 Alan M. O’Doherty
Alan M. O’Doherty David E. MacHugh
David E. MacHugh Charles Spillane
Charles Spillane David A. Magee
David A. Magee- 1UCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Ireland
- 2Animal Genomics Laboratory, UCD School of Agriculture and Food Science, University College Dublin, Belfield, Ireland
- 3Genetics and Biotechnology Laboratory, Plant and AgriBiosciences Research Centre, School of Natural Sciences, National University of Ireland Galway, Galway, Ireland
- 4Department of Animal Science, University of Connecticut, Storrs, CT, USA
Monoallelically expressed genes that exert their phenotypic effect in a parent-of-origin specific manner are considered to be subject to genomic imprinting, the most well understood form of epigenetic regulation of gene expression in mammals. The observed differences in allele specific gene expression for imprinted genes are not attributable to differences in DNA sequence information, but to specific chemical modifications of DNA and chromatin proteins. Since the discovery of genomic imprinting some three decades ago, over 100 imprinted mammalian genes have been identified and considerable advances have been made in uncovering the molecular mechanisms regulating imprinted gene expression. While most genomic imprinting studies have focused on mouse models and human biomedical disorders, recent work has highlighted the contributions of imprinted genes to complex trait variation in domestic livestock species. Consequently, greater understanding of genomic imprinting and its effect on agriculturally important traits is predicted to have major implications for the future of animal breeding and husbandry. In this review, we discuss genomic imprinting in mammals with particular emphasis on domestic livestock species and consider how this information can be used in animal breeding research and genetic improvement programs.
Introduction
Mammals are diploid organisms characterized by the presence of complete sets of paternally- and maternally inherited chromosomes in each somatic cell. Normal mammalian development requires that the paternal and maternal copy of each gene (i.e., parental alleles) is expressed correctly, with each copy having the potential to be expressed equally (e.g., to the same level) in each cell. However, a subset of mammalian autosomal genes has been identified where expression is restricted to one of the two parentally inherited chromosomes in a parent-of-origin specific manner; such genes are said to be imprinted. Imprinted genes on autosomal chromosomes can affect both male and female offspring, and such imprinting effects do not arise as a consequence of sex chromosome inheritance. Rather, ‘classically defined’ autosomal imprinting is a consequence of the parental origin of each allele such that, in general, paternally expressed/maternally imprinted genes are transcriptionally silenced on the maternally inherited chromosome only, while maternally expressed/paternally imprinted genes are silenced solely on the paternally inherited chromosome (Barlow and Bartolomei, 2014). Not all imprinted genes adhere to this classic definition; for some genes transcriptional repression of the ‘imprinted’ parental allele is partial (sometimes termed ‘preferential’ or ‘allele-specific’ gene expression) wherein one allele displays higher levels of expression relative to the other allele in a parent-of-origin manner, while other genes display tissue-and/or temporal-specific imprinting or imprinting patterns that differ between individuals of the same species (Giannoukakis et al., 1996; Prickett and Oakey, 2012).
Importantly, mammalian genes displaying genomic imprinting are distinguishable from genes that display apparent parental-specific expression due to unequal or unique genetic contributions from male and female parents such as the expression of Y-linked genes in XY males, the expression of maternally derived mitochondrial genes, and the expression of X-linked genes that evade the process of X-chromosome inactivation in XX females. X-chromosome inactivation, in particular, has been extensively studied in mammals since it was first described by Lyon (1961). During early female embryonic development, one of the two X-chromosomes is randomly inactivated to equalize the X-linked gene dosage difference between XX females and XY males. This process, called ‘random X-inactivation’, involves the decoration of one X-chromosome with a non-protein coding RNA (termed XIST), which initiates the chromosome-wide gene silencing of the X-chromosome from which the XIST transcript is derived. Interestingly, preferential inactivation of the paternally derived X-chromosome involving XIST transcripts has been reported in the placental tissue of XX female mammals, a process known as ‘imprinted X inactivation’ (Chow and Heard, 2010; Lee and Bartolomei, 2013).
Genomic imprinting was first described ~30 years ago through pronuclear transplantation experiments (Barton et al., 1984; Surani et al., 1984; Cattanach and Kirk, 1985). This work demonstrated that normal murine embryo development requires genetic contributions from both the maternally and paternally inherited haploid genomes. Diploid mouse embryos reconstructed from two maternal or paternal pronuclei with no genetic contributions from paternal or maternal sources (i.e., gynogenetic and androgenetic embryos, respectively) failed to survive. It was hypothesized that a subset of murine genes, expressed solely from the maternal- or paternal-derived haploid genomes, was necessary for normal embryonic growth and development and that these genes carry specific epigenetic marks or ‘imprints’ that control this parent-of-origin, monoallelic expression (Barton et al., 1984; Surani et al., 1984; Cattanach and Kirk, 1985). Gynogenetic and androgenetic embryos have also been generated for cattle, sheep and pigs with results revealing arrested fetal development and lethality, due to aberrant genomic imprinting patterns (Fukui et al., 1992; Lagutina et al., 2004; Zacchini et al., 2011; Sembon et al., 2012).
To date, 132 murine and 79 human imprinted genes (including protein-coding and regulatory non-coding RNA genes) have been documented; however, only 25, 21, and 14 experimentally validated imprinted genes/loci have been reported for cattle, pigs and sheep, respectively (Morison et al., 2001; Jirtle, 2013; Wei et al., 2014). Initial evolutionary studies suggested that imprinted genes were largely conserved across mammalian species (Morison et al., 2005); however, more recent studies have shown that conservation of imprinted genes between primates and rodents is more limited than initially thought (Monk et al., 2006; Khatib et al., 2007). For example, of the 79 imprinted human genes reported in the MetaImprint database (Wei et al., 2014) only 40 of these (51%) are among the 132 imprinted genes reported for mice. Despite this limited conservation, imprinted genes have been shown to share a number of defining features among mammals. For instance, functional analyses have shown that many imprinted genes encode products that regulate a wide range of biological processes—most notably, embryonic and neonatal growth and development, metabolism and behavior—in all mammalian species studied to-date (Plasschaert and Bartolomei, 2014; Tian, 2014). Furthermore, while some imprinted genes map as singletons or as gene pairs, many imprinted genes are organized into clusters (~1 Mb) in which both maternally- and paternally imprinted genes (including protein- and RNA-coding genes) reside and whose expression is regulated by a discrete region [termed ‘the imprinting control region’ (ICR)] located within the clusters (Barlow and Bartolomei, 2014).
The important regulatory roles of ICRs has been highlighted in human biomedical studies, whereby epigenetic or genetic alterations (e.g., DNA sequence changes, deletion of an ICR, loss or gain of an imprint) at these sites result in dysregulated expression of reciprocally imprinted genes leading to developmental disorders (Edwards and Ferguson-Smith, 2007). In cattle, deletion of a 110 kb region proximal to the ICR regulating the expression of the paternally expressed/maternally imprinted PEG3 domain was recently shown to result in the loss of paternal MIMT1 expression in the brain and cotyledon of all carrier fetuses. This mutation is thought to be responsible for late fetal mortality and stillbirth in 85% of the offspring inheriting the causative mutation from the founding sire; it has been postulated that the remaining 15% of progeny inheriting the mutation survive due to incomplete silencing of the maternally inherited MIMT1 allele (Flisikowski et al., 2010, 2012).
The co-localization of imprinted genes has resulted from the processes by which these loci are hypothesized to have evolved. The most credible explanation with significant supporting evidence is the ‘conflict theory’ of genomic imprinting, which states paternally expressed imprinted genes (e.g., IGF2) have evolved to actively promote fetal growth and development, thereby maximizing maternal resources to offspring bearing a particular paternal genome during gestation. In contrast, maternally expressed imprinted genes (e.g., IGF2R) have evolved to suppress fetal growth, thereby causing a more uniform distribution of maternal resources to all offspring carrying a particular maternal genome, despite possessing different paternal genomes (Moore and Haig, 1991; Ashbrook and Hager, 2013).
Genomic Imprinting is a Form of Epigenetic Regulation
Genomic imprinting is an epigenetic mechanism of gene expression regulation, whereby alterations in gene expression do not involve any changes to underlying DNA sequences. Epigenetic regulation is largely characterized by the regional addition or removal of a chemical imprint to either genomic DNA (e.g., DNA methylation) and/or chromatin-associated proteins (e.g., histone acetylation, methylation, ubiquintination). Such epigenetic “imprints” can serve to mediate the local expression of genes, either through transcriptional activation, transcriptional attenuation or complete transcriptional silencing. In mammals, parent-of-origin-specific expression due to genomic imprinting is reliant on the existence of epigenetic differences between the two parental alleles resulting in their differential expression in the same nucleus (Hanna and Kelsey, 2014; Weaver and Bartolomei, 2014).
Genomic imprinting involves the establishment of differential imprints on chromosomes inherited either via the male or the female germ lines during meiosis according to their parent-of-origin. Importantly, such differential imprints are reversible, whereby an imprint established on a chromosome inherited via the female germline will not be established when the same chromosome is inherited via the male germline (or vice versa) in the subsequent generation. These parent-of-origin imprints can be then inherited by all daughter cells through mitosis following fertilization, potentially resulting in imprinted gene expression patterns throughout the lifespan of the animal (Abramowitz and Bartolomei, 2012).
For epigenetic regulation of the imprinted status of genes, the epigenetic imprint must exhibit four major mechanistic attributes: firstly, the imprint must be able to regulate gene product levels; secondly, the imprint must be stably inherited in somatic cells such that the ‘memory’ of parental origin is faithfully transmitted to daughter cells during mitosis; thirdly, the imprint is established independently on either the paternal or maternal genomes when they are not present in the same nucleus (e.g., during meiosis); and fourthly, the imprint must be erased and reset in the germ line such that appropriate parent-of-origin identity is established in the gametes for the subsequent generation (Bartolomei, 2009).
Although many diverse biomolecular mechanisms are now classified as epigenetic (e.g., histone tail modifications and expression of small and long non-coding RNAs), the most extensively studied epigenetic marks associated with genomic imprinting is DNA methylation (Kelsey and Feil, 2013; Plasschaert and Bartolomei, 2014). In mammals, DNA methylation involves the addition of a methyl group (-CH3) by DNA methyltransferase enzymes to the 5′ carbon of cytosine residues that exist primarily in CpG dinucleotides [i.e., cytosine-phosphate-guanine residues that lie adjacent to each other on the same DNA strand] (Bird, 2007). Cytosine methylation at CpG dinucleotides has been shown to be associated with imprinted gene regulation, particularly at genomic regions where CpGs located in promoter-associated and non-promoter-associated ICRs display differential methylation patterns on both the maternally and paternally inherited chromosomes [i.e., differentially methylated regions (DMRs; Henckel et al., 2009; Ito et al., 2013)].
DNA methylation is widely considered as a repressive gene expression mechanism that regulates imprinted gene expression by promoting chromatin condensation, rendering the DNA less accessible to the cell’s transcriptional machinery (Figure 1). Thus, silenced or repressed gene expression is generally observed from the hypermethylated DMR (Hanna and Kelsey, 2014). For example, a recent survey of the allelic methylation profile of human genes in placental tissue revealed that for a panel of known paternally expressed imprinted genes the promoters are methylated on the maternal allele, with corresponding allele-specific expression from the paternal allele (Court et al., 2014). In addition to its classical role in repression of gene expression there is a growing body of evidence demonstrating that DNA methylation, particularly within intragenic regions, may be involved with promoting transcription (Neri et al., 2013; Irwin et al., 2014).
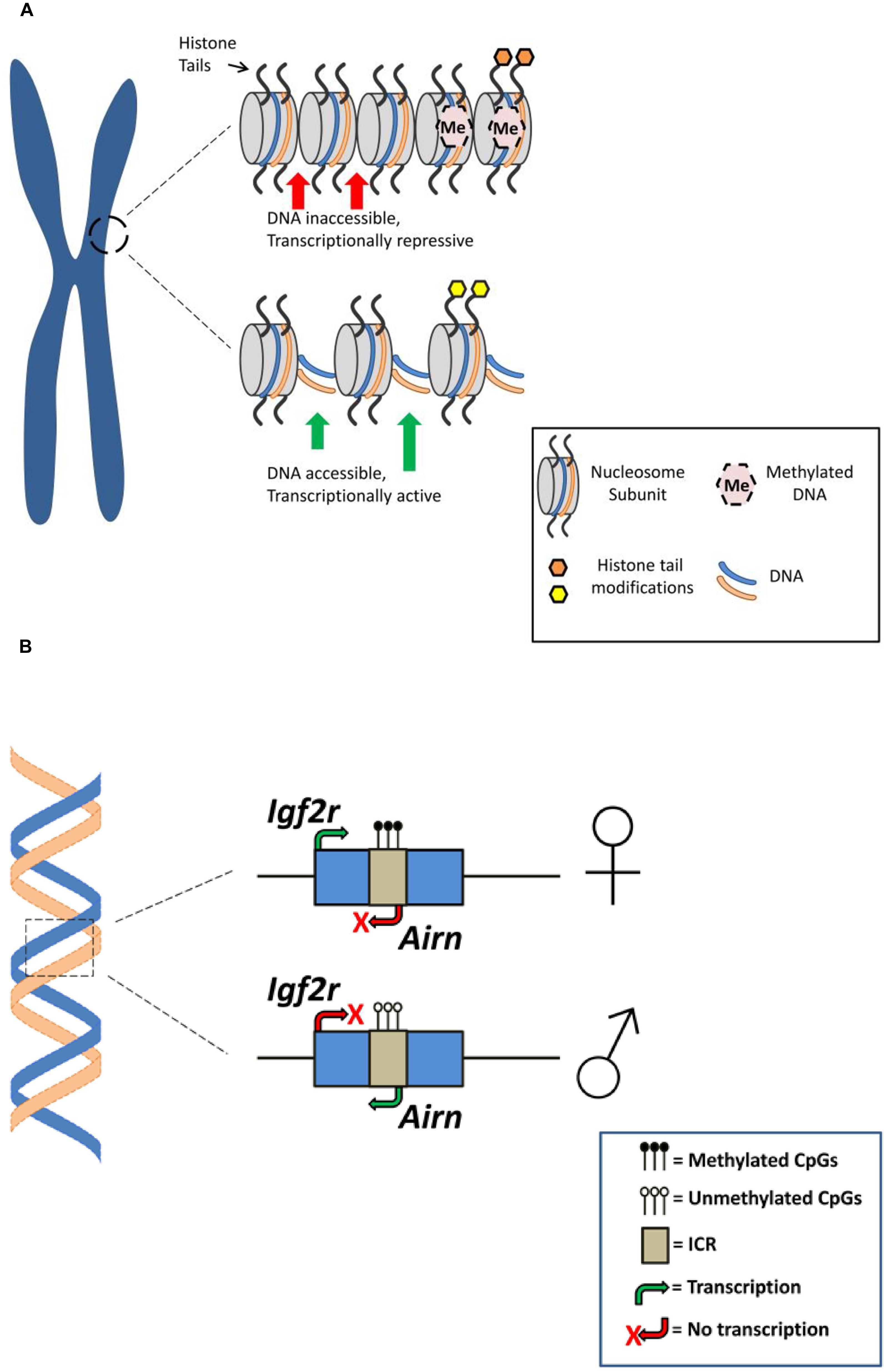
FIGURE 1. Epigenetic mechanisms associated with genomic imprinting. (A) Histone modifications and DNA methylation for different chromatin configurations. Top: Repressive chromatin state associated with histone modification (e.g., histone methylation; orange shading) and dense DNA methylation resulting in gene silencing or attenuated gene expression. Bottom: Active/permissive chromatin state associated with histone modification (e.g., histone acetylation; yellow shading) and reduced DNA methylation rendering DNA accessible for transcription resulting in gene expression (for a comprehensive overview of histone modifications see Bannister and Kouzarides, 2011). (B) Genomic arrangement at an imprinted gene. A simplified schematic of the murine Igf2r locus demonstrating parent-of-origin specific DNA methylation is presented. The imprinting control region (ICR) on the maternal Igf2r allele is methylated, preventing expression of an antisense ncRNA (Airn) and resulting in expression of the maternal Igf2r allele. Alternatively, expression of Airn from the unmethylated paternal allele attenuates paternal Igf2r expression (for a more comprehensive overview of DNA methylation at the Igf2r locus and genomic imprinting see Autuoro et al., 2014).
Post-translational modifications of histone proteins are also recognized as an important epigenetic regulatory mechanism associated with mammalian imprinted genes (Figure 1). In eukaryotic cell nuclei, DNA is tightly packed into chromatin such that the DNA double-helix is wrapped around the histone octameric core to form the basic chromatin unit, the nucleosome. The N-terminal regions of histone proteins that protrude from the nucleosome can undergo various post-translational modifications (e.g., methylation, acetylation, ubiquitination ,and phosphorylation) that can regulate gene expression (Weaver and Bartolomei, 2014). For example, acetylation and methylation of histone lysine residues—typically associated with transcriptionally active and repressed chromatin, respectively—has been associated with several murine imprinted genes including the linked and reciprocally-imprinted H19 and Igf2 genes and the genes located in the Kcnq1 imprinted cluster on chromosome 7 (Pedone et al., 1999; Wagschal et al., 2008; Ciccone et al., 2009).
RNA-mediated gene expression regulation is an additional epigenetic mechanism that is pertinent to understanding the regulation of imprinted gene expression. Epigenetic regulation by long non-coding RNAs (ncRNAs) is well established for X-chromosome inactivation in female mammals (Briggs and Reijo Pera, 2014). Long ncRNAs have also been implicated in the regulation of imprinted loci in mammals, in conjunction with other molecular mechanisms such as insulators, DNA methylation, and histone modifications (Barlow and Bartolomei, 2014). In contrast to trans-acting RNA interference (RNAi)-mediated repression of gene expression, macro ncRNAs, (which are often 100s of kb in length) can elicit cis-regulatory effects on gene expression, and thereby can generate allele-specific imprinting effects on gene expression (Koerner et al., 2009).
In general, studies of imprinted gene regulation and imprinted gene clusters are revealing a complex interplay between DNA methylation, histone modifications, higher-order chromatin structure, RNA-mediated epigenetic effects and transcription, which are all involved in the establishment of the primary genomic “imprint” (Koerner et al., 2009; Adalsteinsson and Ferguson-Smith, 2014).
Epigenetic Dynamics During Mammalian Gametogenesis and Early Development
DNA methylation provides an example of an epigenetic mark that is highly dynamic and that can undergo spatio-temporal changes across cells, tissues and generations (Schneider et al., 2010). Much of what is known regarding DNA methylation dynamics during development comes from studies in mice. Dramatic genome-wide changes in DNA methylation occur during gametogenesis and the early stages of embryo development (Reik and Walter, 2001; Messerschmidt et al., 2014). Primordial germ cells (PGCs) are almost completely ‘erased’ of DNA methylation marks upon entry into the genital ridge (Hajkova et al., 2002; Seisenberger et al., 2012), with some single-copy loci and transposable elements, such as intracisternal A-particles (IAP) and certain endogenous retroviral-derived sequences, retaining moderate levels of methylation (Lane et al., 2003; Guibert et al., 2012). Following this, gamete-specific methylated regions are established during spermatogenesis and oogenesis and these patterns substantially differ depending on which germline they occur. Such methylation differences are most noticeable at imprinted loci whereby specific genomic regions become asymmetrically methylated in sperm and oocytes. In the male germline, imprinted genes can acquire their gamete-specific DNA methylation marks in fetal prospermatagonia prior to birth (Davis et al., 2000; Li et al., 2004). This period of de novo methylation has also been shown to coincide with global changes in histone tail modifications, which are not observed in female germ cells during this period of development (Yoshioka et al., 2009; Singh et al., 2013). Maternal-specific DNA methylation at imprinted genes is acquired in the postnatal growing oocyte (Hiura et al., 2006; O’Doherty et al., 2012).
Following fertilization there is a global cascade of DNA demethylation during the early stages of embryogenesis, whereby the paternal genome is rapidly demethylated in the zygote and the maternal genome is passively demethylated in a replication-dependent manner (Dean et al., 2001; Yang et al., 2007; Iqbal et al., 2011). More recently, it has been hypothesized that both the maternal and paternal genomes undergo global active demethylation and replication-mediated passive demethylation (Gkountela and Clark, 2014; Guo et al., 2014). Irrespective of the mechanisms controlling these genome-wide reprogramming events in the pre-implantation embryo, DNA methylation at imprinted genes is generally considered as being stable until they undergo reprogramming in PGCs (Olek and Walter, 1997; Imamura et al., 2005; Smallwood et al., 2011). However, a study analyzing imprinted DMRs in mouse blastocysts revealed dynamic changes in allelic methylation, suggesting that DMRs are not fully protected from the major reprogramming events in the early embryo (Tomizawa et al., 2011).
Epigenetic Programming and Imprinted Disorders in Domestic Livestock Species
In domesticated species, the importance of establishing appropriate epigenetic marks at imprinted loci has been highlighted largely through assisted reproductive technologies (ART) including somatic cell nuclear transfer (SCNT) cloning studies. ART involves the isolation, handling, manipulation and culture of gametes, and early embryos, usually after hormonal stimulation. As discussed above, major epigenetic reprogramming events occur during gametogenesis and early embryonic development and it has been proposed that ART exposes the epigenome to external factors that may interfere with the correct establishment and maintenance of genome imprints. For example, superovulation, embryo culturing and cryopreservation can affect methylation profiles and gene expression at imprinted loci (Humpherys et al., 2001; DeBaun et al., 2003; Gicquel et al., 2003; Kang et al., 2003; Chang et al., 2005; Ludwig et al., 2005; Sato et al., 2007). Epigenetic perturbations, associated with ART and SCNT, may contribute to developmental issues such as increased abortion rate, perinatal death, enlarged placentomes, enlarged umbilical cords, high-birth weight and large offspring syndrome (LOS; Campbell et al., 1996; Cibelli et al., 1998; Kang et al., 2003; Alexopoulos et al., 2008; Smith et al., 2012).
Another example of an epigenetic-associated developmental disorder is LOS. LOS is an overgrowth disorder in domesticated ruminants bearing phenotypic similarities to Beckwith–Wiedemann syndrome (BWS, an overgrowth disorder in humans), and is characterized by excessive birth weight, enlarged tongue, umbilical hernia, enlarged internal organs and hypoglycemia (Young et al., 1998; Weksberg et al., 2010). Both BWS and LOS can occur naturally; however, there is evidence that these disorders have an increased incidence in individuals generated from ART (Chang et al., 2005).
Previous work has shown that epigenetic changes (also referred to as ‘epimutations’) at two ICRs, that independently regulate the expression of two clusters of reciprocally imprinted genes on human chromosome 11p15, are associated with BWS (Choufani et al., 2010). One imprinting cluster contains the maternally expressed/paternally imprinted ncRNA H19 gene and the paternally expressed/maternally imprinted IGF2 gene, which encodes a fetal mitogen. Studies have shown that both genes are under the control of a single ICR that is unmethylated on the maternal allele and methylated on the paternal allele. In mice, binding of the CCCTC-binding factor (zinc finger protein), CTCF, to the non-methylated ICR inhibits maternal expression of Igf2 by preventing interaction of its promoter with downstream enhancers; however, the H19 promoter has access to the downstream promoters resulting in its maternal expression (Hark et al., 2000; Demars et al., 2010; Poole et al., 2012). The second cluster contains a paternally expressed ncRNA gene, Kcnq1ot1, and several maternally expressed protein-coding genes associated with regulating growth and development, such as Cdkn1c, Kcnq1, and Phlda2. Expression of the genes in this cluster is controlled by a single ICR known as KvDMR1, which is hypomethylated on the paternal copy (Fitzpatrick et al., 2002; Choufani et al., 2010). Paternal expression of Kcnq1ot1 recruits the binding of Polycomb group proteins and initiates histone-tail methylation, which induces a transcriptionally repressive chromatin structure leading to silencing of the protein-coding genes from this locus on the paternal chromosome. Conversely, methylation of the KvDMR1 on the maternal allele prevents Kcnq1ot1 transcription, thus, enabling the protein-coding genes to be expressed from the maternal allele (Fitzpatrick et al., 2002; Pandey et al., 2008; Terranova et al., 2008; Redrup et al., 2009; Choufani et al., 2010). In humans, gain-of-methylation epimutations at the maternal IGF2/H19 ICR, resulting in increased expression of IGF2, can account for 2–7% of all BWS cases, while 50% of cases are due to loss-of-methylation epimutations at the maternal ICR (known as KvDMR1), which is concomitant with biallelic expression of KCNQ1OT1 and downregulation of CDKN1C, a negative regulator of cell proliferation (Weksberg et al., 2001, 2010).
Similarly, studies in ruminants have revealed associations between aberrant methylation at the H19-IGF2 and the KCNQ1OT1-CDKN1C loci and ART-generated fetuses, especially in offspring displaying LOS or which had died shortly after birth (Young et al., 1998; Hiendleder et al., 2004; Farin et al., 2006). For example, investigation of the DNA methylation status within the bovine IGF2-H19 ICR revealed hypomethylation in several cloned animals relative to control animals, which correlated with biallelic expression of H19 in the liver and placenta of these animals (Curchoe et al., 2009). Biallelic expression of bovine IGF2 has also been observed in the brain and spleen tissue of ART-generated animals displaying LOS (Chen et al., 2013). Loss of maternal KvDMR1 methylation has also been associated with biallelic expression of KCNQ1OT1 and reduced expression of CDKN1C in LOS bovine calves and fetuses, suggesting similarities in the epigenetic mechanisms that underlie both BWS and LOS (Hori et al., 2010; Chen et al., 2013). Furthermore, Young et al. (2001) also demonstrated that sheep fetuses displaying LOS has reduced maternal IGF2R mRNA and protein levels relative to control fetuses, which was correlated with a loss of methylation at the IGF2R ICR on the maternally active allele. In mice, the Igf2r ICR contains an antisense ncRNA, Airn, which when expressed from the unmethylated paternal allele attenuates paternal Igf2r expression (Latos et al., 2009, 2012). Thus, for LOS sheep it is conceivable that loss of IGF2R ICR methylation on the maternal chromosome results in increased transcriptional activity from the AIRN promoter leading to a corresponding reduction in IGF2R mRNA and protein expression (Bartolomei, 2009). Also, the bovine IGF2R gene has also been shown to have a maternally methylated DMR located in the second intron (O’Doherty et al., 2012), which displays reduced methylation in both ART- and SCNT-derived samples relative to in vivo samples (Smith et al., 2012); it is possible that this locus, and expression of the ncRNA AIRN, may be disrupted in bovine LOS.
The Complex Interplay between Genetic and Epigenetic Mechanisms in Regulating Gene Expression: the Callipyge Phenotype in Sheep
In the context of genomic imprinting, individual epigenetic regulatory mechanisms do not function independently. Rather, multiple mechanisms tend to work in concert to define the functional states of chromatin that are associated with the regulation of imprinted gene expression (Jones et al., 1998). For example, ICRs displaying differentially methylated DNA regions are often also associated with transcriptionally repressive histone modifications such as methylated lysine residues resulting in chromatin condensation and silenced or repressed gene expression (Yang et al., 2003; Delaval et al., 2007; Meissner et al., 2008). Indeed, data from studies in mouse have led to the proposal that DNA methylation recruits repressive histone modifications at ICRs, thereby suggesting a positive feedback loop for the establishment and maintenance of parental imprints during development (Henckel et al., 2009).
The complex interplay between different epigenetic and genetic mechanisms in regulating mammalian imprinted gene expression is aptly illustrated by the callipyge phenotype in sheep, which is responsible for a ~30% increase in skeletal muscle (most notably at the hindquarters), a corresponding ~8% reduction in fat content and improved feed efficiency (Cockett et al., 1996). This phenotype is observed only in heterozygous individuals that carry the causative mutation on the paternal chromosome (i.e., mat+/patC, where ‘mat’ and ‘pat’ denote maternal and paternal chromosomes, respectively and superscript ‘+’ and ‘C’ represent wild-type and callipyge alleles, respectively)—a mode of non-Mendelian inheritance termed ‘polar overdominance’ (Cockett et al., 1996). The callipyge phenotype is caused by an A-to-G single nucleotide polymorphism (SNP; i.e., the callipyge mutation) located between the paternally expressed/maternally imprinted DLK1 protein-coding gene and the maternally expressed/paternally imprinted MEG3 long non-coding RNA (ncRNA) gene within the imprinted DLK1-DIO3 gene cluster on ovine chromosome 18 (Freking et al., 2002; Smit et al., 2003). This cluster also contains additional paternally expressed/maternally imprinted protein-coding genes such as PEG11, and several maternally expressed/paternally imprinted long ncRNA and microRNA (miRNA) genes (including MEG3, PEG11AS, MEG8, and MIRG (also referred to as MEG9; Freking et al., 2002; Smit et al., 2003; Hagan et al., 2009; Figure 2).
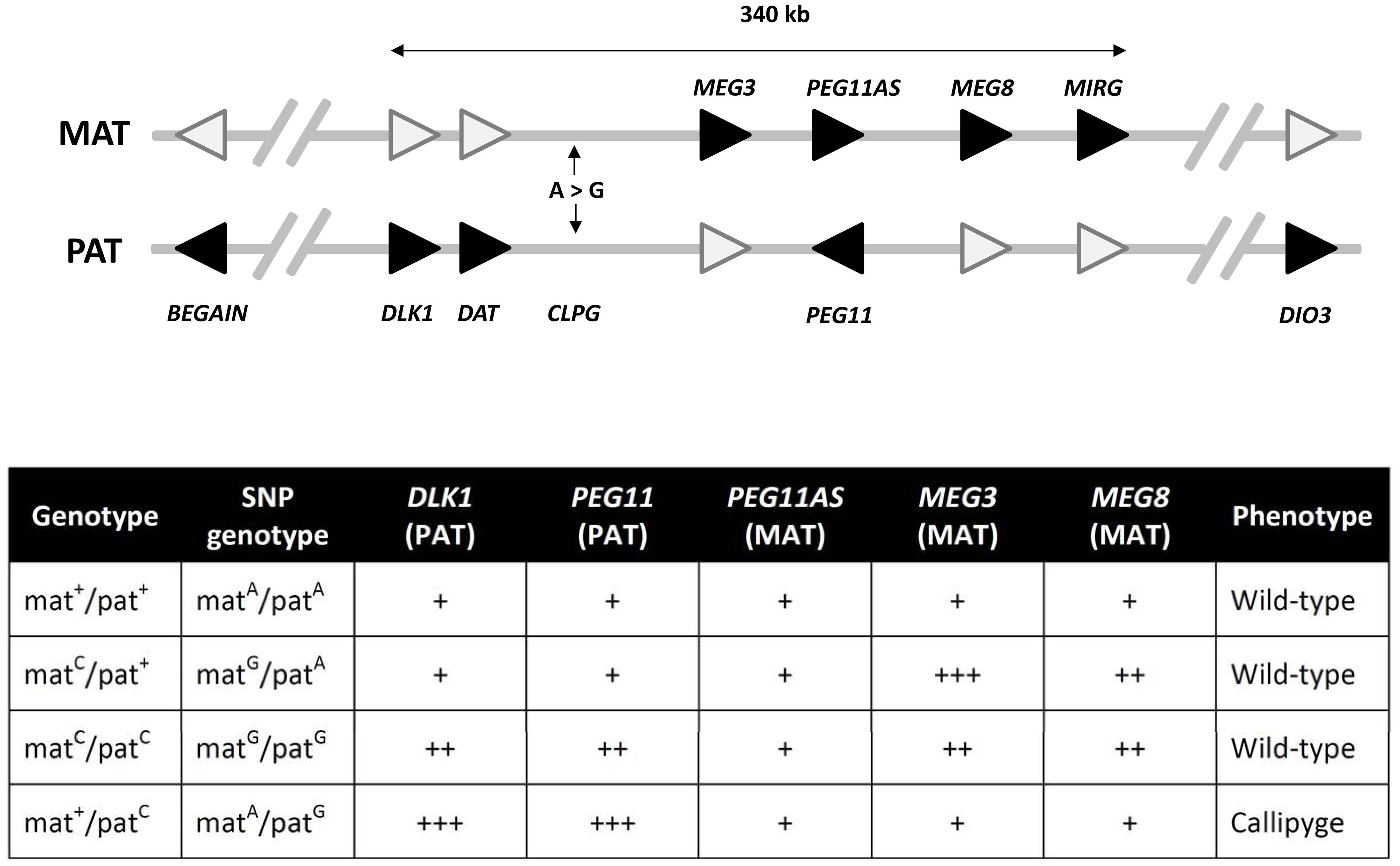
FIGURE 2. The DLK1-DIO3 imprinting domain on ovine chromosome 18. This domain contains the genes whose expression is perturbed upon inheritance of the callipyge mutation (CLPG; an A-to-G SNP). The genes shaded in black represent the expressed imprinted alleles within this domain while white shading indicates the silenced/attenuated imprinted allele on either the maternal (MAT) or paternal (PAT) chromosomes. The arrowhead denotes the direction of transcription of each gene. Genes are not drawn to scale and introns are not shown. The core imprinted genes that have been shown to play a role in the callipyge phenotype occur within a 340 kb region. The expression of the core genes for each of the four possible callipyge genotypes at the CLPG SNP and the observed is summarized in the accompanying table. The relative RNA transcript abundance for the paternally (DLK1, PEG11) and maternally (PEG11AS, MEG3, MEG8, and MIRG) expressed genes are shown (not to scale) for each callipyge genotype. Callipyge animals (mat+/patC) exhibit overexpression of DLK1 and PEG11 and an absence of MEG3 and MEG8 overexpression suggesting that DLK1 and/or PEG11 encodes the primary effector of the callipyge phenotype. Overexpression of the maternal non-coding RNA genes and the absence of muscle hypertophy in matC/patC animals suggest that these transcripts exert their effect via post-transcriptional suppression of the effector. The microRNAs encoded by MIRG have been postulated to also play a role in post-transcriptional suppression of the paternally expressed effector (Georges et al., 2003; Bidwell et al., 2004, 2014; Murphy et al., 2006).
Callipyge individuals (i.e., mat+/patC) display overexpression of the paternally expressed DLK1 and PEG11 protein-coding transcripts in skeletal muscle tissue relative to non-callipyge animals (i.e., mat+/pat+; matC/pat+; matC/patC). In contrast, individuals that inherit the callipyge mutation on the maternal chromosome (i.e., matC/patC or matC/pat+) display upregulation of maternal long ncRNAs and miRNAs in cis relative to wild-type (i.e., mat+/pat+) and callipyge animals (Murphy et al., 2006). The callipyge mutation also causes a muscle tissue-specific reduction of methylation at CpG sites distributed across the DLK1-DIO3 imprinted cluster resulting in increased transcriptional activity from the parental chromosome carrying the mutation (Takeda et al., 2006). Downregulated expression of the histone deacetylase 9 (HDAC9) gene—the encoded product of which removes acetyl groups from histone proteins resulting in increased chromatin condensation and repressed transcription—has also been observed in callipyge animals relative to non-callipyge animals suggesting a role for histone modification in regulation of expression at the DLK1-DIO3 imprinted cluster (Vuocolo et al., 2007). Consequently, the callipyge mutation modifies the chromatin structure of the chromosome on which it is carried, such that the DNA surrounding the callipyge mutation is more permissive for transcription (Murphy et al., 2006).
A recently refined model of the polar overdominance observed for the callipyge phenotype suggests that DLK1 and/or PEG11 are likely to be the primary effectors of the callipyge phenotype—it is possible that the encoded products of DLK1 and PEG11 may act synergistically (Georges et al., 2003; Bidwell et al., 2004, 2014). Inheritance of the callipyge mutation on the paternal chromosome results in chromatin relaxation in the vicinity of the DLK1-DIO3 imprinted cluster leading to increased DLK1 and/or PEG11 expression, which induces the hypertrophy response. Conversely, bi-parental inheritance or maternal inheritance of the callipyge mutation results in the upregulation of the DLK1-DIO3 maternally expressed/paternally imprinted ncRNA and miRNA genes relative to callipyge and wild-type animals. This leads to repression of the phenotypic effects of DLK1 and/or PEG11 and many other genetic loci that regulate the hypertrophy response, thus, giving rise to the normal phenotype (Bidwell et al., 2014). Furthermore, it has been proposed that the repressive activity of the maternal long ncRNAs and miRNAs is achieved by inhibiting the expression of genes and proteins (at the transcriptional and/or post-transcriptional level through RNAi) involved in hypertrophy. In support of this, it has been shown that PEG11AS encodes six miRNAs that promote RNA-induced silencing complex (RISC)-mediated cleavage of PEG11 transcripts (Davis et al., 2005); however, miRNA transcripts generated from the DLK1-DIO3 imprinted cluster have been reported to not mediate the expression of DLK1 (Cheng et al., 2014). It has also been suggested that maternally-derived DLK1-DIO3 miRNAs may act to stabilize in trans the expression of several ncRNA transcripts that regulate hypertrophy (Tellam et al., 2012; Bidwell et al., 2014; Figure 2).
Molecular analysis of the callipyge phenotype highlights the role played by genetic (i.e., the A-to-G SNP that defines the callipyge mutation) and epigenetic mechanisms (i.e., DNA methylation, histone modifications, RNAi mechanisms and chromatin remodeling that regulate the expression of the genes within the DLK1-DIO3 imprinting domain) in regulating complex phenotypes. Indeed, the callipyge phenotype demonstrates that the mammalian ‘hard-wired’ genome is not the single repository of regulatory information that has phenotypic effects, but that the ‘soft-wired’ epigenome—the collective term for epigenetic mechanisms that regulate gene expression—also has an important role in determining phenotype (Hanna and Kelsey, 2014).
Imprinted Genes are Associated with Complex Phenotypic Traits in Mammals
Although analysis of mammalian genomes have shown that <1% of the total number of known mammalian protein-coding genes (~100 genes based on current versions of the human, mouse and bovine genomes in the Ensembl database) are subject to imprinting, several of these have been shown to have major effects on complex mammalian phenotypes. In mice, for example, studies have demonstrated the contribution of imprinted genes to variation in adiposity and body weight, muscle traits, metabolism, and disease susceptibility and resistance to infectious disease (Leighton et al., 1995; York et al., 1997; Clapcott et al., 2000; Lawson et al., 2011). Genetic studies of human phenotypes have also implicated imprinted gene effects in many biomedical conditions including BWS, Prader-Willi and Angelman syndromes, Alzheimer’s disease, cancer and type II diabetes (Bassett et al., 2002; Kong et al., 2009; Bird, 2014; Chaudhry et al., 2014; Eggermann et al., 2014). Similarly, while investigations of the callipyge phenotype have demonstrated a role for imprinting in sheep muscle traits, studies in pigs have identified a single SNP (G-to-A mutation) in the paternally expressed/maternally imprinted porcine IGF2 gene that is responsible for ~30% of the variance for lean meat, 15–30% of the variance for muscle mass and 10–20% of the variance for backfat content (Jeon et al., 1999). This SNP was shown to be located in an evolutionarily conserved CpG island within IGF2 intron 3 that abrogates binding of the zinc finger, BED-type containing 6 (ZBED6) transcriptional repressor. Animals inheriting a sire-derived ‘A’ nucleotide display a three-fold increase in IGF2 expression in post-natal muscle relative to those animals inheriting a sire-derived ‘G’ nucleotide, which results in increased muscle mass and a corresponding reduction in body fat (Van Laere et al., 2003).
Collectively, these studies highlight the important role played by epigenetically regulated loci in contributing to heritable phenotypic variation, making them attractive targets for candidate gene association studies and also inclusion in genome-wide scans that incorporate imprinting/parent-of-origin effects in domestic livestock species.
Imprinted Genes as Candidates for Genotype–Phenotype Association Studies in Domestic Livestock
Since the 1950s, intense selection for economically important production traits (such as feed efficiency, milk production, meat quality, and fertility) has resulted in remarkable rates of genetic improvement and has led to the development of several elite high-performance livestock populations, most notably the Holstein-Friesian dairy breed. Initially, systematic science-based improvement of domestic livestock used quantitative genetic evaluation of phenotypic data generated from managed populations or pedigrees, such that individual animals displaying increased performance (as estimated through breeding values) for desired traits were selected as candidate parents for subsequent generations. In the last two decades, however, there has been a paradigm shift in animal genetic improvement research involving data generated from molecular genetic markers, which has been concomitant with advances in genome sequencing and genotyping technologies, bioinformatics and biostatistics. SNPs and simple tandem repeat (STR) loci represent two of the most abundant DNA sequence polymorphisms within the mammalian genome and are the predominant genetic markers used in genotype-phenotype association studies. The methods that form the basis of these programs involve testing for associations between measured traits (qualitative or quantitative) and genetic marker genotypes. The genetic markers used can be distributed across the whole genome [i.e., genome-wide association (GWA) studies] or be situated within or proximal to genes selected for analysis a priori based on their biological function (i.e., candidate gene association studies; Ron and Weller, 2007; Bush and Moore, 2012). Animals carrying a marker allele(s) or genotype(s) known to associate with a desired complex phenotype (often referred to as ‘quantitative trait loci’) may be selected as parental candidates for subsequent generations; this approach underpinned marker-assisted selection (MAS) strategies that were proposed for the genetic improvement of domestic livestock populations (Weller and Ron, 2011).
There have been a number of genotype–phenotype association studies in domestic livestock that either incorporate imprinting effects in the statistical models used, or which have focused specifically on DNA sequence variation in known imprinted or candidate imprinted genes based on their imprinting status in other species (de Koning et al., 2000; Rattink et al., 2000; Magee et al., 2010a). Early studies based on STR genotypes uncovered parent-of-origin QTL for a series of phenotypic traits in pigs, sheep and cattle. For example, parent-of-origin QTL influencing body composition, carcass and meat quality traits, growth traits and reproductive traits in the F2 progeny of experimental cross-bred pig populations (Nezer et al., 1999; de Koning et al., 2000; Rattink et al., 2000; Holl et al., 2004). Interestingly, a theoretical approach to identifying parent-of-origin effects on body composition data (eye muscle area, rib fat, rump fat, and intramuscular fat percent) collected from ultrasonic measurements revealed that a mean of 28% of the total genetic variance for these traits was due to parent-of-origin effects (Tier and Meyer, 2012).
A recent comprehensive genome-wide scan in cattle that specifically included a parent-of-origin inheritance model identified 24 parent-of-origin QTL (six were significant at the 5% genome-wide level and 18 were significant at the 5% chromosome-wide level) distributed across 15 bovine autosomes influencing growth and carcass traits; two of these QTL encompassed the bovine imprinted GNAS and PEG3 genes (Imumorin et al., 2011). Subsequent studies have revealed associations between SNPs in the bovine PEG3 and GNAS genes and growth-related traits, calving and fertility traits and animal health traits (e.g., somatic cell count, a marker of mastitis infection and susceptibility). Collectively, these results suggest that the GNAS and PEG3 loci play an important role in bovine growth and development, fertility and health (Magee et al., 2010b; Sikora et al., 2011).
Additional studies revealing associations between imprinted loci and livestock production traits include the imprinted bovine IGF2 and IGF2R genes and meat quality, milk production and growth traits in beef and dairy cattle populations (Flisikowski et al., 2007; Goodall and Schmutz, 2007; Bagnicka et al., 2010; Sherman et al., 2010; Berkowicz et al., 2011, 2012), although some authors contend that the IGF2 associations with milk yields may be due to SNP alleles that are in linkage disequilibrium (LD) with neighboring variants in the proximal INS (insulin) gene, which contributes to the regulation of lactation (Akers, 2006; Berkowicz et al., 2011).
Associations between SNPs at the mammalian DLK1-DIO3 imprinted gene cluster and production traits such as growth, fatness and body composition have also been reported in pigs (Kim et al., 2004; Oczkowicz et al., 2011) and cattle (Magee et al., 2011). These findings support the important role of this imprinted cluster in regulating mammalian growth. Notably, a recent survey of SNPs in the imprinted paternally expressed/maternally imprinted DIO3 gene—which is involved in thyroid metabolism and has been shown to be highly expressed in uterine tissues in humans and rodents—was associated with fertility traits in pigs. It has been proposed that DIO3 influences porcine fertility through the regulation of placental and/or fetal growth (Coster et al., 2012).
Examples of imprinted SNP-phenotype associations in pigs, sheep and cattle are listed in Table 1.
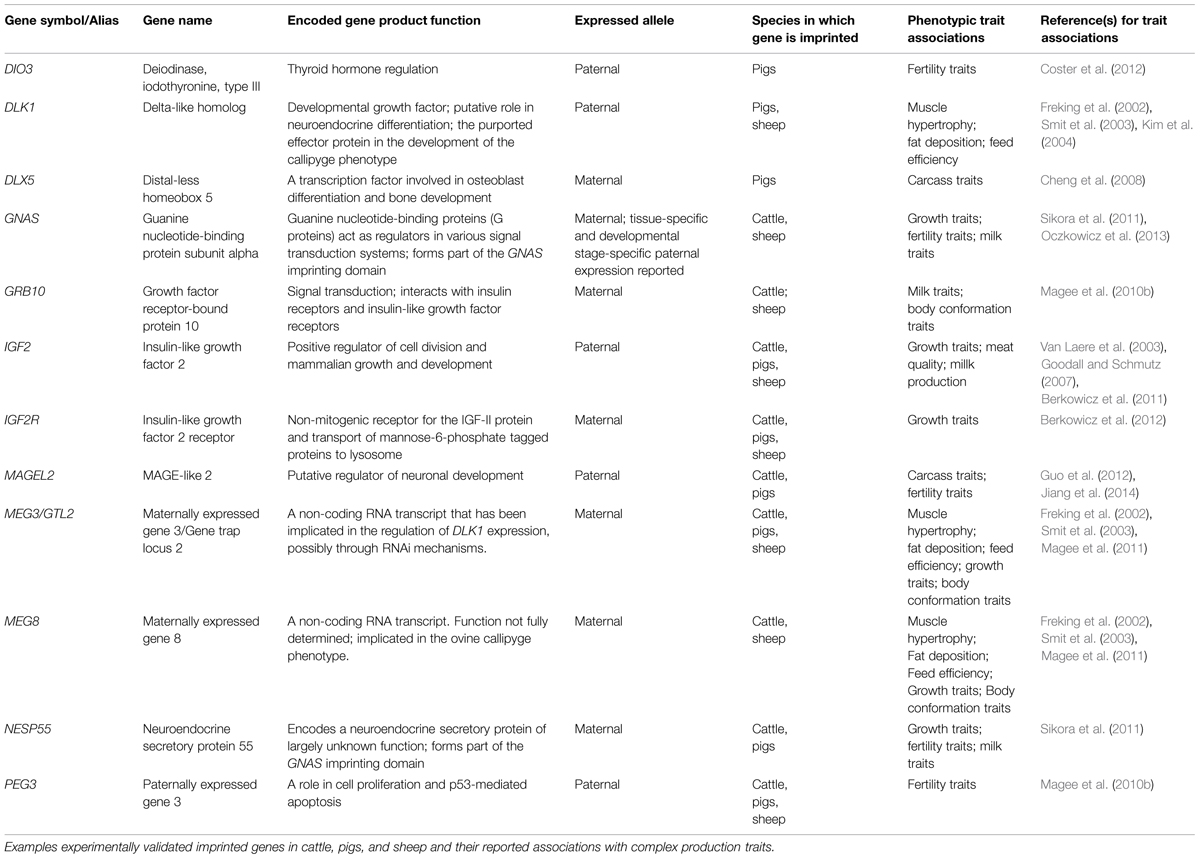
TABLE 1. Examples of associations between DNA sequence polymorphisms in known livestock imprinted genes and phenotypic traits.
The Effects of Imprinted Gene Expression on Phenotype
The documented biological roles of imprinted genes in regulating mammalian growth and development together with the accumulating genotype–phenotype association data in domestic livestock species/populations, suggests that loci subject to genomic imprinting represent an important reservoir of genetic variation that may be exploited in selective breeding programs (Ruvinsky, 1999). However, genomic imprinting raises several interesting theoretical considerations for genotype–phenotype association studies. For example, classic imprinted gene expression (i.e., complete parent-of-origin monoallelic expression) is expected to generate patterns of phenotypic expression whereby phenotype is solely determined by the expressed allele. Consequently, classically defined imprinted loci with two alleles can be regarded as being functionally hemizygous (Bartolomei and Tilghman, 1997). This reduces the number of phenotypic classes at such loci from three (as expected under an additive genetic model) to two such that the heterozygote class is functionally equivalent to one of the two homozygote classes. It is important to note that for loci exhibiting complete imprinting, heterozygous individuals expressing the allele with the greatest phenotypic effect may display similar phenotypic scores to those traits controlled by loci with dominance effects (Figure 3). However, for many imprinted loci, transcriptional silencing is only partial (Khatib, 2007), which can generate functional differences between reciprocal heterozygotes (i.e., heterozygous individuals that have inherited the same allele from different parents) and can lead to four potential phenotypic classes (Spencer, 2000, 2009; Figure 3).
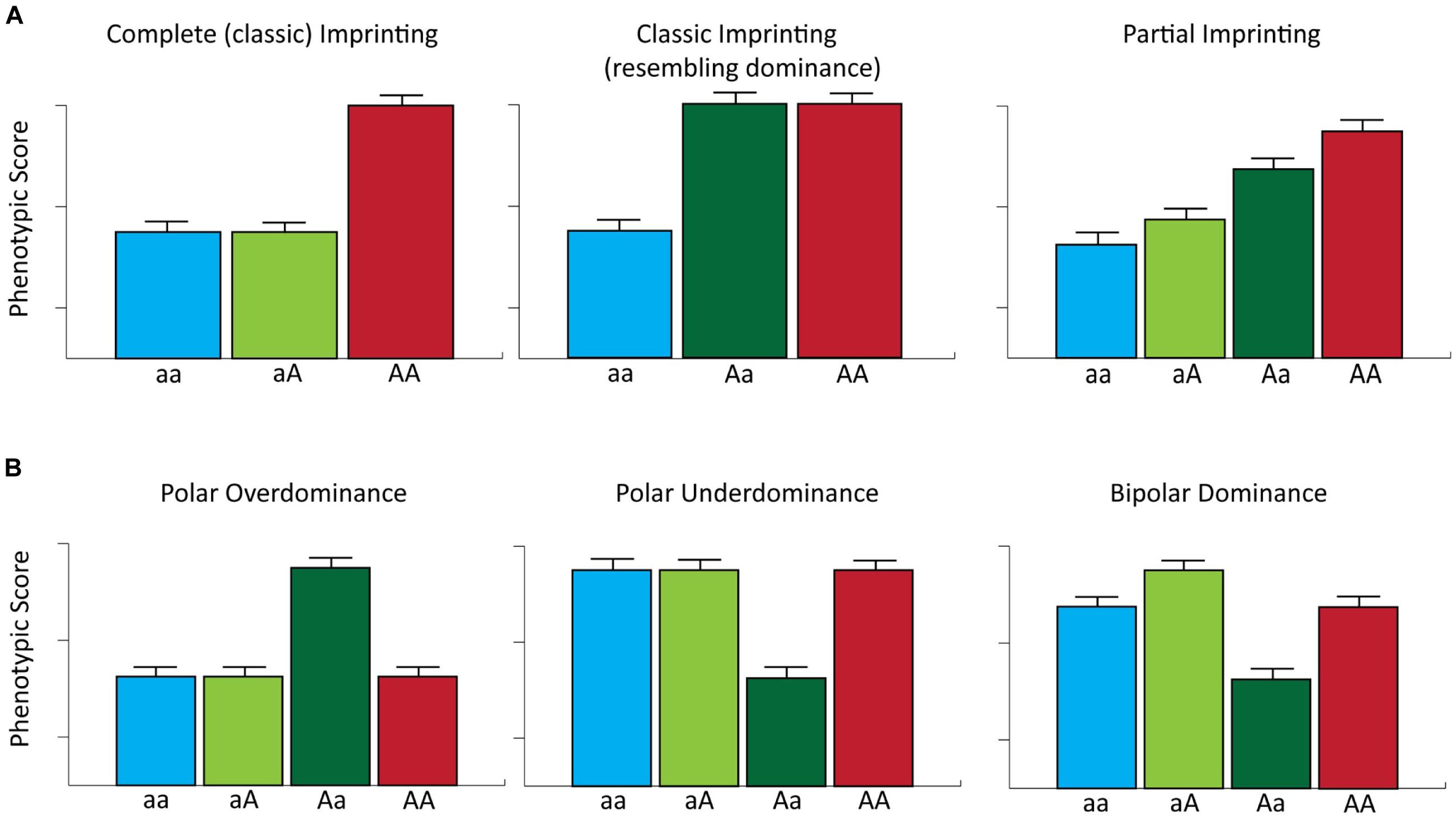
FIGURE 3. Genomic imprinting and parent-of-origin effects on complex phenotypes. (A) The phenotypic effects of complete and partial imprinting are considered for a single locus with two alleles. For complete imprinting, the first listed allele represents the expressed allele and the A allele has a greater effect on phenotype relative to the a allele. Note that in this example the Aa heterozygote displays a phenotypic score that resembles that expected for a locus with a dominance effect. For partial imprinting, aA and Aa represent reciprocal heterozygote genotypes, where the first listed allele is fully expressed and the second listed allele is partially expressed. In addition, the ‘A’ allele has the greatest effect on phenotype. Partial imprinting results in the generation of four potential phenotypic classes. (B) The phenotypic effects of a single locus for which there are two alleles displaying polar overdominance, polar underdominance and bipolar dominance modes of inheritance [modified from Lawson et al. (2011)].
Furthermore, different forms of parent-of-origin effects can also generate phenotypic differences between genotype classes at the same locus. For example, polar overdominance, as exemplified by the callipyge phenotype, can result in phenotypic differences between reciprocal heterozygotes; in addition, under a model of polar overdominance, one of the heterozygous states will display a phenotypic value greater than all three other genotypes, which themselves show no differences in phenotypic values. Conversely, a model of polar underdominance, whereby one of the two reciprocal heterozygotes has a phenotypic value less than all three other phenotypically equivalent genotypes, has been reported in mice (Wolf et al., 2008). Finally, bipolar dominance can exist at imprinted loci such that one heterozygote displays larger phenotypic values and the other heterozygote exhibits lower phenotypic values than both homozygotes, which have the same phenotypic value (Wolf et al., 2008; Figure 3).
Genomic Imprinting in the Era of Genome Selection
For many industrialized countries, the original MAS concept for livestock breeding has been supplanted by ‘genome-wide selection’ or ‘genomic selection’ using 1000s of genetic markers distributed across the genome. Genomic selection in livestock was originally proposed by Meuwissen et al. (2001) more than a decade ago; but has only been applied practically since the advent of high-density, pan-genomic livestock SNP genotyping arrays within the last 8 years. Genomic selection uses a genome-wide panel of dense markers so that all QTL are likely to be in LD (i.e., the non-random association of alleles at different loci) with at least one of the assayed SNPs. The genomic selection process involves the generation of genome-wide genotypic data for a large reference population of animals for which accurate phenotypic data are available. The resulting data serves as a reference for the development of statistical models that estimate the effect of each SNP with the trait(s)-of-interest, leading to the formulation of a predictive equation to estimate a genomic breeding value (GBV; i.e., the additive genetic component that is transmitted to the next generation). The predictive equation can then be used to impute the GBVs of additional animals as required (Goddard and Hayes, 2009; Goddard et al., 2010). This approach has been extremely successful, particularly for genetic improvement of dairy cattle and is rapidly becoming the method of choice for commercial breeding of beef cattle, pigs, and other livestock populations (Varona et al., 2015).
Genomic selection strategies are largely unconcerned with knowledge of the genes and causal variants that directly affect phenotypes (Snelling et al., 2013). However, several authors have recently argued for the refinement of genomic selection methods by incorporating all relevant genetic information that predict future phenotypes (i.e., the performance over its lifetime) rather than GBVs, including epigenetic patterns of gene expression (Gonzalez-Recio, 2011; Hayes et al., 2013). Such refinement might consider weighting imprinted gene-associated SNPs in genomic selection models according to expected effects on gene products during early development and across the whole lifespan of an individual (Snelling et al., 2013).
Conclusion
The phenomena outlined above, demonstrate that imprinting parent-of-origin effects may complicate traditional quantitative genetic models used in phenotypic association studies. This review illustrates that imprinted gene expression can have a major effect on phenotypic traits in domestic livestock populations. Furthermore, imprinting is an important factor to consider in the models used for future the genetic improvement of domestic livestock for those genomic regions where imprinted gene expression is known to occur and to affect economically important traits included in the selection index.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the funding support from the University of Connecticut, the Irish Department of Agriculture, Food and the Marine Research Stimulus Program (project numbers RSF-06-406, RSF-06-0353, and RSF-06-0409) and from Science Foundation Ireland Investigator Grants awarded to DEM (Nos: SFI/01/F.1/B028 and SFI/08/IN.1/B2038) and CS (Nos: 02/IN.1/B49 and 08/IN.1/B1931).
References
Abramowitz, L. K., and Bartolomei, M. S. (2012). Genomic imprinting: recognition and marking of imprinted loci. Curr. Opin. Genet. Dev. 22, 72–78. doi: 10.1016/j.gde.2011.12.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adalsteinsson, B. T., and Ferguson-Smith, A. C. (2014). Epigenetic control of the genome-lessons from genomic imprinting. Genes (Basel) 5, 635–655. doi: 10.3390/genes5030635
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Akers, R. M. (2006). Major advances associated with hormone and growth factor regulation of mammary growth and lactation in dairy cows. J. Dairy Res. 89, 1222–1234. doi: 10.3168/jds.S0022-0302(06)72191-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alexopoulos, N. I., Maddox-Hyttel, P., Tveden-Nyborg, P., D’Cruz, N. T., Tecirlioglu, T. R., Cooney, M. A., et al. (2008). Developmental disparity between in vitro-produced and somatic cell nuclear transfer bovine days 14 and 21 embryos: implications for embryonic loss. Reproduction 136, 433–445. doi: 10.1530/REP-07-0392
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ashbrook, D. G., and Hager, R. (2013). Empirical testing of hypotheses about the evolution of genomic imprinting in mammals. Front. Neuroanat. 7:6. doi: 10.3389/fnana.2013.00006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Autuoro, J. M., Pirnie, S. P., and Carmichael, G. G. (2014). Long noncoding RNAs in imprinting and X chromosome inactivation. Biomolecules 4, 76–100. doi: 10.3390/biom4010076
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bagnicka, E., Siadkowska, E., Strzalkowska, N., Zelazowska, B., Flisikowski, K., Krzyzewski, J., et al. (2010). Association of polymorphisms in exons 2 and 10 of the insulin-like growth factor 2 (IGF2) gene with milk production traits in Polish Holstein-Friesian cattle. J. Dairy Res. 77, 37–42. doi: 10.1017/S0022029909990197
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barlow, D. P., and Bartolomei, M. S. (2014). Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 6, a018382. doi: 10.1101/cshperspect.a018382
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bartolomei, M. S. (2009). Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 23, 2124–2133. doi: 10.1101/gad.1841409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bartolomei, M. S., and Tilghman, S. M. (1997). Genomic imprinting in mammals. Annu. Rev. Genet. 31, 493–525. doi: 10.1146/annurev.genet.31.1.493
Barton, S. C., Surani, M. A., and Norris, M. L. (1984). Role of paternal and maternal genomes in mouse development. Nature 311, 374–376. doi: 10.1038/311374a0
Bassett, S. S., Avramopoulos, D., and Fallin, D. (2002). Evidence for parent of origin effect in late-onset Alzheimer disease. Am. J. Med. Genet. 114, 679–686. doi: 10.1002/ajmg.10648
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Berkowicz, E. W., Magee, D. A., Berry, D. P., Sikora, K. M., Howard, D. J., Mullen, M. P., et al. (2012). Single nucleotide polymorphisms in the imprinted bovine insulin-like growth factor 2 receptor gene (IGF2R) are associated with body size traits in Irish Holstein-Friesian cattle. Anim. Genet. 43, 81–87. doi: 10.1111/j.1365-2052.2011.02211.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Berkowicz, E. W., Magee, D. A., Sikora, K. M., Berry, D. P., Howard, D. J., Mullen, M. P., et al. (2011). Single nucleotide polymorphisms at the imprinted bovine insulin-like growth factor 2 (IGF2) locus are associated with dairy performance in Irish Holstein-Friesian cattle. J. Dairy Res. 78, 1–8. doi: 10.1017/S0022029910000567
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bidwell, C. A., Kramer, L. N., Perkins, A. C., Hadfield, T. S., Moody, D. E., and Cockett, N. E. (2004). Expression of PEG11 and PEG11AS transcripts in normal and callipyge sheep. BMC Biol. 2:17. doi: 10.1186/1741-7007-2-17
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bidwell, C. A., Waddell, J. N., Taxis, T. M., Yu, H., Tellam, R. L., Neary, M. K., et al. (2014). New insights into polar overdominance in callipyge sheep. Anim. Genet. 45(Suppl. 1), 51–61. doi: 10.1111/age.12132
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bird, A. (2007). Perceptions of epigenetics. Nature 447, 396–398. doi: 10.1038/nature05913
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bird, L. M. (2014). Angelman syndrome: review of clinical and molecular aspects. Appl. Clin. Genet. 7, 93–104. doi: 10.2147/TACG.S57386
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Briggs, S. F., and Reijo Pera, R. A. (2014). X chromosome inactivation: recent advances and a look forward. Curr. Opin. Genet. Dev. 28, 78–82. doi: 10.1016/j.gde.2014.09.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bush, W. S., and Moore, J. H. (2012). Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 8:e1002822. doi: 10.1371/journal.pcbi.1002822
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Campbell, K. H., McWhir, J., Ritchie, W. A., and Wilmut, I. (1996). Sheep cloned by nuclear transfer from a cultured cell line. Nature 380, 64–66. doi: 10.1038/380064a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cattanach, B. M., and Kirk, M. (1985). Differential activity of maternally and paternally derived chromosome regions in mice. Nature 315, 496–498. doi: 10.1038/315496a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chang, A. S., Moley, K. H., Wangler, M., Feinberg, A. P., and Debaun, M. R. (2005). Association between Beckwith-Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients. Fertil. Steril. 83, 349–354. doi: 10.1016/j.fertnstert.2004.07.964
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chaudhry, M., Wang, X., Bamne, M. N., Hasnain, S., Demirci, F. Y., Lopez, O. L., et al. (2014). Genetic variation in imprinted genes is associated with risk of late-onset Alzheimer’s disease. J. Alzheimers Dis. 44, 989–994. doi: 10.3233/JAD-142106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Z., Robbins, K. M., Wells, K. D., and Rivera, R. M. (2013). Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics 8, 591–601. doi: 10.4161/epi.24655
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cheng, H., Xu, X., Hadfield, T., Cockett, N., Charlier, C., Georges, M., et al. (2014). Experimental evaluation does not reveal a direct effect of microRNA from the callipyge locus on DLK1 expression. BMC Genomics 15:944. doi: 10.1186/1471-2164-15-944
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cheng, H. C., Zhang, F. W., Jiang, C. D., Li, F. E., Xiong, Y. Z., and Deng, C. Y. (2008). Isolation and imprinting analysis of the porcine DLX5 gene and its association with carcass traits. Anim. Genet. 39, 395–399. doi: 10.1111/j.1365-2052.2008.01740.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Choufani, S., Shuman, C., and Weksberg, R. (2010). Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 343–354. doi: 10.1002/ajmg.c.30267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chow, J. C., and Heard, E. (2010). Nuclear organization and dosage compensation. Cold Spring Harb. Perspect. Biol. 2, a000604. doi: 10.1101/cshperspect.a000604
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cibelli, J. B., Stice, S. L., Golueke, P. J., Kane, J. J., Jerry, J., Blackwell, C., et al. (1998). Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280, 1256–1258. doi: 10.1126/science.280.5367.1256
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ciccone, D. N., Su, H., Hevi, S., Gay, F., Lei, H., Bajko, J., et al. (2009). KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461, 415–418. doi: 10.1038/nature08315
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clapcott, S. J., Teale, A. J., and Kemp, S. J. (2000). Evidence for genomic imprinting of the major QTL controlling susceptibility to trypanosomiasis in mice. Parasite Immunol. 22, 259–263. doi: 10.1046/j.1365-3024.2000.00308.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cockett, N. E., Jackson, S. P., Shay, T. L., Farnir, F., Berghmans, S., Snowder, G. D., et al. (1996). Polar overdominance at the ovine callipyge locus. Science 273, 236–238. doi: 10.1126/science.273.5272.236
Coster, A., Madsen, O., Heuven, H. C., Dibbits, B., Groenen, M. A., van Arendonk, J. A., et al. (2012). The imprinted gene DIO3 is a candidate gene for litter size in pigs. PLoS ONE 7:e31825. doi: 10.1371/journal.pone.0031825
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Court, F., Tayama, C., Romanelli, V., Martin-Trujillo, A., Iglesias-Platas, I., Okamura, K., et al. (2014). Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 24, 554–569. doi: 10.1101/gr.164913.113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Curchoe, C. L., Zhang, S., Yang, L., Page, R., and Tian, X. C. (2009). Hypomethylation trends in the intergenic region of the imprinted IGF2 and H19 genes in cloned cattle. Anim. Reprod. Sci. 116, 213–225. doi: 10.1016/j.anireprosci.2009.02.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, E., Caiment, F., Tordoir, X., Cavaille, J., Ferguson-Smith, A., Cockett, N., et al. (2005). RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr. Biol. 15, 743–749. doi: 10.1016/j.cub.2005.02.060
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, T. L., Yang, G. J., McCarrey, J. R., and Bartolomei, M. S. (2000). The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum. Mol. Genet. 9, 2885–2894. doi: 10.1093/hmg/9.19.2885
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dean, W., Santos, F., Stojkovic, M., Zakhartchenko, V., Walter, J., Wolf, E., et al. (2001). Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc. Natl. Acad. Sci. U.S.A. 98, 13734–13738. doi: 10.1073/pnas.241522698
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DeBaun, M. R., Niemitz, E. L., and Feinberg, A. P. (2003). Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 72, 156–160. doi: 10.1086/346031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Koning, D. J., Rattink, A. P., Harlizius, B., van Arendonk, J. A., Brascamp, E. W., and Groenen, M. A. (2000). Genome-wide scan for body composition in pigs reveals important role of imprinting. Proc. Natl. Acad. Sci. U.S.A. 97, 7947–7950. doi: 10.1073/pnas.140216397
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Delaval, K., Govin, J., Cerqueira, F., Rousseaux, S., Khochbin, S., and Feil, R. (2007). Differential histone modifications mark mouse imprinting control regions during spermatogenesis. EMBO J. 26, 720–729. doi: 10.1038/sj.emboj.7601513
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Demars, J., Shmela, M. E., Rossignol, S., Okabe, J., Netchine, I., Azzi, S., et al. (2010). Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum. Mol. Genet. 19, 803–814. doi: 10.1093/hmg/ddp549
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Edwards, C. A., and Ferguson-Smith, A. C. (2007). Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 19, 281–289. doi: 10.1016/j.ceb.2007.04.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eggermann, T., Binder, G., Brioude, F., Maher, E. R., Lapunzina, P., Cubellis, M. V., et al. (2014). CDKN1C mutations: two sides of the same coin. Trends Mol. Med. 20, 614–622. doi: 10.1016/j.molmed.2014.09.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farin, P. W., Piedrahita, J. A., and Farin, C. E. (2006). Errors in development of fetuses and placentas from in vitro-produced bovine embryos. Theriogenology 65, 178–191. doi: 10.1016/j.theriogenology.2005.09.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fitzpatrick, G. V., Soloway, P. D., and Higgins, M. J. (2002). Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 32, 426–431. doi: 10.1038/ng988
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flisikowski, K., Adamowicz, T., Strabel, T., Jankowski, T., Switonski, M., and Zwierzchowski, L. (2007). An InDel polymorphism in exon 6 of IGF2 associated with the breeding value of Polish Holstein-Friesian bulls. Biochem. Genet. 45, 139–143. doi: 10.1007/s10528-006-9071-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flisikowski, K., Venhoranta, H., Bauersachs, S., Hanninen, R., Furst, R. W., Saalfrank, A., et al. (2012). Truncation of MIMT1 gene in the PEG3 domain leads to major changes in placental gene expression and stillbirth in cattle. Biol. Reprod. 87, 140. doi: 10.1095/biolreprod.112.104240
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flisikowski, K., Venhoranta, H., Nowacka-Woszuk, J., McKay, S. D., Flyckt, A., Taponen, J., et al. (2010). A novel mutation in the maternally imprinted PEG3 domain results in a loss of MIMT1 expression and causes abortions and stillbirths in cattle (Bos taurus). PLoS ONE 5:e15116. doi: 10.1371/journal.pone.0015116
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Freking, B. A., Murphy, S. K., Wylie, A. A., Rhodes, S. J., Keele, J. W., Leymaster, K. A., et al. (2002). Identification of the single base change causing the callipyge muscle hypertrophy phenotype, the only known example of polar overdominance in mammals. Genome Res. 12, 1496–1506. doi: 10.1101/gr.571002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fukui, Y., Sawai, K., Furudate, M., Sato, N., Iwazumi, Y., and Ohsaki, K. (1992). Parthenogenetic development of bovine oocytes treated with ethanol and cytochalasin B after in vitro maturation. Mol. Reprod. Dev. 33, 357–362. doi: 10.1002/mrd.1080330318
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Georges, M., Charlier, C., and Cockett, N. (2003). The callipyge locus: evidence for the trans interaction of reciprocally imprinted genes. Trends Genet. 19, 248–252. doi: 10.1016/S0168-9525(03)00082-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giannoukakis, N., Deal, C., Paquette, J., Kukuvitis, A., and Polychronakos, C. (1996). Polymorphic functional imprinting of the human IGF2 gene among individuals, in blood cells, is associated with H19 expression. Biochem. Biophys. Res. Commun. 220, 1014–1019. doi: 10.1006/bbrc.1996.0524
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gicquel, C., Gaston, V., Mandelbaum, J., Siffroi, J. P., Flahault, A., and Le Bouc, Y (2003). In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am. J. Hum. Genet. 72, 1338–1341. doi: 10.1086/374824
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gkountela, S., and Clark, A. T. (2014). A big surprise in the little zygote: the curious business of losing methylated cytosines. Cell Stem Cell 15, 393–394. doi: 10.1016/j.stem.2014.09.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goddard, M. E., and Hayes, B. J. (2009). Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10, 381–391. doi: 10.1038/nrg2575
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goddard, M. E., Hayes, B. J., and Meuwissen, T. H. (2010). Genomic selection in livestock populations. Genet. Res. (Camb) 92, 413–421. doi: 10.1017/S0016672310000613
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Recio, O. (2011). Epigenetics: a new challenge in the post-genomic era of livestock. Front. Genet. 2:106. doi: 10.3389/fgene.2011.00106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goodall, J. J., and Schmutz, S. M. (2007). IGF2 gene characterization and association with rib eye area in beef cattle. Anim. Genet. 38, 154–161. doi: 10.1111/j.1365-2052.2007.01576.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guibert, S., Forne, T., and Weber, M. (2012). Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 22, 633–641. doi: 10.1101/gr.130997.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, F., Li, X., Liang, D., Li, T., Zhu, P., Guo, H., et al. (2014). Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 15, 447–458. doi: 10.1016/j.stem.2014.08.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, L., Qiao, M., Wang, C., Zheng, R., Xiong, Y. Z., and Deng, C. Y. (2012). Imprinting analysis of porcine MAGEL2 gene in two fetal stages and association analysis with carcass traits. Mol. Biol. Rep. 39, 147–155. doi: 10.1007/s11033-011-0719–710.
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hagan, J. P., O’Neill, B. L., Stewart, C. L., Kozlov, S. V., and Croce, C. M. (2009). At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS ONE 4:e4352. doi: 10.1371/journal.pone.0004352
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hajkova, P., Erhardt, S., Lane, N., Haaf, T., El-Maarri, O., Reik, W., et al. (2002). Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23. doi: 10.1016/S0925-4773(02)00181-8
Hanna, C. W., and Kelsey, G. (2014). The specification of imprints in mammals. Heredity (Edinb) 113, 176–183. doi: 10.1038/hdy.2014.54
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hark, A. T., Schoenherr, C. J., Katz, D. J., Ingram, R. S., Levorse, J. M., and Tilghman, S. M. (2000). CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489. doi: 10.1038/35013106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayes, B. J., Lewin, H. A., and Goddard, M. E. (2013). The future of livestock breeding: genomic selection for efficiency, reduced emissions intensity, and adaptation. Trends Genet. 29, 206–214. doi: 10.1016/j.tig.2012.11.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Henckel, A., Nakabayashi, K., Sanz, L. A., Feil, R., Hata, K., and Arnaud, P. (2009). Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum. Mol. Genet. 18, 3375–3383. doi: 10.1093/hmg/ddp277
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hiendleder, S., Mund, C., Reichenbach, H. D., Wenigerkind, H., Brem, G., Zakhartchenko, V., et al. (2004). Tissue-specific elevated genomic cytosine methylation levels are associated with an overgrowth phenotype of bovine fetuses derived by in vitro techniques. Biol. Reprod. 71, 217–223. doi: 10.1095/biolreprod.103.026062
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hiura, H., Obata, Y., Komiyama, J., Shirai, M., and Kono, T. (2006). Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells 11, 353–361. doi: 10.1111/j.1365-2443.2006.00943.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holl, J. W., Cassady, J. P., Pomp, D., and Johnson, R. K. (2004). A genome scan for quantitative trait loci and imprinted regions affecting reproduction in pigs. J. Anim. Sci. 82, 3421–3429.
Hori, N., Nagai, M., Hirayama, M., Hirai, T., Matsuda, K., Hayashi, M., et al. (2010). Aberrant CpG methylation of the imprinting control region KvDMR1 detected in assisted reproductive technology-produced calves and pathogenesis of large offspring syndrome. Anim. Reprod. Sci. 122, 303–312. doi: 10.1016/j.anireprosci.2010.09.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Humpherys, D., Eggan, K., Akutsu, H., Hochedlinger, K., Rideout, W. M. III, Biniszkiewicz, D., et al. (2001). Epigenetic instability in ES cells and cloned mice. Science 293, 95–97. doi: 10.1126/science.1061402
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imamura, T., Kerjean, A., Heams, T., Kupiec, J. J., Thenevin, C., and Paldi, A. (2005). Dynamic CpG and non-CpG methylation of the Peg1/Mest gene in the mouse oocyte and preimplantation embryo. J. Biol. Chem. 280, 20171–20175. doi: 10.1074/jbc.M501749200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imumorin, I. G., Kim, E. H., Lee, Y. M., de Koning, D. J., van Arendonk, J. A., de Donato, M., et al. (2011). Genome scan of parent-of-origin QTL effects on bovine growth and carcass traits. Front. Genet. 2:44. doi: 10.3389/fgene.2011.00044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iqbal, K., Jin, S. G., Pfeifer, G. P., and Szabo, P. E. (2011). Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 108, 3642–3647. doi: 10.1073/pnas.1014033108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Irwin, R. E., Thakur, A., O’ Neill, K. M., and Walsh, C. P. (2014). 5-Hydroxymethylation marks a class of neuronal gene regulated by intragenic methylcytosine levels. Genomics 104, 383–392. doi: 10.1016/j.ygeno.2014.08.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ito, Y., Nativio, R., and Murrell, A. (2013). Induced DNA demethylation can reshape chromatin topology at the IGF2-H19 locus. Nucleic Acids Res. 41, 5290–5302. doi: 10.1093/nar/gkt240
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jeon, J. T., Carlborg, O., Tornsten, A., Giuffra, E., Amarger, V., Chardon, P., et al. (1999). A paternally expressed QTL affecting skeletal and cardiac muscle mass in pigs maps to the IGF2 locus. Nat. Genet. 21, 157–158. doi: 10.1038/5938
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiang, C., Yang, Y., Huang, C., and Whitelaw, B. (2014). Promoter characterization and functional association with placenta of porcine MAGEL2. Gene 547, 63–69. doi: 10.1016/j.gene.2014.06.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jirtle, R. L., (2013). Geneimprint [Online]. Available at: http://www.geneimprint.com
Jones, P. L., Veenstra, G. J., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191.
Kang, Y. K., Lee, K. K., and Han, Y. M. (2003). Reprogramming DNA methylation in the preimplantation stage: peeping with Dolly’s eyes. Curr. Opin. Cell Biol. 15, 290–295. doi: 10.1016/S0955-0674(03)00031-0
Kelsey, G., and Feil, R. (2013). New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20110336. doi: 10.1098/rstb.2011.0336
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khatib, H. (2007). Is it genomic imprinting or preferential expression? Bioessays 29, 1022–1028. doi: 10.1002/bies.20637
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khatib, H., Zaitoun, I., and Kim, E. S. (2007). Comparative analysis of sequence characteristics of imprinted genes in human, mouse, and cattle. Mamm. Genome 18, 538–547. doi: 10.1007/s00335-007-9039-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, K. S., Kim, J. J., Dekkers, J. C., and Rothschild, M. F. (2004). Polar overdominant inheritance of a DLK1 polymorphism is associated with growth and fatness in pigs. Mamm. Genome 15, 552–559. doi: 10.1007/s00335-004-2341-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koerner, M. V., Pauler, F. M., Huang, R., and Barlow, D. P. (2009). The function of non-coding RNAs in genomic imprinting. Development 136, 1771–1783. doi: 10.1242/dev.030403
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kong, A., Steinthorsdottir, V., Masson, G., Thorleifsson, G., Sulem, P., Besenbacher, S., et al. (2009). Parental origin of sequence variants associated with complex diseases. Nature 462, 868–874. doi: 10.1038/nature08625
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lagutina, I., Lazzari, G., Duchi, R., and Galli, C. (2004). Developmental potential of bovine androgenetic and parthenogenetic embryos: a comparative study. Biol. Reprod. 70, 400–405. doi: 10.1095/biolreprod.103.021972
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lane, N., Dean, W., Erhardt, S., Hajkova, P., Surani, A., Walter, J., et al. (2003). Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 35, 88–93. doi: 10.1002/gene.10168
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Latos, P. A., Pauler, F. M., Koerner, M. V., Senergin, H. B., Hudson, Q. J., Stocsits, R. R., et al. (2012). Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472. doi: 10.1126/science.1228110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Latos, P. A., Stricker, S. H., Steenpass, L., Pauler, F. M., Huang, R., Senergin, B. H., et al. (2009). An in vitro ES cell imprinting model shows that imprinted expression of the Igf2r gene arises from an allele-specific expression bias. Development 136, 437–448. doi: 10.1242/dev.032060
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lawson, H. A., Cady, J. E., Partridge, C., Wolf, J. B., Semenkovich, C. F., and Cheverud, J. M. (2011). Genetic effects at pleiotropic loci are context-dependent with consequences for the maintenance of genetic variation in populations. PLoS Genet. 7:e1002256. doi: 10.1371/journal.pgen.1002256
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, J. T., and Bartolomei, M. S. (2013). X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152, 1308–1323. doi: 10.1016/j.cell.2013.02.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leighton, P. A., Ingram, R. S., Eggenschwiler, J., Efstratiadis, A., and Tilghman, S. M. (1995). Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 375, 34–39. doi: 10.1038/375034a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, J. Y., Lees-Murdock, D. J., Xu, G. L., and Walsh, C. P. (2004). Timing of establishment of paternal methylation imprints in the mouse. Genomics 84, 952–960. doi: 10.1016/j.ygeno.2004.08.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ludwig, M., Katalinic, A., Gross, S., Sutcliffe, A., Varon, R., and Horsthemke, B. (2005). Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J. Med. Genet. 42, 289–291. doi: 10.1136/jmg.2004.026930
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lyon, M. F. (1961). Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190, 372–373. doi: 10.1038/190372a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Magee, D. A., Berkowicz, E. W., Sikora, K. M., Berry, D. P., Park, S. D., Kelly, A. K., et al. (2010a). A catalogue of validated single nucleotide polymorphisms in bovine orthologs of mammalian imprinted genes and associations with beef production traits. Animal 4, 1958–1970. doi: 10.1017/S1751731110001163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Magee, D. A., Sikora, K. M., Berkowicz, E. W., Berry, D. P., Howard, D. J., Mullen, M. P., et al. (2010b). DNA sequence polymorphisms in a panel of eight candidate bovine imprinted genes and their association with performance traits in Irish Holstein-Friesian cattle. BMC Genet. 11:93. doi: 10.1186/1471-2156-11-93
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Magee, D. A., Berry, D. P., Berkowicz, E. W., Sikora, K. M., Howard, D. J., Mullen, M. P., et al. (2011). Single nucleotide polymorphisms within the bovine DLK1-DIO3 imprinted domain are associated with economically important production traits in cattle. J. Hered. 102, 94–101. doi: 10.1093/jhered/esq097
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770. doi: 10.1038/nature07107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Messerschmidt, D. M., Knowles, B. B., and Solter, D. (2014). DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 28, 812–828. doi: 10.1101/gad.234294.113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meuwissen, T. H., Hayes, B. J., and Goddard, M. E. (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829.
Monk, D., Arnaud, P., Apostolidou, S., Hills, F. A., Kelsey, G., Stanier, P., et al. (2006). Limited evolutionary conservation of imprinting in the human placenta. Proc. Natl. Acad. Sci. U.S.A. 103, 6623–6628. doi: 10.1073/pnas.0511031103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moore, T., and Haig, D. (1991). Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 7, 45–49. doi: 10.1016/0168-9525(91)90230-N
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morison, I. M., Paton, C. J., and Cleverley, S. D. (2001). The imprinted gene and parent-of-origin effect database. Nucleic Acids Res. 29, 275–276. doi: 10.1093/nar/29.1.275
Morison, I. M., Ramsay, J. P., and Spencer, H. G. (2005). A census of mammalian imprinting. Trends Genet. 21, 457–465. doi: 10.1016/j.tig.2005.06.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murphy, S. K., Nolan, C. M., Huang, Z., Kucera, K. S., Freking, B. A., Smith, T. P., et al. (2006). Callipyge mutation affects gene expression in cis: a potential role for chromatin structure. Genome Res. 16, 340–346. doi: 10.1101/gr.4389306
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neri, F., Krepelova, A., Incarnato, D., Maldotti, M., Parlato, C., Galvagni, F., et al. (2013). Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 155, 121–134. doi: 10.1016/j.cell.2013.08.056
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nezer, C., Moreau, L., Brouwers, B., Coppieters, W., Detilleux, J., Hanset, R., et al. (1999). An imprinted QTL with major effect on muscle mass and fat deposition maps to the IGF2 locus in pigs. Nat. Genet. 21, 155–156. doi: 10.1038/5935
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oczkowicz, M., Ropka-Molik, K., Piorkowska, K., Rozycki, M., and Rejduch, B. (2011). Frequency of DLK1 c.639C > T polymorphism and the analysis of MEG3/DLK1/PEG11 cluster expression in muscle of swine raised in Poland. Meat Sci. 88, 627–630. doi: 10.1016/j.meatsci.2011.02.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oczkowicz, M., Ropka-Molik, K., and Tyra, M. (2013). Analysis of the associations between polymorphisms in GNAS complex locus and growth, carcass and meat quality traits in pigs. Mol. Biol. Rep. 40, 6419–6427. doi: 10.1007/s11033-013-2756-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O’Doherty, A. M., O’Shea, L. C., and Fair, T. (2012). Bovine DNA methylation imprints are established in an oocyte size-specific manner, which are coordinated with the expression of the DNMT3 family proteins. Biol. Reprod. 86:67. doi: 10.1095/biolreprod.111.094946
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Olek, A., and Walter, J. (1997). The pre-implantation ontogeny of the H19 methylation imprint. Nat. Genet. 17, 275–276. doi: 10.1038/ng1197-275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedone, P. V., Pikaart, M. J., Cerrato, F., Vernucci, M., Ungaro, P., Bruni, C. B., et al. (1999). Role of histone acetylation and DNA methylation in the maintenance of the imprinted expression of the H19 and Igf2 genes. FEBS Lett. 458, 45–50. doi: 10.1016/S0014-5793(99)01124-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Plasschaert, R. N., and Bartolomei, M. S. (2014). Genomic imprinting in development, growth, behavior and stem cells. Development 141, 1805–1813. doi: 10.1242/dev.101428
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poole, R. L., Leith, D. J., Docherty, L. E., Shmela, M. E., Gicquel, C., Splitt, M., et al. (2012). Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur. J. Hum. Genet. 20, 240–243. doi: 10.1038/ejhg.2011.166
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Prickett, A. R., and Oakey, R. J. (2012). A survey of tissue-specific genomic imprinting in mammals. Mol. Genet. Genomics 287, 621–630. doi: 10.1007/s00438-012-0708-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rattink, A. P., De Koning, D. J., Faivre, M., Harlizius, B., van Arendonk, J. A., and Groenen, M. A. (2000). Fine mapping and imprinting analysis for fatness trait QTLs in pigs. Mamm. Genome 11, 656–661. doi: 10.1007/s003350010117
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Redrup, L., Branco, M. R., Perdeaux, E. R., Krueger, C., Lewis, A., Santos, F., et al. (2009). The long noncoding RNA Kcnq1ot1 organises a lineage-specific nuclear domain for epigenetic gene silencing. Development 136, 525–530. doi: 10.1242/dev.031328
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reik, W., and Walter, J. (2001). Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2, 21–32. doi: 10.1038/35047554
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ron, M., and Weller, J. I. (2007). From QTL to QTN identification in livestock-winning by points rather than knock-out: a review. Anim. Genet. 38, 429–439. doi: 10.1111/j.1365-2052.2007.01640.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sato, A., Otsu, E., Negishi, H., Utsunomiya, T., and Arima, T. (2007). Aberrant DNA methylation of imprinted loci in superovulated oocytes. Hum. Reprod. 22, 26–35. doi: 10.1093/humrep/del316
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schneider, E., Pliushch, G., El Hajj, N., Galetzka, D., Puhl, A., Schorsch, M., et al. (2010). Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic Acids Res. 38, 3880–3890. doi: 10.1093/nar/gkq126
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seisenberger, S., Andrews, S., Krueger, F., Arand, J., Walter, J., Santos, F., et al. (2012). The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 48, 849–862. doi: 10.1016/j.molcel.2012.11.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sembon, S., Iwamoto, M., Hashimoto, M., Oishi, T., Fuchimoto, D., Suzuki, S., et al. (2012). Porcine androgenetic embryos develop to fetal stage in recipient mothers. Theriogenology 78, 225–231. doi: 10.1016/j.theriogenology.2012.01.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sherman, E. L., Nkrumah, J. D., and Moore, S. S. (2010). Whole genome single nucleotide polymorphism associations with feed intake and feed efficiency in beef cattle. J. Anim. Sci. 88, 16–22. doi: 10.2527/jas.2008-1759
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sikora, K. M., Magee, D. A., Berkowicz, E. W., Berry, D. P., Howard, D. J., Mullen, M. P., et al. (2011). DNA sequence polymorphisms within the bovine guanine nucleotide-binding protein Gs subunit alpha (Gsα)-encoding (GNAS) genomic imprinting domain are associated with performance traits. BMC Genet. 12:4. doi: 10.1186/1471-2156-12-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singh, P., Li, A. X., Tran, D. A., Oates, N., Kang, E. R., Wu, X., et al. (2013). De novo DNA methylation in the male germ line occurs by default but is excluded at sites of H3K4 methylation. Cell Rep. 4, 205–219. doi: 10.1016/j.celrep.2013.06.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smallwood, S. A., Tomizawa, S., Krueger, F., Ruf, N., Carli, N., Segonds-Pichon, A., et al. (2011). Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 43, 811–814. doi: 10.1038/ng.864
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smit, M., Segers, K., Carrascosa, L. G., Shay, T., Baraldi, F., Gyapay, G., et al. (2003). Mosaicism of solid gold supports the causality of a noncoding A-to-G transition in the determinism of the callipyge phenotype. Genetics 163, 453–456.
Smith, L. C., Suzuki, J. Jr., Goff, A. K., Filion, F., Therrien, J., Murphy, B. D., et al. (2012). Developmental and epigenetic anomalies in cloned cattle. Reprod. Domest. Anim. 47(Suppl. 4), 107–114. doi: 10.1111/j.1439-0531.2012.02063.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Snelling, W. M., Cushman, R. A., Keele, J. W., Maltecca, C., Thomas, M. G., Fortes, M. R., et al. (2013). Breeding and genetics symposium: networks and pathways to guide genomic selection. J. Anim. Sci. 91, 537–552. doi: 10.2527/jas.2012-5784
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spencer, H. G. (2000). Population genetics and evolution of genomic imprinting. Annu. Rev. Genet. 34, 457–477. doi: 10.1146/annurev.genet.34.1.457
Spencer, H. G. (2009). Effects of genomic imprinting on quantitative traits. Genetica 136, 285–293. doi: 10.1007/s10709-008-9300-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Surani, M. A., Barton, S. C., and Norris, M. L. (1984). Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308, 548–550. doi: 10.1038/308548a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takeda, H., Caiment, F., Smit, M., Hiard, S., Tordoir, X., Cockett, N., et al. (2006). The callipyge mutation enhances bidirectional long-range DLK1-GTL2 intergenic transcription in cis. Proc. Natl. Acad. Sci. U.S.A. 103, 8119–8124. doi: 10.1073/pnas.0602844103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tellam, R. L., Cockett, N. E., Vuocolo, T., and Bidwell, C. A. (2012). Genes contributing to genetic variation of muscling in sheep. Front. Genet. 3:164. doi: 10.3389/fgene.2012.00164
Terranova, R., Yokobayashi, S., Stadler, M. B., Otte, A. P., van Lohuizen, M., Orkin, S. H., et al. (2008). Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev. Cell 15, 668–679. doi: 10.1016/j.devcel.2008.08.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tian, X. C. (2014). Genomic imprinting in farm animals. Annu. Rev. Anim. Biosci. 2, 23–40. doi: 10.1146/annurev-animal-022513-114144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tier, B., and Meyer, K. (2012). Analysing quantitative parent-of-origin effects with examples from ultrasonic measures of body composition In Australian beef cattle. J. Anim. Breed Genet. 129, 359–368. doi: 10.1111/j.1439-0388.2012.00996.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tomizawa, S., Kobayashi, H., Watanabe, T., Andrews, S., Hata, K., Kelsey, G., et al. (2011). Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 138, 811–820. doi: 10.1242/dev.061416
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Laere, A. S., Nguyen, M., Braunschweig, M., Nezer, C., Collette, C., Moreau, L., et al. (2003). A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425, 832–836. doi: 10.1038/nature02064
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Varona, L., Munilla, S., Mouresan, E. F., Gonzalez-Rodriguez, A., Moreno, C., and Altarriba, J. (2015). A Bayesian model for the analysis of transgenerational epigenetic variation. G3 (Bethesda) doi: 10.1534/g3.115.016725 [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vuocolo, T., Byrne, K., White, J., McWilliam, S., Reverter, A., Cockett, N. E., et al. (2007). Identification of a gene network contributing to hypertrophy in callipyge skeletal muscle. Physiol. Genomics 28, 253–272. doi: 10.1152/physiolgenomics.00121.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wagschal, A., Sutherland, H. G., Woodfine, K., Henckel, A., Chebli, K., Schulz, R., et al. (2008). G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell. Biol. 28, 1104–1113. doi: 10.1128/MCB.01111-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weaver, J. R., and Bartolomei, M. S. (2014). Chromatin regulators of genomic imprinting. Biochim. Biophys. Acta 1839, 169–177. doi: 10.1016/j.bbagrm.2013.12.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wei, Y., Su, J., Liu, H., Lv, J., Wang, F., Yan, H., et al. (2014). MetaImprint: an information repository of mammalian imprinted genes. Development 141, 2516–2523. doi: 10.1242/dev.105320
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weksberg, R., Nishikawa, J., Caluseriu, O., Fei, Y. L., Shuman, C., Wei, C., et al. (2001). Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum. Mol. Genet. 10, 2989–3000. doi: 10.1093/hmg/10.26.2989
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weksberg, R., Shuman, C., and Beckwith, J. B. (2010). Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18, 8–14. doi: 10.1038/ejhg.2009.106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weller, J. I., and Ron, M. (2011). Invited review: quantitative trait nucleotide determination in the era of genomic selection. J. Dairy Sci. 94, 1082–1090. doi: 10.3168/jds.2010-3793
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wolf, J. B., Cheverud, J. M., Roseman, C., and Hager, R. (2008). Genome-wide analysis reveals a complex pattern of genomic imprinting in mice. PLoS Genet. 4:e1000091. doi: 10.1371/journal.pgen.1000091
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, X., Smith, S. L., Tian, X. C., Lewin, H. A., Renard, J. P., and Wakayama, T. (2007). Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat. Genet. 39, 295–302. doi: 10.1038/ng1973
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Li, T., Vu, T. H., Ulaner, G. A., Hu, J. F., and Hoffman, A. R. (2003). The histone code regulating expression of the imprinted mouse Igf2r gene. Endocrinology 144, 5658–5670. doi: 10.1210/en.2003-0798
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
York, B., Lei, K., and West, D. B. (1997). Inherited non-autosomal effects on body fat in F2 mice derived from an AKR/J x SWR/J cross. Mamm. Genome 8, 726–730. doi: 10.1007/s003359900554
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yoshioka, H., McCarrey, J. R., and Yamazaki, Y. (2009). Dynamic nuclear organization of constitutive heterochromatin during fetal male germ cell development in mice. Biol. Reprod. 80, 804–812. doi: 10.1095/biolreprod.108.072603
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Young, L. E., Fernandes, K., McEvoy, T. G., Butterwith, S. C., Gutierrez, C. G., Carolan, C., et al. (2001). Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 27, 153–154. doi: 10.1038/84769
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Young, L. E., Sinclair, K. D., and Wilmut, I. (1998). Large offspring syndrome in cattle and sheep. Rev. Reprod. 3, 155–163. doi: 10.1530/ror.0.0030155
Zacchini, F., Czernik, M., Iuso, D., Toschi, P., di Egidio, F., Scapolo, P. A., et al. (2011). Efficient production and cellular characterization of sheep androgenetic embryos. Cell Reprogram 13, 495–502. doi: 10.1089/cell.2011.0021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: complex traits, epigenetics, epigenome, genomic imprinting, livestock
Citation: O’Doherty AM, MacHugh DE, Spillane C and Magee DA (2015) Genomic imprinting effects on complex traits in domesticated animal species. Front. Genet. 6:156. doi: 10.3389/fgene.2015.00156
Received: 28 December 2014; Accepted: 06 April 2015;
Published online: 24 April 2015.
Edited by:
Oscar Gonzalez-Recio, Department of Environment and Primary Industries, AustraliaReviewed by:
Dan Nonneman, United States Department of Agriculture, USARomi Pena I. Subirà, University of Lleida, Spain
Copyright © 2015 O’Doherty, MacHugh, Spillane and Magee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Magee, Animal Genomics Laboratory, UCD School of Agriculture and Food Science, University College Dublin, Belfield, Dublin 4, Ireland; Department of Animal Science, University of Connecticut, Storrs, CT 06029, USAZGF2aWQubWFnZWVAdWNkLmll