 Oscar Urtatiz
Oscar Urtatiz Catherine D. Van Raamsdonk
Catherine D. Van Raamsdonk- Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada
In this article, we first briefly outline the function of G protein coupled receptors in cancer, and then specifically examine the roles of the seven transmembrane G protein coupled Endothelin B receptor (Ednrb) and the G proteins, GNAQ and GNA11, in both melanocyte development and melanoma. Ednrb plays an essential role in melanocyte development. GNAQ and GNA11 are oncogenes when mutated in certain types of melanocytic lesions, being extremely frequent in uveal melanoma, which forms from melanocytes located in the eye. Previously, we reported that in mice, Schwann cell precursor derived melanocytes colonize the dermis and hair follicles, while the inter-follicular epidermis is populated by other melanocytes. A pattern has emerged whereby melanocytes whose activities are affected by gain-of-function mutations of the Endothelin 3 ligand and Gαq/11 are the same subset that arise from Schwann cell precursors. Furthermore, the forced expression of the constitutively active human GNAQQ209L oncogene in mouse melanocytes only causes hyper-proliferation in the subset that arise from Schwann cell precursors. This has led us to hypothesize that in Schwann cell precursor derived melanocytes, Ednrb signals through Gαq/11. Ednrb is promiscuous and may signal through other G protein alpha subunits in melanomas located in the inter-follicular epidermis.
Introduction
G-protein coupled receptors (GPCRs) are one of the largest and most diverse membrane protein families, consisting of over 800 members and comprising 30% of drug discovery targets. GPCRs function by detecting a wide spectrum of extracellular signals and ligands, which generate conformational changes in the GPCR structure and cause the activation of signaling networks inside the cell. The non-sensory GPCRs are classified into four main families. These include the rhodopsin-like receptors, which are the most numerous, the secretin-like receptors, the metabotropic glutamate and pheromone receptors, and the frizzled receptors (reviewed in Venkatakrishnan et al., 2013).
The structure of seven transmembrane GPCRs can be divided into three regions: extracellular, transmembrane, and intracellular. The extracellular region consists of the N terminus and three extracellular loops (ECL1–ECL3). The transmembrane (TM) region consists of a core structure of seven alpha helices (TM1–TM7). The intracellular region contains three intracellular loops (ICL1–ICL3), an intracellular amphipathic helix (H8), and the C terminus. The activation of GPCRs begins with the binding of a ligand to the extracellular portions of the receptor, which causes a small conformational change in the TM core. This ultimately leads to larger structural rearrangements at the transmembrane- intracellular domain interface, which alters the interactions of signaling effectors, such as G proteins, GPCR kinases, and arrestins, with the cytoplasmic portions of the receptor. Different ligands produce different conformational states within a GPCR, such as full agonists, partial agonists, inverse agonists, and allosteric modulators. Each of these can bring about different downstream effects. The active state of a GPCR is defined as the conformation of the receptor that couples to and stabilizes an effector molecule [reviewed in (Cabrera-Vera et al., 2003; Venkatakrishnan et al., 2013)].
GPCRs mainly signal through heterotrimeric G proteins, which contain three separate subunits: α, β, and γ. In its inactive state, the α subunit is bound to a guanine diphosphate molecule (GDP) and the β and γ subunits in a complex tethered to the cytosolic side of the cell membrane. When a GPCR is activated by ligand binding, it recruits a heterotrimeric G protein, which triggers the release of GDP and the binding of a guanine triphosphate molecule (GTP) on the Gα subunit. Key residues for GPCR-G protein couplings have been identified within the N termini (McFadzean et al., 1989; Blahos et al., 1998), α2-helices, α2-β4 loop regions (Lee et al., 1995; Onrust et al., 1997), α4-helices, and α4-β6 loop regions (Bae et al., 1997) of the receptor. The GTP-bound Gα subunit dissociates from the receptor and Gβγ subunits to activate downstream effectors. There are five classes of Gα protein (Gαs, Gαq, Gαi, Gα12/13, and the newly discovered Gαv) and the GTP-bound conformations of each class interact with different canonical downstream effectors. Some GPCRs signal through more than one type of Gα protein, which further increases complexity. In addition, the Gβγ subunits can also act on effectors. The activity of Gα is self-limited by the intrinsic GTPase activity of its Ras-like domain, which hydrolyses GTP back to GDP and prevents further interaction of Gα with its effector. This inactivation step is modulated by Regulators of G-protein signaling (Rgs) proteins, which are GTPase accelerating proteins (GAPs). Some effectors also act as GAPs for Gα, such as the phospholipase C effector for Gαq class α subunits [reviewed in (Conklin and Bourne, 1993; Wess, 1997; Yang et al., 1999; Cabrera-Vera et al., 2003; Oka et al., 2009; Kimple et al., 2011; Sánchez-Fernández et al., 2014)].
G Protein Coupled Pathways in Cancer
G-protein coupled receptors regulate many key biological functions, such as differentiation, cell proliferation, cell migration, and metabolic activity, thus it is not surprising that GPCRs play a role in tumorigenesis. In general, there are four mechanisms by which G protein coupled pathways drive tumorigenesis: excess ligand availability, excess GPCR expression, activating mutations in GPCRs, and activating mutations in Gα proteins.
Many potent mitogens such as thrombin, lysophosphatidic acid (LPA), gastrin-releasing peptide (GRP), endothelins, and prostaglandins, stimulate cell proliferation by acting on their cognate GPCR in various cell types (Gutkind, 1998; Marinissen and Gutkind, 2001; Rozengurt et al., 2002; Mills and Moolenaar, 2003). In 1991, it was reported that over-expression of muscarinic cholinergic receptors (mAChRs) in NIH3T3 cells was not sufficient for oncogenic transformation. However, when the cells were treated with an excess of carbachol ligand, foci were readily induced. This result showed that normal GPCRs can promote ligand-dependent neoplastic transformation when stimulated by the unrestricted availability of their ligand (Gutkind et al., 1991). In addition, malignancies such as colon carcinoma (Gao et al., 2006), squamous cell carcinoma (SCC) of the lung (Gugger et al., 2008), basal cell carcinoma (Tanese et al., 2008), hepatocellular carcinoma (Yamamoto et al., 2003), prostate cancer (Weigle et al., 2004), breast cancer (Mihai et al., 2006), and glioblastoma multiforme (Shashidhar et al., 2005) have been reported to over-express GPCRs.
Activating mutations in GPCRs represent another tumorigenic route. Mutations in the thyroid-stimulating hormone receptor are found in ∼30% of thyroid adenomas (Parma et al., 1993). Mutations have also been reported in Smoothened (SMO), glutamate metabotropic receptors (GRMs), members of the adhesion family of GPCRs, and receptors for bioactive lipid mediators such as LPA and sphingosine-1-phosphate (S1P) which tend to accumulate in the tumor microenvironment. In 2010, a ground-breaking study revealed an unexpectedly high frequency of somatic mutations in genes encoding GPCRs in breast, lung, ovarian, and prostate cancer, including LPHN3, GRM8, CMKLR1, MAS1L, AGTRL1, and PTGFR (Kan et al., 2010). It was estimated that 20% of all cancers bear somatic mutations in GPCRs.
Finally, pathway activation can occur through mutations in Gα subunits. Because the Gα subunit must inactivate itself through the hydrolysis of GTP to GDP, mutations that reduce the function of the Ras-like GTPase domain paradoxically lead to constitutive active signaling. In certain cellular contexts, this generates oncogenic transformation. This was first discovered in 1989 when somatic activating mutations in Gαs were found in growth hormone-secreting human pituitary tumors (Landis et al., 1989). Recently, Gαs was reported to be mutated in 3.8% of 28,961 tumor samples, according to the Catalogue of Somatic Mutations in Cancer (COSMIC) database (Forbes et al., 2010). Somatic mutations causing constitutive activity of Gα proteins have been found in other types of cancers as well (Lyons et al., 1990; Van Raamsdonk et al., 2009b, 2010). Common to all these lesions is that the mutations occur in the Ras-like GTPase domain of Gα at the perfectly conserved glutamine and arginine residues that directly contact the gamma phosphate of GTP, stabilizing it for hydrolysis (Landis et al., 1989; Markby et al., 1993; Farfel et al., 1999).
Gαq/11 Subunits in Melanocyte Development and Melanoma
Melanomas arise from pigment producing cells called melanocytes in mammals. Immature melanocytes, melanoblasts, originate in the neural crest, either directly in an early stream, or indirectly from Schwann cell precursors lining developing nerves (Adameyko et al., 2009). During embryogenesis, melanoblasts migrate through the dermis along the dorsal-ventral axis. Some, but not all of the melanoblasts, will enter the epidermis. There they can choose to migrate into hair follicles or persist in between hair follicles in the “inter-follicular” epidermis. Other melanoblasts never enter the epidermis. These non-epithelial melanocytes remain in the dermis or migrate into the uveal tract of the eye or the leptomeninges of the central nervous system.
The majority of human melanomas arise from melanocytes located in the inter-follicular epidermis. The rest arise from non-epithelial melanocytes, which as a group are characterized by frequent oncogenic mutations in the heterotrimeric G protein alpha subunits, GNAQ and GNA11, which encode Gαq and Gα11 (Van Raamsdonk et al., 2009b, 2010; Küsters-Vandevelde et al., 2010) The importance of these two proteins in melanocytes first became apparent during a study of a set of mouse mutants with a darker dermis (Dsk1, Dsk7, Dsk10), obtained during an ENU (N-ethyl-N-nitrosourea) mutagenesis screen of 30,000 mice (Hrabé de Angelis et al., 2000; Fitch et al., 2003; Van Raamsdonk et al., 2004). These mice carried hyper-active, but not constitutively active, single amino acid substitution mutations in either Gnaq or Gna11 at isoleucine 63, valine 179, or phenylalanine 335. These mutations increased the number of melanoblasts in the embryo beginning immediately after the first commitment of these cells to the melanocyte lineage. The increased melanocytes persisted in the dermis throughout the life of the mice, but did not cause tumors. The Gnaq and Gna11 dark dermis mutations acted additively, darkening the dermis in a quantitative and step-wise fashion as the number of mutant Gnaq and Gna11 alleles increased, indicating that the read-out of Gαq/11 signaling can be quantitative. Strikingly, the inter-follicular epidermis of the tail, which is pigmented, was unaffected by the mutations, even when all four Gnaq and Gna11 alleles were replaced with gain-of-function Dsk versions. The gain-of-function alleles partially rescued the reduction in melanoblast numbers caused by heterozygous loss of the c-Kit tyrosine kinase receptor, Pax3 transcription factor, and endothelin B receptor, Ednrb. However, homozygous loss of Ednrb prevented all Gαq and Gα11 generated skin darkening. Based upon previous in vitro work (Okamoto et al., 1997; Doi et al., 1999; Imamura et al., 2000), this led to the hypothesis that Endothelin receptor B, a G protein coupled receptor, signals through Gαq/11 in melanocytes.
The mutations in GNAQ and GNA11 in human melanocytic lesions are somatic, mutually exclusive, and occur at two hotspots, glutamine 209 and arginine 183, which causes constitutive activity. 50%–85% of non-epithelial melanocytic lesions are affected by these mutations, and include lesions in the dermis, called blue nevi, leptomeningeal melanocytomas, uveal nevi, and uveal melanomas. The two genes are mutated with unequal frequency in each type of lesion. GNAQ mutations are 7.9 times more frequent in dermal lesions, 4.6 times more frequent in leptomeningeal melanocytomas, and 1.4 times more frequent in primary uveal melanomas, compared with GNA11 mutations (Küsters-Vandevelde et al., 2010; Van Raamsdonk et al., 2010; Gessi et al., 2012; Küsters-Vandevelde et al., 2014). Conversely, GNA11 mutations are 2.6 times more frequent than GNAQ mutations in uveal melanoma metastases (Van Raamsdonk et al., 2010). Given their presence in uveal nevi, GNAQ and GNA11 mutations are hypothesized to be early events in uveal melanomagenesis (Van Raamsdonk et al., 2010). Q209 mutations are much more frequent than R183 mutations, and are predicted to have a greater inhibitory effect on the GTPase activity of Gαq/11 (Berman et al., 1996; Orth et al., 2009; Van Raamsdonk et al., 2010) (Figure 1A).
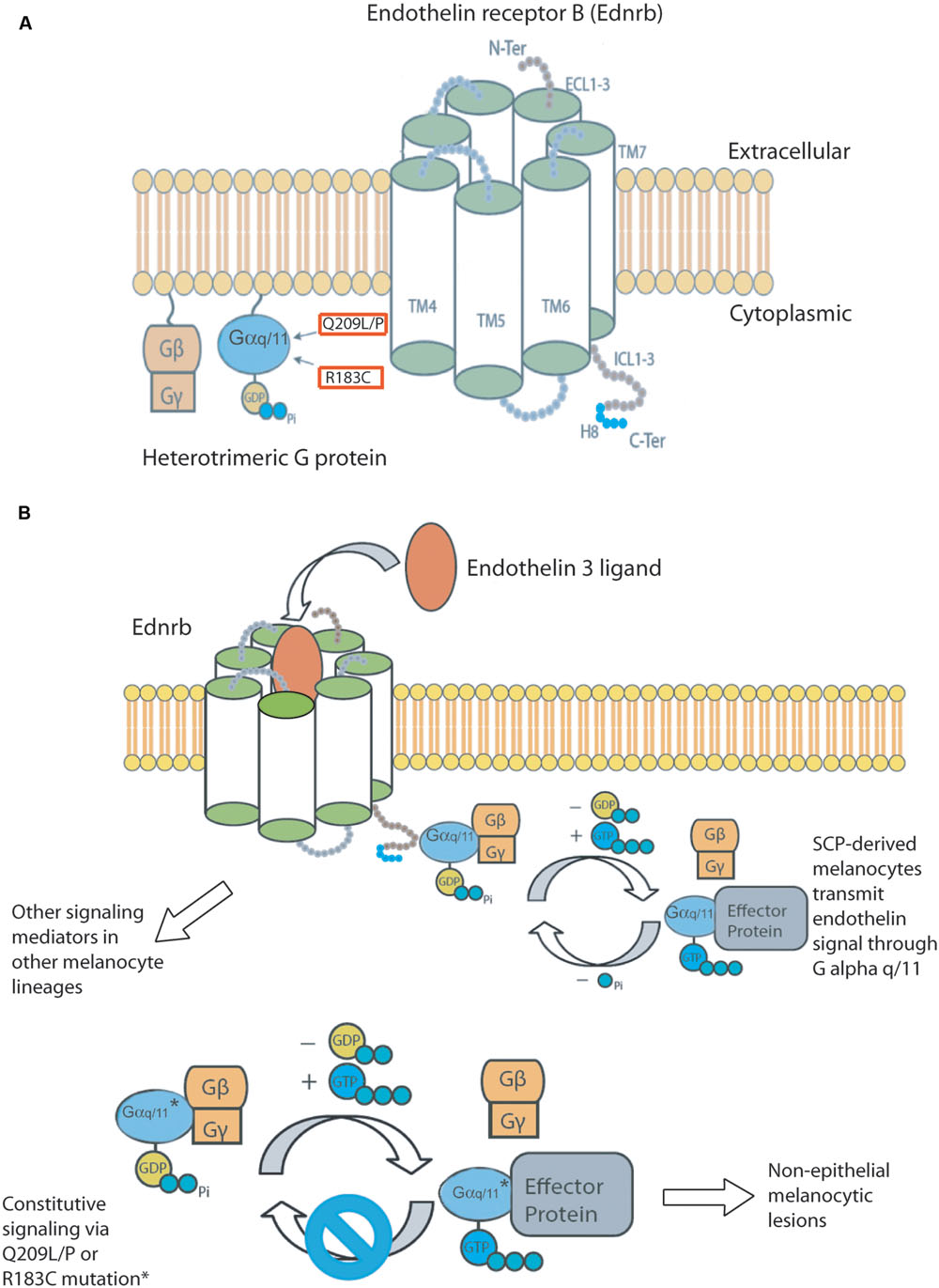
FIGURE 1. (A) Components of the hypothesized Endothelin signaling pathway. Constitutively active mutations found in GNAQ and GNA11 in human melanocytic lesions are boxed in red. (B) We hypothesize that Schwann cell precursor derived melanocytes transmit endothelin signals through Gαq/11. Schwann cell precursor derived melanocytes are also susceptible to developing melanocytic lesions when either GNAQ or GNA11 are mutated to their constitutively active forms. This melanomagenic pathway gives rise to non-epithelial associated lesions. Other G protein alpha subunits may transmit endothelin signals in epithelial associated melanomas.
Mutations in either gene are extremely rare in human melanomas located in the epidermis. The COSMIC database v72 reported four patients with GNAQ mutations among 1,696 entries (0.2%) for superficial spreading, lentigo maligna, nodular, and otherwise unspecified malignant melanomas of the skin. Consistent with this, the forced expression of GNAQQ209L in mouse melanocytes caused cell loss in the inter-follicular epidermis, in contrast to the hyper-proliferation observed in the dermis, hair follicles, leptomeninges, and uveal tract (Huang et al., 2015). Fate mapping during mouse development has shown that Schwann cell precursor derived melanoblasts migrate to the dermis and hair follicles, but not the inter-follicular epidermis of the tail (Deo et al., 2013). Therefore, we hypothesize that the developmental lineage of a melanocyte determines whether constitutive activity of Gαq and Gα11 is oncogenic (Figure 1B).
The first effector of Gαq class α subunits to be identified was phospholipase C (PLC). PLC cleaves phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 diffuses in the cytoplasm and binds to receptors such as calcium channels in the endoplasmic reticulum, releasing calcium as a second messenger. Calcium and DAG also activate protein kinase C, which can phosphorylate RAF in the MAP kinase pathway. One of the primary effects of oncogenic Gαq and Gα11 is the activation of the MAP kinase pathway and the phosphorylation of MEK and ERK (Van Raamsdonk et al., 2009b, 2010). In a randomized, phase II trial of the MEK inhibitor, selumetinib, treatment improved progression-free survival, and the response rate to chemotherapy, but unfortunately did not lead to improvement in overall survival (Carvajal et al., 2014). PKC activity may have additional effects, because PKC inhibition decreased NFkB signaling (Wu et al., 2012) and combined PKC and MEK inhibition is more efficacious at inhibiting MAP-kinase pathway activation, halting proliferation, and inducing apoptosis in vitro (Chen et al., 2014). A phase Ib/II combination trial of a MEK inhibitor, MEK162, plus the PKC inhibitor, AEB071, has been initiated (NCT01801358).
MEK inhibition has been reported to lead to PI3K/AKT up-regulation, which may contribute to tumor resistance (Babchia et al., 2010). A phase II trial will compare the MEK inhibitor, trametinib, alone or trametinib plus the AKT inhibitor, GSK2141795 (NCT01979523). Additionally, a phase 1b trial will be launched using the PKC inhibitor sotrastaurin plus the PI3K-alpha inhibitor, BYL719 (Shoushtari and Carvajal, 2014). Alternative effectors mediate Gαq activity independently of PLC, revealing unanticipated complexity. Oncogenic Gαq stimulates the transcriptional co-activator, YAP, through a Trio-Rho/Rac signaling circuitry that promotes actin polymerization, in a pathway that is independent of both PLC and the canonical Hippo pathway (Feng et al., 2014; Yu et al., 2014). This provides yet another rational therapeutic avenue for uveal melanoma. YAP dephosphorylation and nuclear translocation may stimulate melanoma by increasing Notch signaling, through up-regulation of the Notch ligand, JAG-1 (Liu et al., 2015).
Endothelin Signaling in Melanocyte Development
Endothelin receptors are GPCRs that belong to the rhodopsin-like class. In mammals, there are two Endothelin receptors, type A and B, and three 21 amino acid long Endothelin ligands, Edn1, Edn2, and Edn3, which are processed to their mature forms by Endothelin converting enzymes 1 and 2 (Ece-1, Ece-2). The loss of Endothelin receptor B (Ednrb) in mice causes a severe reduction in melanoblast numbers, while Endothelin receptor A (Ednra) knockout does not produce a pigmentation phenotype (Baynash et al., 1994; Hosoda et al., 1994; Clouthier et al., 1998). Thus, the effect of endothelin signaling in melanocyte development appears to be transduced by Ednrb. Ednrb has an equal affinity for all three of the Endothelin ligands (Sakurai et al., 1990). Edn3 and Ece-1 mutant mice have a very similar phenotype compared to Ednrb mutant mice, neatly linking the three proteins during development (Baynash et al., 1994; Yanagisawa et al., 1998).
During mouse embryogenesis, Ednrb is expressed in melanoblasts by the time they are committed to the melanocyte cell fate (Lee et al., 2003). In Ednrb null embryos, melanoblasts appear in the mesenchyme directly adjacent to the dorsal neural tube, sometimes referred to as the melanoblast staging area, but then are lost in an anterior to posterior progression (Hosoda et al., 1994; Lee et al., 2003). A similar result was found studying Schwann cell precursor derived melanoblasts around cranial nerves IX–X in Ednrb null mouse embryos. Melanoblasts appeared in the mutants, but were greatly reduced in number and did not break contact with the nerve (Adameyko et al., 2012). A temporally regulated Ednrb expression system in mice demonstrated that melanoblasts that have migrated into the epidermis do not require Ednrb signaling to persist there (Shin et al., 1999). Thus, the primary role of Ednrb during mouse development seems to be to support and direct newly created melanoblasts when they are migrating in the dermis, which all melanoblasts must do. It is important to note that a few melanoblasts can survive without endothelin signaling in the head and lower body, leading to small patches of pigmented fur in those areas on an otherwise white background (Hosoda et al., 1994). Interestingly, melanoblasts in the midbrain region, which arise independently of nerves, are affected less by Ednrb loss and therefore may be a source of pigmented head spots in Ednrb mutants (Adameyko et al., 2012). The lack of any pigmentation in the trunk indicates that both Schwann cell precursor derived and other lineages of melanoblasts in that region require Endothelin for development.
In chickens, the function of the endothelin receptor is shared by two different homologs. Ednrb is expressed by neural crest precursors prior to commitment to the melanocytic lineage and Ednrb2 is expressed in migrating melanoblasts (Nataf et al., 1996; Lecoin et al., 1998). Thus, the role of avian Ednrb2 seems more homologous to mammalian Ednrb (Pla et al., 2005). In chick experiments, Ednrb2 was required for melanoblasts to enter the dorsal-lateral pathway and the ectopic expression of Ednrb2 in neuronal neural crest cell precursors caused them to switch from the ventral pathway to the dorsal-lateral pathway (Harris et al., 2008). An inverted duplication present in the chicken genome in several breeds leads to the over-expression of Edn3 (Dorshorst et al., 2012). In these chick embryos, melanoblasts migrate aberrantly along the ventral pathway, in addition to the expected dorsal-lateral pathway (Faraco et al., 2001). This demonstrates that endothelin signaling directs melanoblast migration.
It is not known exactly why melanoblasts fail to persist in Ednrb null mouse embryos. Apoptotic melanoblasts were not found in the melanocyte staging area of Ednrb mutants, but these cells may have been difficult to detect (Lee et al., 2003) and it is likely that endothelins do stimulate melanocyte survival/proliferation. Many different in vitro experiments have shown that Edn3 increases cell proliferation of neural crest cell progenitors and melanoblasts in culture (Reid et al., 1996; Lahav et al., 1998; Dupin et al., 2000; Real et al., 2006). In addition, transgenic over-expression of the Edn3 ligand in the skin increased dermal and epidermal melanoblast numbers during embryogenesis (Garcia et al., 2008). However, melanoblasts in the dermis were more strongly stimulated than melanoblasts in the epidermis. Similarly, the over-expression of Edn3 in chickens causes intense hyper-pigmentation of the dermis (Dorshorst et al., 2012). In mice, and most other mammals besides humans, melanocytes persist in the post-natal dermis in the tail and ears and, in smaller numbers, in the trunk (Fitch et al., 2003; Van Raamsdonk et al., 2004). These cells probably require on-going endothelin signaling for support (Garcia et al., 2008; Hyter et al., 2013).
The Relationship Between Ednrb and Gαq/11 During Mouse Development
Because homozygous loss of Ednrb prevented all Gαq and Gα11 generated skin darkening in the Dsk mice, it was hypothesized that Ednrb signals through Gαq/11 in melanocytes (Van Raamsdonk et al., 2004). Direct data concerning the coupling of Ednrb to a specific Gα subunit in melanocytes is limited. GPCRs can couple to multiple Gα or Gβγ subunits [reviewed in (Hermans, 2003)]. In reconstituted phospholipid vesicles (Doi et al., 1999) and Chinese hamster ovary cells (Okamoto et al., 1997), Ednrb stimulated phospholipase C and inhibited adenylyl cyclase, through Gαq and Gαi, respectively. A deletion in the C terminus of Ednrb, and the EdnrbG57S and EdnrbR319W mutations, impair Gαi signaling, but not Gαq (Okamoto et al., 1997; Fuchs et al., 2001). In contrast, an EdnrbW276C mutation impairs Gαq coupling, but not Gαi (Imamura et al., 2000). In human kidney 293 cells, EDN3 increased GTP binding of Gα13 (Kitamura et al., 1999). In the cellular context of melanocytes, the treatment of a cell line expressing EDNRB with EDN3 increased inositol 1,4,5-triphosphate and intra-cytoplasmic calcium concentrations, which supports a role for Gαq (Kang et al., 1998). These data high-light the promiscuity of Ednrb.
If all of the effects of Ednrb activation in melanocytes were generated through Gαq/11, then the complete knockout of Gnaq and Gna11 would be expected to have the same phenotype as Ednrb null mice, i.e., severe hypo-pigmentation. A double knockout of Gnaq and Gna11 is lethal in mid-gestation in mice, however, the knockout of a single allele of Gnaq is enough to reduce the pigmentation of the adult dermis (Van Raamsdonk et al., 2004). The presence of melanoblasts was noted at E18.5 in a conditional knockout of Gnaq and Gna11 made specifically in the neural crest lineage, but the number and distribution of these melanoblasts was not described (Dettlaff-Swiercz et al., 2005). A similar conditional knockout of Ednrb in the neural crest lineage eliminated melanocytes in the trunk (Druckenbrod et al., 2008).
There are several possible explanations for why Gnaq and Gna11 conditional knockout in the neural crest lineage did not completely eliminate melanoblasts. One is that Ednrb does not couple to Gαq/11 in melanoblasts and the similarities in the effects of Edn3 over-expression and Gαq/11 hyper-activity are just coincidental. We think this is unlikely. Another explanation is that in the absence of Gαq/11, Ednrb receptors activate other G protein alpha subunits, which buffers the system. A third possibility is that with four alleles of Gnaq and Gna11, Cre is not as effective at eliminating all Gαq/11 as compared to Ednrb. There is no data yet to distinguish between these possibilities.
However, in yet another striking similarity, both the over-expression of Edn3 and the hyper-active Dsk alleles of Gαq/11 darken the mouse coat through increased post-natal pigment production in hair follicles, using a mechanism that is independent of melanocyte cell number (Garcia et al., 2008; Van Raamsdonk et al., 2009a). Edn3 expression also correlates with light and dark areas of cat coat color (Kaelin et al., 2012). We point out that Schwann cell precursor derived melanocytes localize to the dermis and hair follicles, and these are both places where Edn3 and Gαq/11 gain-of-function mutations generate developmental phenotypes. Meanwhile, the inter-follicular epidermis is virtually unchanged as a result of hyper-active Gαq/11 (Van Raamsdonk et al., 2004) and Schwann cell precursor derived melanocytes do not colonize this part of the epidermis (Deo et al., 2013). Thus, there is a correlation between melanocyte lineage, response to Edn3 and Gαq/11 gain-of-function mutations, and melanocyte location during development (Figure 1B).
The Relationship Between Ednrb and Gαq/11 in Melanomagenesis
Gαq/11 mutations play an obvious role in the development of non-epithelial associated melanocytic lesions. There are specific hotspot oncogenic mutations that are very frequently found in a specific subset of melanocytic lesions, which do not involve the epidermis. The role of Ednrb in melanoma is more difficult to interpret. In fact, much of the data concerns the effects of Ednrb on melanomas located in the epithelium, which implies that the effects of Ednrb are not a result of Gαq/11 activation. As Ednrb is probably able to couple to multiple G alpha subunits, this is perhaps not surprising. In addition, signaling pathways do not play the exact same roles in development as they do in cancer, even while cancer often reactivates innate developmental pathways. We will first summarize what has been reported about Ednrb signaling in melanomas associated with the epithelium, then discuss findings in uveal melanoma.
In general, EDNRB expression is positively correlated with melanoma progression in cutaneous melanoma (Demunter et al., 2001). Over-expression of EDNRB increased melanoma brain metastases in mice orthotopically transplanted with human melanoma cell lines (Cruz-Muñoz et al., 2012). The activation of EDNRB by endothelins induces the expression of HIF-1α, which leads to the up-regulation of vascular endothelial growth factor and its receptor in primary and metastatic melanoma cell lines, resulting in MAP kinase and AKT activation (Spinella et al., 2012). These pathways up-regulate MCAM, a melanoma cell adhesion molecule that promotes invasion and metastasis (Williams et al., 2014).
The EDNRB inhibitors, BQ788, A192621, and Bonsentan inhibit melanoma cell growth in vitro, in xenografts of human melanomas in nude mice, and in some human patients, and restore cell morphology to a more normal appearance (Bagnato et al., 2004; Lahav et al., 2004; Berger et al., 2006; Kefford et al., 2007; Cruz-Muñoz et al., 2012; Asundi et al., 2014; Wouters et al., 2015). One recent study found that using a combination of an antibody-drug conjugate targeting EDNRB, together with small-molecule inhibitors of the MAP kinase pathway, increased anti-tumor activity in BRAF/NRAS mutant cell lines and tumor models (Asundi et al., 2014).
In contrast to these studies, putative loss-of-function germline variants in EDNRB were associated with cyclin-dependent kinase inhibitor 2A, CDKN2A, variants in familial melanoma cases in a French population, but not in an Italian population (Spica et al., 2011). EDNRB variants were not associated with sporadic melanoma in either the French or Italian populations. Larger studies and other populations are necessary to validate these findings. Endothelin signaling has also been suggested to enhance nucleotide excision repair following UV damage, and so a deficiency in this process could possibly act synergistically with a weak checkpoint (D’Orazio, 2015; von Koschembahr et al., 2015).
Next, we will consider uveal melanoma. Although EDNRB mutations have not been identified in uveal melanoma, the loss of EDNRB expression in uveal melanoma has been correlated with a worse prognosis in two different studies (Smith et al., 2002; Onken et al., 2004). Because oncogenic Gαq/11 mutations increase signaling, this is seems unexpected and contradictory if one assumes that EDNRB signals through Gαq/11 in uveal melanoma cells. However, there are two important points to consider. One, it is not known yet whether Gαq/11 proteins with constitutively active mutations require a receptor for activation or, in fact, will even couple to a receptor given that they are fixedly bound to GTP. EDNRB and Gαq/11 may become unlinked in the situation of uveal melanoma. If this is true, then it is possible that a decrease in the expression of EDNRB may reduce activation of the wildtype Gαq/11 proteins, which could somehow enhance the effect of the mutant Gαq/11 proteins through a lack of competition for downstream effectors.
The second point is that uveal melanoma patients without GNAQ or GNA11 mutations tend to have a worse prognosis than those with GNAQ or GNA11 mutations (Van Raamsdonk et al., 2010). In a completely different hypothesis, GNAQ and GNA11 mutations could be early events that boost melanocyte cell growth, but eventually it is necessary to down-regulate Gαq/11 signaling to achieve metastasis. This could be accomplished by down-regulating EDNRB expression. Melanocytes without GNAQ and GNA11 mutations would not need to go through this step, having arisen through a different mechanism, and thus progress more rapidly. In the future, more information on the interactions of EDNRB with different mutant and wildtype G protein alpha subunits is needed. In addition, it would be very interesting to correlate tumor progression with both EDNRB expression and the presence or absence of GNAQ and GNA11 mutations in uveal melanoma.
Conclusion
During development, both Ednrb and Gαq/11 regulate melanoblasts migrating in the dermis with strikingly similar effects. This suggests that they are closely linked together in the same signaling pathway. In support of this, Ednrb is required for the hyper-proliferative effects of the hyper-active Dsk Gnaq and Gna11 alleles. Ednrb is also required for the development of almost all melanocytes in the trunk, because all melanoblasts must migrate through the dermis, even if their eventual destination is the epidermis. Gain-of-function mutations in Edn3 and Gαq/11 increase melanocytes in the dermis and have little effect on those in the inter-follicular epidermis. In addition, gain-of-function mutations in Edn3 and Gαq/11 darken coat color using a mechanism that is independent of cell number in hair follicles. Thus, a pattern has emerged whereby melanocytes whose activities are affected by gain-of-function mutations of the Endothelin 3 ligand and Gαq/11 are the same subset that arise from Schwann cell precursors. Furthermore, the forced expression of the human GNAQQ209L oncogene in mouse melanocytes only caused hyper-proliferation in the subset that arise from Schwann cell precursors. This has led us to hypothesize that in Schwann cell precursor derived melanocytes, Ednrb signals through Gαq/11. Ednrb is promiscuous and may signal through other G protein alpha subunits in melanomas located in the inter-follicular epidermis.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (MOP-79511) and the Michael Smith Foundation for Health Research.
References
Adameyko, I., Lallemend, F., Aquino, J. B., Pereira, J. A., Topilko, P., Müller, T., et al. (2009). Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 139, 366–379. doi: 10.1016/j.cell.2009.07.049
Adameyko, I., Lallemend, F., Furlan, A., Zinin, N., Aranda, S., Kitambi, S. S., et al. (2012). Sox2 and Mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 139, 397–410. doi: 10.1242/dev.065581
Asundi, J., Lacap, J. A., Clark, S., Nannini, M., Roth, L., and Polakis, P. (2014). MAPK pathway inhibition enhances the efficacy of an anti-endothelin B receptor drug conjugate by inducing target expression in melanoma. Mol. Cancer Ther. 13, 1599–1610. doi: 10.1158/1535-7163.MCT-13-0446
Babchia, N., Calipel, A., Mouriaux, F., Faussat, A. M., and Mascarelli, F. (2010). The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: interaction with B-Raf/ERK. Invest. Ophthalmol. Vis. Sci. 51, 421–429. doi: 10.1167/iovs.09-3974
Bae, H., Anderson, K., Flood, L. A., Skiba, N. P., Hamm, H. E., and Graber, S. G. (1997). Molecular determinants of selectivity in 5-hydroxytryptamine1B receptor-G protein interactions. J. Biol. Chem. 272, 32071–32077. doi: 10.1074/jbc.272.51.32071
Bagnato, A., Rosanò, L., Spinella, F., Di Castro, V., Tecce, R., and Natali, P. G. (2004). Endothelin B receptor blockade inhibits dynamics of cell interactions and communications in melanoma cell progression. Cancer Res. 64, 1436–1443. doi: 10.1158/0008-5472.CAN-03-2344
Baynash, A. G., Hosoda, K., Giaid, A., Richardson, J. A., Emoto, N., Hammer, R. E., et al. (1994). Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 79, 1277–1285. doi: 10.1016/0092-8674(94)90018-3
Berger, Y., Bernasconi, C. C., and Juillerat-Jeanneret, L. (2006). Targeting the endothelin axis in human melanoma: combination of endothelin receptor antagonism and alkylating agents. Exp. Biol. Med. (Maywood) 231, 1111–1119.
Berman, D. M., Wilkie, T. M., and Gilman, A. G. (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86, 445–452. doi: 10.1016/S0092-8674(00)80117-8
Blahos, J. II, Mary, S., Perroy, J., de Colle, C., Brabet, I., Bockaert, J., et al. (1998). Extreme C terminus of G protein alpha-subunits contains a site that discriminates between Gi-coupled metabotropic glutamate receptors. J. Biol. Chem. 273, 25765–25769. doi: 10.1074/jbc.273.40.25765
Cabrera-Vera, T. M., Vanhauwe, J., Thomas, T. O., Medkova, M., Preininger, A., Mazzoni, M. R., et al. (2003). Insights into G protein structure, function, and regulation. Endocr. Rev. 24, 765–781. doi: 10.1210/er.2000-0026
Carvajal, R. D., Sosman, J. A., Quevedo, J. F., Milhem, M. M., Joshua, A. M., Kudchadkar, R. R., et al. (2014). Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA 311, 2397–2405. doi: 10.1001/jama.2014.6096
Chen, X., Wu, Q., Tan, L., Porter, D., Jager, M. J., Emery, C., et al. (2014). Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 33, 4724–4734. doi: 10.1038/onc.2013.418
Clouthier, D. E., Hosoda, K., Richardson, J. A., Williams, S. C., Yanagisawa, H., Kuwaki, T., et al. (1998). Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development 125, 813–824.
Conklin, B. R., and Bourne, H. R. (1993). Structural elements of G alpha subunits that interact with G beta gamma, receptors, and effectors. Cell 73, 631–641. doi: 10.1016/0092-8674(93)90245-L
Cruz-Muñoz, W., Jaramillo, M. L., Man, S., Xu, P., Banville, M., Collins, C., et al. (2012). Roles for endothelin receptor B and BCL2A1 in spontaneous CNS metastasis of melanoma. Cancer Res. 72, 4909–4919. doi: 10.1158/0008-5472.CAN-12-2194
Demunter, A., De Wolf-Peeters, C., Degreef, H., Stas, M., and Van Den Oord, J. J. (2001). Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch. 438, 485–491. doi: 10.1007/s004280000362
Deo, M., Huang, J. L., Fuchs, H., De Angelis, M. H., and Van Raamsdonk, C. D. (2013). Differential effects of neurofibromin gene dosage on melanocyte development. J. Invest. Dermatol. 133, 49–58. doi: 10.1038/jid.2012.240
Dettlaff-Swiercz, D. A., Wettschureck, N., Moers, A., Huber, K., and Offermanns, S. (2005). Characteristic defects in neural crest cell-specific Galphaq/Galpha11- and Galpha12/Galpha13-deficient mice. Dev. Biol. 282, 174–182. doi: 10.1016/j.ydbio.2005.03.006
Doi, T., Sugimoto, H., Arimoto, I., Hiroaki, Y., and Fujiyoshi, Y. (1999). Interactions of endothelin receptor subtypes A and B with Gi, Go, and Gq in reconstituted phospholipid vesicles. Biochemistry 38, 3090–3099. doi: 10.1021/bi981919m
D’Orazio, J. A. (2015). Melanocyte UV resistance: feelin’ the endothelin. Exp. Dermatol. 24, 414–415. doi: 10.1111/exd.12663
Dorshorst, B., Molin, A. M., Rubin, C. J., Johansson, A. M., Strömstedt, L., Pham, M. H., et al. (2012). A complex genomic rearrangement involving the endothelin 3 locus causes dermal hyperpigmentation in the chicken. PLoS Genet. 7:e1002412. doi: 10.1371/journal.pgen.1002412
Druckenbrod, N. R., Powers, P. A., Bartley, C. R., Walker, J. W., and Epstein, M. L. (2008). Targeting of endothelin receptor-B to the neural crest. Genesis 46, 396–400. doi: 10.1002/dvg.20415
Dupin, E., Glavieux, C., Vaigot, P., and Le Douarin, N. M. (2000). Endothelin 3 induces the reversion of melanocytes to glia through a neural crest-derived glial-melanocytic progenitor. Proc. Natl. Acad. Sci. U.S.A. 97, 7882–7887. doi: 10.1073/pnas.97.14.7882
Faraco, C. D., Vaz, S. A., Pastor, M. V., and Erickson, C. A. (2001). Hyperpigmentation in the Silkie fowl correlates with abnormal migration of fate-restricted melanoblasts and loss of environmental barrier molecules. Dev. Dyn. 220, 212–225. doi: 10.1002/1097-0177(20010301)220:3<212::AID-DVDY1105>3.0.CO;2-9
Farfel, Z., Bourne, H. R., and Iiri, T. (1999). The expanding spectrum of G protein diseases. N. Engl. J. Med. 340, 1012–1020. doi: 10.1056/NEJM199904013401306
Feng, X., Degese, M. S., Iglesias-Bartolome, R., Vaque, J. P., Molinolo, A. A., Rodrigues, M., et al. (2014). Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25, 831–845. doi: 10.1016/j.ccr.2014.04.016
Fitch, K. R., McGowan, K. A., van Raamsdonk, C. D., Fuchs, H., Lee, D., Puech, A., et al. (2003). Genetics of dark skin in mice. Genes Dev. 17, 214–228. doi: 10.1101/gad.1023703
Forbes, S. A., Bindal, N., Bamford, S., Cole, C., Kok, C. Y., Beare, D., et al. (2010). COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950. doi: 10.1093/nar/gkq929
Fuchs, S., Amiel, J., Claudel, S., Lyonnet, S., Corvol, P., and Pinet, F. (2001). Functional characterization of three mutations of the endothelin B receptor gene in patients with Hirschsprung’s disease: evidence for selective loss of Gi coupling. Mol. Med. 7, 115–124.
Gao, Y., Kitagawa, K., Hiramatsu, Y., Kikuchi, H., Isobe, T., Shimada, M., et al. (2006). Up-regulation of GPR48 induced by down-regulation of p27Kip1 enhances carcinoma cell invasiveness and metastasis. Cancer Res. 66, 11623–11631. doi: 10.1158/0008-5472.CAN-06-2629
Garcia, R. J., Ittah, A., Mirabal, S., Figueroa, J., Lopez, L., Glick, A. B., et al. (2008). Endothelin 3 induces skin pigmentation in a keratin-driven inducible mouse model. J. Invest. Dermatol. 128, 131–142. doi: 10.1038/sj.jid.5700948
Gessi, M., Hammes, J., Lauriola, L., Dörner, E., Kirfel, J., Kristiansen, G., et al. (2012). GNA11 and N-RAS mutations: alternatives for MAPK pathway activating GNAQ mutations in primary melanocytic tumours of the central nervous system. Neuropathol. Appl. Neurobiol. 39, 417–425. doi: 10.1111/j.1365-2990.2012.01288.x
Gugger, M., White, R., Song, S., Waser, B., Cescato, R., Rivière, P., et al. (2008). GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis. Markers 24, 41–50. doi: 10.1155/2008/857474
Gutkind, J. S. (1998). Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene 17, 1331–1342. doi: 10.1038/sj.onc.1202186
Gutkind, J. S., Novotny, E. A., Brann, M. R., and Robbins, K. C. (1991). Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc. Natl. Acad. Sci. U.S.A. 88, 4703–4707. doi: 10.1073/pnas.88.11.4703
Harris, M. L., Hall, R., and Erickson, C. A. (2008). Directing pathfinding along the dorsolateral path – the role of EDNRB2 and EphB2 in overcoming inhibition. Development 135, 4113–4122. doi: 10.1242/dev.023119
Hermans, E. (2003). Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol. Ther. 99, 25–44. doi: 10.1016/S0163-7258(03)00051-2
Hosoda, K., Hammer, R. E., Richardson, J. A., Baynash, A. G., Cheung, J. C., Giaid, A., et al. (1994). Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 79, 1267–1276. doi: 10.1016/0092-8674(94)90017-5
Hrabé de Angelis, M. H., Flaswinkel, H., Fuchs, H., Rathkolb, B., Soewarto, D., Marschall, S., et al. (2000). Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 25, 444–447. doi: 10.1038/78146
Huang, J. L., Urtatiz, O., and Van Raamsdonk, C. D. (2015). Oncogenic G protein GNAQ induces uveal melanoma and intravasation in mice. Cancer Res. 75, 3384–3397. doi: 10.1158/0008-5472.CAN-14-3229
Hyter, S., Coleman, D. J., Ganguli-Indra, G., Merrill, G. F., Ma, S., Yanagisawa, M., et al. (2013). Endothelin-1 is a transcriptional target of p53 in epidermal keratinocytes and regulates ultraviolet-induced melanocyte homeostasis. Pigment Cell Melanoma Res. 26, 247–258. doi: 10.1111/pcmr.12063
Imamura, F., Arimoto, I., Fujiyoshi, Y., and Doi, T. (2000). W276 mutation in the endothelin receptor subtype B impairs Gq coupling but not Gi or Go coupling. Biochemistry 39, 686–692. doi: 10.1021/bi991981z
Kaelin, C. B., Xu, X., Hong, L. Z., David, V. A., McGowan, K. A., Schmidt-Küntzel, A., et al. (2012). Specifying and sustaining pigmentation patterns in domestic and wild cats. Science 337, 1536–1541. doi: 10.1126/science.1220893
Kan, Z., Jaiswal, B. S., Stinson, J., Janakiraman, V., Bhatt, D., Stern, H. M., et al. (2010). Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873. doi: 10.1038/nature09208
Kang, H. Y., Kang, W. H., and Lee, C. (1998). Endothelin-B receptor-mediated Ca2+ signaling in human melanocytes. Pflugers Arch. 435, 350–356. doi: 10.1007/s004240050522
Kefford, R., Beith, J. M., Van Hazel, G. A., Millward, M., Trotter, J. M., Wyld, D. K., et al. (2007). A phase II study of bosentan, a dual endothelin receptor antagonist, as monotherapy in patients with stage IV metastatic melanoma. Invest. New Drugs 25, 247–252. doi: 10.1007/s10637-006-9014-7
Kimple, A. J., Bosch, D. E., Giguere, P. M., and Siderovski, D. P. (2011). Regulators of G-protein signaling and their Galpha substrates: promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 63, 728–749. doi: 10.1124/pr.110.003038
Kitamura, K., Shiraishi, N., Singer, W. D., Handlogten, M. E., Tomita, K., and Miller, R. T. (1999). Endothelin-B receptors activate Galpha13. Am. J. Physiol. 276, C930–C937.
Küsters-Vandevelde, H. V., Klaasen, A., Küsters, B., Groenen, P. J., van Engen-van Grunsven, I. A., van Dijk, M. R., et al. (2010). Activating mutations of the GNAQ gene: a frequent event in primary melanocytic neoplasms of the central nervous system. Acta Neuropathol. 119, 317–323. doi: 10.1007/s00401-009-0611-3
Küsters-Vandevelde, H. V., van Engen-van Grunsven, I. A., Coupland, S. E., Lake, S. L., Rijntjes, J., Pfundt, R., et al. (2014). Mutations in g protein encoding genes and chromosomal alterations in primary leptomeningeal melanocytic neoplasms. Pathol. Oncol. Res. 21, 439–447. doi: 10.1007/s12253-014-9841-3
Lahav, R., Dupin, E., Lecoin, L., Glavieux, C., Champeval, D., Ziller, C., et al. (1998). Endothelin 3 selectively promotes survival and proliferation of neural crest-derived glial and melanocytic precursors in vitro. Proc. Natl. Acad. Sci. U.S.A. 95, 14214–14219. doi: 10.1073/pnas.95.24.14214
Lahav, R., Suva, M. L., Rimoldi, D., Patterson, P. H., and Stamenkovic, I. (2004). Endothelin receptor B inhibition triggers apoptosis and enhances angiogenesis in melanomas. Cancer Res. 64, 8945–8953. doi: 10.1158/0008-5472.CAN-04-1510
Landis, C. A., Masters, S. B., Spada, A., Pace, A. M., Bourne, H. R., and Vallar, L. (1989). GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340, 692–696. doi: 10.1038/340692a0
Lecoin, L., Sakurai, T., Ngo, M. T., Abe, Y., Yanagisawa, M., and Le, Douarin NM (1998). Cloning and characterization of a novel endothelin receptor subtype in the avian class. Proc. Natl. Acad. Sci. U.S.A. 95, 3024–3029. doi: 10.1073/pnas.95.6.3024
Lee, C. H., Katz, A., and Simon, M. I. (1995). Multiple regions of G alpha 16 contribute to the specificity of activation by the C5a receptor. Mol. Pharmacol. 47, 218–223.
Lee, H. O., Levorse, J. M., and Shin, M. K. (2003). The endothelin receptor-B is required for the migration of neural crest-derived melanocyte and enteric neuron precursors. Dev. Biol. 259, 162–175. doi: 10.1016/S0012-1606(03)00160-X
Liu, H., Lei, C., Long, K., Yang, X., Zhu, Z., Zhang, L., et al. (2015). Mutant GNAQ promotes cell viability and migration of uveal melanoma cells through the activation of Notch signaling. Oncol. Rep. 34, 295–301. doi: 10.3892/or.2015.3949
Lyons, J., Landis, C. A., Harsh, G., Vallar, L., Grünewald, K., Feichtinger, H., et al. (1990). Two G protein oncogenes in human endocrine tumors. Science 249, 655–659. doi: 10.1126/science.2116665
Marinissen, M. J., and Gutkind, J. S. (2001). G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 22, 368–376. doi: 10.1016/S0165-6147(00)01678-3
Markby, D. W., Onrust, R., and Bourne, H. R. (1993). Separate GTP binding and GTPase activating domains of a G alpha subunit. Science 262, 1895–1901. doi: 10.1126/science.8266082
McFadzean, I., Mullaney, I., Brown, D. A., and Milligan, G. (1989). Antibodies to the GTP binding protein, Go, antagonize noradrenaline-induced calcium current inhibition in NG108-15 hybrid cells. Neuron 3, 177–182. doi: 10.1016/0896-6273(89)90030-5
Mihai, R., Stevens, J., McKinney, C., and Ibrahim, N. B. (2006). Expression of the calcium receptor in human breast cancer–a potential new marker predicting the risk of bone metastases. Eur. J. Surg. Oncol. 32, 511–515. doi: 10.1016/j.ejso.2006.02.009
Mills, G. B., and Moolenaar, W. H. (2003). The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 3, 582–591. doi: 10.1038/nrc1143
Nataf, V., Lecoin, L., Eichmann, A., and Le Douarin, N. M. (1996). Endothelin-B receptor is expressed by neural crest cells in the avian embryo. Proc. Natl. Acad. Sci. U.S.A. 93, 9645–9650. doi: 10.1073/pnas.93.18.9645
Oka, Y., Saraiva, L. R., Kwan, Y. Y., and Korsching, S. I. (2009). The fifth class of Galpha proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 1484–1489. doi: 10.1073/pnas.0809420106
Okamoto, Y., Ninomiya, H., Tanioka, M., Sakamoto, A., Miwa, S., and Masaki, T. (1997). Palmitoylation of human endothelinB. Its critical role in G protein coupling and a differential requirement for the cytoplasmic tail by G protein subtypes. J. Biol. Chem. 272, 21589–21596. doi: 10.1074/jbc.272.34.21589
Onken, M. D., Worley, L. A., Ehlers, J. P., and Harbour, J. W. (2004). Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 64, 7205–7209. doi: 10.1158/0008-5472.CAN-04-1750
Onrust, R., Herzmark, P., Chi, P., Garcia, P. D., Lichtarge, O., Kingsley, C., et al. (1997). Receptor and betagamma binding sites in the alpha subunit of the retinal G protein transducin. Science 275, 381–384. doi: 10.1126/science.275.5298.381
Orth, J. H., Preuss, I., Fester, I., Schlosser, A., Wilson, B. A., and Aktories, K. (2009). Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc. Natl. Acad. Sci. U.S.A. 106, 7179–7184. doi: 10.1073/pnas.0900160106
Parma, J., Duprez, L., Van Sande, J., Cochaux, P., Gervy, C., Mockel, J., et al. (1993). Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365, 649–651. doi: 10.1038/365649a0
Pla, P., Alberti, C., Solov’eva, O., Pasdar, M., Kunisada, T., and Larue, L. (2005). Ednrb2 orients cell migration towards the dorsolateral neural crest pathway and promotes melanocyte differentiation. Pigment Cell Res. 18, 181–187. doi: 10.1111/j.1600-0749.2005.00230.x
Real, C., Glavieux-Pardanaud, C., Le Douarin, N. M., and Dupin, E. (2006). Clonally cultured differentiated pigment cells can dedifferentiate and generate multipotent progenitors with self-renewing potential. Dev. Biol. 300, 656–669. doi: 10.1016/j.ydbio.2006.09.032
Reid, K., Turnley, A. M., Maxwell, G. D., Kurihara, Y., Kurihara, H., Bartlett, P. F., et al. (1996). Multiple roles for endothelin in melanocyte development: regulation of progenitor number and stimulation of differentiation. Development 122, 3911–3919.
Rozengurt, E., Guha, S., and Sinnett-Smith, J. (2002). Gastrointestinal peptide signalling in health and disease. Eur. J. Surg. Suppl. 587, 23–38.
Sakurai, T., Yanagisawa, M., Takuwa, Y., Miyazaki, H., Kimura, S., Goto, K., et al. (1990). Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 348, 732–735. doi: 10.1038/348732a0
Sánchez-Fernández, G., Cabezudo, S., García-Hoz, C., Benincá, C., Aragay, A. M., Mayor, F. Jr., et al. (2014). Galphaq signalling: the new and the old. Cell. Signal. 26, 833–848. doi: 10.1016/j.cellsig.2014.01.010
Shashidhar, S., Lorente, G., Nagavarapu, U., Nelson, A., Kuo, J., Cummins, J., et al. (2005). GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 24, 1673–1682. doi: 10.1038/sj.onc.1208395
Shin, M. K., Levorse, J. M., Ingram, R. S., and Tilghman, S. M. (1999). The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature 402, 496–501. doi: 10.1038/990040
Shoushtari, A. N., and Carvajal, R. D. (2014). GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 24, 525–534. doi: 10.1097/CMR.0000000000000121
Smith, S. L., Damato, B. E., Scholes, A. G., Nunn, J., Field, J. K., and Heighway, J. (2002). Decreased endothelin receptor B expression in large primary uveal melanomas is associated with early clinical metastasis and short survival. Br. J. Cancer 87, 1308–1313. doi: 10.1038/sj.bjc.6600620
Spica, T., Fargnoli, M. C., Hetet, G., Bertrand, G., Formicone, F., Descamps, V., et al. (2011). EDNRB gene variants and melanoma risk in two southern European populations. Clin. Exp. Dermatol. 36, 782–787. doi: 10.1111/j.1365-2230.2011.04062.x
Spinella, F., Caprara, V., Di Castro, V., Rosanò, L., Cianfrocca, R., Natali, P. G., et al. (2012). Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J. Mol. Med. (Berl.) 91, 395–405. doi: 10.1007/s00109-012-0956-2
Tanese, K., Fukuma, M., Yamada, T., Mori, T., Yoshikawa, T., Watanabe, W., et al. (2008). G-protein-coupled receptor GPR49 is up-regulated in basal cell carcinoma and promotes cell proliferation and tumor formation. Am. J. Pathol. 173, 835–843. doi: 10.2353/ajpath.2008.071091
Van Raamsdonk, C. D., Barsh, G. S., Wakamatsu, K., and Ito, S. (2009a). Independent regulation of hair and skin color by two G protein-coupled pathways. Pigment Cell Melanoma Res. 22, 819–826. doi: 10.1111/j.1755-148X.2009.00609.x
Van Raamsdonk, C. D., Bezrookove, V., Green, G., Bauer, J., Gaugler, L., O’Brien, J. M., et al. (2009b). Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602. doi: 10.1038/nature07586
Van Raamsdonk, C. D., Fitch, K. R., Fuchs, H., De Angelis, M. H., and Barsh, G. S. (2004). Effects of G-protein mutations on skin color. Nat. Genet. 36, 961–968. doi: 10.1038/ng1412
Van Raamsdonk, C. D., Griewank, K. G., Crosby, M. B., Garrido, M. C., Vemula, S., Wiesner, T., et al. (2010). Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 363, 2191–2199. doi: 10.1056/NEJMoa1000584
Venkatakrishnan, A. J., Deupi, X., Lebon, G., Tate, C. G., Schertler, G. F., and Babu, M. M. (2013). Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194. doi: 10.1038/nature11896
von Koschembahr, A. M., Swope, V. B., Starner, R. J., and Abdel-Malek, Z. A. (2015). Endothelin-1 protects human melanocytes from UV-induced DNA damage by activating JNK and p38 signalling pathways. Exp. Dermatol. 24, 269–274. doi: 10.1111/exd.12638
Weigle, B., Fuessel, S., Ebner, R., Temme, A., Schmitz, M., Schwind, S., et al. (2004). D-GPCR: a novel putative G protein-coupled receptor overexpressed in prostate cancer and prostate. Biochem. Biophys. Res. Commun. 322, 239–249. doi: 10.1016/j.bbrc.2004.07.106
Wess, J. (1997). G-protein-coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J. 11, 346–354.
Williams, B., Schneider, R. J., and Jamal, S. (2014). Akt and PI3K-dependent but CREB-independent upregulation of MCAM by endothelin-3 in human melanocytes. Melanoma Res. 24, 404–407. doi: 10.1097/CMR.0000000000000077
Wouters, J., Hunger, R. E., Garrod, T., Dubuis, B., Hunziker, T., van den Oord, J. J., et al. (2015). First-in-human proof-of-concept study: intralesional administration of BQ788, an endothelin receptor B antagonist, to melanoma skin metastases. Oncologist 20, 1121–1122. doi: 10.1634/theoncologist.2015-0139
Wu, X., Li, J., Zhu, M., Fletcher, J. A., and Hodi, F. S. (2012). Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-kappaB pathways. Mol. Cancer Ther. 11, 1905–1914. doi: 10.1158/1535-7163.MCT-12-0121
Yamamoto, Y., Sakamoto, M., Fujii, G., Tsuiji, H., Kenetaka, K., Asaka, M., et al. (2003). Overexpression of orphan G-protein-coupled receptor, Gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology 37, 528–533. doi: 10.1053/jhep.2003.50029
Yanagisawa, H., Yanagisawa, M., Kapur, R. P., Richardson, J. A., Williams, S. C., Clouthier, D. E., et al. (1998). Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development 125, 825–836.
Yang, C. S., Skiba, N. P., Mazzoni, M. R., and Hamm, H. E. (1999). Conformational changes at the carboxyl terminus of Galpha occur during G protein activation. J. Biol. Chem. 274, 2379–2385. doi: 10.1074/jbc.274.4.2379
Keywords: GNAQ, GNA11, EDNRB, endothelin, melanoma, melanocyte, GPCR, Schwann cell precursors
Citation: Urtatiz O and Van Raamsdonk CD (2016) Gnaq and Gna11 in the Endothelin Signaling Pathway and Melanoma. Front. Genet. 7:59. doi: 10.3389/fgene.2016.00059
Received: 10 February 2016; Accepted: 01 April 2016;
Published: 20 April 2016.
Edited by:
Suzie Chen, Rutgers University, USAReviewed by:
Bin Zheng, Massachusetts General Hospital and Harvard Medical School, USAAparna Ranganathan Sertil, University of Arizona, USA
Copyright © 2016 Urtatiz and Van Raamsdonk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine D. Van Raamsdonk, Yy52ckB1YmMuY2E=