 Karen Usdin
Karen Usdin- Section on Gene Structure and Disease, Laboratory of Cell and Molecular Biology, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA
The fragile X-related disorders are members of the Repeat Expansion Diseases, a group of genetic conditions resulting from an expansion in the size of a tandem repeat tract at a specific genetic locus. The repeat responsible for disease pathology in the fragile X-related disorders is CGG/CCG and the repeat tract is located in the 5′ UTR of the FMR1 gene, whose protein product FMRP, is important for the proper translation of dendritic mRNAs in response to synaptic activation. There are two different pathological FMR1 allele classes that are distinguished only by the number of repeats. Premutation alleles have 55–200 repeats and confer risk of fragile X-associated tremor/ataxia syndrome and fragile X-associated primary ovarian insufficiency. Full mutation alleles on the other hand have >200 repeats and result in fragile X syndrome, a disorder that affects learning and behavior. Different symptoms are seen in carriers of premutation and full mutation alleles because the repeat number has paradoxical effects on gene expression: Epigenetic changes increase transcription from premutation alleles and decrease transcription from full mutation alleles. This review will cover what is currently known about the mechanisms responsible for these changes in FMR1 expression and how they may relate to other Repeat Expansion Diseases that also show repeat-mediated changes in gene expression.
Introduction
The fragile X-related disorders are members of 20+ human genetic conditions known as the Repeat Expansion Diseases (Mirkin, 2007). The disease-causing mutation in all cases is an expansion or increase in the number of repeats in a specific tandem repeat tract. In the case of the fragile X-related disorders, the repeat unit is CGG/CCG and the repeat tract is located in the 5′ untranslated region of the fragile X mental retardation 1 (FMR1) gene (Fu et al., 1991; Verkerk et al., 1991). The FMR1 gene encodes FMRP, a protein important for the regulation of translation of dendritic mRNAs in response to synaptic activation (Weiler et al., 1997; Weiler and Greenough, 1999). Two different pathological FMR1 allele size classes are distinguished. Premutation alleles have 55–200 repeats, while alleles with >200 repeats are referred to as full mutation alleles. These two size classes are distinguished because they confer risk of different clinical conditions. Carriers of premutation alleles are at risk of an adult-onset neurodegenerative disorder known as fragile X-associated tremor/ataxia syndrome (reviewed, in Hagerman, 2013). Female carriers are also at risk of fragile X-associated primary ovarian insufficiency, a condition that is associated with fertility problems and an earlier than normal menopause (reviewed in, Sullivan et al., 2011; Sherman et al., 2014). In contrast, full mutation alleles are associated with fragile X syndrome, the leading heritable cause of intellectual disability and the major monogenic cause of autism (Oberle et al., 1991; Fu et al., 1991; Verkerk et al., 1991).
Premutation and full mutation carrier symptoms are different, in part, because the repeat has paradoxical effects on gene expression. Premutation alleles are hyper-expressed (Tassone et al., 2000a) and disease symptoms result from the deleterious consequences of high levels of expression of the transcript with large numbers of CGG-repeats (reviewed, in Sellier et al., 2014). In contrast, the symptoms of fragile X syndrome result from repeat-mediated heterochromatin formation that causes transcriptional silencing and a subsequent deficiency of FMRP (Pieretti et al., 1991; Sutcliffe et al., 1992). Interestingly, it is becoming increasingly apparent that heterochromatin formation is a common feature of other Repeat Expansion Diseases that involve large repeat tracts. This includes myotonic dystrophy type 1, Friedreich ataxia and amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD; Otten and Tapscott, 1995; Steinbach et al., 1998; Herman et al., 2006; Greene et al., 2007; Belzil et al., 2013; Xi et al., 2013).
How the repeats cause hyper-expression of premutation alleles and silencing of full mutation alleles is not well understood. This review will cover recent findings that suggest how the repeats are able to have such different effects on gene expression.
The CGG-repeat Tract Forms Stable Secondary Structures
The CGG-repeat that is present in both the FMR1 gene and its transcript can form a variety of secondary structures. In vitro the DNA repeats form a stem-loop/hairpin and a folded-hairpin-like structure known as a G-tetraplex or quadruplex (Fry and Loeb, 1994; Kettani et al., 1995; Mitas et al., 1995; Usdin and Woodford, 1995; Patel et al., 2000). The hairpin contains a mixture of Watson-Crick G:C base pairs and Hoogsteen G:G base pairs in a 2:1 ratio. The quadruplex is stabilized primarily by G-quartets. The RNA forms a similar set of structures (Handa et al., 2003; Zumwalt et al., 2007; Malgowska et al., 2014). Similar structures are also formed by many of the repeats responsible for other members of the Repeat Expansion Disease family that show repeat-mediated epigenetic changes (reviewed, in Usdin et al., 2015), and quadruplex formation has been reported for the very GC-rich ALS/FTD repeats (Fratta et al., 2012; Reddy et al., 2013; Haeusler et al., 2014) and the repeats responsible for progressive myoclonus epilepsy type 1 (EPM1; Saha and Usdin, 2001). There is evidence for the formation of stem-loop structures in vivo by the CGG-strand of the fragile X repeat (Loomis et al., 2014) and the CAG- and CTG-repeats responsible for diseases like Myotonic dystrophy type 1 and Huntington disease (Liu et al., 2010). Transient unpairing of the repeat region during DNA replication, DNA repair or transcription is thought to provide the opportunity for these structures to form.
In addition to the above-mentioned structures, many disease-associated repeats including the fragile X repeats (Groh et al., 2014; Loomis et al., 2014), the Friedreich ataxia repeat (Groh et al., 2014), the ALS/FTD repeat (Haeusler et al., 2014) and the CTG/CAG-repeats responsible for a number of Repeat Expansion Diseases (Lin et al., 2010) form stable R loops during transcription. This R loop contains an RNA:DNA hybrid formed between the nascent transcript and the transcribed strand. This leaves the displaced non-template strand unpaired. Such R loops form in regions with strand-asymmetry with regard to the distribution of purines and pyrimidines, particularly when the pyrimidine-rich strand is being transcribed. This R loop may be facilitated by hairpin formation by the non-template strand that would reduce the likelihood of reannealing of the duplex behind the advancing transcription complex. Conversely, the persistence of this hybrid might also favor the formation of the hairpin on the non-template strand. R loops are the only structures identified to date that are formed by all repeats that become heterochromatinized. In bacteria and yeast R loops can also be formed in trans (e.g., by a distally transcribed mRNA) in the presence of RecA and Rad51 respectively (Zaitsev and Kowalczykowski, 2000; Wahba and Koshland, 2013), but evidence for such R loops in mammalian cells is lacking.
The number of repeats required to form an R loop on the FMR1 gene and the timing of R loop formation is the subject of some debate. One study suggests that they are only formed on full mutation alleles relatively late in neuronal differentiation (Colak et al., 2014), while other work suggests that R loops at the FMR1 locus also form on normal and premutation alleles in a number of different cell types (Groh et al., 2014; Loomis et al., 2014). This discrepancy may reflect differences in the stability of the structures being measured. Formation of these structures in vivo likely reflects some combination of the effect of repeat number, transcription rate (Groh et al., 2014; Loomis et al., 2014) and the expression of proteins that affect R loop stability (Colak et al., 2014).
Repeat-mediated Epigenetic Effects on the Premutation Allele
There is a direct relationship between the repeat number and FMR1 mRNA levels in humans and mice carrying the premutation allele, such that premutation carriers have 2–10 times more FMR1 mRNA than individuals with repeat numbers in the normal range (Tassone et al., 2000b; Entezam et al., 2007; Brouwer et al., 2008). The increased RNA levels are the result of increased transcription initiation rather than increased transcript stability (Tassone et al., 2007). The promoter of premutation alleles is enriched for acetylated histones (Todd et al., 2010) and premutation alleles initiate transcription from upstream start sites more frequently than is seen in normal cells (Beilina et al., 2004). While hyper-expression has not been reported for other Repeat Expansion Diseases, a similar increased usage of more 5′ start sites has been reported in individuals with ALS (Sareen et al., 2013).
Very little is currently known about why premutation alleles are overexpressed. CGG/CCG-repeats exclude nucleosomes in vitro (Wang et al., 1996). In principle, this could result in the 5′ end of the FMR1 gene being more accessible to transcription factors or to the transcription complex. However, to date there is no evidence for repeat-mediated nucleosome exclusion on FMR1 alleles in vivo. The FMR1 promoter is very CpG rich and has many of the hallmarks of a CpG-island promoter. CpG-island promoters act as transcription-independent nucleation sites for the zinc finger CxxC domain-containing chromatin modifying proteins like CxxC finger protein (CFP1; Thomson et al., 2010). CFP1 is a component of the SET1A/B-containing methyltransferase complex that facilitates H3K4 trimethylation (Clouaire et al., 2012), a histone mark typically associated with the 5′ ends of active genes. This has led to the suggestion that such proteins provide a self-reinforcing loop of unmethylated CpG recognition and subsequent protection from DNA methylation (Blackledge et al., 2013). In this view, hyper-expression of premutation alleles would be related to the high density of CpGs in the repeat that favors recruitment of factors that inhibit gene silencing. Alternatively, proteins like ATRX, a member of the SNF2 family of helicases/ATPases, could also contribute to hyper-expression of premutation alleles. ATRX colocalizes with G-rich regions that, like the FMR1 locus (Fry and Loeb, 1994; Kettani et al., 1995; Usdin and Woodford, 1995; Patel et al., 2000), have quadruplex-forming potential. ATRX facilitates transcription elongation through these regions by reducing transcription stalling (Levy et al., 2015).
R loops are a characteristic feature of unmethylated CpG-islands where they have been suggested to play a role in preventing gene silencing (Ginno et al., 2012). There is evidence to suggest that R loops provide some protection from de novo methylation by DNMT3B1, the primary de novo DNA methyltransferase active during early development (Ginno et al., 2012). How they do so is currently unknown, but one possibility is that the single stranded region of the R loop is a preferential binding site for a number of epigenetic modifiers that are positive regulators of transcription. These modifiers include members of the H3K4 methyltransferase family (Krajewski et al., 2005), whose activity is thought to inhibit de novo methylation (Ooi et al., 2007). It also includes the activation-induced cytosine deaminase (Chaudhuri et al., 2003) that is thought to be important for DNA demethylation (Popp et al., 2010). R loops may also be less capable of properly binding nucleosomes (Dunn and Griffith, 1980) and there is evidence to suggest that R loop formation causes chromosome decondensation (Powell et al., 2013). R loops formed on longer repeat tracts have been shown to extend further into the flanking regions (Loomis et al., 2014). This could perhaps favor transcription initiation by increasing the likelihood that promoter melting will occur or by facilitating the binding of additional transcription factors or chromatin modifiers to the promoter that in turn promote transcription initiation.
Repeat-mediated Gene Silencing of Full Mutation Alleles
In contrast to the marks of active chromatin found at the 5′ end of the FMR1 gene in premutation carriers, full mutation alleles are hypermethylated and associated with hypoacetylated histones (Chiurazzi et al., 1998, 1999; Coffee et al., 1999; Pietrobono et al., 2002, 2005; Biacsi et al., 2008). They are also hypomethylated on H3K4, enriched for marks typical of facultative heterochromatin like H3K9me2 and H3K27me3, as well as histone modifications associated with constitutive heterochromatin- H3K9me3 and H4K20me3 (Kumari and Usdin, 2010). H3K9 dimethylation has also been reported on Myotonic dystrophy type 1 alleles (Cho et al., 2005), H3K9 trimethylation has been reported on Friedreich ataxia alleles (Herman et al., 2006) and H3K9, H3K27, and H4K20 trimethylation have been reported for heterochromatinized ALS alleles (Belzil et al., 2013).
Treatment of fragile X patient cells with DNA methylation inhibitors leads to partial gene reactivation that is not associated with the loss of H3K9me2 or H3K9me3 (Kumari and Usdin, 2014). This suggests that DNA methylation occurs downstream, or is independent of, the deposition of these chromatin modifications. This would be consistent with the observation that in rare full mutation carriers in which gene silencing does not occur, the FMR1 gene is enriched for H3K9me2 but shows no CpG methylation (Tabolacci et al., 2008). The FMR1 gene can also be reactivated by inhibition of Sirtuin 1 (SIRT1), the enzyme responsible for the deacetylation of H3K9 and H4K16 on fragile X alleles (Biacsi et al., 2008). Since DNA demethylation leads to acetylation of H4K16 but not H3K9 (Biacsi et al., 2008), it would be consistent with the idea that deacetylation of H3K9 precedes DNA methylation while deacetylation of H4K16 is a late event in the silencing process acting downstream of DNA methylation.
The earliest epigenetic change associated with silencing of the fragile X allele is unknown. However, the fact that the histone marks H3K9me3 and H4K20me3 show a peak of enrichment in the region of the fragile X repeat would be consistent with the idea that the repeats themselves are the site of nucleation for the silencing process (Kumari and Usdin, 2010). FMR1 mRNA knockdown blocks FMR1 gene silencing during neuronal differentiation in fragile X embryonic stem cells (Colak et al., 2014) and decreases the recruitment of polycomb repressive complex 2 (PRC2) to full mutation alleles that have been reactivated with 5-azadeoxycytidine (Kumari and Usdin, 2014). These findings suggest that the FMR1 transcript plays a key role in gene silencing by facilitating, either directly or indirectly, the recruitment of repressive histone modifying complexes like PRC2.
The FMR1 transcript may facilitate gene silencing by recruiting repressive histone modifiers as many long non-coding RNAs do. Examples are known where such RNA acts in cis or trans to recruit PRC2 along with either the LSD1-CoREST complexes responsible for H3K4me2 demethylation (Tsai et al., 2010), or the polycomb repressive complex 1 (Yap et al., 2010), or the H3K9 methylase G9a (Pandey et al., 2008). Long non-coding RNAs are also involved in recruiting the H4K20 trimethylase Suv4-20h (Bierhoff et al., 2014). The FMR1 transcript may be acting like these RNAs to recruit repressors to the fragile X locus. These factors may in turn act as a molecular scaffold to which other chromatin modifiers bind to ultimately generate the histone modification signature found on fragile X alleles (Kumari and Usdin, 2010). Of interest in this regard is the fact that the Fmr1 transcript is one of the transcripts most commonly associated with PRC2 in normal mouse embryonic stem cells (Zhao et al., 2010). This may be related to the ability of the 5′ end of the transcript to form stem-loop structures as assessed by the RNAfold algorithm (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). Such structures are thought to be important for PRC2 binding (Zhao et al., 2008). The human FMR1 transcript is predicted to form similar structures and may thus also recruit PRC2 to even to normal and premutation alleles. However, not only would the amount of PRC2 recruited to the FMR1 locus be limited by the relatively low stability of the R loop on small repeat tracts, but any activity of PRC2 would be also inhibited by the presence of nascent RNA (Cifuentes-Rojas et al., 2014) or H3K36me3 (Schmitges et al., 2011), a histone modification deposited co-transcriptionally by the SETD2 protein. Even should some trimethylation of H3K27 occur, it’s spread would be limited by the high levels of H3K36me3 present in the region (Yuan et al., 2011).
In some cases targeting of the long non-coding RNA is thought to be accomplished by a bivalent protein that binds both the RNA and the target locus (Jeon and Lee, 2011), whilst in others the RNA is tethered to its target gene via the formation of a RNA:DNA:DNA triplex (Schmitz et al., 2010), or an RNA: DNA hybrid (Rinn and Chang, 2012). Tethering via the formation of an RNA:RNA hybrid has also been suggested (Rinn and Chang, 2012). Recruitment of repressive complexes via an RNA:DNA hybrid may be relevant for FMR1 gene silencing given the persistent R loop that is found coincident with the repeat on FMR1 alleles (Groh et al., 2014; Loomis et al., 2014) and the effect of R loop depletion on gene silencing in fragile X ESC-derived neurons (Colak et al., 2014). R loops have also been implicated in gene silencing in Friedreich ataxia (Groh et al., 2014). In principle, either a cis or trans-association of the transcript with the FMR1 locus could tether the transcript to the promoter allowing it to recruit repressive epigenetic modifiers like PRC2 that ultimately result in FMR1 gene silencing.
Gene silencing may also be triggered by the R loops themselves in ways that are independent of the FMR1 transcript per se. The free DNA strand in the fragile X R loop forms secondary structures (Loomis et al., 2014) that have been suggested to directly recruit DNA methyltransferases (Smith et al., 1994). However, our data suggests that DNA methylation is not the first step in the fragile X gene silencing process (Biacsi et al., 2008; Kumari and Usdin, 2014). Furthermore, Friedreich ataxia repeats do not form such secondary structures and methylatable CpG residues are not present in either the Friedreich ataxia repeats or the myotonic dystrophy type 1 repeats. Thus if gene silencing in these disorders has a common molecular basis, the use of this pathway to initiate gene silencing seems unlikely. R loops also facilitate Pol II pausing and termination (Skourti-Stathaki et al., 2011; Groh et al., 2014). In the β-actin locus pausing results in the production of an antisense transcript. The sense-antisense RNA pair leads to the RNAi-dependent deposition of H3K9me2 and the recruitment of heterochromatin protein 1γ (HP1γ; Skourti-Stathaki et al., 2014). However, knockdown of Dicer, Ago1, and Ago2, genes important for RNAi did not prevent FMR1 gene silencing in neurons differentiated from fragile X embryonic stem cells (Colak et al., 2014). This would seem to rule out a role for RNAi in silencing of full mutation alleles, whether the RNAi is R loop-mediated or mediated independently of R loops by the interaction of the FMR1 sense and antisense transcript (Ladd et al., 2007) or CGG-RNA hairpins that are Dicer substrates (Handa et al., 2003). R loops are also prone to chromatin compaction via a H3S10 phosphorylation-dependent mechanism that is still unknown (Castellano-Pozo et al., 2013). R loops have also been shown to result in double-strand breaks (Sordet et al., 2010) that could lead to the recruitment of a variety of epigenetic modifiers including DNA methyltransferase 1 and SIRT1 (O’Hagan et al., 2008).
Unresolved Questions and Future Directions
More work is needed before we understand the epigenetic regulation of the FMR1 gene. Until we do the question of why shorter repeat tracts favor increased gene expression while longer ones favor gene silencing remains unanswered. The paradoxical effect of the repeats presumably reflects the equilibrium between interactions of the repeat tract with factors that favor the accumulation of positive chromatin modifications and those that favor the generation of repressive chromatin. For example, as illustrated in Figure 1, smaller repeat tracts may be more likely to form RNA: DNA hybrids that have a short half-life and that may leave the non-template strand single-stranded just long enough to bind transcriptional activators or DNA demethylases that have a preference for single-stranded regions. In contrast, longer repeat tracts would be more likely to form stable RNA:DNA hybrids that were better able to recruit complexes like G9a and PRC2 that deposit repressive chromatin marks. The longer hybrids may also be more prone to transcription termination that could result in the loss of H3K36me3 and H3K4me3 and thus the protection from encroachment of repressive chromatin (Yuan et al., 2011). The non-template strand may also be more likely to form stable secondary structures that reduce binding of transcription activators with a preference for single-stranded regions.
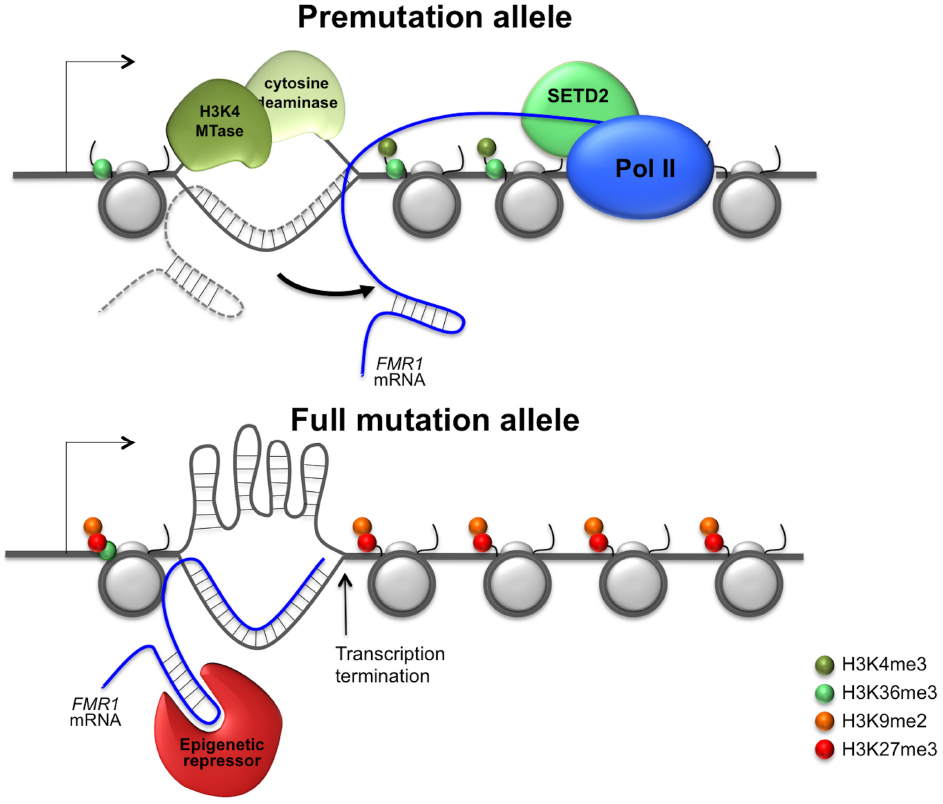
Figure 1. Model for repeat-mediated gene dysregulation in fragile X premutation and full mutation carriers. A metastable R loop formed by the premutation allele would leave the non-template strand transiently unpaired and thus able to recruit transcription activators that show a preferential binding to single-stranded regions (Chaudhuri et al., 2003; Krajewski et al., 2005). However, since the RNA:DNA hybrid is relatively short, transcription termination is low and the transcript is not tethered to the FMR1 locus long enough to recruit transcriptional repressors that might bind the hairpins formed by the 5′ end of the FMR1 transcript. The net result is that the premutation allele would be associated with elevated levels of active histone modifications and thus hyper-expressed. In contrast, on full mutation alleles the RNA:DNA hybrid is likely to be more stable . It may thus be able to effectively recruit repressive chromatin modifiers to the 5′ end of the FMR1 gene that result in the deposition of repressive histone marks. The hybrid may also be long enough to cause significant transcription termination (Skourti-Stathaki et al., 2011; Groh et al., 2014). This would result in a drop in the levels of co-transcriptionally deposited active chromatin modifications. This could result in the loss of the protective effect that these histone marks provide against the deposition of repressive histone marks (Schmitges et al., 2011; Yuan et al., 2011). The non-template strand in the R loop may also be more likely to form secondary structures and thus less likely to bind transcription activators with a preference for single-stranded regions. The net result would be transcriptional silencing of the full mutation allele.
In addition, while the idea that a co-transcriptionally formed R loop is responsible for silencing is appealing, the ability of siRNA or shRNA to the FMR1 transcript to reduce repression of fragile X alleles (Colak et al., 2014; Kumari and Usdin, 2014) suggests that the situation may not be quite so simple. Despite reports of active RNAi factors in the nucleus (Gagnon et al., 2014), the ability of RNAi to deplete RNAs that are exclusively nuclear is still controversial. An alternative explanation consistent with the available data is that the R loop forms in trans from a transcript that has transitioned through the cytoplasm. If so, then R loop formation may be facilitated by the ability of the CGG-strand of the DNA to form secondary structures during transcription thus leaving the CCG-DNA strand free.
The question of the timing of gene silencing in the fragile X embryo is also still unresolved. This is significant for our understanding as to whether FMRP is present during early embryonic development in individuals with fragile X syndrome. Silencing has been reported to be a relatively late event in embryonic development, occurring >45 days after differentiation of neurons from embryonic stem cells is initiated (Colak et al., 2014). However, in many fragile X ESC lines significant gene silencing has already occurred (Avitzour et al., 2014). This includes the ESC lines used in Colak et. al. study as evidenced by the fact that a significant fraction of the fragile X alleles in these cells are resistant to digestion by Eag I, a methylation-sensitive restriction enzyme (Supplementary Figure 1; Colak et al., 2014). How much silencing occurs in the early embryo and how this silencing compares to the silencing observed in neurons are thus important open questions.
There is also the question of what, if anything, can be done to prevent or reverse the repeat-mediated epigenetic changes in premutation and full mutation carriers. A number of compounds that are able to inhibit repressive epigenetic modifications are already in clinical trials for treatment of Friedreich ataxia. It is of interest that Vitamin B3 (nicotinamide), an inhibitor of SIRT1, has shown promise in Phase I trials for Friedreich ataxia (Libri et al., 2014). The identification of FMR1 transcription as the trigger for silencing opens up a whole new potential approach to gene reactivation. A small molecule that blocks hybrid formation and prevents silencing (Colak et al., 2014) was unable to reactivate a silenced allele. However, it may able to do so in combination with other epigenetic modifiers. Of course, any benefit to be gained by reactivation of the fragile X allele would have to be offset by the risk posed by expression of FMR1 mRNA with long CGG-repeat tracts (Loesch et al., 2012). It may also be possible to ameliorate the symptoms seen in premutation carriers by using epigenetic modifying compounds that reduce the FMR1 hyper-expression. For example, since SIRT1 is involved in down regulation of FMR1 expression, SIRT1 activators may help normalize the levels of the toxic transcript produced from premutation alleles.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Drs Ann Dean, Kai Ge and Bruce Hayward for their careful reading of the manuscript. This work was made possible by a grant to KU from the Intramural Research Program of the NIDDK, NIH (DK057808).
References
Avitzour, M., Mor-Shaked, H., Yanovsky-Dagan, S., Aharoni, S., Altarescu, G., Renbaum, P., et al. (2014). FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports 3, 699–706. doi: 10.1016/j.stemcr.2014.09.001
Beilina, A., Tassone, F., Schwartz, P. H., Sahota, P., and Hagerman, P. J. (2004). Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum. Mol. Genet. 13, 543–549. doi: 10.1093/hmg/ddh053
Belzil, V. V., Bauer, P. O., Prudencio, M., Gendron, T. F., Stetler, C. T., Yan, I. K., et al. (2013). Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neupathol. 126, 895–905. doi: 10.1007/s00401-013-1199-1
Biacsi, R., Kumari, D., and Usdin, K. (2008). SIRT1 inhibition alleviates gene silencing in fragile X mental retardation syndrome. PLoS Genet. 4:e1000017. doi: 10.1371/journal.pgen.1000017
Bierhoff, H., Dammert, M. A., Brocks, D., Dambacher, S., Schotta, G., and Grummt, I. (2014). Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell 54, 675–682. doi: 10.1016/j.molcel.2014.03.032
Blackledge, N. P., Thomson, J. P., and Skene, P. J. (2013). CpG island chromatin is shaped by recruitment of ZF-CxxC proteins. Cold Spring Harb. Perspect. Biol. 5, a018648. doi: 10.1101/cshperspect.a018648
Brouwer, J. R., Huizer, K., Severijnen, L. A., Hukema, R. K., Berman, R. F., Oostra, B. A., et al. (2008). CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J. Neurochem. 107, 1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x
Castellano-Pozo, M., Santos-Pereira, J. M., Rondon, A. G., Barroso, S., Andujar, E., Perez-Alegre, M., et al. (2013). R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 52, 583–590. doi: 10.1016/j.molcel.2013.10.006
Chaudhuri, J., Tian, M., Khuong, C., Chua, K., Pinaud, E., and Alt, F. W. (2003). Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 422, 726–730. doi: 10.1038/nature01574
Chiurazzi, P., Pomponi, M. G., Pietrobono, R., Bakker, C. E., Neri, G., and Oostra, B. A. (1999). Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 8, 2317–2323. doi: 10.1093/hmg/8.12.2317
Chiurazzi, P., Pomponi, M. G., Willemsen, R., Oostra, B. A., and Neri, G. (1998). In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 7, 109–113. doi: 10.1093/hmg/7.1.109
Cho, D. H., Thienes, C. P., Mahoney, S. E., Analau, E., Filippova, G. N., and Tapscott, S. J. (2005). Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 20, 483–489. doi: 10.1016/j.molcel.2005.09.002
Cifuentes-Rojas, C., Hernandez, A. J., Sarma, K., and Lee, J. T. (2014). Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 55, 171–185. doi: 10.1016/j.molcel.2014.05.009
Clouaire, T., Webb, S., Skene, P., Illingworth, R., Kerr, A., Andrews, R., et al. (2012). Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 26, 1714–1728. doi: 10.1101/gad.194209.112
Coffee, B., Zhang, F., Warren, S. T., and Reines, D. (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 22, 98–101.
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W. Y., Gerhardt, J., et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. doi: 10.1126/science.1245831
Dunn, K., and Griffith, J. D. (1980). The presence of RNA in a double helix inhibits its interaction with histone proteins. Nucl. Acids Res. 11, 555–566.
Entezam, A., Biacsi, R., Orrison, B., Saha, T., Hoffman, G. E., Grabczyk, E., et al. (2007). Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene 395, 125–134. doi: 10.1016/j.gene.2007.02.026
Fratta, P., Mizielinska, S., Nicoll, A. J., Zloh, M., Fisher, E. M., Parkinson, G., et al. (2012). C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2, 1016. doi: 10.1038/srep01016
Fry, M., and Loeb, L. A. (1994). The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. U.S.A. 91, 4950–4954.
Fu, Y. H., Kuhl, D. P., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S., et al. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058.
Gagnon, K. T., Li, L., Chu, Y., Janowski, B. A., and Corey, D. R. (2014). RNAi factors are present and active in human cell nuclei. Cell Rep. 6, 211–221. doi: 10.1016/j.celrep.2013.12.013
Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., and Chedin, F. (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 45, 814–825. doi: 10.1016/j.molcel.2012.01.017
Greene, E., Mahishi, L., Entezam, A., Kumari, D., and Usdin, K. (2007). Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 35, 3383–3390. doi: 10.1093/nar/gkm271
Groh, M., Lufino, M. M., Wade-Martins, R., and Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10:e1004318. doi: 10.1371/journal.pgen.1004318
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A., Shaw, P. G., Kim, M. S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
Hagerman, P. (2013). Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 126, 1–19. doi: 10.1007/s00401-013-1138-1
Handa, V., Saha, T., and Usdin, K. (2003). The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 31, 6243–6248. doi: 10.1093/nar/gkg818
Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S. L., and Gottesfeld, J. M. (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2, 551–558. doi: 10.1038/nchembio815
Jeon, Y., and Lee, J. T. (2011). YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146, 119–133. doi: 10.1016/j.cell.2011.06.026
Kettani, A., Kumar, R. A., and Patel, D. J. (1995). Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 254, 638–656.
Krajewski, W. A., Nakamura, T., Mazo, A., and Canaani, E. (2005). A motif within SET-domain proteins binds single-stranded nucleic acids and transcribed and supercoiled DNAs and can interfere with assembly of nucleosomes. Mol. Cell. Biol. 25, 1891–1899. doi: 10.1128/MCB.25.5.1891-1899.2005
Kumari, D., and Usdin, K. (2010). The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 19, 4634–4642. doi: 10.1093/hmg/ddq394
Kumari, D., and Usdin, K. (2014). Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 23, 6575–6583. doi: 10.1093/hmg/ddu378
Ladd, P. D., Smith, L. E., Rabaia, N. A., Moore, J. M., Georges, S. A., Hansen, R. S., et al. (2007). An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187. doi: 10.1093/hmg/ddm293
Levy, M. A., Kernohan, K. D., Jiang, Y., and Berube, N. G. (2015). ATRX promotes gene expression by facilitating transcriptional elongation through guanine-rich coding regions. Hum. Mol. Genet. 24, 1824–1835. doi: 10.1093/hmg/ddu596
Libri, V., Yandim, C., Athanasopoulos, S., Loyse, N., Natisvili, T., Law, P. P., et al. (2014). Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: an exploratory, open-label, dose-escalation study. Lancet 384, 504–513. doi: 10.1016/S0140-6736(14)60382-2
Lin, Y., Dent, S. Y., Wilson, J. H., Wells, R. D., and Napierala, M. (2010). R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. U.S.A. 107, 692–697. doi: 10.1073/pnas.0909740107
Liu, G., Chen, X., Bissler, J. J., Sinden, R. R., and Leffak, M. (2010). Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat. Chem. Biol. 6, 652–659. doi: 10.1038/nchembio.416
Loesch, D. Z., Sherwell, S., Kinsella, G., Tassone, F., Taylor, A., Amor, D., et al. (2012). Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin. Genet. 82, 88–92. doi: 10.1111/j.1399-0004.2011.01675.x
Loomis, E. W., Sanz, L. A., Chedin, F., and Hagerman, P. J. (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10:e1004294. doi: 10.1371/journal.pgen.1004294
Malgowska, M., Gudanis, D., Kierzek, R., Wyszko, E., Gabelica, V., and Gdaniec, Z. (2014). Distinctive structural motifs of RNA G-quadruplexes composed of AGG, CGG and UGG trinucleotide repeats. Nucleic Acids Res. 42, 10196–10207. doi: 10.1093/nar/gku710
Mirkin, S. M. (2007). Expandable DNA repeats and human disease. Nature 447, 932–940. doi: 10.1038/nature05977
Mitas, M., Yu, A., Dill, J., and Haworth, I. S. (1995). The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 34, 12803–12811.
O’Hagan, H. M., Mohammad, H. P., and Baylin, S. B. (2008). Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 4:e1000155. doi: 10.1371/journal.pgen.1000155
Oberle, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi: 10.1126/science.252.5009.1097
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. doi: 10.1038/nature05987
Otten, A. D., and Tapscott, S. J. (1995). Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc. Natl. Acad. Sci. U.S.A. 92, 5465–5469.
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
Patel, P. K., Bhavesh, N. S., and Hosur, R. V. (2000). Cation-dependent conformational switches in d-TGGCGGC containing two triplet repeats of Fragile X Syndrome: NMR observations. Biochem. Biophys. Res. Commun. 278, 833–838. doi: 10.1006/bbrc.2000.3878
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822.
Pietrobono, R., Pomponi, M. G., Tabolacci, E., Oostra, B., Chiurazzi, P., and Neri, G. (2002). Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 30, 3278–3285. doi: 10.1093/nar/gkf434
Pietrobono, R., Tabolacci, E., Zalfa, F., Zito, I., Terracciano, A., Moscato, U., et al. (2005). Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 14, 267–277. doi: 10.1093/hmg/ddi024
Popp, C., Dean, W., Feng, S., Cokus, S. J., Andrews, S., Pellegrini, M., et al. (2010). Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463, 1101–1105. doi: 10.1038/nature08829
Powell, W. T., Coulson, R. L., Gonzales, M. L., Crary, F. K., Wong, S. S., Adams, S., et al. (2013). R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. U.S.A. 110, 13938–13943. doi: 10.1073/pnas.1305426110
Reddy, K., Zamiri, B., Stanley, S. Y., Macgregor, R. B. Jr., and Pearson, C. E. (2013). The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 288, 9860–9866. doi: 10.1074/jbc.C113.452532
Rinn, J. L., and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. doi: 10.1146/annurev-biochem-051410-092902
Saha, T., and Usdin, K. (2001). Tetraplex formation by the progressive myoclonus epilepsy type-1 repeat: implications for instability in the repeat expansion diseases. FEBS Lett. 491, 184–187. doi: 10.1016/S0014-5793(01)02190-1
Sareen, D., O’rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5, 208ra149. doi: 10.1126/scitranslmed.3007529
Schmitges, F. W., Prusty, A. B., Faty, M., Stutzer, A., Lingaraju, G. M., Aiwazian, J., et al. (2011). Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell. 42, 330–341. doi: 10.1016/j.molcel.2011.03.025
Schmitz, K. M., Mayer, C., Postepska, A., and Grummt, I. (2010). Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 24, 2264–2269. doi: 10.1101/gad.590910
Sellier, C., Usdin, K., Pastori, C., Peschansky, V. J., Tassone, F., and Charlet-Berguerand, N. (2014). The multiple molecular facets of fragile X-associated tremor/ataxia syndrome. J. Neurodev. Disord. 6, 23. doi: 10.1186/1866-1955-6-23
Sherman, S. L., Curnow, E. C., Easley, C. A., Jin, P., Hukema, R. K., Tejada, M. I., et al. (2014). Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). J. Neurodev. Disord. 6, 26. doi: 10.1186/1866-1955-6-26
Skourti-Stathaki, K., Kamieniarz-Gdula, K., and Proudfoot, N. J. (2014). R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 516, 436–439. doi: 10.1038/nature13787
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 42, 794–805. doi: 10.1016/j.molcel.2011.04.026
Smith, S. S., Laayoun, A., Lingeman, R. G., Baker, D. J., and Riley, J. (1994). Hypermethylation of telomere-like foldbacks at codon 12 of the human c-Ha-ras gene and the trinucleotide repeat of the FMR-1 gene of fragile X. J. Mol. Biol. 243, 143–151. doi: 10.1006/jmbi.1994.1640
Sordet, O., Nakamura, A. J., Redon, C. E., and Pommier, Y. (2010). DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle 9, 274–278. doi: 10.4161/cc.9.2.10506
Steinbach, P., Glaser, D., Vogel, W., Wolf, M., and Schwemmle, S. (1998). The DMPK gene of severely affected myotonic dystrophy patients is hypermethylated proximal to the largely expanded CTG repeat. Am. J. Hum. Genet. 62, 278–285. doi: 10.1086/301711
Sullivan, S. D., Welt, C., and Sherman, S. (2011). FMR1 and the continuum of primary ovarian insufficiency. Semin. Reprod. Med. 29, 299–307. doi: 10.1055/s-0031-1280915
Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400.
Tabolacci, E., Moscato, U., Zalfa, F., Bagni, C., Chiurazzi, P., and Neri, G. (2008). Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 16, 1487–1498. doi: 10.1038/ejhg.2008.130
Tassone, F., Beilina, A., Carosi, C., Albertosi, S., Bagni, C., Li, L., et al. (2007). Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 13, 555–562. doi: 10.1261/rna.280807
Tassone, F., Hagerman, R. J., Loesch, D. Z., Lachiewicz, A., Taylor, A. K., and Hagerman, P. J. (2000a). Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am. J. Med. Genet. 94, 232–236. doi: 10.1002/1096-8628(20000918)94:3<232::AID-AJMG9>3.0.CO;2-H
Tassone, F., Hagerman, R. J., Taylor, A. K., Gane, L. W., Godfrey, T. E., and Hagerman, P. J. (2000b). Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 66, 6–15. doi: 10.1086/302720
Thomson, J. P., Skene, P. J., Selfridge, J., Clouaire, T., Guy, J., Webb, S., et al. (2010). CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464, 1082–1086. doi: 10.1038/nature08924
Todd, P. K., Oh, S. Y., Krans, A., Pandey, U. B., Di Prospero, N. A., Min, K. T., et al. (2010). Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 6:e1001240. doi: 10.1371/journal.pgen.1001240
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., et al. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. doi: 10.1126/science.1192002
Usdin, K., House, N. C., and Freudenreich, C. H. (2015). Repeat instability during DNA repair: insights from model systems. Crit. Rev. Biochem. Mol. Biol. 50, 142–167. doi: 10.3109/10409238.2014.999192
Usdin, K., and Woodford, K. J. (1995). CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 23, 4202–4209.
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914.
Wahba, L., and Koshland, D. (2013). The Rs of biology: R-loops and the regulation of regulators. Mol. Cell 50, 611–612. doi: 10.1016/j.molcel.2013.05.024
Wang, Y. H., Gellibolian, R., Shimizu, M., Wells, R. D., and Griffith, J. (1996). Long CCG triplet repeat blocks exclude nucleosomes: a possible mechanism for the nature of fragile sites in chromosomes. J. Mol. Biol. 263, 511–516. doi: 10.1006/jmbi.1996.0593
Weiler, I. J., and Greenough, W. T. (1999). Synaptic synthesis of the Fragile X protein: possible involvement in synapse maturation and elimination. Am. J. Med. Genet. 83, 248–252.
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., et al. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400.
Xi, Z., Zinman, L., Moreno, D., Schymick, J., Liang, Y., Sato, C., et al. (2013). Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 92, 981–989. doi: 10.1016/j.ajhg.2013.04.017
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., et al. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674. doi: 10.1016/j.molcel.2010.03.021
Yuan, W., Xu, M., Huang, C., Liu, N., Chen, S., and Zhu, B. (2011). H3K36 methylation antagonizes PRC2 mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989. doi: 10.1074/jbc.M110.194027
Zaitsev, E. N., and Kowalczykowski, S. C. (2000). A novel pairing process promoted by Escherichia coli RecA protein: inverse DNA and RNA strand exchange. Genes Dev. 14, 740–749.
Zhao, J., Ohsumi, T. K., Kung, J. T., Ogawa, Y., Grau, D. J., Sarma, K., et al. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40, 939–953. doi: 10.1016/j.molcel.2010.12.011
Zhao, J., Sun, B. K., Erwin, J. A., Song, J. J., and Lee, J. T. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. doi: 10.1126/science.1163045
Keywords: fragile X-associated tremor/ataxia syndrome, fragile X-associated primary ovarian insufficiency, fragile X syndrome, Repeat-mediated gene silencing, R loops, polycomb repressive complex 2
Citation: Usdin K and Kumari D (2015) Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front. Genet. 6:192. doi: 10.3389/fgene.2015.00192
Received: 03 March 2015; Accepted: 13 May 2015;
Published: 03 June 2015.
Edited by:
Marguerite Evans-Galea, Murdoch Childrens Research Institute, AustraliaReviewed by:
Clotilde Lagier-Tourenne, University of California, San Diego, USAElisabetta Soragni, The Scripps Research Institute, USA
Copyright © 2015 Usdin and Kumari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen Usdin, Section on Gene Structure and Disease, Laboratory of Cell and Molecular Biology, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Building 8, Room 2A19, 8 Center Drive MSC 0830, Bethesda, MD 20892-0830, USA,a3VAaGVsaXgubmloLmdvdg==