 Joanna Moreton
Joanna Moreton Abril Izquierdo
Abril Izquierdo Richard D. Emes
Richard D. Emes- 1Advanced Data Analysis Centre, Sutton Bonington Campus, University of Nottingham, Leicestershire, UK
- 2School of Veterinary Medicine and Science, Sutton Bonington Campus, University of Nottingham, Leicestershire, UK
De novo assembly of a complete transcriptome without the need for a guiding reference genome is attractive, particularly where the cost and complexity of generating a eukaryote genome is prohibitive. The transcriptome should not however be seen as just a quick and cheap alternative to building a complete genome. Transcriptomics allows the understanding and comparison of spatial and temporal samples within an organism, and allows surveying of multiple individuals or closely related species. De novo assembly in theory allows the building of a complete transcriptome without any prior knowledge of the genome. It also allows the discovery of alternate splice forms of coding RNAs and also non-coding RNAs, which are often missed by proteomic approaches, or are incompletely annotated in genome studies. The limitations of the method are that the generation of a truly complete assembly is unlikely, and so we require some methods for the assessment of the quality and appropriateness of a generated transcriptome. Whilst no single consensus pipeline or tool is agreed as optimal, various algorithms, and easy to use software do exist making transcriptome generation a more common approach. With this expansion of data, questions still exist relating to how do we make these datasets fully discoverable, comparable and most useful to understand complex biological systems?
Introduction
It is desirable to fully understand the complexity of an organism and the diversity of cell types arising from a single genome, or to compare the compliment of genes between evolutionary groups. This requires a capability to view and catalog the changes in gene expression of a cell or tissue. The transcriptome is the complete set of transcripts (RNA molecules) within a cell including protein-coding and non-coding RNAs. Additionally, the transcriptome encompasses all alternative splice forms, alternatively polyadenylated, and RNA-edited transcripts. Together, these reflect the genes that are actively expressed in a particular tissue (Grobe et al., 2002; Lu et al., 2013). Understanding the complete transcriptome is a technical challenge requiring technologies for capturing an accurate representation of the RNA in a cell or tissue. The dominant technology for the assessment of gene expression was microarrays which use printed or synthesized probes corresponding to mRNAs (Fu et al., 2009). Whilst these technologies are robust and offer a more mature framework for data analysis, they require an already annotated complete genome to design the probes. Microarrays are also limited by inaccurate hybridization of sequences to probes, which is difficult to model and hence account for (Wang et al., 2009; Compeau et al., 2011). In the case of model organisms, microarrays are still hugely useful to measure and compare gene expression. However, where high quality annotation and appropriate arrays do not exist, DNA sequencing offers the best method to understand the transcriptome. With the advent of Next Generation Sequencing (NGS) technologies and improved extraction methods to accurately purify RNA from smaller amounts of tissue or even single cells (Islam et al., 2011), the possibility to catalog and measure gene expression from a wider range of organisms has become possible.
KEY CONCEPT 1. Annotation
The process of assigning functional information to transcripts, such as gene ontology terms, in order to characterize the sequences and allow understanding of the system studied.
Transcriptome assembly is the process of identifying transcripts and their variants that are expressed in a determined sample (Lu et al., 2013). The simple premise is to reconstruct the complete sequences of all transcripts in the transcriptome. It is uncommon to achieve this in practice as most of the time the sequencing depth is not sufficient to cover all full-length transcripts, particularly the ones of low abundance. A transcriptome is therefore a set of contiguous (contig) sequences that represent transcript regions (Li et al., 2014). Generally the strategies for transcriptome assembly fall into two categories: reference-based and de novo (Figure 1), although a combination of both can be used (Chen et al., 2011; Garber et al., 2011; Martin and Wang, 2011; Haas et al., 2013; Lu et al., 2013). Whilst a comprehensive set of tools is unrealistic, we have compiled a set of commonly used, freely available tools for de novo assembly and assessment (Supplementary Table 1).

Figure 1. An overview of the two transcriptome assembly pipelines. The key parts of two transcriptome assembly pipelines are shown depending on whether a reference genome is available. This review is focused on de novo transcriptome assembly; more information on the pipeline for reference-based transcriptome assembly can be found in review papers such as Martin and Wang (2011).
Transcriptome Assembly Methods
Reference-Based Transcriptome Assembly Method
Reference-based transcriptome assembly is widely used when a model organism, with a sequenced genome for the target transcriptome, is accessible. Thus, the transcriptome is reconstructed by mapping to previously known sequences (Martin and Wang, 2011). The short reads are aligned to the reference genome allowing the overlapping regions to be assembled into transcripts. Where a good quality reference exists, the reference-based strategy is highly sensitive and it has become the basic method for many RNA sequencing (RNA-seq) studies. However, the accuracy of reference-based transcriptome assembly depends on correct read alignment, and issues such as alternative splicing and sequencing errors increase the difficulty of this task (Grabherr et al., 2011). In a referenced-based assembly approach, the sequence reads are aligned to the genome using a tool such as TopHat2 (Kim et al., 2013), which takes splicing into consideration. This is necessary as copies of mature spliced RNA have been sequenced, but these need to be mapped to a genome containing introns. All alternative splicing events are then captured in a graph for each given locus. Different paths are traversed in the graph to find transcript variants (Martin and Wang, 2011). Two transcriptome assemblers that are commonly used for graph building and traversal are Cufflinks (Trapnell et al., 2010) and Scripture (Guttman et al., 2010). The computational requirements of reference-based transcriptome assembly are significantly less compared to de novo transcriptome assembly. Furthermore, the presence of artifacts or sequencing contamination does not represent a major issue since these can often be resolved when aligning the reads to the genome. However, the quality of the results depends largely on the quality of the genome model used.
KEY CONCEPT 2. Reference-based transcriptome assembly
A method which is used to reconstruct transcript sequences by aligning RNA sequencing reads to a reference genome.
KEY CONCEPT 3. De novo transcriptome assembly
A process by which overlapping RNA sequencing reads are combined without a reference genome to reconstruct transcript sequences.
The transcriptome assembly can also be complicated by reads that align to multiple sites in the genome; these are known as multi-mapped reads. This problem is increased if the reads are short, therefore large complex transcriptomes are not easily assembled from very short reads (Martin and Wang, 2011). If there is insufficient unique information in the read sequences, then it is difficult to assign the reads to the correct location during alignment to the reference genome. If multi-mapped reads are discarded, then information for non-unique regions will be lost including gene families where gene sequences can be highly similar (Robert and Watson, 2015). If they are retained, it can be a challenge to accurately estimate gene or transcript abundances (Patro et al., 2014). Recently, Robert and Watson (2015) proposed a method for dealing with multi-mapped reads. They suggest taking all of the reads that cannot not be aligned to a unique gene and instead allocating them to a “multi-mapped group.” These groups are determined from the RNA-seq data rather than relying on existing annotation. By performing differential expression analysis on multi-mapped gene groups, rather than individual genes, important biological information can be examined that would have otherwise been filtered out (Robert and Watson, 2015).
Once reads are mapped and transcripts are identified, there are tools that can be used to quantitate gene expression such as Cufflinks (Trapnell et al., 2010), DESeq2 (Love et al., 2014), or EdgeR (Robinson et al., 2010). Thus, for organisms with an accurate, complete and well annotated genome, the measurement of genes expressed in a sample is becoming commonplace with robust methods for mapping transcript fragments to the genome and measuring the transcriptome content. However, where an annotated genome does not exist, or the number of alternate transcript isoforms is high, the problem of generating an accurate representation of the complete transcriptome remains. It is in these situations that de novo transcriptome assembly is particularly attractive as it provides an alternative option for assessing a non-model transcriptome (Zhao et al., 2011). De novo transcriptome assembly works without a reference to attempt to directly reconstruct overlapping reads into transcripts (Grabherr et al., 2011; Martin and Wang, 2011; Clarke et al., 2013; Lu et al., 2013). The complexities of this approach make it more computationally demanding, however a range of software tools exist including Oases (Schulz et al., 2012), Trans-ABySS (Robertson et al., 2010), MIRA (Chevreux et al., 2004), and Trinity (Grabherr et al., 2011). Several studies have been carried out to evaluate the execution of transcript assemblers (e.g., Clarke et al., 2013), and although they all differ in performance, currently there is no single transcriptome assembler categorized to be the best option for every condition (Grabherr et al., 2011; Clarke et al., 2013; Góngora-Castillo and Buell, 2013; Lu et al., 2013). With these specialist comparisons of performance available, it is not the objective of this review to describe nuances of different approaches or to promote a single method as optimal. In many cases the use of multiple approaches and subsequent merging of assemblies to generate a consensus single or set of assemblies might be appropriate. For example, incorporating sequences from different assemblers and parameters to generate a consensus transcriptome, by using transcripts present in multiple original transcriptome assemblies (Moreton et al., 2014).
De novo Transcriptome Assembly Method
De novo transcriptome assemblers commonly use a strategy which involves constructing de Bruijn graphs (e.g., Grabherr et al., 2011; Schulz et al., 2012). In this approach all subsequences of length k are found in the reads and these are known as “k-mers.” A de Bruijn graph is created using all unique k-mers as nodes, with connecting edges representing immediately overlapping k-mers (Figure 2). That is if a k-mer substring is shifted by one sequence base, and it overlaps another k-mer (by k-1 bases), then an edge is drawn between the nodes associated with those k-mers (Martin and Wang, 2011). A linear chain of k-mer nodes is compressed into a single node where possible (where the two nodes are joined by a single unique edge). Transcript variants can then be assembled by traversing the paths of the graph. Figure 2 shows a toy example of a de Bruijn graph constructed from two 7 bp sequence reads and k-mers of length 5. In this example two paths can be found from the graph representing two possible transcript isoforms.
KEY CONCEPT 4. k-mers
A subsequence of specified length k. They are often used by de novo assemblers to allow sequence information to be compacted, which makes reconstruction of transcripts easier computationally.
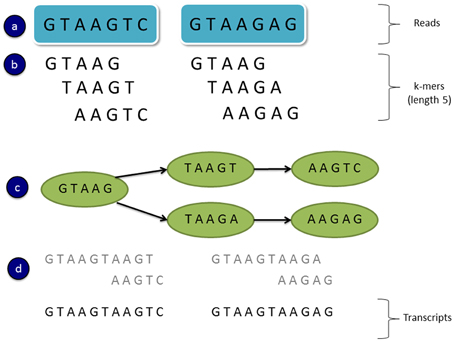
Figure 2. An example of a simple de Bruijn graph. (A) Read sequences (B) All subsequence k-mers of length 5 from the reads (C) A de Bruijn graph constructed from unique k-mers as the nodes and overlapping k-mers connected by edges (a k-mer shifted by one base overlaps another k-mer by k-1 bases) (D) Assembled transcripts by traversing the two paths in the graph.
Before the introduction of de Bruijn graphs, assemblers used the overlap-layout-consensus algorithm where overlap information between read sequences is added to a mathematical graph to find a consensus sequence (Li et al., 2012b). In this strategy, each graph node corresponds to a read and if two reads overlap, their nodes are joined by an edge on the graph. The overlap-layout-consensus alignment step is computationally intensive when assembling a huge number of short reads, so a de Bruijn graph algorithm is preferred for generating de novo assemblies. By compacting the sequence information into k-mers, the graph theory method for finding a path in the graph becomes easier computationally (Pevzner et al., 2001; Li et al., 2012b). One disadvantage in using the de Bruijn graph approach is the generation of misassembled contigs which occurs because of the use of k-mers (Clarke et al., 2013). If two transcripts from different genes have the same k-mer sequence they could be erroneously connected. The computational proficiency of the de Bruijn graph strategy is clearly beneficial, but it is an ongoing problem to balance this with assembly accuracy (Clarke et al., 2013).
There are a number of difficulties that are encountered by the de novo transcriptome assembly strategy. For example, it is challenging to discriminate between transcript variants that are produced from processes such as alternative splicing or sequences transcribed from paralogous genes (Grabherr et al., 2011; Vijay et al., 2013). These sorts of sequences will share k-mer sequences and hence it is difficult to tease them apart into separate transcripts. Software tools have been designed to distinguish transcript variants using paired-end read data and read coverage (Góngora-Castillo and Buell, 2013). For instance, the Trinity assembler (Grabherr et al., 2011) reconstructs alternatively spliced transcripts and paralogous sequences by clustering overlapping contigs and generating a de Bruijn graph for each cluster of sequences independently. These graphs are then supplemented with the read and paired-end information to generate all possible transcript variants. Despite the challenges, the transcriptomes of many different organisms have been assembled using the de novo approach (e.g., Kumar and Blaxter, 2010; Robertson et al., 2010; Zhao et al., 2011; Price et al., 2015). These complexities are additionally compounded when mixed samples are included, for example in pathogen and host, or when transcripts may not form distinct entities due to dense or overlapping transcripts, as seen in prokaryote organisms. In the case of bacterial de novo assembly, tools such as Rockhopper (McClure et al., 2013; Tjaden, 2015) have been specifically developed.
Assessment of Generated De novo Assemblies
Whilst a number of studies have focused on transcriptome assembly, the assessment of the overall quality of the derived assemblies is less well defined. A number of different measures are commonly used to evaluate assembled transcriptomes. Commonly used metrics when there is no close reference include the number of contigs (transcripts) assembled, summed contig length, mean transcript length, N50 value, and the proportion of reads that could be mapped back to the assembled transcripts (RMBT; e.g., Zhao et al., 2011). These measures can be used to compare and select optimal assemblies, for example the N50 value can be maximized whilst keeping the total assembly length as long as possible (Zerbino, 2010). It is also important to consider the time taken to generate the assemblies (Kumar and Blaxter, 2010). When reference sequences of closely related species are available, the assembled contigs can be compared using a sequence similarity tool such as BLAST (McGinnis and Madden, 2004) to assess the validity of the assembly (e.g., Arun-Chinnappa and McCurdy, 2015; Ghaffari et al., 2015). However, this approach is biased by the appropriateness of the choice of related species for comparison and will be biased toward available “model” genomes.
Assessment of the completeness of an assembled transcriptome is more problematic. This is due to the impossibility of knowing a priori what the complete transcriptome for a previously unsequenced cell, or collection of cells, at a particular time point is. However, the theoretical completeness can also be assessed, using methods to determine the assembly of transcripts that are expected to be present in all cells at all times, such as the Core Eukaryotic Genes Mapping Approach (CEGMA) tool by Parra et al. (2007). Although not developed specifically for this purpose, many studies have used this approach to determine if a collection of newly assembled transcripts encode one or more of a set of core genes conserved across a wide range of eukaryotic species, thus providing a percentage “completeness” score (e.g., Chauhan et al., 2014; Moreton et al., 2014; Frías-López et al., 2015; Powell et al., 2015; Price et al., 2015). A recent web-based tool “TRUFA,” developed by Kornobis et al. (2015), incorporates CEGMA into its pipeline as part of the assessment stage of de novo assemblies. As of May 2015 CEGMA is no longer being supported, however a new tool “BUSCO” has been published by Simão et al. (2015), to assess assembly and annotation completeness using sets of Benchmarking Universal Single-Copy Orthologs (BUSCO), selected from OrthoDB (Kriventseva et al., 2015). When comparing the completeness of genome assemblies and gene sets across 40 species, the BUSCO assessments were more consistent than CEGMA, the run-times were much faster and the software can also be used to assess gene sets and transcriptomes (Simão et al., 2015).
Some authors have suggested that evaluation measures such as N50 might be misleading and uninformative for evaluating transcriptome assemblies (e.g., O'Neil and Emrich, 2013; Li et al., 2014; Chen et al., 2015). For example, Chen et al. (2015) found that the transcriptome assemblies with the highest N50 values, did not make a significant contribution to the best assembled transcript set based on coding potential. Li et al. (2014) developed the “DETONATE” (DE novo TranscriptOme rNa-seq Assembly with or without the Truth Evaluation) software, which includes both reference-free (RSEM-EVAL) and reference-based (REF-EVAL) methods. The reference-free approach is based on a probabilistic model that uses only the read and assembly data. When reference transcripts are available, the REF-EVAL component can be used to generate scores based on different reference-based measures. DETONATE is currently only designed to evaluate assemblies generated from Illumina data, although there are plans to update the package to handle data from other sequencing platforms. O'Neil and Emrich (2013) assessed a number of metrics for de novo transcriptome assemblies including unique annotations and “ortholog hit ratio” from their earlier work (O‘Neil et al., 2010). The correlation between the REF-EVAL score and the ortholog hit ratio measure was found to be low, although the number of unique proteins matched had good correlation to REF-EVAL (Li et al., 2014).
There are a number of errors that can occur in de novo transcriptome assembly, for example two transcripts may be combined into a single false chimeric transcript, or contigs might be incomplete or mis-assembled (Smith-Unna et al., 2015). These errors can be detected using read evidence. The TransRate tool (Smith-Unna et al., 2015) aligns the paired-end reads that were used to generate the assembly, back to the assembled contigs. The alignments are then evaluated and each contig is assigned a score based on properties such as how well the nucleotides in the aligned reads matched to the assembled contigs, the coverage of the contig nucleotides, and the order of the contig nucleotides based on the paired-end read orientations. TransRate also calculates an assembly score which is generated from the individual contig scores, and the proportion of input reads that were incorporated into the de novo assembly. As mentioned before, RSEM-EVAL is another reference-free evaluation method; however it does not focus on the evaluation of individual contigs. The RSEM-EVAL tool is also limited to assemblies generated from Illumina data, but TransRate is not restricted in this way. The TransRate tool is also useful because it allows the filtration of individual contigs based on their scores. Furthermore, the authors used 155 previously published de novo assemblies in a meta-analysis to allow users to analyze their assemblies in comparison with others. In summary, assembly assessments are essential and will be increasingly important for evaluation of new methods, or in the combination of assemblies as part of optimization strategies.
Annotation of Transcriptome Assembly
Annotation of function is required to characterize transcripts and allow understanding of the system studied. Most approaches to annotation of protein coding transcripts use one or more homology based approaches to identify related sequences of known function, and hence transfer this annotation to the new transcript (Emes, 2008). There are however limitations to these approaches. The problem of transfer of inappropriate or inaccurate annotation from one dataset to another, leading to the propagation of annotation error, is the most concerning. A preferred method is the use of protein domain architecture to drive the annotation. Searching for conserved domains using hidden Markov model search tools, such as HMMER3 (Finn et al., 2011), is a relatively simple process. These tools search comprehensive libraries of domains such as Pfam (Finn et al., 2014) or InterPro (Mitchell et al., 2015). Databases such as Pfam2GO, from the gene ontology consortium (Gene Ontology Consortium, 2015), allow the domain content to generate restricted descriptors of each transcript. Pipeline tools to automate this process using both sequence similarity and domain composition, such as the Trinotate pipeline (https://trinotate.github.io/), are available but are currently relatively slow or computationally intense to use. Another consideration for the annotation process is searching for repeat elements using programs such as RepeatMasker (http://www.repeatmasker.org) or the Tandem Repeats Finder (Benson, 1999). For example, RepeatMasker can be used with the Repbase database (Bao et al., 2015) to identify transposable elements and other types of repeats (Gillard et al., 2014; Kumar et al., 2014; Cokus et al., 2015; Richardson and Sherman, 2015).
De novo Transcriptome Assembly Availability
Whilst most journals require raw sequencing reads to be made publicly available in a database such as the Sequence Read Archive (SRA; Kodama et al., 2012), often the assembled transcripts and annotations are not made available. This results in lack of clarity and wasted effort to redo the analysis. The SRA is part of the International Nucleotide Sequence Database Collaboration (Kodama et al., 2012). This repository is available at the National Center for Biotechnology Information (NCBI, www.ncbi.nlm.nih.gov/sra), European Bioinformatics Institute (EBI, www.ebi.ac.uk/ena), and DNA Data Bank of Japan (DDBJ, http://trace.ddbj.nig.ac.jp/dra). There are support pages and handbooks to help with submitting data, and these are available at the NCBI, EBI, and DDBJ websites. As well as raw sequence data, alignment files in BAM (Li et al., 2009) format can also be submitted to the SRA. With reducing costs of sequencing and availability of software for transcriptome assembly, the making of transcriptome assembly open and available is a key problem in bioinformatics. Often generic genome browsers are difficult to set up and are not well-suited for transcriptome data (Jones and Blaxter, 2013), and so a number of software solutions to host and visualize transcriptome assemblies have been developed. Jones and Blaxter (2013) developed the web application “afterParty” which enables users to make a transcriptome publicly available. The application can take as input either Roche 454 reads, or assembled contigs (putative transcripts) from any platform. If raw 454 sequencing reads are used as an input, then afterParty can assemble them using MIRA (Chevreux et al., 2004) and then annotate the resulting contigs using BLASTX (Altschul et al., 1997), UniProt (Uniprot Consortium, 2012), and InterProScan (Zdobnov and Apweiler, 2001). In the other afterParty workflows, contigs generated by the user from any sequencing platform can be uploaded with or without annotation. AfterParty can also be used to browse transcriptomes and visualize data sets in a web browser. For example, all contigs with annotation matching a particular search term can be used to generate a scatter plot of GC content against coverage in a comparison to the full assembly (Jones and Blaxter, 2013). Different contig sets, chart types, and displays can be selected. In addition to filtering by annotation, a DNA or protein sequence can be used to find contigs with sequence similarity. The contigs can also be searched by properties such as length, quality, coverage, and GC content. A number of studies have already used the afterParty website as a means of hosting and distributing transcriptome data (e.g., Heitlinger et al., 2014; Short et al., 2014; McTaggart et al., 2015). For users running afterParty locally, the source code, and dependencies can be installed. However, the more convenient method would be to use the virtual disk image (available on GitHub), which contains all the required dependencies to run the software using a virtual machine. Alternatively, afterParty is also available through a public server.
RNAbrowse is an alternative package with a web interface that can be used to store and visualize de novo transcriptome data (Mariette et al., 2014). It is based on the BioMart (Smedley et al., 2015) software and in addition to the web interface it includes a command line tool for administration which requires a unix server and MySQL database. The project introduction page of the web interface contains useful information such as the software and parameters used to generate the alignment, annotation, assembly, and variant analysis. The contig and variant overview pages show general statistics and related figures such as a bar chart of contig length distribution. There is a blast query form to search the contigs using an input sequence, and the BioMart search page can also be used to filter the data based on criteria such as contig name, length, or annotation. In the sequence view, the longest open reading frame can be identified. It is also possible to view the sequences and annotations in JBrowse (Skinner et al., 2009) and compare read coverage between samples in the contig depth view. The figures produced using the interface can be easily printed or downloaded and there is also a dedicated download page to enable users to save some or all of the data (Mariette et al., 2014). In its simplest form, RNAbrowse can be set up using the assembled contig sequences (FASTA format) alongside the annotation and alignment files. Again, installation requires a number of prerequisite tools and the setup process can be quite time consuming (Mariette et al., 2014). This may therefore be better attempted in collaboration with a bioinformatics group or local support. However, there is a project website with lots of information about RNAbrowse including guides, demonstrations, example datasets and a configuration file template for larger projects. Different schedulers can also be selected to address any time issues (Mariette et al., 2014). As an example of a practical use, RNAbrowse has been used to display and distribute beech tree de novo transcriptome data (Lesur et al., 2015).
Apart from more complete packages such as afterParty and RNAbrowse, there are limited tools with web interfaces that are available for analysis of transcriptome data. CBrowse (Li et al., 2012a) is a web browser which takes assembled contig sequences and BAM/SAM alignment files as input, and enables the user to identify polymorphisms and view the contigs in the web interface. Its focus is not on annotation, however CBrowse can be used to disseminate assembled transcriptome data (Li et al., 2012a). As a less permanent solution, some research groups have used individual online resources to make their data available. For example, Aya et al. (2015) developed a transcriptome database as a public web resource for downloading and browsing fern de novo transcriptome assembly data, where both BLAST and keyword searches can be performed. Another research group released their axolotl read and transcriptome assembly data on a website with a keyword search facility (Stewart et al., 2013). However, the risk of non-specialist solutions is that repositories are not maintained or, with the movement of personnel, that the skill to maintain repositories is lost. As an interim solution, we and others have simply made transcriptome assembly data available to download by partnering with appropriate journals (Moreton et al., 2014; Ghaffari et al., 2015). Given these considerations, and the enhanced ability to query, filter and visualize transcriptome data, tools like afterParty, and RNAbrowse make the most ideal options.
Conclusion
As the desire to catalog and compare the varied transcriptomes of complex organisms continues, de novo transcriptome assembly is an important tool in the bioinformatician's arsenal. Whilst rapid progress in single molecule sequencing is being made, it is currently not mature and so assembly, annotation and assessment of transcriptomes from relatively short reads will continue to be essential. To make these methods truly useful, assemblies that are accurately assembled and annotated are essential, but also the availability and openness of assembled transcriptomes not simply raw data must become expected practice.
Author Contributions
JM, AI, and RE wrote the paper, prepared figures, and reviewed drafts of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AI was supported by an international PhD studentship from Consejo Nacional de Ciencia y Tecnologia (CONACYT) Mexico. JM and RE were funded by the University of Nottingham.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fgene.2015.00361
Author Biography
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Arun-Chinnappa, K. S., and McCurdy, D. W. (2015). De novo assembly of a genome-wide transcriptome map of Vicia faba (L.) for transfer cell research. Front. Plant Sci. 6:217. doi: 10.3389/fpls.2015.00217
Aya, K., Kobayashi, M., Tanaka, J., Ohyanagi, H., Suzuki, T., Yano, K., et al. (2015). De novo transcriptome assembly of a fern, Lygodium japonicum, and a web resource database, Ljtrans DB. Plant Cell Physiol. 56, e5. doi: 10.1093/pcp/pcu184
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11. doi: 10.1186/s13100-015-0041-9
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Chauhan, P., Hansson, B., Kraaijeveld, K., De Knijff, P., Svensson, E. I., and Wellenreuther, M. (2014). De novo transcriptome of Ischnura elegans provides insights into sensory biology, colour and vision genes. BMC Genomics 15:808. doi: 10.1186/1471-2164-15-808
Chen, G., Wang, C., and Shi, T. (2011). Overview of available methods for diverse RNA-Seq data analyses. Sci. China Life Sci. 54, 1121–1128. doi: 10.1007/s11427-011-4255-x
Chen, S., Mcelroy, J. S., Dane, F., and Peatman, E. (2015). Optimizing transcriptome assemblies for leaf and seedling by combining multiple assemblies from three de novo assemblers. Plant Genome 8:1. doi: 10.3835/plantgenome2014.10.0064
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A. J., Müller, W. E., Wetter, T., et al. (2004). Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14, 1147–1159. doi: 10.1101/gr.1917404
Clarke, K., Yang, Y., Marsh, R., Xie, L., and Zhang, K. K. (2013). Comparative analysis of de novo transcriptome assembly. Sci. China Life Sci. 56, 156–162. doi: 10.1007/s11427-013-4444-x
Cokus, S. J., Gugger, P. F., and Sork, V. L. (2015). Evolutionary insights from de novo transcriptome assembly and SNP discovery in California white oaks. BMC Genomics 16:552. doi: 10.1186/s12864-015-1761-4
Compeau, P. E., Pevzner, P. A., and Tesler, G. (2011). How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 29, 987–991. doi: 10.1038/nbt.2023
Emes, R. D. (2008). Inferring function from homology. Methods Mol. Biol. 453, 149–168. doi: 10.1007/978-1-60327-429-6_6
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Frías-López, C., Almeida, F. C., Guirao-Rico, S., Vizueta, J., Sánchez-Gracia, A., Arnedo, M. A., et al. (2015). Comparative analysis of tissue-specific transcriptomes in the funnel-web spider Macrothele calpeiana (Araneae, Hexathelidae). PeerJ 3:e1064. doi: 10.7717/peerj.1064
Fu, X., Fu, N., Guo, S., Yan, Z., Xu, Y., Hu, H., et al. (2009). Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 10:161. doi: 10.1186/1471-2164-10-161
Garber, M., Grabherr, M. G., Guttman, M., and Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 8, 469–477. doi: 10.1038/nmeth.1613
Gene Ontology Consortium (2015). Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. doi: 10.1093/nar/gku1179
Ghaffari, N., Arshad, O. A., Jeong, H., Thiltges, J., Criscitiello, M. F., Yoon, B.-J., et al. (2015). Examining De Novo transcriptome assemblies via a quality assessment pipeline. Comput. Biol. Bioinformatics IEEE/ACM Trans. 99:1. doi: 10.1109/TCBB.2015.2446478
Gillard, G. B., Garama, D. J., and Brown, C. M. (2014). The transcriptome of the NZ endemic sea urchin Kina (Evechinus chloroticus). BMC Genomics 15:45. doi: 10.1186/1471-2164-15-45
Góngora-Castillo, E., and Buell, C. R. (2013). Bioinformatics challenges in de novo transcriptome assembly using short read sequences in the absence of a reference genome sequence. Nat. Prod. Rep. 30, 490–500. doi: 10.1039/c3np20099j
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grobe, K., Esko, J., Aikawa, J., Grobe, K., Tsujimoto, M., and Esko, J. (2002). Analysis of the mouse transcriptome based on functional annotation. Nature 420, 563–573. doi: 10.1038/nature01266
Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., et al. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510. doi: 10.1038/nbt.1633
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Heitlinger, E., Taraschewski, H., Weclawski, U., Gharbi, K., and Blaxter, M. (2014). Transcriptome analyses of Anguillicola crassus from native and novel hosts. PeerJ 2:e684. doi: 10.7717/peerj.684
Islam, S., Kjällquist, U., Moliner, A., Zajac, P., Fan, J.-B., Lönnerberg, P., et al. (2011). Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 21, 1160–1167. doi: 10.1101/gr.110882.110
Jones, M., and Blaxter, M. (2013). afterParty: turning raw transcriptomes into permanent resources. BMC Bioinformatics 14:301. doi: 10.1186/1471-2105-14-301
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kodama, Y., Shumway, M., and Leinonen, R. (2012). The Sequence read archive: explosive growth of sequencing data. Nucleic Acids Res. 40, D54–D56. doi: 10.1093/nar/gkr854
Kornobis, E., Cabellos, L., Aguilar, F., Frías-López, C., Rozas, J., Marco, J., et al. (2015). TRUFA: a user-friendly web server for de novo RNA-seq analysis using cluster computing. Evol. Bioinform. Online 11, 97–104. doi: 10.4137/EBO.S23873
Kriventseva, E. V., Tegenfeldt, F., Petty, T. J., Waterhouse, R. M., Simão, F. A., Pozdnyakov, I. A., et al. (2015). OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 43, D250–D256. doi: 10.1093/nar/gku1220
Kumar, M., Gantasala, N. P., Roychowdhury, T., Thakur, P. K., Banakar, P., Shukla, R. N., et al. (2014). De Novo transcriptome sequencing and analysis of the cereal cyst nematode, Heterodera avenae. PLoS ONE 9:e96311. doi: 10.1371/journal.pone.0096311
Kumar, S., and Blaxter, M. L. (2010). Comparing de novo assemblers for 454 transcriptome data. BMC Genomics 11:571. doi: 10.1186/1471-2164-11-571
Lesur, I., Bechade, A., Lalanne, C., Klopp, C., Noirot, C., Leple, J. C., et al. (2015). A unigene set for European beech (Fagus sylvatica L.) and its use to decipher the molecular mechanisms involved in dormancy regulation. Mol. Ecol. Resour. 15, 1192–1204. doi: 10.1111/1755-0998.12373
Li, B., Fillmore, N., Bai, Y., Collins, M., Thomson, J. A., Stewart, R., et al. (2014). Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 15, 553. doi: 10.1186/s13059-014-0553-5
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, P., Ji, G., Dong, M., Schmidt, E., Lenox, D., Chen, L., et al. (2012a). CBrowse: a SAM/BAM-based contig browser for transcriptome assembly visualization and analysis. Bioinformatics 28, 2382–2384. doi: 10.1093/bioinformatics/bts443
Li, Z., Chen, Y., Mu, D., Yuan, J., Shi, Y., Zhang, H., et al. (2012b). Comparison of the two major classes of assembly algorithms: overlap–layout–consensus and de-bruijn-graph. Brief. Funct. Genomics 11, 25–37. doi: 10.1093/bfgp/elr035
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lu, B., Zeng, Z., and Shi, T. (2013). Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq. Sci. China Life Sci. 56, 143–155. doi: 10.1007/s11427-013-4442-z
Mariette, J., Noirot, C., Nabihoudine, I., Bardou, P., Hoede, C., Djari, A., et al. (2014). RNAbrowse: RNA-Seq de novo assembly results browser. PLoS ONE 9:e96821. doi: 10.1371/journal.pone.0096821
Martin, J. A., and Wang, Z. (2011). Next-generation transcriptome assembly. Nat. Rev. Genet. 12, 671–682. doi: 10.1038/nrg3068
McClure, R., Balasubramanian, D., Sun, Y., Bobrovskyy, M., Sumby, P., Genco, C. A., et al. (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41, e140. doi: 10.1093/nar/gkt444
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
McTaggart, S. J., Hannah, T., Bridgett, S., Garbutt, J. S., Kaur, G., and Boots, M. (2015). Novel insights into the insect trancriptome response to a natural DNA virus. BMC Genomics 16:310. doi: 10.1186/s12864-015-1499-z
Mitchell, A., Chang, H. Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Moreton, J., Dunham, S. P., and Emes, R. D. (2014). A consensus approach to vertebrate de novo transcriptome assembly from RNA-seq data: assembly of the duck (Anas platyrhynchos) transcriptome. Front. Genet. 5:190. doi: 10.3389/fgene.2014.00190
O‘Neil, S. T., Dzurisin, J. D., Carmichael, R. D., Lobo, N. F., Emrich, S. J., and Hellmann, J. J. (2010). Population-level transcriptome sequencing of nonmodel organisms Erynnis propertius and Papilio zelicaon. BMC Genomics 11:310. doi: 10.1186/1471-2164-11-310
O'Neil, S. T., and Emrich, S. J. (2013). Assessing de novo transcriptome assembly metrics for consistency and utility. BMC Genomics 14:465. doi: 10.1186/1471-2164-14-465
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Patro, R., Mount, S. M., and Kingsford, C. (2014). Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 32, 462–464. doi: 10.1038/nbt.2862
Pevzner, P. A., Tang, H., and Waterman, M. S. (2001). An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. U.S.A. 98, 9748–9753. doi: 10.1073/pnas.171285098
Powell, D., Knibb, W., Remilton, C., and Elizur, A. (2015). De-novo transcriptome analysis of the banana shrimp (Fenneropenaeus merguiensis) and identification of genes associated with reproduction and development. Mar. Genomics 22, 71–78. doi: 10.1016/j.margen.2015.04.006
Price, S. J., Garner, T. W. J., Balloux, F., Ruis, C., Paszkiewicz, K. H., Moore, K., et al. (2015). A de novo Assembly of the Common Frog (Rana temporaria) transcriptome and comparison of transcription following exposure to Ranavirus and Batrachochytrium dendrobatidis. PLoS ONE 10:e0130500. doi: 10.1371/journal.pone.0130500
Richardson, M. F., and Sherman, C. D. H. (2015). De Novo assembly and characterization of the invasive northern pacific seastar transcriptome. PLoS ONE 10:e0142003. doi: 10.1371/journal.pone.0142003
Robert, C., and Watson, M. (2015). Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol. 16, 1–16. doi: 10.1186/s13059-015-0734-x
Robertson, G., Schein, J., Chiu, R., Corbett, R., Field, M., Jackman, S. D., et al. (2010). De novo assembly and analysis of RNA-seq data. Nat. Methods 7, 909–912. doi: 10.1038/nmeth.1517
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Schulz, M. H., Zerbino, D. R., Vingron, M., and Birney, E. (2012). Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092. doi: 10.1093/bioinformatics/bts094
Short, S., Yang, G., Guler, Y., Green Etxabe, A., Kille, P., and Ford, A. T. (2014). Crustacean intersexuality is feminization without demasculinization: implications for environmental toxicology. Environ. Sci. Technol. 48, 13520–13529. doi: 10.1021/es5050503
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J., and Holmes, I. H. (2009). JBrowse: a next-generation genome browser. Genome Res. 19, 1630–1638. doi: 10.1101/gr.094607.109
Smedley, D., Haider, S., Durinck, S., Pandini, L., Provero, P., Allen, J., et al. (2015). The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 43, W589–W598. doi: 10.1093/nar/gkv350
Smith-Unna, R. D., Boursnell, C., Patro, R., Hibberd, J. M., and Kelly, S. (2015). TransRate: reference free quality assessment of de-novo transcriptome assemblies. BioRxiv 021626. doi: 10.1101/021626
Stewart, R., Rascón, C. A., Tian, S., Nie, J., Barry, C., Chu, L.-F., et al. (2013). Comparative RNA-seq Analysis in the unsequenced axolotl: the oncogene burst highlights early gene expression in the blastema. PLoS Comput. Biol. 9:e1002936. doi: 10.1371/journal.pcbi.1002936
Tjaden, B. (2015). De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 16, 1. doi: 10.1186/s13059-014-0572-2
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Uniprot Consortium (2012). Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75. doi: 10.1093/nar/gkr981
Vijay, N., Poelstra, J. W., Künstner, A., and Wolf, J. B. (2013). Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol. Ecol. 22, 620–634. doi: 10.1111/mec.12014
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zerbino, D. R. (2010). Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinformatics Chapter 11, Unit 11:15. doi: 10.1002/0471250953.bi1105s31
Keywords: de novo transcriptome assembly, high-throughput sequencing, assessment, availability, annotation
Citation: Moreton J, Izquierdo A and Emes RD (2016) Assembly, Assessment, and Availability of De novo Generated Eukaryotic Transcriptomes. Front. Genet. 6:361. doi: 10.3389/fgene.2015.00361
Received: 20 September 2015; Accepted: 19 December 2015;
Published: 11 January 2016.
Edited by:
Chun Liang, Miami University, USAReviewed by:
Shizuka Uchida, Goethe University Frankfurt, GermanyMadavan Vasudevan, Bionivid Technology Pvt. Ltd., India
Copyright © 2016 Moreton, Izquierdo and Emes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sm9hbm5hLk1vcmV0b25Abm90dGluZ2hhbS5hYy51aw==