 Stephanie A. Cohen
Stephanie A. Cohen Christopher A. Tan
Christopher A. Tan Ryan Bisson
Ryan Bisson- 1Cancer Genetics Risk Assessment Program, St. Vincent Health, Indianapolis, IN, USA
- 2Invitae Corporation, San Francisco, CA, USA
- 3Cancer Genetics Center, UF Health Cancer Center–Orlando Health, Orlando, FL, USA
The utilization of next-generation sequencing technology to interrogate multiple genes simultaneously is being utilized more frequently in hereditary cancer testing. While this has benefits of reducing cost and allowing clinicians to cast a wide net in the elucidation of their patient's cancer, panel testing has the potential to reveal unexpected information. We report on a proband with pathogenic variants resulting in two different hereditary colon cancer syndromes. A 39-year-old male with a history of colon cancer, more than 20 colon polyps and a family history of colon cancer presented for genetic counseling. Testing with a 7-gene high-risk hereditary colon cancer panel identified a homozygous pathogenic variant, c.1187G>A (p.Gly396Asp) in MUTYH, and a likely pathogenic duplication of exon 7 in MSH2. Since this test result, the proband's mother was diagnosed with colon cancer; subsequent genetic testing confirmed she also carries the likely pathogenic duplication in the MSH2 gene. Although the cancer risk in individuals who carry multiple pathogenic variants has not been established for combined biallelic MUTYH-associated polyposis and Lynch syndrome, the identification of multiple pathogenic variants does allow for screening for cancers associated with both syndromes and has implications for cancer risk for family members. In particular, this has significant impact on those who test negative for a known familial pathogenic variant, yet could be still be at risk for cancer due to a second pathogenic variant in a family. More information is needed on the frequency of occurrence of multiple pathogenic variants, as well as the phenotypic spectrum when multiple pathogenic variants are present.
Introduction
A 39-year-old male who works as a general manager presented to the Cancer Genetics Risk Assessment program for genetic counseling due to a personal history of rectal cancer at the age of 38. In addition to the rectal cancer, more than 20 adenomatous polyps were found in his colon. He underwent a laparoscopic total proctocolectomy with ileoanal J-pouch anastomosis and diverting loop ileostomy. The tumor was a superficially invasive, moderately differentiated adenocarcinoma, arising within a background of adenoma and multifocal high-grade dysplasia. Immunohistochemistry staining as part of the hospital's universal screening program for Lynch syndrome displayed intact MLH1, MSH2, MSH6, and PMS2 proteins; microsatellite instability (MSI) testing was not performed. The proband's father had a history of basal cell carcinoma at the age of 58 and five colon polyps at the age of 61. Colon cancer was reported in the proband's paternal uncle at the age of 70 and a paternal great-grandmother in her 50s. Finally, the proband reported that his mother had a hysterectomy at a young age due to an unspecified gynecologic cancer.
The proband underwent genetic testing with a 7-gene high-risk hereditary colon cancer panel which screened for variants in the APC, EPCAM, MLH1, MSH2, MSH6, MUTYH, and PMS2 genes. The proband was found to carry a homozygous pathogenic variant, c.1187G>A (p.Gly396Asp) in MUTYH, and a likely pathogenic duplication of exon 7 in MSH2 (Figure 1). The c.1187G>A (p.Gly396Asp) variant is a common cause of MUTYH-associated polyposis in individuals of Northern European ancestry and experimental studies have shown that this missense variant disrupts MUTYH protein function (Aretz et al., 2006; Ali et al., 2008). While the exact position of the exon 7 duplication in MSH2 was not conclusively determined, the most likely explanation is that it occurs in tandem, which would likely result in a frameshift leading to a premature translational stop signal and an absent or disrupted MSH2 protein. A similar duplication of exon 7 in the MSH2 gene has been reported previously in a patient affected with colorectal cancer (Casey et al., 2005). Both MUTYH and MSH2 variants were confirmed by using an appropriate orthogonal method.
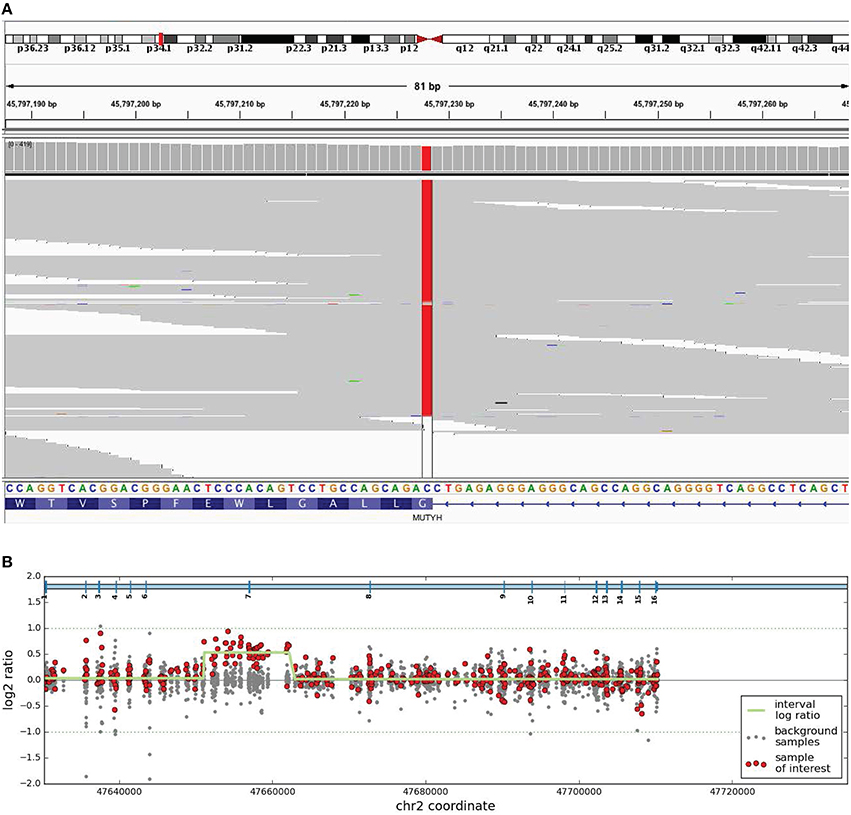
Figure 1. Biallelic MUTYH mutation and likely pathogenic deletion in MSH2. (A) Integrative Genomics Viewer images of next generation sequencing data of homozygous c.1187G>A (p.Gly396Asp) sequence variant with reference MUTYH nucleotide and amino acid nomenclature indicated. (B) Array CGH image. Red dots indicate the relevant clinical sample's log2 ratio confirming duplication of exon 7 in MSH2. Gray dots indicate the log2 ratios for seven other control samples. The green line indicates the segmentation by the DNA copy R package.
The proband's parents subsequently underwent genetic counseling. The proband's mother clarified that she had a diagnosis of vulvar cancer at the age of 34 and five colon polyps at the age of 50. She reported a family history of colorectal cancer in her maternal grandmother at an unknown age, her maternal aunt at the age of 50 and a maternal first cousin at the age of 53; all of this was unknown at the time of the proband's initial genetic counseling appointment (Figure 2). While the proband's mother was waiting for her genetic test results, she was diagnosed with an invasive, well-differentiated mucinous adenocarcinoma of the cecum at the age of 63. The MSH2 and MSH6 proteins were absent by immunohistochemistry staining on the tumor, while MLH1 and PMS2 were preserved; MSI was not performed.
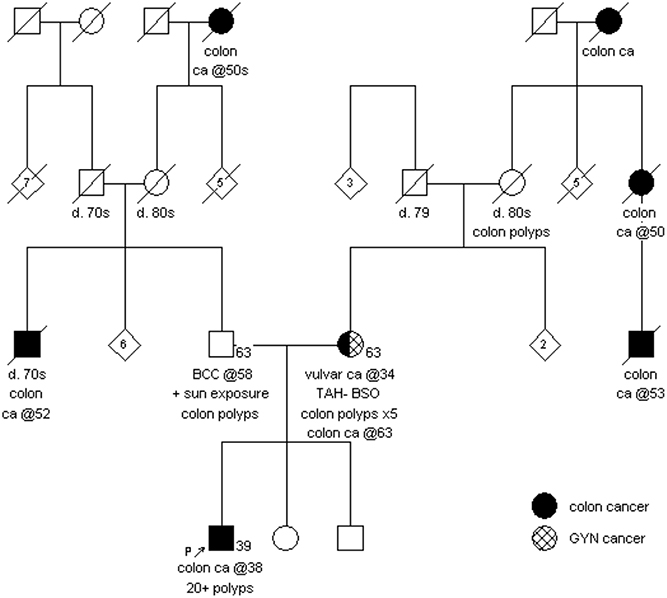
Figure 2. Family history as reported by proband and his mother.
Genetic testing confirmed that each parent was heterozygous for the pathogenic variant c.1187G>A (p.Gly396Asp) in the MUTYH gene. The proband's mother was also found to carry the likely pathogenic duplication in the MSH2 gene.
Background
Lynch syndrome is an autosomal dominant condition caused by a pathogenic variant in one of four mismatch repair (MMR) genes or in EPCAM, a gene involved in epithelial cell adhesion. Lynch syndrome is characterized by an increased risk for colorectal cancer, as well as cancers of the endometrium, ovary, stomach, small intestine, hepatobiliary tract, urinary tract, and brain (Kohlmann and Gruber, 2004/2014; Cohen and Leininger, 2014). The lifetime risk for cancer among individuals with Lynch syndrome ranges widely, depending on the specific mismatch repair gene that is involved. The lifetime risk for colorectal cancer associated with a pathogenic variant in MSH2 approaches 60%, and females with a pathogenic variant in MSH2 have up to a 40% lifetime risk for endometrial cancer.
MUTYH-associated polyposis (MAP), an autosomal recessive condition caused by biallelic pathogenic variants in MUTYH, is characterized by multiple colorectal adenomas arising in adulthood and increased colorectal cancer risk. Lifetime risk for colorectal cancer among patients with biallelic pathogenic variants in MUTYH is estimated to be at least 43 to almost 100% in the absence of timely surveillance (Maartje Nielsen et al., 2012/2015; Lubbe et al., 2009). Biallelic pathogenic variants in MUTYH have also been described in Lynch and Lynch-like families in the absence of pathogenic variants in the MMR genes (Castillejo et al., 2014). Whether monoallelic pathogenic variants in MUTYH also increase cancer risk is unclear, although there is some evidence suggesting a slight increase, especially when there is a family history of colon cancer (Win et al., 2014). The risk for colorectal cancers for carriers of a pathogenic variant in one of the MMR genes in addition to a single MUTYH pathogenic variant was substantially higher than that for carriers of a single MUTYH pathogenic variant alone, but not different from that for carriers of a MMR pathogenic variant alone (Win et al., 2015).
Cases of multiple hereditary cancer syndromes in a family caused by pathogenic variants in different genes related to two hereditary cancer syndromes have been previously reported in the literature (Thiffault et al., 2004; Gong et al., 2012). The cumulative cancer risks for patients who have both MAP and Lynch syndrome are not well characterized. There is one previous report of a patient identified with compound heterozygous pathogenic variants in MUTYH who also carried a pathogenic variant in MSH6; this individual exhibited a relatively mild clinical presentation (van Puijenbroek et al., 2007). To our knowledge, biallelic MUTYH pathogenic variants in addition to a pathogenic variant in MSH2, as described in our patient, have not been previously reported.
Discussion
This case demonstrates several points about genetic testing in the era of next generation sequencing (NGS) panel testing. First, had an immunohistochemistry-guided approach to testing been followed for the proband, the likely pathogenic variant in MSH2 would have been missed. It appears in this case that the proband's rectal cancer was a result of the biallelic pathogenic variants in the MUTYH gene, which would not have resulted in absent MSH2 staining in his tumor. Conversely, his mother's tumor appears to be a result of a MMR pathway defect that did result in absent MSH2/MSH6 staining. A panel was chosen for the proband in this case due to his diagnosis of colon cancer at a very young age, the presence of colon polyps, the presence of colorectal cancer in his father's family and the possible gynecologic (at the time unknown) cancer in his mother. There is some recent evidence that the incidence of pathogenic variants among individuals diagnosed with colon cancer at a very young age is high, with as many as 14–35% of individuals with colon cancer under age 35 found to carry a pathogenic variant in a variety of different genes (Chubb et al., 2015; Mork et al., 2015). Even considering a broader age range with diagnoses of colon cancer up to the age of 50, as many as 15% had an underlying pathogenic variant in a hereditary cancer gene (Pearlman et al., submitted).
The inheritance patterns for these two genetic conditions are different. Lynch syndrome is autosomal dominant and MAP is autosomal recessive. This has significant implications to family members. Had only the biallelic pathogenic variants in the MUTYH gene been identified, the proband's children would have been falsely reassured that they were just carriers of a single MUTYH variant. Surveillance recommendations for individuals who are carriers of a single MUTYH pathogenic variant are not well defined, but typically would include colonoscopy every 5 years, similar to recommendations for individuals with a first-degree relative with colorectal cancer (Stoffel et al., 2014; Provenzale et al., 2015). Surveillance for individuals with a pathogenic variant in MSH2 is very different, with annual colonoscopy recommended beginning at age 25 (Provenzale et al., 2015; Syngal et al., 2015). In addition, surveillance for other cancers may be considered, such as upper endoscopy, due to the risks for other cancers associated with Lynch syndrome. Finally, females are at increased risk for endometrial and ovarian cancers with Lynch syndrome, so hysterectomy after childbearing should be considered. As there is variable expression of MAP and Lynch syndrome in general, the identification of additional patients with combined diagnoses would be necessary to make more definitive conclusions regarding cumulative cancer risks.
The proband's extended family is also impacted by this complex test result. The determination of the MSH2 duplication as maternally inherited has implications for maternal family members, while the cause of the colon cancer in paternal family members remains unexplained, which may prompt family members to undergo further analysis. Communication among family members is very important, especially in cases where multiple pathogenic variants are identified. There are at least three affected family members for every person identified with Lynch syndrome (Hampel et al., 2008). If this is true when there are multiple pathogenic variants present, then many more relatives could potentially be impacted. Sharing of test results among family members and their doctors is critical to identification of other family members at risk and recommendations for appropriate surveillance.
As more genetic testing is completed with NGS panels, we anticipate a rise in the identification of cases of multiple pathogenic variants. It is also important for clinicians to be mindful of the possibility of a second pathogenic variant in a family where one pathogenic variant has already been described, particularly in cases where the inheritance pattern may not match the known pathogenic variant (for example, if the inheritance appears to be autosomal dominant, but a recessive condition like MAP is documented), or if the clinical picture doesn't match the known pathogenic variant.
Concluding Remarks
Although the cumulative cancer risk in individuals with a combined diagnosis of MAP and Lynch syndrome is unknown, the identification of multiple pathogenic variants does allow for screening for cancers associated with both genes and has implications for cancer risk and surveillance recommendations for family members. In particular, this has significant impact on those who test negative for a known familial pathogenic variant, yet could be still be at risk for cancer due to a second pathogenic variant in a family. More information is needed to understand the frequency of occurrence of multiple pathogenic variants, as well as the phenotypic spectrum when more than one syndrome is present.
Author Contributions
SC, CT, and RB contributed to the acquisition of the information for this case report. SC drafted the manuscript, and it was reviewed and approved by all three authors.
Ethics
Informed written consent was obtained from the proband, his mother and father for genetic testing and from the proband and his mother for inclusion in this case report.
Conflict of Interest Statement
CT is an employee of Invitae Corporation, a testing laboratory that furnished the 7-gene panel test results used in this study.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Authors would like to thank the family, who provided written consent to allow us to report this case. The Authors also acknowledge the following individuals: Marina Rabideau and Federico Monzon for assistance with clinical interpretation of data; Michael Kennemer and Scott Fay for technical assistance with figure images.
References
Ali, M., Kim, H., Cleary, S., Cupples, C., Gallinger, S., and Bristow, R. (2008). Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology 135, 499–507. doi: 10.1053/j.gastro.2008.04.035
Aretz, S., Uhlhaas, S., Goergens, H., Siberg, K., Vogel, M., Pagenstecher, C., et al. (2006). MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int. J. Cancer 119, 807–814. doi: 10.1002/ijc.21905
Casey, G., Lindor, N. M., Papadopoulous, N., Thibodeau, S. N., Moskow, J., Steelman, S., et al. (2005). Conversion analysis for mutation detection in MLH1 and MSH2 in patients with colorectal cancer. JAMA 293, 799–809. doi: 10.1001/jama.293.7.799
Castillejo, A., Vargas, G., Castillejo, M. I., Navarro, M., Barberá, V. M., González, S., et al. (2014). Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur. J. Cancer 50, 2241–2250. doi: 10.1016/j.ejca.2014.05.022
Chubb, D., Broderick, P., Frampton, M., Kinnersley, B., Sherborne, A., Penegar, S., et al. (2015). Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J. Clin. Oncol. 33, 426–432. doi: 10.1200/JCO.2014.56.5689
Cohen, S. A., and Leininger, A. (2014). The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 7, 147–158. doi: 10.2147/TACG.S51483
Gong, P., Charles, S., Rosenblum, N., Wang, Z., and Witkiewicz, A. K. (2012). A case of endometrial cancer in the context of a BRCA2 mutation and double heterozygosity for Lynch syndrome. Gynecol. Oncol. Case Rep. 2 69–72. doi: 10.1016/j.gynor.2012.03.001
Hampel, H., Frankel, W. L., Martin, E., Arnold, M., Khanduja, K., Kuebler, P., et al. (2008). Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J. Clin. Oncol. 26, 5783–5788. doi: 10.1200/JCO.2008.17.5950
Kohlmann, W., and Gruber, S. (2004/2014). Lynch Syndrome. Seattle, WA: University of Washington. Updated May 22, 2014; cited 2015 December 15.
Lubbe, S. J., Di Bernardo, M. C., Chandler, I. P., and Houlston, R. S. (2009). Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J. Clin. Oncol. 27, 3975–3980. doi: 10.1200/JCO.2008.21.6853
Maartje Nielsen, M. D., Henry Lynch, M. D., and Elena Infante, M. S., CGC, Randall Brand, M. D. (2012/2015). MUTYH-Associated Polyposis. Seattle, WA: University of Washington, Seattle. [Updated 2015 Sep 24] [cited 2015 11/24/2015].
Mork, M. E., You, Y. N., Ying, J., Bannon, S. A., Lynch, P. M., Rodriguez-Bigas, M. A., et al. (2015). High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J. Clin. Oncol. 33, 3544–3549. doi: 10.1200/JCO.2015.61.4503
Provenzale, D., Gupta, S., Ahnen, D. J., Aslanian, H., and Bray, T. (2015). Genetic/Familial High-Risk Assessment: Colorectal. [updated 10/7/2015; cited 2015 11/24/2015]. Available online at: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#colorectal_screening
Stoffel, E. M., Mangu, P. B., Gruber, S. B., Hamilton, S. R., Kalady, M. F., Lau, M. W. Y., et al. (2014). Hereditary colorectal cancer syndromes: American society of clinical oncology clinical practice guideline endorsement of the familial risk–colorectal cancer: European society for medical oncology clinical practice guidelines. J. Clin. Oncol. 33, 209–217. doi: 10.1200/JCO.2014.58.1322
Syngal, S., Brand, R. E., Church, J. M., Giardiello, F. M., Hampel, H. L., and Burt, R. W. (2015). ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 110, 223–262. doi: 10.1038/ajg.2014.435
Thiffault, I., Hamel, N., Pal, T., McVety, S., Marcus, V. A., Farber, D., et al. (2004). Germline truncating mutations in both MSH2 and BRCA2 in a single kindred. Br. J. Cancer 90, 483–491. doi: 10.1038/sj.bjc.6601424
van Puijenbroek, M., Nielsen, M., Reinards, T. C. M., Weiss, M., Wagner, A., Hendriks, Y. C., et al. (2007). The natural history of a combined defect in MSH6 and MUTYH in a HNPCC family. Fam. Cancer 6, 43–51. doi: 10.1007/s10689-006-9103-y
Win, A. K., Dowty, J. G., Cleary, S. P., Kim, H., Buchanan, D. D., Young, J. P., et al. (2014). Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 146, 1208–1211e1-5. doi: 10.1053/j.gastro.2014.01.022
Keywords: next-generation sequencing, hereditary cancer, genetic counseling, MUTYH-associated polyposis, Lynch syndrome, case report
Citation: Cohen SA, Tan CA and Bisson R (2016) An Individual with Both MUTYH-Associated Polyposis and Lynch Syndrome Identified by Multi-Gene Hereditary Cancer Panel Testing: A Case Report. Front. Genet. 7:36. doi: 10.3389/fgene.2016.00036
Received: 28 December 2015; Accepted: 29 February 2016;
Published: 16 March 2016.
Edited by:
Logan Walker, University of Otago, New ZealandReviewed by:
Bryony Thompson, University of Utah, USAMark Clendenning, The University of Melbourne, Australia
Copyright © 2016 Cohen, Tan and Bisson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie A. Cohen, c2Fjb2hlbkBzdHZpbmNlbnQub3Jn