.1120976
Goyal, R., Reinhardt, R., and Jeltsch, A. (2006). Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucl. Acids Res. 34, 1182–1188. doi: 10.1093/nar/gkl002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, J. U., Su, Y., Shin, J. H., Shin, J., Li, H., Xie, B.,et al. (2014). Distribution, recognition, and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222. doi: 10.1038/nn.3607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hata, K., Okano, M., Lei, H., and Li, E. (2002). Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993.
Hermann, A., Goyal, R., and Jeltsch, A. (2004). The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 279, 48350–48359. doi: 10.1074/jbc.M403427200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hermann, A., Schmitt, S., and Jeltsch, A. (2003). The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. J. Biol. Chem. 278, 31717–31721. doi: 10.1074/jbc.M305448200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hojo, K., Imamura, T., Takanashi, M., Ishii, K., Sasaki, M., Imura, S.,et al. (1999). Hereditary sensory neuropathy with deafness and dementia: a clinical and neuroimaging study. Eur. J. Neurol. 6, 357–361. doi: 10.1046/j.1468-1331.1999.630357.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holliday, R., and Pugh, J. E. (1975). DNA modification mechanisms and gene activity during development. Science 187, 226–232. doi: 10.1126/science.1111098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, E. E., Okitsu, C. Y., Smith, A. D., and Hsieh, C. L. (2013). Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell. Biol. 33, 2683–2690. doi: 10.1128/MCB.00220-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hor, H., Bartesaghi, L., Kutalik, Z., Vicário, J. L., de Andrés, C., Pfister, C.,et al. (2011). A missense mutation in myelin oligodendrocyte glycoprotein as a cause of familial narcolepsy with cataplexy. Am. J. Hum. Genet. 89, 474–479. doi: 10.1016/j.ajhg.2011.08.007
Howard, G., Eiges, R., Gaudet, F., Jaenisch, R., and Eden, A. (2008). Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 27, 404–408. doi: 10.1038/sj.onc.1210631
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iacobazzi, V., Castegna, A., Infantino, V., and Andria, G. (2013). Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 110, 25–34. doi: 10.1016/j.ymgme.2013.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iacobuzio-Donahue, C. A. (2009). Epigenetic changes in cancer. Annu. Rev. Pathol. 4, 229–249. doi: 10.1146/annurev.pathol.3.121806.151442
Infantino, V., Castegna, A., Iacobazzi, F., Spera, I., Scala, I., Andria, G.,et al. (2011). Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol. Genet. Metab. 102, 378–382. doi: 10.1016/j.ymgme.2010.11.166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishida, M. I., and Moore, G. E. (2013). The role of imprinted genes in humans. Mol. Aspects Med. 34, 826–840. doi: 10.1016/j.mam.2012.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jakovcevski, M., and Akbarian, S. (2012). Epigenetic mechanisms in neurodevelopmental and neurodegenerative disease. Nat. Med. 18, 1194–1204. doi: 10.1038/nm.2828
Jeltsch, A. (2002). Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 3, 274–293. doi: 10.1002/1439-7633(20020402)3:4274::AID-CBIC2743.0.CO;2-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeltsch, A. (2006). Molecular enzymology of mammalian DNA methyltransferases. Curr. Top Microbiol. Immunol. 301, 203–225. doi: 10.1007/3-540-31390-7_7
Jeltsch, A., and Jurkowska, R. Z. (2014). New concepts in DNA methylation. Trends Biochem. Sci. 39, 310–318. doi: 10.1016/j.tibs.2014.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jia, D., Jurkowska, R. Z., Zhang, X., Jeltsch, A., and Cheng, X. (2007). Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251. doi: 10.1038/nature06146
Jones, P. A., and Baylin, S. B. (2007). The epigenomics of cancer. Cell 128, 683–692. doi: 10.1016/j.cell.2007.01.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, P. A., and Liang, G. (2009). Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 10, 805–811. doi: 10.1038/nrg2651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jowaed, A., Schmitt, I., Kaut, O., and Wüllner, U. (2010). Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 30, 6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jurkowska, R. Z., Jurkowski, T. P., and Jeltsch, A. (2011a). Structure and function of mammalian DNA methyltransferases. Chembiochem 12, 206–222. doi: 10.1002/cbic.201000195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jurkowska, R. Z., Rajavelu, A., Anspach, N., Urbanke, C., Jankevicius, G., Ragozin, S.,et al. (2011b). Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J. Biol. Chem. 286, 24200–24207. doi: 10.1074/jbc.M111.254987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jurkowski, T. P., Meusburger, M., Phalke, S., Helm, M., Nellen, W., Reuter, G.,et al. (2008). Human DNMT2 methylates tRNA(Asp) molecules using a DNA methyltransferase-like catalytic mechanism. RNA 14, 1663–1670. doi: 10.1261/rna.970408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaneda, M., Okano, M., Hata, K., Sado, T., Tsujimoto, N., Li, E.,et al. (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903. doi: 10.1038/nature02633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanherkar, R. R., Bhatia-Dey, N., and Csoka, A. B. (2014a). Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2:49. doi: 10.3389/fcell.2014.00049
Kanherkar, R. R., Bhatia-Dey, N., Makarev, E., and Csoka, A. B. (2014b). Cellular reprogramming for understanding and treating human disease. Front. Cell Dev. Biol. 2:67. doi: 10.3389/fcell.2014.00067
Kar, S., Deb, M., Sengupra, D., Shilpi, A., Parbin, S., Torrisani, J.,et al. (2012). An insight into the various regulatory mechanisms modulating human DNA methyltransferase 1 stability and function. Epigenetics 9, 994–1007. doi: 10.4161/epi.21568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelly, R. D., Mahmud, A., McKenzie, M., Trounce, I. A., and St John, J. C. (2012). Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 40, 10124–10138. doi: 10.1093/nar/gks770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kimura, F., Seifert, H. H., Florl, A. R., Santourlidis, S., Steinhoff, C., Swiatkowski, S.,et al. (2003). Decrease of DNA methyltransferase 1 expression relative to cell proliferation in transitional cell carcinoma. Int. J. Cancer 104, 568–578. doi: 10.1002/ijc.10988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klein, C. J., Bird, T., Ertekin-Taner, N., Lincoln, S., Hjorth, R., Wu Y.,et al. (2013). DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 80, 824–828. doi: 10.1212/WNL.0b013e318284076d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klein, C. J., Botuyan, M. V., Wu, Y., Ward, C. J., Nicholson, G. A., Hammans, S.,et al. (2011). Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 43, 595–600. doi: 10.1038/ng.830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kondilis-Mangum, H. D., and Wade, P. A. (2013). Epigenetics and the adaptive immune response. Mol. Aspects Med. 34, 813–825. doi: 10.1016/j.mam.2012.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kornum, B. R., Kawashima, M., Faraco, J., Lin, L., Rico, T. J., Hesselson, S.,et al. (2011). Common variants in P2RY11 are associated with narcolepsy. Nat. Genet. 43, 66–71. doi: 10.1038/ng.734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krishnakumar, R., and Blelloch, R. H. (2013). Epigenetics of cellular reprogramming. Curr. Opin. Genet. Dev. 23, 548–555. doi: 10.1016/j.gde.2013.06.005
Landan, G., Cohen, N. M., Mukamel, Z., Bar, A., Molchadsky, A., Brosh, R.,et al. (2012). Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 44, 1207–1214. doi: 10.1038/ng.2442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lande-Diner, L., Zhang, J., Ben-Porath, I., Amariglio, N., Keshet, I., Hecht, M.,et al. (2007). Role of DNA methylation in stable gene repression. J. Biol. Chem. 282, 12194–12200. doi: 10.1074/jbc.M607838200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lascaro, D., Castellana, S., Gasparre, G., Romeo, G., Saccone, C., and Attimonelli, M. (2008). The RHNumtS compilation: features and bioinformatics approaches to locate and quantify Human NumtS. BMC Genomics 9:267. doi: 10.1186/1471-2164-9-267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lashley, T., Gami, P., Valizadeh, N., Li, A., Revesz, T., and Balazs, R. (2014). Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. doi: 10.1111/nan.12183 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laurent, L., Wong, E., Li, G., Huynh, T., Tsirigos, A., Ong, C. T.,et al. (2010). Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331. doi: 10.1101/gr.101907.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, P. P., Fitzpatrick, D. R., Beard, C., Jessup, H. K., Lehar, S., Makar, K. W.,et al. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774. doi: 10.1016/S1074-7613(01)00227-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, T., Zhai, J., and Meyers, B. C. (2010). Conservation and divergence in eukaryotic DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 107, 9027–9028. doi: 10.1073/pnas.1005440107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leonhardt, H., Page, A. W., Weier, H. U., and Bestor, T. H. (1992). A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71, 865–873. doi: 10.1016/0092-8674(92)90561-P
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, E., Bestor, T. H., and Jaenisch, R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. doi: 10.1016/0092-8674(92)90611-F
Li, G., Zan, H., Xu, Z., and Casali, P. (2013). Epigenetics of the antibody response. Trends Immunol. 34, 460–470. doi: 10.1016/j.it.2013.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loughery, J. E., Dunne, P. D., O’Neill, K. M., Meehan, R. R., McDaid, J. R., and Walsh, C. P. (2011). DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum. Mol. Genet. 20, 3241–3255. doi: 10.1093/hmg/ddr236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, H., Liu, X., Deng, Y., and Qing, H. (2013). DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 5:85. doi: 10.3389/fnagi.2013.00085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maekawa, M., Taniguchi, T., Higashi, H., Sugimura, H., Sugano, K., and Kanno, T. (2004). Methylation of mitochondrial DNA is not a useful marker for cancer detection. Clin. Chem. 50, 1480–1481. doi: 10.1373/clinchem.2004.035139
Mastroeni, D., McKee, A., Grover, A., Rogers, J., and Coleman, P. D. (2009). Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 4:e6617. doi: 10.1371/journal.pone.0006617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsumoto, L., Takuma, H., Tamaoka, A., Kurisaki, H., Date, H., Tsuji, S.,et al. (2010). CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 5:e15522. doi: 10.1371/journal.pone.0015522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Melberg, A., Dahl, N., Hetta, J., Valind, S., Nennesmo, I., Lundberg, P. O., and Raininko, R. (1999). Neuroimaging study in autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Neurology 53, 2190–2192. doi: 10.1212/WNL.53.9.2190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Melberg, A., Hetta, J., Dahl, N., Nennesmo, I., Bengtsson, M., Wibom, R.,et al. (1995). Autosomal dominant cerebellar ataxia deafness and narcolepsy. J. Neurol. Sci. 134, 119–129. doi: 10.1016/0022-510X(95)00228-0
Moghadam, K. K., Pizza, F., La Morgia, C., Franceschini, C., Tonon, C., Lodi, R.,et al. (2014). Narcolepsy is a common phenotype in HSAN IE and ADCA-DN. Brain 137, 1643–1655. doi: 10.1093/brain/awu069
Mortusewicz, O., Schermelleh, L., Walter, J., Cardoso, M. C., and Leonhardt, H. (2005). Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. U.S.A. 102, 8905–8909. doi: 10.1073/pnas.0501034102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nass, M. M. (1973). Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells. In vivo and in vitro methylation. J. Mol. Biol. 80, 155–175. doi: 10.1016/0022-2836(73)90239-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
Okano, M., Xie, S., and Li, E. (1998). Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 19, 219–220. doi: 10.1038/890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Keefe, R. T., Henderson, S. C., and Spector, D. L. (1992). Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J. Cell. Biol. 116, 1095–1110. doi: 10.1083/jcb.116.5.1095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z.,et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. doi: 10.1038/nature05987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Otani, J., Nankumo, T., Arita, K., Inamoto, S., Ariyoshi, M., and Shirakawa, M. (2009). Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 10, 1235–1241. doi: 10.1038/embor.2009.218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Papp, B., and Plath, K. (2013). Epigenetics of reprogramming to induced pluripotency. Cell 152, 1324–1343. doi: 10.1016/j.cell.2013.02.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pedroso, J. L., Povoas Barsottini, O. G., Lin, L., Melberg, A., Oliveira, A. S., and Mignot, E. (2013). A novel de novo exon 21 DNMT1 mutation causes cerebellar ataxia, deafness, and narcolepsy in a Brazilian patient. Sleep 36, 1257–1259.
Peyron, C., Faraco, J., Rogers, W., Ripley, B., Overeem, S., Charnay, Y.,et al. (2000). A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 6, 991–997. doi: 10.1038/79690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeifer, G. P., Tang, M., and Denissenko, M. F. (2000). Mutation hotspots and DNA methylation. Curr. Top Microbiol. Immunol. 249, 1–19. doi: 10.1007/978-3-642-59696-4_1
Pirola, C. J., Gianotti, T. F., Burgueño, A. L., Rey-Funes, M., Loidl, C. F., Mallardi, P.,et al. (2013). Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 62, 1356–1363. doi: 10.1136/gutjnl-2012-302962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pollack, Y., Kasir, J., Shemer, R., Metzger, S., and Szyf, M. (1984). Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 12, 4811–4824. doi: 10.1093/nar/12.12.4811
Pradhan, M., Estève, P. O., Chin, H. G., Samaranayke, M., Kim, G. D., and Pradhan, S. (2008). CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 47, 10000–100009. doi: 10.1021/bi8011725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qin, W., Leonhardt, H., and Pichler, G. (2011). Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2, 392–402. doi: 10.4161/nucl.2.5.17928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, C., Sawada, K., Zhang, X., and Cheng, X. (2002). The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 9, 217–224.
Qureshi, I. A., and Mehler, M. F. (2013). Epigenetic mechanisms governing the process of neurodegeneration. Mol. Aspects Med. 34, 875–882. doi: 10.1016/j.mam.2012.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rao, J. S., Keleshian, V. L., Klein, S., and Rapoport, S. I. (2012). Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2:e132. doi: 10.1038/tp.2012.55
Rebelo, A. P., Williams, S. L., and Moraes, C. T. (2009). In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 37, 6701–6715. doi: 10.1093/nar/gkp727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reik, W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432. doi: 10.1038/nature05918
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Riggs, A. D. (1975). X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet.14, 9–25. doi: 10.1159/000130315
Rivera, C. M., and Ren, B. (2013). Mapping human epigenomes. Cell 155, 39–55. doi: 10.1016/j.cell.2013.09.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robertson, K. D., Uzvolgyi, E., Liang, G., Talmadge, C., Sumegi, J., Gonzales, F. A.,et al. (1999). The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 27, 2291–2298. doi: 10.1093/nar/27.11.2291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rountree, M. R., Bachman, K. E., and Baylin, S. B. (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277. doi: 10.1038/77023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaefer, M., and Lyko, F. (2010). Solving the Dnmt2 enigma. Chromosoma 119, 35–40. doi: 10.1007/s00412-009-0240-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaefer, M., Steringer, J. P., and Lyko, F. (2008). The Drosophila cytosine-5 methyltransferase Dnmt2 is associated with the nuclear matrix and can access DNA during mitosis. PLoS ONE 3:e1414. doi: 10.1371/journal.pone.0001414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schon, E. A., and Przedborski, S. (2011). Mitochondria: the next (neurode) generation. Neuron 70, 1033–1053. doi: 10.1016/j.neuron.2011.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sha, K. Y. (2008). A mechanistic view of genomic imprinting. Annu. Rev. Genom. Hum. Genet. 9, 197–216. doi: 10.1146/annurev.genom.122007.110031
Shmookler, R. J., and Goldstein, S. (1983). Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J. Biol. Chem. 258, 9078–9085.
Shock, L. S., Thakkar, P. V., Peterson, E. J., Moran, R. G., and Taylor, S. M. (2011). DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 3630–3635. doi: 10.1073/pnas.1012311108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, J., Rechkoblit, O., Bestor, T. H., and Patel, D. J. (2011). Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–1040. doi: 10.1126/science.1195380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Straub, T., and Becker, P. B. (2007). Dosage compensation: the beginning and end of generalization. Nat. Rev. Genet. 8, 47–57. doi: 10.1038/nrg2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Subramaniam, D., Thombre, R., Dhar, A., and Anant, S. (2014). DNA methyltransferases: a novel target for prevention and therapy. Front. Oncol. 4:80. doi: 10.3389/fonc.2014.00080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, Z., Wu, Y., Ordog, T., Baheti, S., Nie, J., Duan, X.,et al. (2014). Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics 9, 1184–1193. doi: 10.4161/epi.29676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sung, H. Y., Choi, E. N., AhnJo, S., Oh, S., and Ahn, J. H. (2011). Amyloid protein mediated differential DNA methylation status regulates gene expression in Alzheimer’s disease model cell line. Biochem. Biophys. Res. Commun. 414, 700–705. doi: 10.1016/j.bbrc.2011.09.136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Syeda, F., Fagan, R. L., Wean, M., Avvakumov, G. V., Walker, J. R., Xue, S.,et al. (2011). The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 286, 15344–15351. doi: 10.1074/jbc.M110.209882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tafti, M., Hor, H., Dauvilliers, Y., Lammers, G. J., Overeem, S., Mayer, G.,et al. (2014). DQB1 locus alone explains most of the risk and protection in narcolepsy with cataplexy in Europe. Sleep 37, 19–25.
Takai, D., and Jones, P. A. (2004). Origins of bidirectional promoters: computational analyses of intergenic distance in the human genome. Mol. Biol. Evol. 21, 463–467. doi: 10.1093/molbev/msh040
Takeshita, K., Suetake, I., Yamashita, E., Suga, M., Narita, H., Nakagawa, A.,et al. (2011). Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. U.S.A. 108, 9055–9059. doi: 10.1073/pnas.1019629108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tatton-Brown, K., Seal, S., Ruark, E., Harmer, J., Ramsay, E., Del Vecchio Duarte, S.,et al. (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 46, 385–388. doi: 10.1038/ng.2917
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valenti, D., Manente, G. A., Moro, L., Marra, E., and Vacca, R. A. (2011). Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. Biochem. J. 435, 679–688. doi: 10.1042/BJ20101908
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vanyushin, B. F., and Kirnos, M. D. (1974). The nucleotide composition and pyrimidine clusters in DNA from beef heart mitochondria. FEBS Lett. 39, 195–199. doi: 10.1016/0014-5793(74)80049-9
Vanyushin, B. F., and Kirnos, M. D. (1977). Structure of animal mitochondrial DNA (base composition, pyrimidine clusters, character of methylation). Biochim. Biophys. Acta 475, 323–336. doi: 10.1016/0005-2787(77)90023-5
Vanyushin, B. F., Kiryanov, G. I., Kudryashova, I. B., and Belozersky, A. N. (1971). DNA-methylase in loach embryos (Misgurnus fossilis). FEBS Lett. 15, 313–316. doi: 10.1016/0014-5793(71)80646-4
Vilkaitis, G., Suetake, I., Klimasauskas, S., and Tajima, S. (2005). Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J. Biol. Chem. 280, 64–72. doi: 10.1074/jbc.M411126200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, L., Liu, Y., Beier, U. H., Han, R., Bhatti, T. R., Akimova, T.,et al. (2013). Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood 121, 3631–3639. doi: 10.1182/blood-2012-08-451765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weemaes, C. M., van Tol, M. J., Wang, J., van Ostaijen-ten Dam, M. M., van Eggermond, M. C., Thijssen, P. E.,et al. (2013). Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur. J. Hum. Genet. 21, 1219–1225. doi: 10.1038/ejhg.2013.40
Weissman, J., Naidu, S., and Bjornsson, H. T. (2014). Abnormalities of the DNA methylation mark and its machinery: an emerging cause of neurologic dysfunction. Semin. Neurol. 34, 249–257. doi: 10.1055/s-0034-1386763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wijsman, E. M., Daw, E. W., Yu, C. E., Payami, H., Steinbart, E. J., Nochlin, D.,et al. (2004). Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am. J. Hum. Genet. 75, 398–409. doi: 10.1086/423393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Winkelmann, J., Lin, L., Schormair, B., Kornum, B. R., Faraco, J., Plazzi, G.,et al. (2012). Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 21, 2205–2210. doi: 10.1093/hmg/dds035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, M., Gertz, B., Chestnut, B. A., and Martin, L. J. (2013). Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell Neurosci. 7:279. doi: 10.3389/fncel.2013.00279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, B. K., Mei, S. C., and Brenner, C. (2014). RFTS-deleted DNMT1 enhances tumorigenicity with focal hypermethylation and global hypomethylation. Cell Cycle 13, 3222–3231. doi: 10.4161/15384101.2014.950886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, H., Zeng, H., Lam, R., Tempel, W., Amaya, M. F., Xu, C.,et al. (2011). Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 6:e18919. doi: 10.1371/journal.pone.0018919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, H., and Zhang, Y. (2014). Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68. doi: 10.1016/j.cell.2013.12.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, G. L., Bestor, T. H., Bourc’his, D., Hsieh, C. L., Tommerup, N., Bugge, M.,et al. (1999). Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191. doi: 10.1038/46214
Yu, L., Chibnik, L. B., Srivastava, G. P., Pochet, N., Yang, J., Xu, J.,et al. (2014). Association of Brain DNA Methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 With Pathological Diagnosis of Alzheimer Disease. JAMA Neurol. 72, 15–24. doi: 10.1001/jamaneurol.2014.3049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, J., Higuchi, Y., Nagado, T., Nozuma, S., Nakamura, T., Matsuura, E.,et al. (2013). Novel mutation in the replication focus targeting sequence domain of DNMT1 causes hereditary sensory and autonomic neuropathy IE. J. Peripher. Nerv. Syst. 18, 89–93. doi: 10.1111/jns5.12012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Jurkowska, R., Soeroes, S., Rajavelu, A., Dhayalan, A., Bock, I.,et al. (2010). Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–53. doi: 10.1093/nar/gkq147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: DNMT1 mutations, mtDNA methylation, ADCA-DN, HSN1E, mitochondrial dysfunction
Citation: Maresca A, Zaffagnini M, Caporali L, Carelli V and Zanna C (2015) DNA methyltransferase 1 mutations and mitochondrial pathology: is mtDNA methylated? Front. Genet. 6:90. doi: 10.3389/fgene.2015.00090
Received: 30 December 2014; Accepted: 19 February 2015;
Published online:12 March 2015.
Edited by:
Babajan Banganapalli, King Abdulaziz University, Saudi ArabiaReviewed by:
Khalid Moghem AL Harbi, Taibah University, Saudi ArabiaXusheng Wang, St. Jude Children’s Research Hospital, USA
Copyright © 2015 Maresca, Zaffagnini, Caporali, Carelli and Zanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Zanna, Department of Pharmacy and Biotechnology, University of Bologna, Via Irnerio 42, 40126 Bologna, Italy claudia.emFubmE3NkBnbWFpbC5jb20=