 Tony Shen
Tony Shen Stefan Hans Pajaro-Van de Stadt1
Stefan Hans Pajaro-Van de Stadt1 Nai Chien Yeat
Nai Chien Yeat Jimmy C.-H. Lin
Jimmy C.-H. Lin- 1Rare Genomics Institute, Bethesda, MD, USA
- 2School of Medicine, Washington University, Saint Louis, MO, USA
This article will review recent impact of massively parallel next-generation sequencing (NGS) in our understanding and treatment of cancer. While whole exome sequencing (WES) remains popular and effective as a method of genetically profiling different cancers, advances in sequencing technology has enabled an increasing number of whole-genome based studies. Clinically, NGS has been used or is being developed for genetic screening, diagnostics, and clinical assessment. Though challenges remain, clinicians are in the early stages of using genetic data to make treatment decisions for cancer patients. As the integration of NGS in the study and treatment of cancer continues to mature, we believe that the field of cancer genomics will need to move toward more complete 100% genome sequencing. Current technologies and methods are largely limited to coding regions of the genome. A number of recent studies have demonstrated that mutations in non-coding regions may have direct tumorigenic effects or lead to genetic instability. Non-coding regions represent an important frontier in cancer genomics.
Introduction
Cancer, in its many forms, accounted for 8.2 million deaths in 2012 (GLOBOCAN, 2012). The rapid development of DNA sequencing technologies has driven a revolution in our understanding of this highly complex and diverse group of diseases (Devita and Rosenberg, 2012). This review fulfills two purposes. First, this article summarizes the history of massively parallel next-generation sequencing (NGS) in the context of cancer genomics and reviews recent research and clinical applications. Second, we highlight the importance and potential of complete or 100% genome sequencing, i.e., the ability to sequence highly repetitive non-coding sequences beyond the reach of current NGS technologies.
Background and History
Sequencing the First Cancer Exomes and Genomes
The first cancer exomes were sequenced soon after the completion of the Human Genome Project in 2001. Compared to whole genome sequencing (WGS), exome sequencing covers only the 1% of the genome that is translated into protein, greatly reducing the technical burden of data collection and analysis. Ley et al. piloted the use of NGS to study the exomes of 140 samples of human acute myeloid leukemia (AML) cells in 2003, identifying 6 previously described and 7 undescribed mutations relevant for AMP pathogenesis. The investigation searched for mutations capable of altering gene function and identified the FLT3 gene as a distinguishing mutant in AML patients (Ley et al., 2003).
The first solid tumor exomes to be investigated were from 11 breast and 11 colorectal cancer tissue samples. Sjoblom et al. identified 189 frequently mutated genes associated with these cancers, most of which were not previously known. Additionally, the investigators found that the average tumor accumulates an average of ~90 mutations over its course of development, though only a subset of mutations contributes to the formation of neoplasms (Sjöblom et al., 2006). A follow-up study revealed varying mutation frequencies for individual genes with few but commonly mutated gene “mountains” and numerous but infrequently mutated gene “hills” (Wood et al., 2007). Despite similarities in the number of mutations for each cancer, the types and locations of these mutations result in distinct cancer subtypes (Wood et al., 2007).
Ley et al. performed the first whole-genome sequencing study on AML cells collected from a single patient. The patient's skin cells were used as a control. The study identified 2 genes known to contribute to tumor progression and 8 known to be present in tumor cells but which have unknown functions. As a proof-of-concept, the study demonstrated the feasibility of WGS as an unbiased tool for the molecular profiling of individual cancers (Ley et al., 2008). Table 1 provides a summary of cancers and gene mutations.
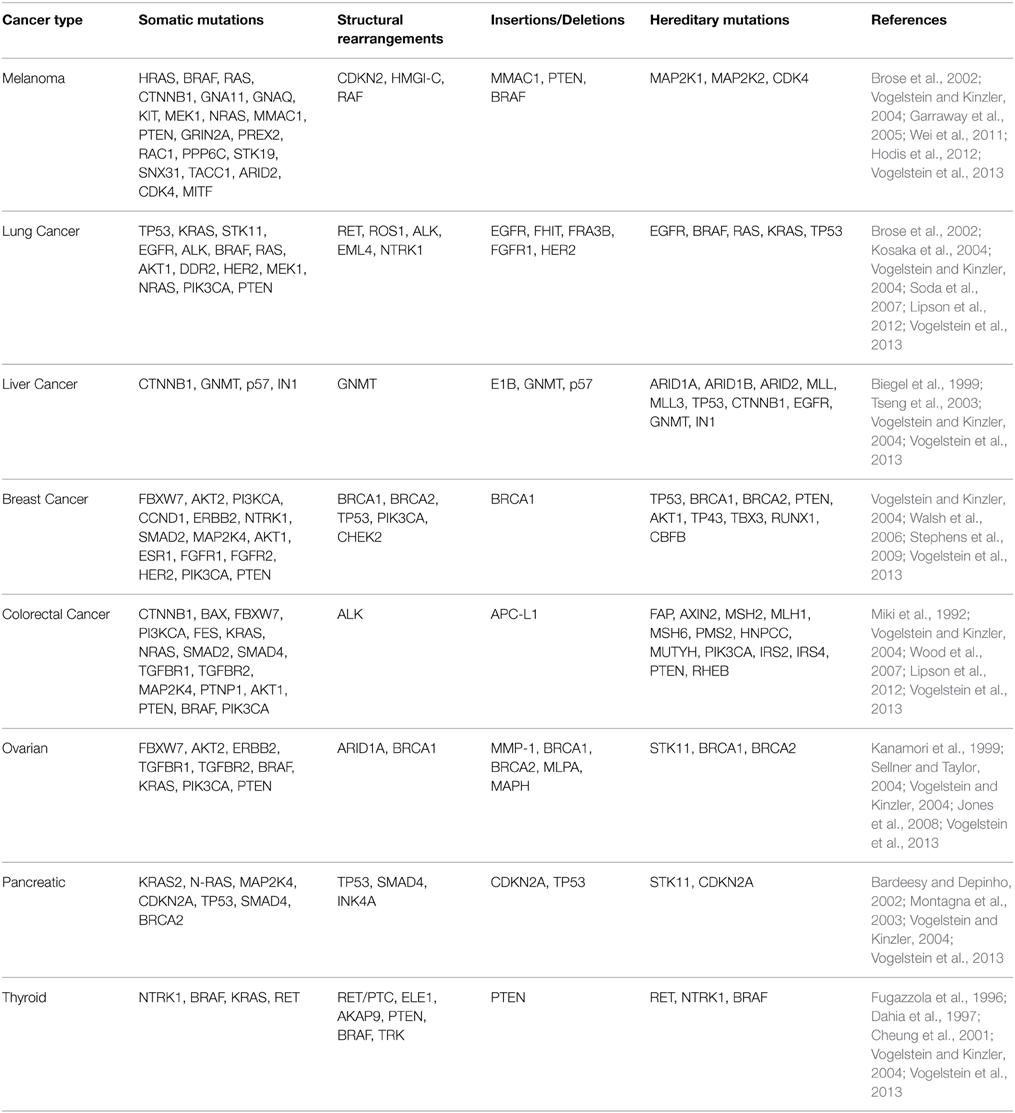
Table 1. Summary of genetic mutations associated with selected cancers.
Discoveries
Melanoma
Analyzing genomic data from melanoma samples is uniquely challenging because of the high number of passenger mutations caused by ultraviolet light exposure (Hodis et al., 2012). To overcome this, Hodis et al. controlled for UV-induced mutational load by comparing mutated genes-of-interest against a baseline level of intronic mutation. They identified six novel genes: PPP6C, RAC1, SNX31, TACC1, STK19, and ARID2. Of these genes, PPP6C, RAC1, and STK19 are thought to be potentially targetable. In a WES study of 147 melanomas, Krauthammer et al. identified a P29S mutation RAC1 in 9.2% of sun-exposed exposed melanomas. Mutations in the active site of PPP6C, a serine/threonine phosphatase, were found in 12% of sun-exposed melanomas that already possessed BRAF or NRAS mutations (Krauthammer et al., 2012).
Mutations in BRAF, NRAS, and KIT are known to be involved in the pathogenesis of metastatic melanoma. BRAF inhibitors are a class of targeted therapeutics approved for treating metastatic melanoma and have demonstrated significantly improved overall survival (Kunz et al., 2013).
Breast Cancer
The Cancer Genome Atlas network sequenced 510 breast cancers exomes, identifying 4 distinct subtypes: luminal A (ER+ and/or PR+, HER2−), luminal B (ER+ and/or PR+, HER2+), HER2-enriched (ER−, PR−, HER2+), and basal-like (ER−, PR−, HER2−). 40% of luminal A tumors possessed a mutated PIK3CA gene. TP53 and PIK3CA were mutated in 29% of luminal B tumors. Compared to luminal A or luminal B subtypes, the basal subtype exhibited a more consistent pattern of mutation, with TP53 mutated in 80% of cases. The HER2-enriched subtype is characterized by HER2 amplification, found in 80% of these tumors (Koboldt et al., 2012). Basal type tumors are characterized by the highest amount of mutations, while luminal A types generally contain the lowest frequencies of mutations (Wang et al., 2014b). Another study investigated CAG repeat lengths of breast cancer tumor samples to determine the significance of intratumor genetic heterogeneity (ITGH) (Gottlieb et al., 2013). The findings between differing repeat lengths showed that shorter CAG repeats may play protective roles against breast cancer, as opposed to longer repeat lengths, which may contribute to cancer development (Gottlieb et al., 2013). The study demonstrates that merely identifying genetic variations does not provide sufficient understanding of cancer etiology; rather it is necessary to determine frequency and distribution of mutations between cancerous and normal tissues (Gottlieb et al., 2013; Riahi et al., 2014).
Clinical Utility
The information generated by next generation sequencing (NGS) technologies enables clinicians to make improved diagnostic and treatment decisions. For example, breast cancers have traditionally been diagnosed by mammogram, physical exam, and histology. The discovery of BRCA1, BRCA2, and other biomarkers introduced genetics as an important consideration (Van De Vijver et al., 2002; Van't Veer et al., 2002). Today, commercially available micro-array-based tests such as OncoType DX and MammaPrint allow more accurate profiling of breast cancers based on genetic biomarkers such as HER2, ER, and PR, each with their own treatment protocols. In parallel with improved diagnostics, identification of cancer-associated genes has led to the development of molecularly targeted therapies such as trastuzumab, which was among the first therapies specifically targeted to HER2+ breast cancers. Moreover, sequencing of hundreds of breast tumors has also revealed significant intratumor heterogeneity, reflecting an additional level of complexity for the development of new treatments (Desmedt et al., 2012; Shah et al., 2012).
Genetic Screening
As the cost of NGS continues to approach the $1000 threshold, population-wide genomic screening becomes more likely (Brunicardi et al., 2011). Already, NGS may improve genetic testing in families with histories of high penetrance cancer genes such as BRCA1, BRCA2, APC, and TP53 (Meldrum et al., 2011). Several investigators have tested the Illumina HiSeq platform in detecting BRCA1, BRCA2, and TP53 from a tumor cell line (Morgan et al., 2010; Schroeder et al., 2010). In these studies, NGS analysis identified all known variants in the tumor cell line with sensitivity and specificity greater than traditional diagnostic methods, demonstrating the effectiveness of NGS as a diagnostic tool. More important than the improvement in sensitivity/specificity, the data generated by NGS allows for more sophisticated analysis of gene interactions.
Economical NGS screening will also benefit patients with de novo mutations who would not otherwise undergo genetic screening based on family history. In the case of BRCA mutations, family history only accounts for 30–50% of mutations (Moller et al., 2007). Additionally, NGS testing allows for testing of genes with a wide range in frequency (Meldrum et al., 2011). Several companies and institutions offer cancer gene panels that screen for over 70 genes (Washington University in St. Louis, University of Washington, Baylor College of Medicine, Ambrygen, Genewiz). While NGS remains too costly for routine sequencing of all individuals, we expect the prevalence of screening to continue increasing as prices decrease.
Diagnostics and Assessment
Currently, NGS-based gene panels are regularly used for cancer diagnostics. For example, the 2015 National Comprehensive Cancer Network guidelines recommend NGS gene panels for patients with hereditary and ovarian cancer who have tested negative for high-penetrance genes (Park et al., 2014). In a study of 141 patients who tested negative for BRCA1/2, evaluation by an NGS panel of 40 genes identified 16 patients who have pathogenic variants in 9 non-BRCA genes (Kurian et al., 2014). The use of NGS in clinical diagnostics may be separated into three approaches: gene panels, whole exome sequencing (WES), and whole genome sequencing. Among these, gene panels have been in use for the longest period, with more than 16 laboratories in the United States panels for hereditary cancer (Wang et al., 2014a). The diagnostic yields of these panels range from 20 to 51%, which is comparable to that of exome or genome-based methods (Wang et al., 2014a). Recent exome-based diagnostics for mitochondrial respiratory disease and intellectual disability demonstrated diagnostic yields of 60 and 16%, respectively (De Ligt et al., 2012; Taylor et al., 2014). Other evaluations of exome-based diagnostics demonstrated diagnostic yields between 25 and 30% (Yang et al., 2013, 2014; Lee et al., 2014). Genome-based diagnostics are comparatively newer and there are fewer studies that evaluate their use. Studies that evaluated the use of genome sequencing to diagnose intellectual disability and early-onset epilepsy demonstrated a diagnostic yield of 50 and 24%, respectively (Gilissen et al., 2014; Martin et al., 2014). As our understanding of the genome grows, in-depth genome-based diagnostics will have higher diagnostic yield.
Clinical Decision-making and Treatment
NGS has enabled investigators to discover and elucidate hundreds of genes involved in cancer, advances that will inevitably reveal novel therapeutic targets. Targeted therapies, a growing group of therapeutic agents with molecular-level specificity have greatly changed the treatment and management for many cancers. Notable examples include imatinib (BCR-ABL) and trastuzumab (HER2). Targeted therapies have the potential to be more effective and less toxic than traditional chemotherapies for patients suffering from cancer (Tsimberidou et al., 2014). Treatments like EGFR-targeted plasmonic magnetic particles have been shown to be more effective in suppressing lung cancer development and tumor growth by abrogating the G2/M cell cycle phase and inducing apoptosis (Kuroda et al., 2014). Another promising development in targeted cancer therapy is signaling-directed androgen receptor treatment for castration-resistant prostate cancer, which deactivates tumor proliferation pathways and inhibits cancer progression (Bastos et al., 2014). Currently, discoveries on the diagnostics and assessment outpace the development of targeted therapies. As NGS technology matures, the selection of targeted therapies stands to expand greatly as more targets become known.
Due to the easy accessibility of circulating tumor DNA (ctDNA), sequencing ctDNA is another attractive method for analyzing tumor load and treatment effectiveness (Wang and Wheeler, 2014). The mechanism by which ctDNA enters the bloodstream is not well understood, though Gormally et al. propose two mechanisms: release of whole cells followed by lysis and apoptosis (Gormally et al., 2007). Diehl et al. demonstrated that ctDNA measurements in 18 patients with colorectal cancer were a reliable indicator of tumor dynamics (Diehl et al., 2007). In 2014, Lohr et al. developed computational methods to isolate ctDNA sequences from serum. A comparison of their ctDNA sequences to previously sequenced tumor samples revealed 90% of early trunk mutations and 73% of metastatic trunk mutations in the tumor were also found in ctDNA (Lohr et al., 2014). In another study by Murtaza et al. sequencing of ctDNA from patients with breast, lung, and ovarian cancers allowed investigators to track mutations in the tumor genome in a non-invasive manner (Murtaza et al., 2013). Additionally, Dawson et al. demonstrated that the sensitivity of ctDNA surpassed that of other circulating biomarkers (Dawson et al., 2013). These studies suggest that NGS analysis of ctDNA holds great potential for screening, diagnosis, and clinical decision-making.
There remain many challenges for investigators and clinicians. With massive amounts of data generated, healthcare infrastructure needs new strategies for data curation. Additionally, the economic implications of widespread sequencing are not yet known given the complex interplay between healthcare providers, the biomedical industry, insurance companies, and academic research (Buchanan et al., 2013).
Commercialization
Today, patients may choose between many services and technologies offered by gene sequencing companies for assistance in patient diagnosis and treatment decision processes. Some companies, such as Foundation Medicine and Personal Genome Diagnostics, offer clinical tests like FoundationOne and CancerComplete to help identify the genetic variants in a patient's DNA sample that may be contributing to cancer development.
A number of companies and institutions provide individual gene panel tests for different types of cancers. Cancer-specific kits such as AmbryGen's OvaNext and ColoNext selectively enrich genes-of-interest for ovarian and colorectal cancers, respectively. Other companies and institutions offer similar cancer-specific testing, such as Neogenomics Laboratories, GPS@WUSTL, Emory Genetics Laboratory, ARUP Laboratories, Myriad Genetics, and GeneDx. WES-based tests such as the FoundationOne or FoundationOne-Heme have been developed to be highly sensitive and specific (Frampton et al., 2013). Table 2 provides a summary of commercial services using NGS.
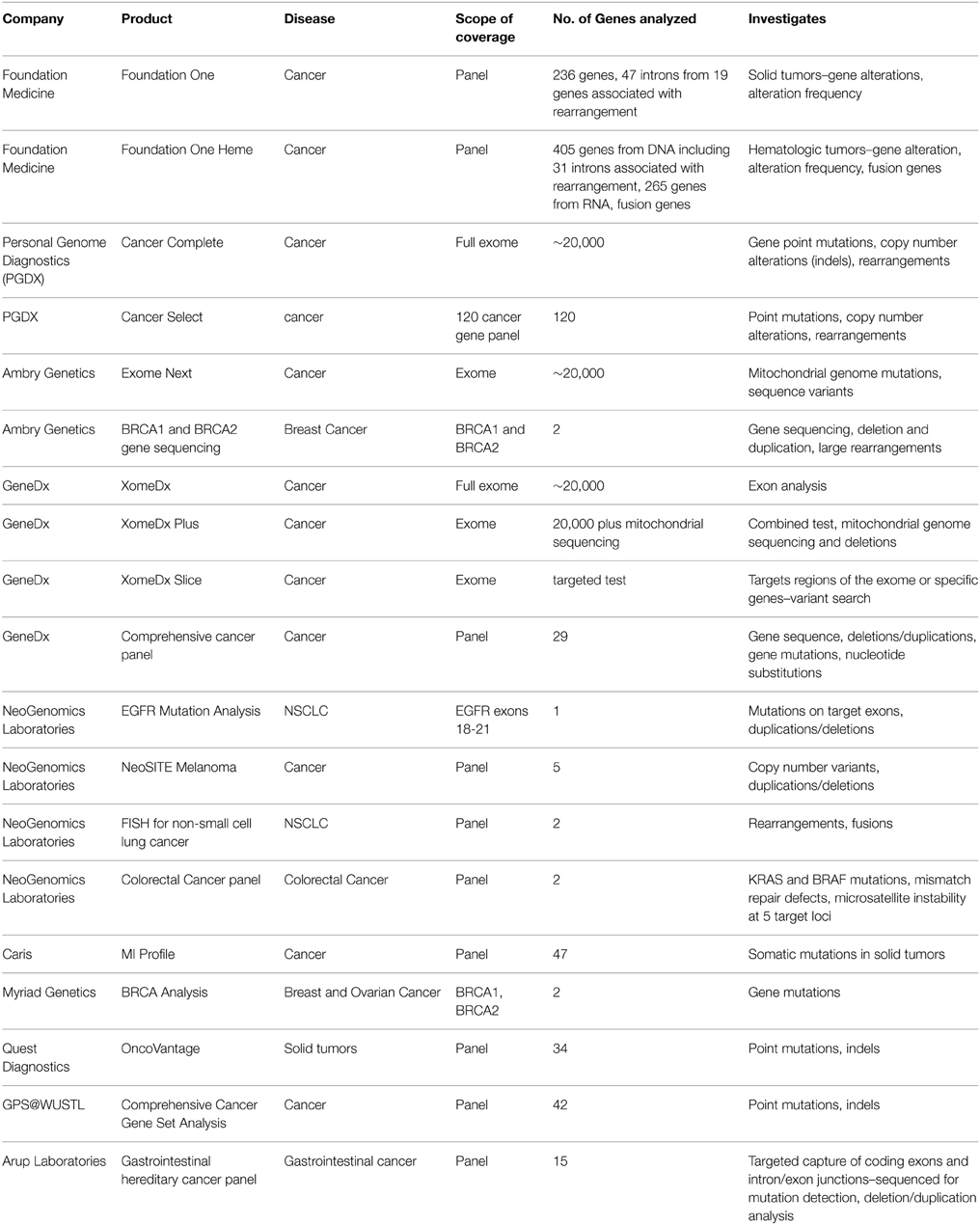
Table 2. Summary and comparison of NGS diagnostic services.
Future Directions
Single Molecule Sequencing
While NGS has undoubtedly driven a revolution in genetics, current technologies face certain limitations. First, the amplification of DNA libraries may result in differential expansion of certain regions of the genome. This introduces the problem of biased reads. Second, epigenetic modifications are lost during amplification. Third, the short read lengths generated from current NGS platforms requires a reliance on a reference genome and robust alignment algorithms to produce sequences from raw NGS data (Mardis, 2013). Lastly, the entire sequencing process from sample preparation to computational construction of the sequence takes days to weeks (Liu et al., 2012).
Single molecule or third generation sequencing systems address shortcomings present in current NGS platforms. Single-molecule real-time sequencing (SMRT, Pacific Biosciences) and nanopore sequencing (Oxford Nanopore) are perhaps the most well-known. Conceptually, both of these systems detect nucleotide incorporation onto a single DNA strand in real time. SMRT uses a system of fluorescent signals to monitor nucleotide incorporation while nanopore detects voltage across a lipid bilayer as as DNA is ratcheted across an α-hemolysin nanopore (Eid et al., 2009; Liu et al., 2012). Sequencing a single strand eliminates the need for amplification, reducing sequencing bias present in NGS platforms. Monitoring systems can also be tuned to detect epigenetic changes. Read lengths are also significantly longer compared to current NGS systems, with SMRT reads in the range of ~5000–6000 bp (English et al., 2012; Chin et al., 2013). Long read lengths allow for rapid de novo sequencing of organisms without reference genomes as well as the sequencing of highly repetitive regions. Additionally, simplified preparation and sequencing in real time with nucleotide incorporation significantly shortens run times. SMRT runs can be completed in just 1 day (Liu et al., 2012).
Single molecule sequencing systems are beginning to make an impact in cancer genomics and are poised to play a larger role moving forward. While SMRT has been available since 2011, nanopore sequencing has yet to see widespread use. SMRT has been used to resequence AML patients to identify a marker (FLT3 with internal tandem duplication) associated with poor prognosis (Smith et al., 2012). Kleinman et al. used SMRT to elucidate a distinct DNA methylation pattern and epigenetic alteration program in embryonal brain tumors (Kleinman et al., 2014). In addition to the ability to study genetic and epigenetic changes undetectable by NGS, single molecule sequencing also holds great potential for sensitive diagnostics. Sequencing results comparable to NGS reads may be obtained from as little as 500 pg of starting DNA (Raley et al., 2014). In late 2013, Pacific Biosciences announced a large-scale collaboration with Roche to produce diagnostics tools based on SMRT (Pacific Biosciences Investor Relations, 2013).
Diagnostic Yield
As the field moves forward to understand a wider range of genetic abnormalities, there must be a concurrent shift toward 100% genome sequencing for diagnostics. However, current genome-based diagnostics still face several challenges. Dewey et al. analyzed 12 healthy genomes for 56 clinically significant genes and found that the best platform (Illumina) was only able to sequence 51 out of 56 genes with adequate coverage (Dewey et al., 2014). This is a technical hurdle that we hope will be overcome with improved technologies and bioinformatics. Genome-based diagnostics are also inherently limited by the inability to sequence the whole genome, with approximately 8% of the genome not sequenced. This challenge may only be overcome with 100% genome sequencing. Improvement in the diagnostic yield of genetic testing can only occur given a solid foundation of complete gene coverage. Given that the relatively low diagnostic yields of current strategies, we submit that the goal of 100% genome sequencing will be clinically significant.
100% Genome Sequencing
Current findings suggest that a variety of defects in non-coding regions can contribute to neoplastic pathogenesis (Chmielecki and Meyerson, 2014). Huang et al. identified a pattern of mutations in the promoter region of telomerase reverse transcriptase, demonstrating that somatic mutations in non-coding regions can have a direct tumorigenic effect (Huang et al., 2013). Zhu et al. demonstrated that genomic instability following the loss of Brca1 in mice may be due to de-repression of satellite DNA transcription. The authors propose that tumor suppressor functions of BRCA1 primarily stem from its role as a regulator of heterochromatin (Zhu et al., 2011). Ting et al. identified a pattern of satellite transcript overexpression in epithelial cancers of the lung, kidney, ovary, colon, and prostate, proposing that heterochromatic alterations may serve as a biomarker for cancer (Ting et al., 2011). These findings suggest that non-coding regions may be a rich source of new insights into tumorigenesis.
Complete genomic sequencing may also reveal epigenetic changes in cancer that were previously inaccessible due to limitations in the coverage and resolution of last-generation technologies The epigenetics of nucleosomes for example, have eluded investigation in the past, but new protocols utilizing NGS which enable genome-wide studies of nucleosome positioning at high sensitivity might allow networks of chromatin modulation to finally be probed (Wei et al., 2012). Using NGS, Kim et al. characterized the instability of microsatellite regions in colorectal and endometrial cancers, showing that this genetic instability affects nucleosome positioning (Kim et al., 2013). Studies of the epigenetics of cancer have already produced surprising findings, such as the discovery that regions of DNA hypomethylation in breast cancer cells are paradoxically, silenced by the formation of repressive chromatin domains (Hon et al., 2012), and the discovery that histone modifications drive pediatric glioblastoma (Rheinbay et al., 2012). Still more protocols that utilize NGS for studying epigenetic changes are in the pipeline: A recently published method of deciphering histone post-translational modifications (PTMs) using DNA-barcoded nucleosome libraries, is enabled in large part by the speed and low-cost of high throughput DNA sequencing (Nguyen et al., 2014). A thorough understanding of chromatin regulation however, will require not just knowledge of DNA base sequences, but also an understanding of how DNA segments interact with histones and their regulatory elements. To this end, biophysical models, such as a recently published model by Chertsvy and Teif on the electrostatics of histone-DNA interactions (Cherstvy and Teif, 2014), are essential. In another modeling study, Beshnova et al. demonstrate that although major satellite repeats in mice encode preferences for nucleosome positioning, nucleosome positioning is not rigid, and can alternate between two separate types of positioning (Beshnova et al., 2014).
Final Comments
In this article, we discussed the past, present, and future of DNA sequencing in cancer genomics. As advancements in sequencing technology enabled investigators to study cancer genomes in greater breadth and depth, the field has produced novel insights into tumor pathogenesis, identified clinically useful biomarkers, and developed increasingly precise diagnostics and targeted therapeutics. However, it is important to note that our understanding of the genome is limited, with <5000 of 23,000 known (Park et al., 2014). The clinical usefulness of genomic sequencing requires advancement in our knowledge of the genome and bioinformatic systems to process genetic data—advancements that would be built on a foundation of 100% genome sequencing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bardeesy, N., and Depinho, R. A. (2002). Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2, 897–909. doi: 10.1038/nrc949
Bastos, D. A., Dzik, C., Rathkopf, D., and Scher, H. I. (2014). Expanding androgen- and androgen receptor signaling-directed therapies for castration-resistant prostate cancer. Oncology (Williston Park, NY) 28, 693–699.
Beshnova, D. A., Cherstvy, A. G., Vainshtein, Y., and Teif, V. B. (2014). Regulation of the nucleosome repeat length in vivo by the DNA sequence, protein concentrations and long-range interactions. PLoS Comput. Biol. 10:e1003698. doi: 10.1371/journal.pcbi.1003698
Biegel, J. A., Zhou, J.-Y., Rorke, L. B., Stenstrom, C., Wainwright, L. M., and Fogelgren, B. (1999). Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 59, 74–79.
Brose, M. S., Volpe, P., Feldman, M., Kumar, M., Rishi, I., Gerrero, R., et al. (2002). BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 62, 6997–7000.
Brunicardi, F. C., Gibbs, R. A., Wheeler, D. A., Nemunaitis, J., Fisher, W., Goss, J., et al. (2011). Overview of the development of personalized genomic medicine and surgery. World J. Surg. 35, 1693–1699. doi: 10.1007/s00268-011-1056-0
Buchanan, J., Wordsworth, S., and Schuh, A. (2013). Issues surrounding the health economic evaluation of genomic technologies. Pharmacogenomics 14, 1833–1847. doi: 10.2217/pgs.13.183
Cherstvy, A. G., and Teif, V. B. (2014). Electrostatic effect of H1-histone protein binding on nucleosome repeat length. Phys. Biol. 11:044001. doi: 10.1088/1478-3975/11/4/044001
Cheung, C. C., Carydis, B., Ezzat, S., Bedard, Y. C., and Asa, S. L. (2001). Analysis of ret/PTC gene rearrangements refines the fine needle aspiration diagnosis of thyroid cancer. J. Clin. Endocrinol. Metab. 86, 2187–2190. doi: 10.1210/jcem.86.5.7504
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Chmielecki, J., and Meyerson, M. (2014). DNA sequencing of cancer: what have we learned? Annu. Rev. Med. 65, 63–79. doi: 10.1146/annurev-med-060712-200152
Dahia, P. L. M., Marsh, D. J., Zheng, Z., Zedenius, J., Komminoth, P., Frisk, T., et al. (1997). Somatic deletions and mutations in the cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 57, 4710–4713.
Dawson, S.-J., Tsui, D. W., Murtaza, M., Biggs, H., Rueda, O. M., Chin, S.-F., et al. (2013). Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209. doi: 10.1056/NEJMoa1213261
De Ligt, J., Willemsen, M. H., Van Bon, B. W., Kleefstra, T., Yntema, H. G., Kroes, T., et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. doi: 10.1056/NEJMoa1206524
Desmedt, C., Voet, T., Sotiriou, C., and Campbell, P. J. (2012). Next-generation sequencing in breast cancer: first take home messages. Curr. Opin. Oncol. 24, 597–604. doi: 10.1097/CCO.0b013e328359554e
Devita, V. T., and Rosenberg, S. A. (2012). Two hundred years of cancer research. N. Engl. J. Med. 366, 2207–2214. doi: 10.1056/NEJMra1204479
Dewey, F. E., Grove, M. E., Pan, C., Goldstein, B. A., Bernstein, J. A., Chaib, H., et al. (2014). Clinical interpretation and implications of whole-genome sequencing. JAMA 311, 1035–1045. doi: 10.1001/jama.2014.1717
Diehl, F., Schmidt, K., Choti, M. A., Romans, K., Goodman, S., Li, M., et al. (2007). Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990. doi: 10.1038/nm.1789
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., et al. (2012). Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7:e47768. doi: 10.1371/journal.pone.0047768
Frampton, G. M., Fichtenholtz, A., Otto, G. A., Wang, K., Downing, S. R., He, J., et al. (2013). Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031. doi: 10.1038/nbt.2696
Fugazzola, L., Pierotti, M. A., Vigano, E., Pacini, F., Vorontsova, T. V., and Bongarzone, I. (1996). Molecular and biochemical analysis of RET/PTC4, a novel oncogenic rearrangement between RET and ELE1 genes, in a post-Chernobyl papillary thyroid cancer. Oncogene 13, 1093–1097.
Garraway, L. A., Widlund, H. R., Rubin, M. A., Getz, G., Berger, A. J., Ramaswamy, S., et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. doi: 10.1038/nature03664
Gilissen, C., Hehir-Kwa, J. Y., Thung, D. T., van de Vorst, M., van Bon, B. W. M., Willemsen, M. H., et al. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347. doi: 10.1038/nature13394
GLOBOCAN (2012). Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Available online at: http://globocan.iarc.fr/Pages/fact_sheets_population.aspx
Gormally, E., Caboux, E., Vineis, P., and Hainaut, P. (2007). Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutat. Res. 635, 105–117. doi: 10.1016/j.mrrev.2006.11.002
Gottlieb, B., Alvarado, C., Wang, C., Gharizadeh, B., Babrzadeh, F., Richards, B., et al. (2013). Making sense of intratumor genetic heterogeneity: altered frequency of androgen receptor CAG repeat length variants in breast cancer tissues. Hum. Mutat. 34, 610–618. doi: 10.1002/humu.22287
Hodis, E., Watson, I. R., Kryukov, G. V., Arold, S. T., Imielinski, M., Theurillat, J. P., et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. doi: 10.1016/j.cell.2012.06.024
Hon, G. C., Hawkins, R. D., Caballero, O. L., Lo, C., Lister, R., Pelizzola, M., et al. (2012). Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 22, 246–258. doi: 10.1101/gr.125872.111
Huang, F. W., Hodis, E., Xu, M. J., Kryukov, G. V., Chin, L., and Garraway, L. A. (2013). Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959. doi: 10.1126/science.1229259
Jones, S., Zhang, X., Parsons, D. W., Lin, J. C.-H., Leary, R. J., Angenendt, P., et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. doi: 10.1126/science.1164368
Kanamori, Y., Matsushima, M., Minaguchi, T., Kobayashi, K., Sagae, S., Kudo, R., et al. (1999). Correlation between expression of the matrix metalloproteinase-1 gene in ovarian cancers and an insertion/deletion polymorphism in its promoter region. Cancer Res. 59, 4225–4227.
Kim, T.-M., Laird, P. W., and Park, P. J. (2013). The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 155, 858–868. doi: 10.1016/j.cell.2013.10.015
Kleinman, C. L., Gerges, N., Papillon-Cavanagh, S., Sin-Chan, P., Pramatarova, A., Quang, D.-A. K., et al. (2014). Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat. Genet. 46, 39–44. doi: 10.1038/ng.2849
Koboldt, D. C., Fulton, R. S., Mclellan, M. D., Schmidt, H., Kalicki-Veizer, J., Mcmichael, J. F., et al. (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. doi: 10.1038/nature11412
Kosaka, T., Yatabe, Y., Endoh, H., Kuwano, H., Takahashi, T., and Mitsudomi, T. (2004). Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 64, 8919–8923. doi: 10.1158/0008-5472.CAN-04-2818
Krauthammer, M., Kong, Y., Ha, B. H., Evans, P., Bacchiocchi, A., McCusker, J. P., et al. (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014. doi: 10.1038/ng.2359
Kunz, M., Dannemann, M., and Kelso, J. (2013). High-throughput sequencing of the melanoma genome. Exp. Dermatol. 22, 10–17. doi: 10.1111/exd.12054
Kurian, A. W., Hare, E. E., Mills, M. A., Kingham, K. E., Mcpherson, L., Whittemore, A. S., et al. (2014). Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J. Clin. Oncol. 32, 2001–2009. doi: 10.1200/JCO.2013.53.6607
Kuroda, S., Tam, J., Roth, J. A., Sokolov, K., and Ramesh, R. (2014). EGFR-targeted plasmonic magnetic nanoparticles suppress lung tumor growth by abrogating G2/M cell-cycle arrest and inducing DNA damage. Int. J. Nanomed. 9, 3825. doi: 10.2147/IJN.S65990
Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., et al. (2014). Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA 312, 1880–1887. doi: 10.1001/jama.2014.14604
Ley, T. J., Mardis, E. R., Ding, L., Fulton, B., Mclellan, M. D., Chen, K., et al. (2008). DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456, 66–72. doi: 10.1038/nature07485
Ley, T. J., Minx, P. J., Walter, M. J., Ries, R. E., Sun, H., Mclellan, M., et al. (2003). A pilot study of high-throughput, sequence-based mutational profiling of primary human acute myeloid leukemia cell genomes. Proc. Natl. Acad. Sci. U.S.A. 100, 14275–14280. doi: 10.1073/pnas.2335924100
Lipson, D., Capelletti, M., Yelensky, R., Otto, G., Parker, A., Jarosz, M., et al. (2012). Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 18, 382–384. doi: 10.1038/nm.2673
Liu, L., Li, Y., Li, S., Hu, N., He, Y., Pong, R., et al. (2012). Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 11. doi: 10.1155/2012/251364
Lohr, J. G., Adalsteinsson, V. A., Cibulskis, K., Choudhury, A. D., Rosenberg, M., Cruz-Gordillo, P., et al. (2014). Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat. Biotechnol. 32, 479–484. doi: 10.1038/nbt.2892
Mardis, E. R. (2013). Next-generation sequencing platforms. Annu. Rev. Anal. Chem. (Palo. Alto Calif). 6, 287–303. doi: 10.1146/annurev-anchem-062012-092628
Martin, H. C., Kim, G. E., Pagnamenta, A. T., Murakami, Y., Carvill, G. L., Meyer, E., et al. (2014). Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 23, 3200–3211. doi: 10.1093/hmg/ddu030
Meldrum, C., Doyle, M. A., and Tothill, R. W. (2011). Next-generation sequencing for cancer diagnostics: a practical perspective. Clin. Biochem. Rev. 32, 177–195.
Miki, Y., Nishisho, I., Horii, A., Miyoshi, Y., Utsunomiya, J., Kinzler, K. W., et al. (1992). Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 52, 643–645.
Moller, P., Evans, D. G., Reis, M. M., Gregory, H., Anderson, E., Maehle, L., et al. (2007). Surveillance for familial breast cancer: differences in outcome according to BRCA mutation status. Int. J. Cancer 121, 1017–1020. doi: 10.1002/ijc.22789
Montagna, M., Palma, M. D., Menin, C., Agata, S., De Nicolo, A., Chieco-Bianchi, L., et al. (2003). Genomic rearrangements account for more than one-third of the BRCA1 mutations in northern Italian breast/ovarian cancer families. Hum. Mol. Genet. 12, 1055–1061. doi: 10.1093/hmg/ddg120
Morgan, J. E., Carr, I. M., Sheridan, E., Chu, C. E., Hayward, B., Camm, N., et al. (2010). Genetic diagnosis of familial breast cancer using clonal sequencing. Hum. Mutat. 31, 484–491. doi: 10.1002/humu.21216
Murtaza, M., Dawson, S.-J., Tsui, D. W., Gale, D., Forshew, T., Piskorz, A. M., et al. (2013). Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112. doi: 10.1038/nature12065
Nguyen, U. T. T., Bittoval, L., Muller, M. M., Fierz, B., David, Y., Houck-Loomis, B., et al. (2014). Accelerated chromatin biochemistry using DNA-barcoded nucteosome libraries. Nat. Methods 11, 834–840. doi: 10.1038/nmeth.3022
Pacific Biosciences Investor Relations. (2013). Pacific Biosciences Announces Agreement With Roche Diagnostics to Develop and Supply DNA Sequencing-Based Products for Clinical Diagnostics. Available online at: http://investor.pacificbiosciences.com/releasedetail.cfm?ReleaseID=793199
Park, J. Y., Kricka, L. J., Clark, P., Londin, E., and Fortina, P. (2014). Clinical genomics: when whole genome sequencing is like a whole-body CT scan. Clin. Chem. 60, 1390–1392. doi: 10.1373/clinchem.2014.230276
Raley, C., Munroe, D., Jones, K., Tsai, Y.-C., Guo, Y., Tran, B., et al. (2014). Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: application to single-molecule, real-time (SMRT) Sequencing on the pacific biosciences RS II. bioRxiv. doi: 10.1101/003566
Rheinbay, E., Louis, D. N., Bernstein, B. E., and Suvà, M. L. (2012). A tell-tail sign of chromatin: histone mutations drive pediatric glioblastoma. Cancer Cell 21, 329–331. doi: 10.1016/j.ccr.2012.03.001
Riahi, A., Kharrat, M., Ghourabi, M. E., Khomsi, F., Gamoudi, A., Lariani, I., et al. (2014). Mutation spectrum and prevalence of BRCA1 and BRCA2 genes in patients with familial and early-onset breast/ovarian cancer from Tunisia. Clin. Genet. 87, 155–160. doi: 10.1111/cge.12337
Schroeder, C., Stutzmann, F., Weber, B. H., Riess, O., and Bonin, M. (2010). High-throughput resequencing in the diagnosis of BRCA1/2 mutations using oligonucleotide resequencing microarrays. Breast Cancer Res. Treat. 122, 287–297. doi: 10.1007/s10549-009-0639-z
Sellner, L. N., and Taylor, G. R. (2004). MLPA and MAPH: new techniques for detection of gene deletions. Hum. Mutat. 23, 413–419. doi: 10.1002/humu.20035
Shah, S. P., Roth, A., Goya, R., Oloumi, A., Ha, G., Zhao, Y., et al. (2012). The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399. doi: 10.1038/nature10933
Sjöblom, T., Jones, S., Wood, L. D., Parsons, D. W., Lin, J., Barber, T. D., et al. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274. doi: 10.1126/science.1133427
Smith, C. C., Wang, Q., Chin, C.-S., Salerno, S., Damon, L. E., Levis, M. J., et al. (2012). Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 485, 260–263. doi: 10.1038/nature11016
Soda, M., Choi, Y. L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., et al. (2007). Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566. doi: 10.1038/nature05945
Stephens, P. J., Mcbride, D. J., Lin, M.-L., Varela, I., Pleasance, E. D., Simpson, J. T., et al. (2009). Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462, 1005–1010. doi: 10.1038/nature08645
Taylor, R. W., Pyle, A., Griffin, H., Blakely, E. L., Duff, J., He, L., et al. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77. doi: 10.1001/jama.2014.7184
Ting, D. T., Lipson, D., Paul, S., Brannigan, B. W., Akhavanfard, S., Coffman, E. J., et al. (2011). Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331, 593–596. doi: 10.1126/science.1200801
Tseng, T.-L., Shih, Y.-P., Huang, Y.-C., Wang, C.-K., Chen, P.-H., Chang, J.-G., et al. (2003). Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res. 63, 647–654.
Tsimberidou, A.-M., Wen, S., Hong, D. S., Wheler, J. J., Falchook, G. S., Fu, S., et al. (2014). Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin. Cancer Res. 20, 4827–4836. doi: 10.1158/1078-0432.CCR-14-0603
Van De Vijver, M. J., He, Y. D., van ‘t Veer, L. J., Dai, H., Hart, A. A. M., Voskuil, D. W., et al. (2002). A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347, 1999–2009. doi: 10.1056/NEJMoa021967
Van't Veer, L. J., Dai, H., Van De Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536. doi: 10.1038/415530a
Vogelstein, B., and Kinzler, K. W. (2004). Cancer genes and the pathways they control. Nat. Med. 10, 789–799. doi: 10.1038/nm1087
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., and Kinzler, K. W. (2013). Cancer genome landscapes. Science 339, 1546–1558. doi: 10.1126/science.1235122
Walsh, T., Casadei, S., Coats, K., Swisher, E., Stray, S. M., Higgins, J., et al. (2006). Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295, 1379–1388. doi: 10.1001/jama.295.12.1379
Wang, J., Gotway, G., Pascual, J. M., and Park, J. Y. (2014a). DIagnostic yield of clinical next-generation sequencing panels for epilepsy. JAMA Neurol. 71, 650–651. doi: 10.1001/jamaneurol.2014.405
Wang, L., and Wheeler, D. A. (2014). Genomic sequencing for cancer diagnosis and therapy. Annu. Rev. Med. 65, 33–48. doi: 10.1146/annurev-med-120811-171056
Wang, Y., Waters, J., Leung, M. L., Unruh, A., Roh, W., Shi, X., et al. (2014b). Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160. doi: 10.1038/nature13600
Wei, G., Hu, G., Cui, K., and Zhao, K. (2012). “Chapter thirteen - genome-wide mapping of nucleosome occupancy, histone modifications, and gene expression using next-generation sequencing technology,” in Methods in Enzymology, eds W. Carl and C. D. Allis (San Diego, CA: Academic Press), 297–313.
Wei, X., Walia, V., Lin, J. C., Teer, J. K., Prickett, T. D., Gartner, J., et al. (2011). Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 43, 442–446. doi: 10.1038/ng.810
Wood, L. D., Parsons, D. W., Jones, S., Lin, J., Sjöblom, T., Leary, R. J., et al. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113. doi: 10.1126/science.1145720
Yang, Y., Muzny, D. M., Reid, J. G., Bainbridge, M. N., Willis, A., Ward, P. A., et al. (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511. doi: 10.1056/NEJMoa1306555
Yang, Y., Muzny, D. M., Xia, F., Niu, Z., Person, R., Ding, Y., et al. (2014). Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879. doi: 10.1001/jama.2014.14601
Keywords: next-generation sequencing (NGS), exome sequencing, cancer genomics, clinical genomics, non-coding DNA
Citation: Shen T, Pajaro-Van de Stadt SH, Yeat NC and Lin JC-H (2015) Clinical applications of next generation sequencing in cancer: from panels, to exomes, to genomes. Front. Genet. 6:215. doi: 10.3389/fgene.2015.00215
Received: 26 February 2015; Accepted: 02 June 2015;
Published: 17 June 2015.
Edited by:
Yih-Horng Shiao, US Patent Trademark Office, USAReviewed by:
Andrey Cherstvy, Research Center Juelich, GermanyParvin Mehdipour, Tehran University of Medical Sciences, Iran
Copyright © 2015 Shen, Pajaro-Van de Stadt, Yeat and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jimmy C.-H. Lin, Rare Genomics Institute, 5225 Pooks Hills Road, Suite 1701N, Bethesda, MD 20814, USA,amltbXkubGluQHJhcmVnZW5vbWljcy5vcmc=