Abstract
The melanocortin 1 receptor (MC1R) is a melanocytic Gs protein coupled receptor that regulates skin pigmentation, UV responses, and melanoma risk. It is a highly polymorphic gene, and loss of function correlates with a fair, UV-sensitive, and melanoma-prone phenotype due to defective epidermal melanization and sub-optimal DNA repair. MC1R signaling, achieved through adenylyl cyclase activation and generation of the second messenger cAMP, is hormonally controlled by the positive agonist melanocortin, the negative agonist agouti signaling protein, and the neutral antagonist β-defensin 3. Activation of cAMP signaling up-regulates melanin production and deposition in the epidermis which functions to limit UV penetration into the skin and enhances nucleotide excision repair (NER), the genomic stability pathway responsible for clearing UV photolesions from DNA to avoid mutagenesis. Herein we review MC1R structure and function and summarize our laboratory’s findings on the molecular mechanisms by which MC1R signaling impacts NER.
Melanocortin Receptors
The melanocortin (MC) receptor family is the smallest member of the class A (rhodopsin-like) family of G-protein coupled receptors (GPCRs) (Gether, 2000; Montero-Melendez, 2015) and consists of five members: MC1R, MC2R, MC3R, MC4R, and MC5R with varying tissue expression and functions. MC1R is found on both melanocytes and leukocytes and its activation promotes UV resistance and anti-inflammatory signaling, respectively, (Mountjoy et al., 1992). MC2R, cloned Mountjoy et al. (1992), is found in the adrenal cortex. MC3R, cloned Desarnaud et al. (1994), and MC4R, cloned Gantz et al. (1993), are both found primarily in the CNS regulating food intake and sexual function. MC5R, located in skeletal muscle and brain, has an exocrine function (Gantz et al., 1994a). Sequence homology between the five receptors is only 40–60% which accounts for the lack of ligand specificity between receptors (Gantz et al., 1993; Yang et al., 2003). This review will focus on the role of MC1R in melanocytes with an emphasis on ligands, signaling pathways, structure, and function.
Melanocortin 1 Receptor (MC1R)
The human MC1R is 317 amino acids (Garcia-Borron et al., 2005), and it was originally identified and cloned by two independent groups Chhajlani and Wikberg (1992) and Mountjoy et al. (1992) and mapped to chromosome 16q24.3 Gantz et al. (1994b). The receptor is primarily located on melanocytes and transformed melanoma cells (Ghanem et al., 1988; Siegrist et al., 1989, 1994; Donatien et al., 1992). MC1R protein expression is typically low, with an estimated 700 protein units expressed per melanocyte and somewhat higher numbers on melanoma cells (Donatien et al., 1992; Roberts et al., 2006). The 315 amino acid murine homolog, Mc1r, was also cloned and identified Mountjoy et al. (1992) and mapped to the extension locus Robbins et al. (1993). Mice with a mutated extension locus display a reddish blonde coat color instead of the darkly pigmented black coat color typically found on the C57BL/6 background, thus providing the first genetic evidence that MC1R may play an important role in the regulation of pigment (Searle, 1968).
MC1R Structure
Like other GPCRs, MC1R is made up of 7 α-helical transmembrane (TM) domains with a DRY motif at the junction of the third TM domain, an intracellular C-terminus with a palmitoylation site, and an extracellular N-terminus with an N-linked glycosylation site (Yang, 2011) (Figure 1). Unique to the MC receptor subfamily compared to other GPCRs is the lack of one or two cysteines in the first and second extracellular domains and lack of proline in the fourth and fifth TM domains (Yang, 2011).
FIGURE 1
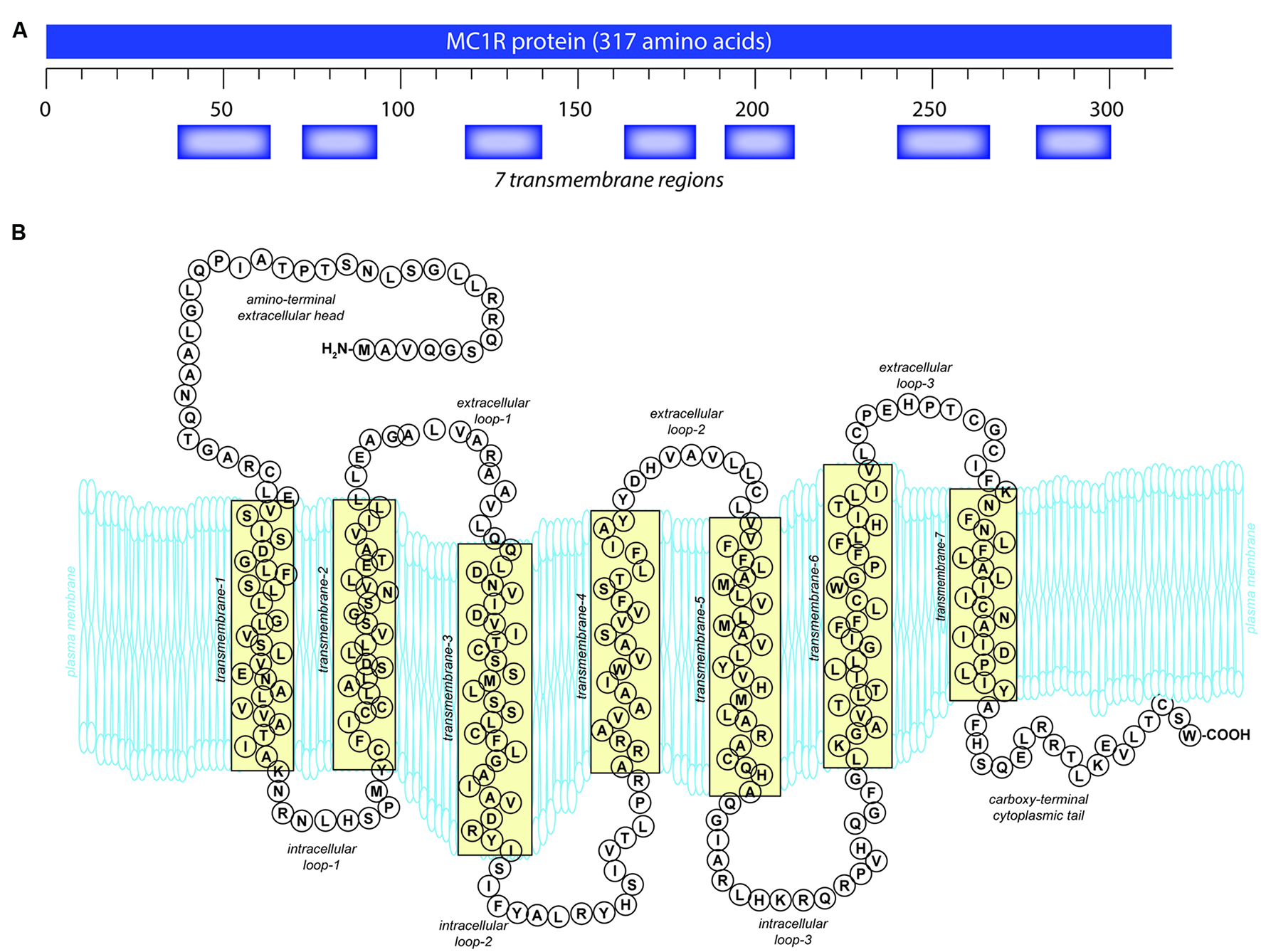
Melanocortin 1 receptor (MC1R) gene and protein structures. (A) The human MC1R locus (cytogenetic location: 16q24.3) encodes a seven transmembrane protein that is highly polymorphic. (B) The mature MC1R protein is a Gs-protein coupled receptor (GPCR) that spans the membrane seven times. Extracellular and transmembrane domains engage MC1R ligands while intracellular and transmembrane domains regulate adenylyl cyclase interactions and signaling.
N-terminus and c-terminus
The extracellular N-terminal tail (Figure 1) functions both for ligand affinity (Chhajlani et al., 1996) and as a signal anchor (Wallin and von Heijne, 1995; Garcia-Borron et al., 2005). There is a conserved cysteine residue located at the junction of the N-terminus and the first TM domain which is absolutely required for receptor function (Frandberg et al., 2001; Sanchez-Laorden et al., 2006b). The C-terminus in GPCR often plays a role in protein trafficking from the endoplasmic reticulum to the plasma membrane (Schulein et al., 1998; Qanbar and Bouvier, 2003) and also in receptor interactions with the G protein at the plasma membrane (Strader et al., 1994). Similar to the other MC receptors, MC1R has a characteristically short C-terminal tail that is only 14 amino acids in length (Figure 1). A pentapeptide present on the C-terminal tail contains the invariant tripeptide sequence T314, C315, and W317 present in all MC receptors. The pentapeptide, and specifically the invariant tripeptide, are required for translocation of the receptor to the plasma membrane (Sanchez-Mas et al., 2005b). Mutations which disrupt the pentapeptide or specifically the invariant tripeptide such as premature termination at R306 (Newton et al., 2000) or deletion of the terminal pentapeptide (Sanchez-Mas et al., 2005b), result in decreased plasma membrane MC1R expression. Although C-terminal deletions have detrimental effects, additional amino acids on the end of the C-terminus do not appear to affect MC1R function. A splice variant exists with an additional 65 amino acids that displays similar pharmacology to the unspliced protein (Tan et al., 1999). In addition to affecting receptor localization to the plasma membrane, the C-terminus also plays a role in desensitization and internalization (Pitcher et al., 1998; Luttrell and Lefkowitz, 2002).
Intracellular and Extracellular Loops
Melanocortin 1 receptor’s intracellular and extracellular loops (ils and els, respectively) are found between the transmembrane regions (Figure 1) and have conserved sequences found across many MC receptors. MC1R els are small compared to most GPCRs but are critical for basal constitutive signaling activity (Holst and Schwartz, 2003). Because the els of MC1R interact with ligands, mutations in this region impact binding affinity (Chhajlani et al., 1996). El3 in particular appears to play a critical role in melanocortin affinity through conserved proline and cysteine residues (Holst and Schwartz, 2003). El3 interacts with TM6 and TM7 which are also required for ligand-receptor binding, and it is believed that C267 and C275 in el3 form disulfide bonds between TM6 and TM7 affecting the tertiary structure of the receptor (Frandberg et al., 2001; Holst and Schwartz, 2003; Garcia-Borron et al., 2005).
Similarly, MC1R ils are important for binding of the Gs protein and have sites for phosphorylation that affect signal regulation, internalization, and receptor cycling (Strader et al., 1994). Il1 is important for normal activation, and mutations of this domain increase MC1R signaling activity as seen in the tobacco mutation (S69L in mouse; S71 in human). Six mutations have been reported in il1, four of which cause a loss of signaling function (Robbins et al., 1993). As is characteristic for class A GPCRs, the tripeptide 141 DRY 143 located at the interface of il2 and TM3 is required for MC1R function (Schioth et al., 1999). Additionally, there are putative protein kinase A (PKA) and protein kinase C phosphorylation sites in il2 of both human and mouse (and il3 in mouse), however, neither PKA nor PKC has been shown to phosphorylate MC1R to date (Garcia-Borron et al., 2005).
MC1R Oligomerization
Like many other GPCRs, oligomerization of MC1R is functionally important for modulation of ligand binding, coupling efficiency, desensitization, and trafficking through the endoplasmic reticulum (Sanchez-Laorden et al., 2006a). MC1R undergoes constitutive dimerization without a ligand binding requirement (Mandrika et al., 2005; Sanchez-Laorden et al., 2006a) at the level of the ER (Sanchez-Laorden et al., 2006a). MC1R homo-dimerization is dependent upon both covalent and non-covalent interactions rather than a coiled-coil mechanism, mediated by four inter-subunit disulfide bonds at C35, C267, C273, and C275 or by domain swapping. Disruption of any disulfide bond abolishes MC1R function, however, only C35 is required for MC1R to travel from the ER to the plasma membrane. Although mutation of C35 prevents translocation of MC1R to the plasma membrane, the protein can still dimerize, therefore dimerization is not sufficient for ER to plasma membrane transport (Sanchez-Laorden et al., 2006a).
Dimerization of heterogeneous receptors can have a dramatic effect on MC1R signaling. Dimerization between mutant and wild-type MC1R proteins can cause a dominant negative effect (Sanchez-Laorden et al., 2006a) similar to that reported in dimerization of mutant MC4R with wild-type MC4R (Biebermann et al., 2003). Similarly, dimerization between wild type and a mutant MC1R unable to translocate to the surface of the cell also resulted in dose-dependent dominant negative inhibition of wild-type MC1R cell surface localization (Sanchez-Laorden et al., 2006a). Conversely, coupling with wild type GPCR can partially rescue function of mutant GPCR via exchange of defective domains (Breitwieser, 2004). Co-expression of two MC1R mutants with mutations in different domains may similarly rescue function through complementation, however, rescue was not observed if mutations were in the same domain (Sanchez-Laorden et al., 2006a). MC1R dimerization characteristics could therefore impact melanocortin signaling and play an important role in individuals with inherited polymorphisms in the MC1R protein.
MC1R Desensitization
Like other GPCRs, MC1R desensitization and internalization represent major mechanisms whereby its function can be regulated (Pitcher et al., 1998). Multiple members of the melanocortin receptor family undergo homologous desensitization including murine Mc2R (Baig et al., 2001), murine Mc4r (Shinyama et al., 2003), and both murine Mc1R and human MC1R (Sanchez-Mas et al., 2005a). MC1R undergoes homologous desensitization following short exposure to its positive agonist, α-MSH in a PKA independent and G protein coupled receptor kinase (GRK) dependent manner. Following agonist stimulation, GRKs phosphorylate GPCRs resulting in receptor decoupling from the G protein and subsequent internalization (Benovic et al., 1985; Hausdorff et al., 1990). MC1R desensitization is dependent upon GRK2 and GRK6, however, internalization only requires GRK6 phosphorylation of T308 and S316 on the C-terminus (Sanchez-Laorden et al., 2007). Studies conducted in primary human melanocytes, however, demonstrated that GRK expression varies between individuals (Swope et al., 2012). In addition, MC1R desensitization is further mediated via β-arrestins (ARRB). ARRBs bind the phosphorylated receptor and prevent the receptor from coupling to the G protein and target the receptor for internalization (Attramadal et al., 1992). Recently, ARRB2 but not ARRB1 has been shown to play a role in receptor desensitization and internalization (Abrisqueta et al., 2013).
Melanocortin Signaling and cAMP
Melanocortin 1 receptor is complexed to the heterotrimeric G protein. Following activation with agonistic ligands the Gαs protein dissociates from MC1R and stimulates adenylyl cyclase activity which cleaves ATP to generate the second messenger cAMP (Figure 2). In melanocytes, increased cAMP levels lead to a host of downstream signaling events including activation of effector proteins such as cAMP-dependent protein kinase (PKA) (Neves et al., 2002; Dorsam and Gutkind, 2007). In this manner MC1R signaling activates various signaling cascades within the cell. In melanocytes, cAMP induction leads to increased melanin synthesis (Suzuki et al., 1997; Abdel-Malek et al., 2014) and resistance to UV injury through enhanced antioxidant defenses and acceleration of nucleotide excision repair (NER) (Kokot et al., 2009; Song et al., 2009; Kadekaro et al., 2010, 2012; Abdel-Malek et al., 2014; Jarrett et al., 2014). The dissociated Gαβ protein can also modify intracellular signaling including the mitogen-activated protein kinase family which affects a multitude of signaling pathways (further reviewed by Dorsam and Gutkind, 2007). MC1R, like other GPCRs, displays some degree of ligand independent basal signaling (Chalmers and Behan, 2002; Milligan, 2003). This has been demonstrated for both human MC1R (Mas et al., 2003) and murine Mc1r (Jackson et al., 2007), human MC3R, human MC4R, and murine Mc5r (Nijenhuis et al., 2001). Genetic proof of basal MC1R signaling is evident in Pomc1 knockout mice which are incapable of generating melanocortins which are the major agonists of MC1R. In contrast to Mc1r-defective strains such as extension, POMC-null mice maintain a dark coat color, suggesting Mc1r has constitutive ligand independent activity (Bennett and Lamoreux, 2003).
FIGURE 2
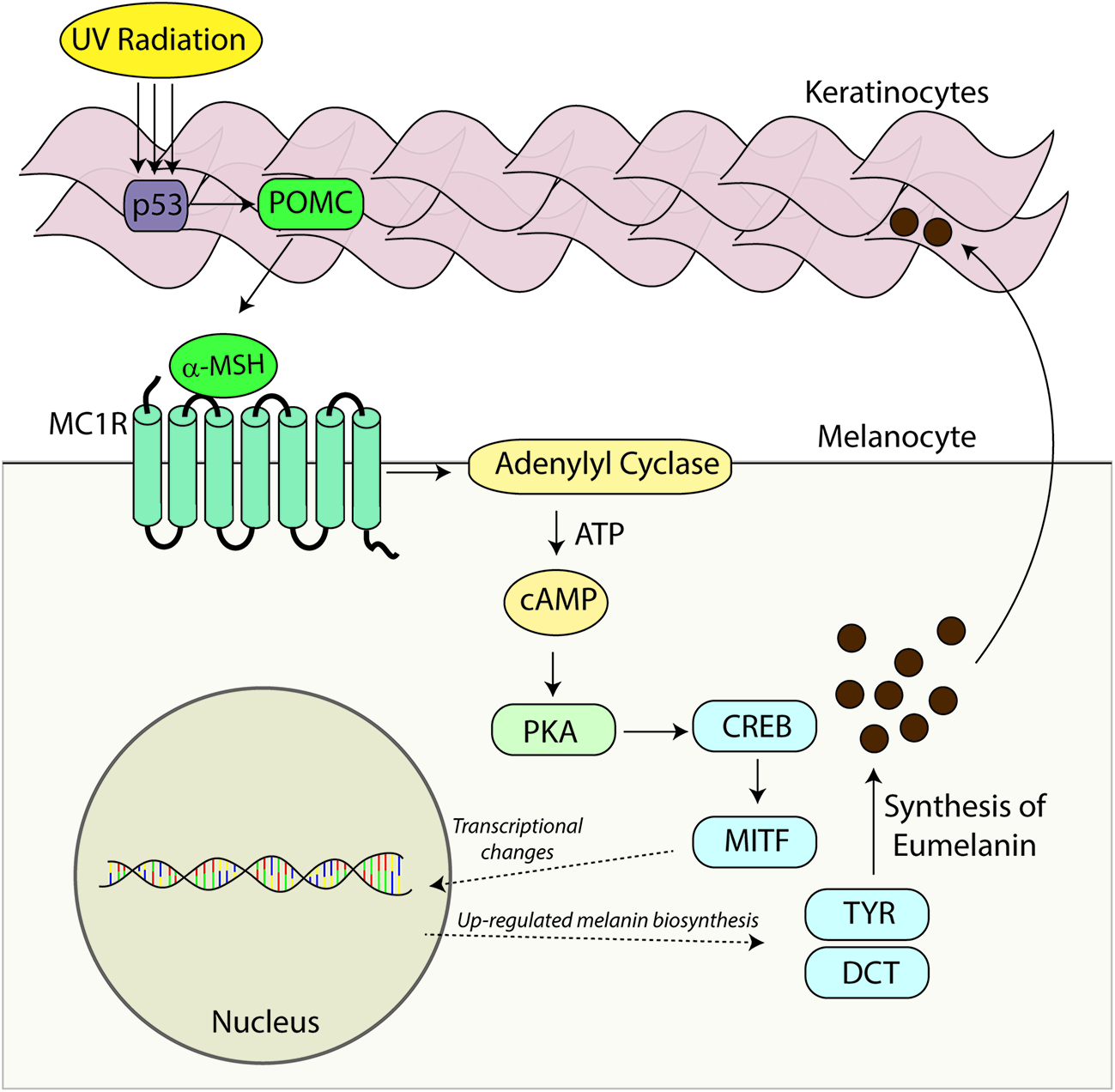
Melanocortin – MC1R signaling axis. Melanocortins (α-MSH, ACTH) are produced basally by the pituitary and induced in the skin after UV injury. Binding of these melanocortin ligands to MC1R promotes critical UV-resistance physiologic changes in melanocytes and protects the skin from UV damage. Upon binding melanocortins, MC1R activates adenylyl cyclase and stimulates cAMP production. In turn, a variety of downstream effector pathways including induction of the CREB and Mitf transcription factor networks and an increase in the activity of PKA takes place. Expression of a variety of enzymes including tyrosinase (TYR) and dopachrome tautomerase (DCT) involved in melanin biosynthesis is increased and melanin production is up-regulated. Melanin produced in organelles termed melanosomes, is transferred to neighboring keratinocytes and in this way a UV-protective layer of pigment in the epidermis is established to enhance the skin’s ability to resist further UV injury. Importantly, melanocytic genomic stability is also enhanced through improved DNA repair. In the absence of a functional melanocortin signaling axis, these pathways are blunted, the skin is under-melanized and melanocytes accumulate more UV mutations as a result of ineffective DNA repair. In this way, individuals with inherited defects in MC1R signaling are at heightened risk for melanoma.
MC1R and Pigmentation
There are two major types of pigment present in the skin, the darkly pigmented eumelanin and the red/yellow sulfated pheomelanin. Eumelanin is chemically inert and is highly photoprotective by absorbing UV radiation (Kaidbey et al., 1979; Scherer and Kumar, 2010) and oxidants (Hoogduijn et al., 2004). In contrast, pheomelanin is much less efficient at blocking penetration of UV radiation into the skin and can promote UV-induced cellular damage by contributing to free radical and oxidative injury (Thody et al., 1991; Mitra et al., 2012). MC1R signaling is a major determinant for the amount and type of melanin pigments synthesized by melanocytes, regulating both basal pigmentation and the UV induced tanning response (D’Orazio et al., 2006). MC1R signaling increases eumelanin synthesis, the ratio of eumelanin-to-pheomelanin (Hunt et al., 1995), and enhances melanosome transfer to enhance melanin deposition in keratinocytes (Virador et al., 2002).
Both eumelanin and pheomelanin derive from the sequential cyclization and oxidation of the amino acid tyrosine (Figure 3) (Ito, 2003). The first two biosynthetic steps are shared between the two pathways: the conversion of tyrosine to DOPA and then to DOPAquinone by the enzyme tyrosinase. Eumelanogenesis and pheomelanogenesis diverge after formation of DOPAquinone. Other enzymes beside tyrosinase are needed for melanin synthesis including dopachrome tautomerase and tyrosinase-related protein 1. Defects in many pigment enzymes yield hypomelanotic phenotypes such as albinism (Baxter and Pavan, 2013). Pheomelanin production is dependent upon the incorporation of a cysteine and retention of sulfur after the synthesis of DOPAquinone, which may explain why mature pheomelanin pigments are reddish/yellow rather than dark brown/black as eumelanin is. Although the control of the pigment switch between eumelanin and pheomelanin is regulated by multiple factors including the pH of the cellular milieu and the levels of tyrosinase (Burchill and Thody, 1986; Ancans et al., 2001), the presence of a functional MC1R is required for effective synthesis of eumelanin. Since eumelanin absorbs UV radiation, the more eumelanin the skin has, the more protected it is from UV damage.
FIGURE 3
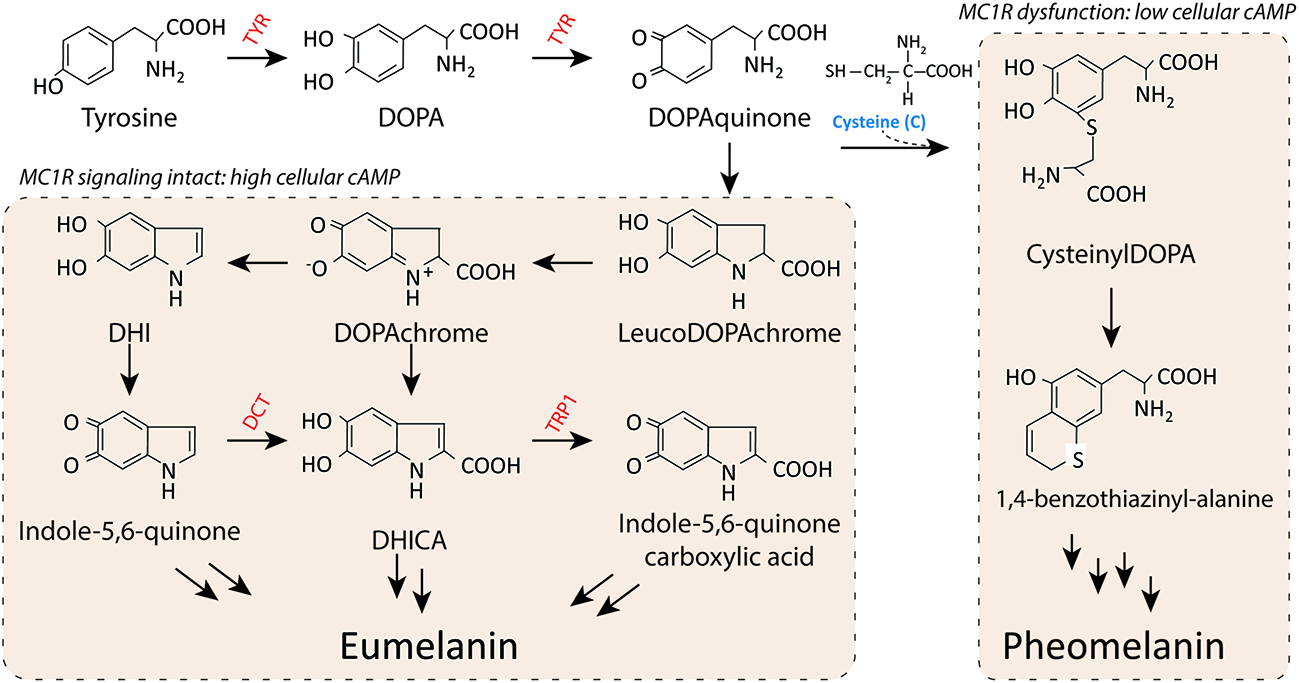
Melanin synthesis. There are two main types of melanin: the dark brown/black UV-protective eumelanin and the red/blonde sulfated pheomelanin pigment. Each is derived from progressive cyclization and oxidation of the amino acid tyrosine. Tyrosinase, the rate-limiting enzyme for melanogenesis, catalyzes the first two stages of melanin biosynthesis. When MC1R is functional and cAMP levels are high, melanocytes produce eumelanin preferentially. In contrast, when MC1R is dysfunctional and cAMP levels are low, cysteine is incorporated and pheomelanin is made instead. Important pigment enzymes which cause hypopigmentary phenotypes when defective include tyrosinase (TYR), dopachrome tautomerase (DCT), and tyrosinase related protein-1 (TRP1). Complexion and UV resistance are mainly determined by the amount of eumelanin in the skin.
MC1R Variants
Melanocortin 1 receptor is a highly polymorphic protein, and in humans many of the loss-of-function variants are associated with the “red hair color” (RHC) phenotype (Valverde et al., 1995; Box et al., 1997; Smith et al., 1998; Abdel-Malek et al., 2014). Individuals with a dysfunctional MC1R may have decreased eumelanin synthesis leading to fair skin and an increased sensitivity to UV exposure (Smith et al., 1998; Palmer et al., 2000; Landi et al., 2005). Degree of MC1R function correlates with the extent of pigmentation phenotype in individuals with RHC variants (Beaumont et al., 2007), and the effects of MC1R on basal pigmentation can be seen in both humans and murine models. Murine coat color is heavily influenced by MC1R signaling as clearly evident by variations in coat color associated with MC1R mutations such as the extension locus (Figure 4). Mice with the recessive yellow mutation (mutation of the extension locus) produce a non-functional MC1R and exhibit a blonde pheomelanotic coat color as a result (Robbins et al., 1993). Conversely, an increase in MC1R activity found in either the somber (constitutive active receptor) or tobacco (hyperactive receptor) mutation is associated with an increase in eumelanin synthesis and a darker coat color (Robbins et al., 1993) although no gain of function mutations have been identified in the human MC1R gene. The effect of MC1R signaling on basal pigmentation can be further seen in the lethal yellow mutation which affects an MC1R ligand rather than MC1R directly. Mice with the lethal yellow mutation have a blonde pheomelanotic coat color due to the ubiquitous overexpression of the negative agonist murine homolog of ASIP (ASP) inhibiting which diminishes basal Mc1r activity (Lovett et al., 1987).
FIGURE 4
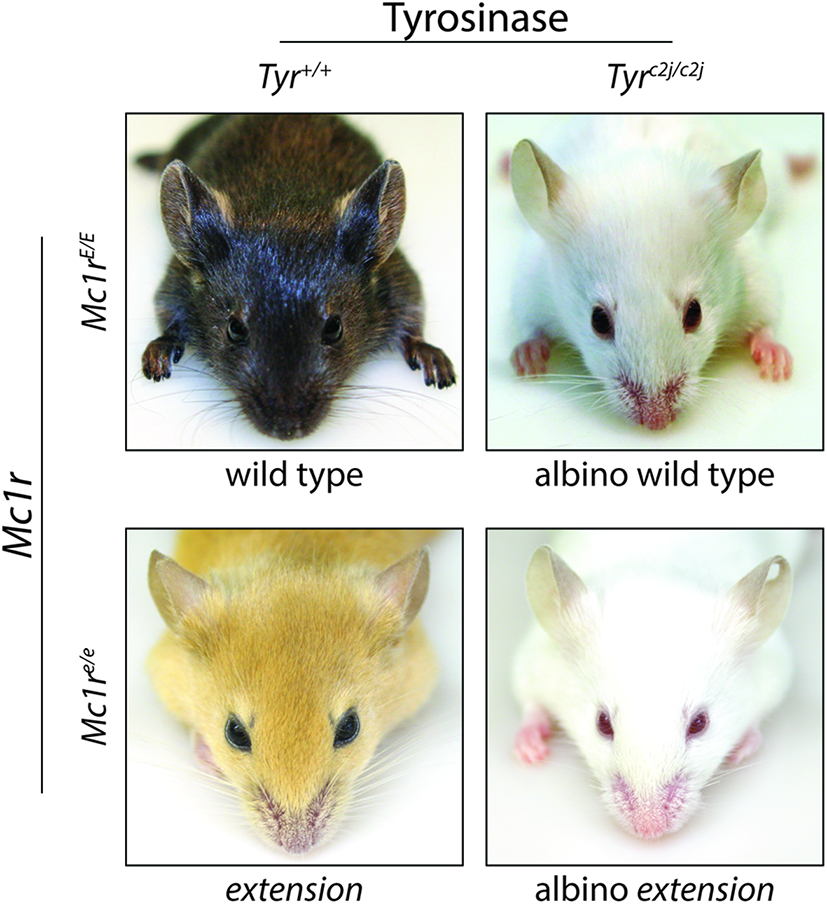
Melanocortin 1 receptor controls coat color in C57BL/6 mice. C57BL/6 mice with wild type (functional) Mc1r exhibit a eumelanotic phenotype, whereas congenic animals defective for Mc1r, such as extension mice, exhibit a pheomelanotic coat color. Shown are Mc1rE/E (wt) and Mc1re/e (extension) age-matched animals along with C57BL/6 Tyrc2j/c2j animals in either the Mc1rE/E (wt) or Mc1re/e (extension) background in which neither eumelanin nor pheomelanin can be made because of defective tyrosinase function (Jarrett et al., 2014). Note that these animals express the K14-Scf transgene resulting in retention of interfollicular epidermal melanocytes and pigmentation of the skin in addition to the fur.
Adaptive Tanning
The ability of the skin to respond to UV radiation by increasing melanin production is dependent on the functionality of MC1R. The adaptive pigmentation pathway represents a major innate protective mechanism by which the skin prevents further damage from ultraviolet radiation and is dependent upon MC1R signaling. UV radiation causes DNA damage to keratinocytes in the epidermis of the skin and the subsequent increased expression of the POMC protein in a p53 dependent manner (Cui et al., 2007). Cleavage of POMC by proconvertase 1 and 2 leads to the generation of the positive agonist α-MSH (Benjannet et al., 1991). Binding of α-MSH to MC1R leads to the activation of adenylyl cyclase and promotes the generation of cAMP (Kadekaro et al., 2003; Millington, 2006). cAMP accumulation promotes the activation of PKA leading to the phosphorylation of the cAMP responsive binding element (CREB). CREB functions as a transcription factor causing the upregulation of microphthalmia transcription factor (MITF). MITF functions as a master transcription factor and leads to the increased expression of multiple pigment dependent enzymes including tyrosinase (Levy et al., 2006).
MC1R and Pigment-independent UV Protection
Besides its role in regulating melanocyte pigment production, MC1R is a critical determinant of cellular genome maintenance pathways (Abdel-Malek et al., 2014). Melanocortins augment melanocyte anti-oxidant defense mechanisms and diminish free radical injury in melanocytes (Kokot et al., 2009; Song et al., 2009; Kadekaro et al., 2012). MC1R signaling also plays a critical role in the ability of melanocytes to recover from UV damage, particularly with respect to damage to genomic DNA. UV causes direct damage to pyrimidine bases within DNA that promote mutagenesis if not cleared from the genome. The NER pathway is the major means by which cells remove UV photoproducts from genomic DNA (reviewed in Scharer, 2013 and Shah and He, 2015) (Scharer, 2013; Shah and He, 2015).
Nucleotide excision repair
Nucleotide excision repair is the major genome maintenance pathway by which cells remove bulky DNA lesions that distort the DNA double helical structure including UV induced photoproducts (6,4 photoproducts and cyclobutane dimers). The NER pathway involves the coordinated action of multiple factors that recognize, excise, and repair damaged nucleotides. There are two types of NER - global genome nucleotide excision repair (GG-NER) and transcription coupled nucleotide excision repair (TC-NER). They differ in the initial stages of damage recognition, ultimately converging on a common repair pathway. TC-NER is invoked when transcription is stalled by nucleotide lesions in actively transcribed genes (Mellon et al., 1987; Mu and Sancar, 1997; Sugasawa et al., 1998, 2001). TC-NER is mediated by Cockayne syndrome B (CSB) and Cockayne syndrome A (CSA) proteins which are recruited to helical distortions that have caused RNA polymerase to stall (Mellon et al., 1987; Venema et al., 1991; Donahue et al., 1994; Mu and Sancar, 1997; Kamiuchi et al., 2002). In GG-NER, xeroderma pigmentosum complementation group C (XPC) and HR23B heterodimerize and scan the genome for helical distortions (Sugasawa et al., 1998, 2001). After damage recognition, TFIIH, a multiprotein complex composed of nine proteins, is recruited. TFIIH contains the helicases XPB and XPD which function in the 3′-5′ and 5′-3′ directions, respectively, (Gerard et al., 1991). The helicases unwind approximately 20–30 nucleotides surrounding the DNA lesion creating two unprotected single strand sequences. RPA and XPA are recruited to stabilize the open DNA conformation (Tapias et al., 2004; Park and Choi, 2006) followed by the endonucleases ERCC1-XPF and XPG which remove the damaged base (Mu and Sancar, 1997; Houtsmuller et al., 1999). Polymerase δ and 𝜀 in combination with proliferating cell nuclear antigen replace the gap using the undamaged complementary strand for fidelity (Shivji et al., 1995; Cleaver, 2005; Shah and He, 2015). In this way, NER efficiently repairs UV photoproducts and prevents UV mutagenesis.
MC1R signaling and NER
Individuals with defective MC1R signaling are prone to UV induced skin cancer not only because they have decreased pigmentation but also because they have a blunted DNA repair response. We and others have reported that activation of MC1R and the subsequent cAMP signaling cascade are major regulators of NER kinetics and efficiency independent of pigmentation (Figure 5) (Kadekaro et al., 2005, 2010; Smith et al., 2008; Jagirdar et al., 2013; Jarrett et al., 2014; Swope et al., 2014). cAMP signaling directly impacts how long UV photodamage persists in melanocytes (Hauser et al., 2006; Abdel-Malek et al., 2009), and repair of photodamage in the skin of K14-Scf mice is much more robust when Mc1r is functional or when pharmacologic agents are topically applied to the skin that induce cAMP signaling (Jarrett et al., 2014).
FIGURE 5
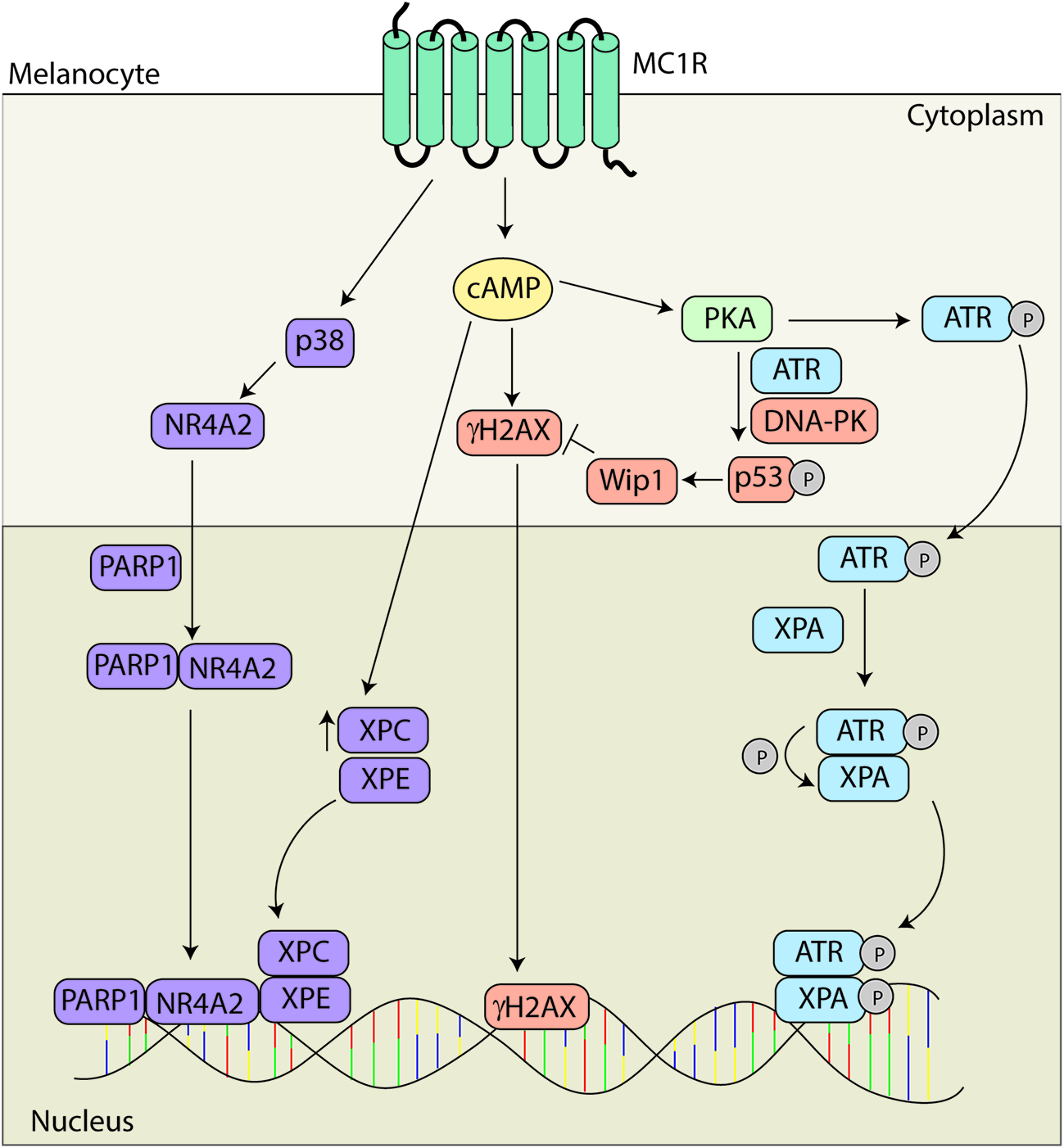
Melanocortin 1 receptor signaling and melanocyte genomic integrity. MC1R signaling promotes genomic stability through multiple mechanisms. MC1R activation induces translocation of NR4A2 to the nucleus in a p38 and PARP1 dependent manner where it co-localizes with XPC and XPE at sites of UV induced DNA damage. MC1R activation also leads to elevated levels of XPC and γH2Ax promoting the formation of DNA repair complexes. Levels of γH2AX are regulated by Wip1 downstream of ATR and DNA-PK mediated phosphorylation of p53 at S15. In addition, PKA activation promotes the phosphorylation of ATR at S435. pS435-ATR complexes with XPA in the nucleus. Following phosphorylation of XPA, the complex translocates to sites of UV induced DNA damage to accelerate and enhance nucleotide excision repair (NER).
Although it has been known for a decade that MC1R signaling accelerates NER kinetics (Abdel-Malek et al., 2006), the molecular mechanisms by which the phenomenon occurs have only recently begun to be elucidated and appear to be complex. Acceleration of repair of photo damage has been shown to be dependent upon both the nuclear receptor subfamily 4 group A member 2 (NR4A2) and ataxia telangiectasia mutated and Rad3 related (ATR) signaling pathways (Smith et al., 2008; Jagirdar et al., 2013; Jarrett et al., 2014). MC1R signaling leads to the induction of the NR4A2 which translocates to sites of photodamage in a p38 and poly ADP ribose polymerase (PARP) dependent manner. The NR4A2/PARP complex colocalizes with the DNA damage proteins XPC and XPE at sites of DNA damage. Data suggests that NRFA2 may play a role in promoting chromatin relaxation to promote DNA repair (Smith et al., 2008; Jagirdar et al., 2013).
Recently, we reported that enhancement of NER by cAMP is dependent on a post-translational modification of ATR protein. Specifically, we found that MC1R stimulation by melanocortins or pharmacological cAMP induction caused PKA to phosphorylate ATR at S435. This event promoted enhanced binding to the NER factor XPA in the nucleus and together pS435-ATR and XPA localized to UV photodamage in an accelerated and robust manner. Without PKA-mediated phosphorylation of ATR on S435, we observed no enhancement of NER in melanocytes. Therefore we concluded that MC1R signaling regulates genomic stability through this ATR-dependent mechanism (Jarrett et al., 2014). In subsequent work, we found that the MC1R agonists α-MSH or ACTH stimulated PKA-mediated ATR phosphorylation and NER whereas the MC1R negative agonist ASIP and the neutral antagonist βD3 inhibited the pathway, suggesting that melanocyte genomic stability and susceptibility to UV mutagenesis may be hormonally influenced (Jarrett et al., 2015).
Activation of MC1R has also been shown to facilitate repair via an increase in DNA damage response proteins. Treatment with α-MSH leads to an increase in XPC and γH2AX levels promoting formation of DNA repair complexes in primary human melanocytes (Swope et al., 2014). There is also a concomitant increase in DNA repair gene expression dependent upon MC1R signaling (Kadekaro et al., 2010). In addition, MC1R signaling also promotes the return to homeostasis via p53 signaling. MC1R activation promotes the phosphorylation of p53 at S15 in an ATR and DNA-PK dependent fashion leading to activation of wild-type p53 induced phosphatase 1 and degradation of γH2AX (Kadekaro et al., 2012; Swope et al., 2014).
Hormonal Regulation of MC1R
Melanocortin 1 receptor signaling is complex and dynamic, with signaling heavily influenced by receptor interactions with melanocortins, agouti signaling protein (ASIP), or β-defensin 3 (βD3). Melanocortins enhance MC1R signaling, ASIP inhibits MC1R signaling directly, and the neutral antagonist βD3 blunts melanocortin-induced signaling by competing with MC1R agonists for MC1R binding (Figure 6). The major melanocortin for MC1R, α-MSH, functions to increase cAMP levels after binding to MC1R whereas binding of ASIP to MC1R competes with α-MSH binding to prevent melanocortin activation and results in a decrease in basal cAMP levels. Binding of βD3 to MC1R does not affect basal cAMP levels, however, it functions as a competitive inhibitor and interferes with binding of either α-MSH or ASIP.
FIGURE 6
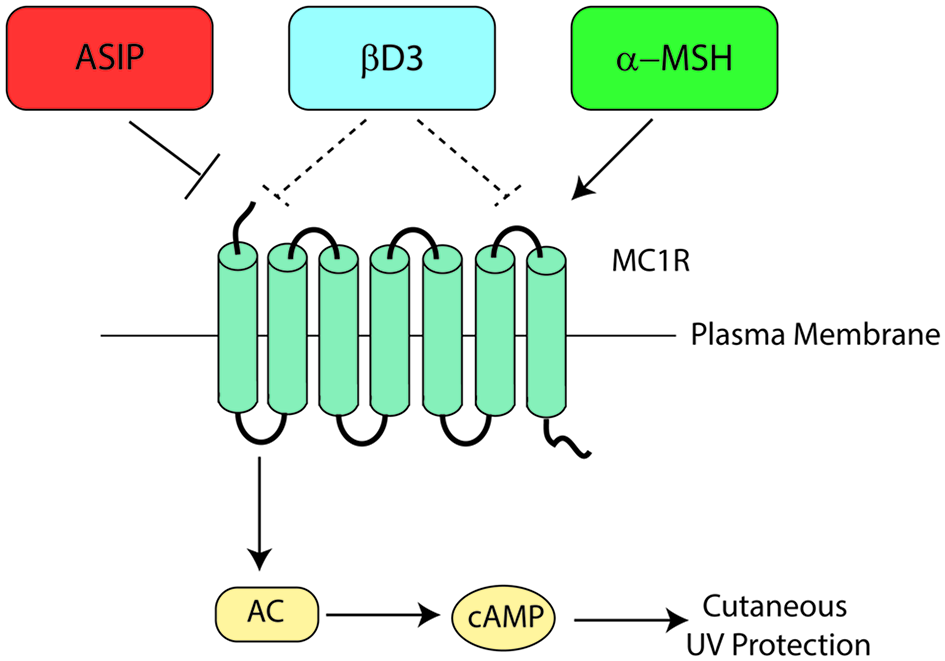
Melanocortin 1 receptor ligand interactions. Three major MC1R ligands are α-MSH, ASIP, and βD3. The melanocortin α-MSH functions as a positive agonist to increase cAMP levels downstream of MC1R. ASIP is a potent negative agonist that decreases basal MC1R signaling and interferes with melanocortin-induced cAMP upregulation. βD3 is thought to act as a neutral antagonist of MC1R, blocking the interactions with either melanocortins or ASIP but having little signaling impact independently. Each ligand appears to function as a competitive inhibitor for the others with only one able to bind to MC1R at any given time. Please note that although not shown in this diagram, ACTH’s ability to agonize MC1R is roughly the same as that of α-MSH.
Melanocortins
There are four endogenous melanocortin ligands: α-MSH, β-MSH, γ-MSH, and adrenocorticotropic hormone (ACTH). Melanocortins are derived as cleavage products of the pro-opiomelanocortin (POMC) protein (Wintzen and Gilchrest, 1996) (Figure 7), and each has varying receptor binding affinities across the MC family. The POMC gene contains three exons and two introns with the bioactive peptides present in exon 3 (Wintzen and Gilchrest, 1996). POMC is processed by proconvertase 1 to generate ACTH, the N-terminal fragment, and beta-lipotropin (β-LPH). Further cleavage of ACTH by proconvertase 2 generates corticotropin-like intermediate peptide (CLIP) and α-MSH. Cleavage of the N-terminal fragment by proconvertase 2 generates γ-MSH. Cleavage of β-LPH generates γ-LPH and β-endorphin (Wintzen and Gilchrest, 1996) which is active at opiate receptors in the skin. The UV-dependent β-endorphin production and the resultant opiate “high” is believed to contribute to UV-seeking behavior and an increase in analgesic threshold due to opioid dependence (Fell et al., 2014). POMC expression and processing is induced in both keratinocytes and melanocytes following UV exposure, leading to the secretion of MC1R ligands α-MSH and ACTH (Schauer et al., 1994; Chakraborty et al., 1996) in a p53 dependent manner (Cui et al., 2007).
FIGURE 7

Melanocortin synthesis through proopiomelanocortin (POMC) processing. Melanocortins are derived from the proopiomelanocortin (POMC) precursor peptide that is cleaved into functional peptide fragments by a pair of serine protease pro-protein convertases, PC1 and PC2. PC1 cleaves POMC into four subunits including an n-terminal region from which γ-MSH is derived, adrenocorticotropic hormone (ACTH) and β-lipotropin (β-LPH) from which γ-LPH, β-MSH and β-endorphin are generated by PC2 cleavage. α-MSH represents the first 13 amino acids of ACTH and is dependent on PC2 activity for its synthesis.
ACTH and α-MSH are the two major melanocortins for MC1R (Abdel-Malek et al., 2000). ACTH was first identified in Smith (1930), and α-MSH (first termed simply MSH prior to the discovery of β- and γ-MSH) was identified in the porcine pituitary Lerner and Lee (1955). α-MSH is expressed in both human melanocytes and keratinocytes (Chakraborty et al., 1996), and was first shown to bind to murine and human melanoma cells via a high affinity receptor Siegrist et al. (1989) and Solca et al. (1989). α-MSH binding to normal human melanocytes was demonstrated Donatien et al. (1992) and De Luca et al. (1993) by two independent investigators.
All melanocortin ligands have an HFRW motif required for receptor binding that contributes to the generalized ability for melanocortins to interact with more than one melanocortin receptor, albeit with different affinities. Binding to MC2R, however, also requires an additional motif, KKRRP, which is only present in ACTH. Therefore only ACTH can bind to MC2R (Mountjoy et al., 1992; Dores, 2009). Both MC1R (Abdel-Malek et al., 2000; Sanchez-Mas et al., 2004) and MC4R (Gantz et al., 1993) bind ACTH and α-MSH with similar affinities and each binds β- and γ-MSH with lower affinity. Similarly, MC5R binds α-MSH with the highest affinity followed by β-MSH, ACTH, and γ-MSH in order of decreasing affinity (Gantz et al., 1994a). Agonists interact with extracellular loops of melanocortin receptors as well as charged and aromatic residues in the TM fragments (Garcia-Borron et al., 2005). Specifically, the arginine of the melanocortin HFRW pharmacophore core interacts with E94 (TM2), D117, and D121 (TM3). Similarly, aromatic residues near the extracellular side of melanocortin receptor TM regions 4, 5, and 6 interact with aromatic residues of the melanocortin pharmacophore (Yang et al., 1997).
Agouti Signaling Protein
The human agouti signaling protein is a 132 amino acid MC1R ligand encoded by the agouti locus on chromosome 20q11.2 with 85% homology to the murine protein (Miller et al., 1993; Kwon et al., 1994; Wilson et al., 1995). ASIP functions as a competitive MC1R inhibitor, efficiently preventing α-MSH binding to MC1R and inhibiting MC1R activation (Blanchard et al., 1995; Suzuki et al., 1997). Binding of ASIP and α-MSH to MC1R are mutually exclusive (Ollmann et al., 1998). In addition, ASIP functions as an inverse agonist to decrease basal MC1R signaling and inhibit eumelanogenesis (Lu et al., 1994; Wilson et al., 1995). Although ASIP’s primary sequence has no similarities to either ACTH or α-MSH, it binds to MC1R with almost equal affinity (Siegrist et al., 1996). ASIP’s cysteine rich C-terminal region binds to MC1R via an octaloop structure with four residues that are homologous to other melanocortin ligands (Ollmann et al., 1998; Tota et al., 1999; McNulty et al., 2005). The C-terminal region of ASIP is both necessary and sufficient for its effect at MC1R, and the C-terminus alone can function as a competitive antagonist to melanocortins at melanocortin receptors (Ollmann and Barsh, 1999).
Agouti signaling protein was known to promote a pheomelanotic coat phenotype before it was determined to be a direct ligand of MC1R. It is expressed in the dermal papilla of the hair follicle where it functions as a paracrine signal to regulate hair color (Millar et al., 1995). In the fur of a variety of animals, the agouti locus is expressed transiently to create alternating bands of pheomelanin and eumelanin on the hair shaft which results in improved camouflage (Tamate and Takeuchi, 1984). Canonically, ASP (the murine homolog of the human ASIP) is secreted locally only at the hair follicle, however, there are multiple murine models with varying coat phenotypes dependent upon altered agouti expression. For example, mice with the lethal yellow mutation (Ay), a dominant gain of function mutation resulting in ectopic and ubiquitous ASP expression, have a yellow/blonde coat color and are obese with hyperinsulinemia due to binding of the overexpressed ASP to MC4R (Takeuchi et al., 1989; Bultman et al., 1992; Robbins et al., 1993; Duhl et al., 1994a,b; Michaud et al., 1994).
The ability of ASIP to promote a pheomelanotic phenotype is dependent upon a functional MC1R, and ASIP had no effect on MC1R with constitutive activity or loss of function (Ollmann et al., 1998; Abdel-Malek et al., 2001). Treatment with ASIP inhibits α-MSH binding to MC1R preventing α-MSH induced cAMP production and tyrosinase, tyrosine related proteins 1 and 2, and MITF expression suppressing melanogenesis (Aberdam et al., 1998). ASIP’s inhibitory effect on pigment production, however, is seen with or without concomitant stimulation with α-MSH suggesting that the effects of ASIP at MC1R are not completely explained via preventing α-MSH binding (Graham et al., 1997). Treatment of primary murine or human melanocytes or melanoma cell lines with ASIP caused a decrease in eumelanosomes and an inhibition of pigmentation (Hunt and Thody, 1995; Sakai et al., 1997), and binding of ASIP to wild-type MC1R leads to decreased basal tyrosinase activity and decreased tyrosinase and tyrosinase related protein 1, 2, and 3 levels preventing eumelanogenesis (Sakai et al., 1997; Abdel-Malek et al., 2001).
Treatment with agouti also affects additional MC1R signaling pathways including NER, proliferation, and migration. Concomitant stimulation with ASIP and α-MSH abrogated α-MSH’s acceleration of NER following UV treatment (Jarrett et al., 2014, 2015), decreased basal DNA repair kinetics in a dose dependent manner and decreased basal ATR phosphorylation at S435 in primary melanocytes (Jarrett et al., 2015). Consistent with inhibition of MC1R signaling, treatment with ASIP leads to a decrease in proliferation in a mouse melanoma cell line (Siegrist et al., 1996) and an increase in melanocyte migration (Le Pape et al., 2009).
ASP functions with two major accessory proteins: attractin and mahogunin. Attractin is encoded by the Atrn gene and is a large single transmembrane protein (Gunn et al., 1999; Nagle et al., 1999) with an ectodomain that binds the amino terminus of ASP (He et al., 2001) and is absolutely required for ASP signaling in vivo. Although it is unknown whether attractin, ASP, and Mc1r form a complex, mice without a functional attractin protein are unresponsive to ASP signaling suggesting attractin is required for ASP function at Mc1r. Mahogunin is encoded by the Mgrn1 gene and is an intracellular protein that functions as an E3 ubiquitin ligase (Phan et al., 2002; He et al., 2003a,b).
Mutations in murine Atrn or Mgrn1 (formerly the mahogany and mahoganoid mutations, respectively) are associated with eumelanotic phenotypes similar to that seen in either a mutation causing loss of function Agouti mutations or a gain of function Mc1r mutation (Lane and Green, 1960). Mutations in Atrn or Mgrn1 have no effect on plasma α-MSH or ACTH levels, however, they do interfere with agouti signaling and prevent it from binding to MC1R effectively thereby resulting in a darkened coat color (Miller et al., 1997). The interaction between attractin, mahogunin, and ASP are unclear, however, a recent study suggests that attractin and mahogunin mediated ASIP signaling is cAMP independent, however, the cAMP dependent ASIP signaling requires neither attractin nor mahogunin (Hida et al., 2009).
Beta Defensin 3 (βD3)
The defensins are a group of antimicrobial peptides that link innate and adaptive immune responses. They are small cationic amphiphilic proteins composed of 30–40 amino acids (Boman, 1995; McCray and Bentley, 1997; De Lucca and Walsh, 1999; Schneider et al., 2005) (Kesting et al., 2010) that target gram positive and gram negative bacteria in addition to fungi (Garcia et al., 2001; Schneider et al., 2005; Arnett and Seveau, 2011) by binding to negatively charged membrane components (Schneider et al., 2005). Although there are two major classes of defensins, α and β (Schneider et al., 2005), only the β-defensins have been identified in the skin (Bensch et al., 1995; Weinberg et al., 1998; Huttner and Bevins, 1999; Schroder and Harder, 1999). Although βD1, βD2, and βD3 are all expressed in the skin, βD3 appears to be unique in its ability to bind the MC1R and influence melanocyte physiology (e.g., mammalian coat color). βD3 is synthesized by keratinocytes in the spinous and granular layers (Sawamura et al., 2005) and functions in a paracrine manner by binding to MC1R on interdigitating melanocytes (Harder et al., 2001; Garcia-Borron et al., 2005; Candille et al., 2007) via electrostatic interactions (Nix et al., 2013).
Clarence Cook Little was the first to demonstrate that the dominant inheritance of black coat color in dogs was independent of MC1R mutations and suggested that the color was due to an unusual allele of agouti (Little, 1957; Candille et al., 2007). However, Candille et al. (2007) identified the locus responsible for Little’s observation as CBD103 (β defensin 103), the canine homolog of βD3, rather than an agouti variant. Indeed, overexpression of β defensin 103 and its subsequent binding to MC1R inhibited interactions between ASIP and MC1R, preventing the dogs from developing a blonde coat (Candille et al., 2007). The ability of βD3 homologs to promote mammalian black coat color requires a functional MC1R protein, showing β defensin 103’s effects are mediated by interactions with MC1R rather than other melanocyte surface receptors (Nix et al., 2013).
Although regulation of βD3 following inflammatory stimuli is established, its regulation in keratinocytes following exposure to UV radiation is not well-characterized. βD3 expression is induced in keratinocytes in the setting of inflammation or wound formation (Kesting et al., 2010) and is mediated by inflammatory cytokines including TNFα, IL-7, and IL-1β (Harder et al., 2001). In contrast, neither TNFα nor IL-17 influence POMC or ASIP expression, suggesting βD3 may be independently regulated compared to the other melanocortins and that inflammatory-mediated pigment changes may be βD3-dependent (Wang et al., 2013). UVB radiation, which induces POMC gene expression both in vivo and in vitro, has variable effects on βD3 induction, depending upon the experimental model. In vivo exposure of adult skin to UVB radiation led to increases in βD3 gene and protein expression (Glaser et al., 2009), however, UV exposure of ex vivo neonatal skin explants did not induce βD3 expression suggesting that βD3 up-regulation following UVB exposure may require recruitment of additional cell types, potentially cytokine-producing immune cells, to the skin (Wolf Horrell and D’Orazio, 2014).
Multiple reports have demonstrated that βD3 binds MC1R. In MC1R-transfected human embryonic kidney cells (HEK293), βD3 functioned as a weak agonist and promoted cAMP accumulation and MAP kinase activation (Beaumont et al., 2012). However, in MC1Rwt primary melanocytes or in human melan-a melanocytes, βD3 acted as a neutral MC1R antagonist, exerting little influence on cellular cAMP levels independently but preventing MSH and ASIP binding to MC1R (Swope et al., 2012; Nix et al., 2013). Similarly, Candille et al. (2007) found evidence that the canine homolog CBD103 functioned as a neutral MC1R antagonist. Finally, we found that βD3 inhibited α-MSH-induced generation of pS435-ATR and enhancement of DNA repair (Jarrett et al., 2015). Thus most available data support the hypothesis that βD3 acts as a neutral MC1R antagonist. Importantly, regardless of experimental system, βD3 is unable prevent the induction of cAMP and downstream melanocyte differentiation pathways (e.g., pigment induction) by the direct adenylyl cyclase activating drug forskolin confirming that βD3 functions at the level of the MC1R rather than by inhibiting the adenylyl cyclase enzyme or other downstream MC1R signaling event (Swope et al., 2012).
Targeting the Melanocortin-MC1R Axis
The MC1R gene is a highly polymorphic genetic locus and inherited defects in MC1R function are common among fair-skinned, UV-sensitive and melanoma-prone persons. Indeed, there may be up to 6–8 million Americans harboring double allele polymorphisms and millions more being hemizygous for MC1R (Kennedy et al., 2001). Individuals with defective MC1R signaling are prone to melanoma and other UV induced skin cancers not only because their skin is under-melanized but also because they have a blunted melanocytic DNA repair response. Because MC1R-mediated UV protection and melanoma resistance is proportional to the robustness of the cAMP response downstream of MC1R signaling and a variety of pharmacologic strategies exist to impact cAMP, it might be possible to exploit MC1R signaling as a UV- and melanoma-preventive strategy. For example, topical application of either forskolin, a direct activator of adenylyl cyclase, or rolipram, a phosphodiesterase inhibitor, potently rescued eumelanin production in pheomelanotic Mc1r-defective C57BL/6 animals and protected the skin against UV damage (D’Orazio et al., 2006; Khaled et al., 2010). Similarly, topical forskolin promoted clearance of UV photoproducts in the skin (Jarrett et al., 2014). These proof-of-principle studies indicate that beneficial effects of MC1R signaling can be induced by pharmacologic manipulation cAMP levels in the skin. Topical application of agents that induce cAMP production or prevent its clearance, while effective, lacks melanocyte specificity and off-target effects must be considered before this approach can be deemed safe or appropriate for translational use. Melanocortin analogs under development offer a more targeted approach (Abdel-Malek et al., 2006), but their efficacy is dependent on expression of functional MC1R and therefore individuals with homozygous or compound heterozygous MC1R defects may not benefit from these agents. Although there is clearly a need for much more investigation into the mechanisms, feasibility and consequences of pharmacologic MC1R targeting, the melanocortin-MC1R signaling axis may prove to be a useful target for rational development of novel UV-resistance and melanoma-preventive strategies.
Therapeutic Implications and Perspectives
Taking into account the abundance of data linking MC1R function to the vigor of melanocytic UV physiologic responses, it is clear that the MSH-MC1R signaling axis represents a critical innate UV-protective mechanism in the skin. Whereas its importance was first appreciated based on its contribution to melanin biosynthesis, more recent studies have documented a direct link between cAMP signaling and melanocyte genomic stability. It is now well-established that melanoma ranks among the highest of human tumors with respect to somatic mutational load (Hodis et al., 2012; Lawrence et al., 2014) and that UV signature mutations account for the majority of melanoma-associated mutations (Shain et al., 2015). Epidermal melanocytes, the precursor cells that give rise to melanoma, are long-lived cells that because of their position in the skin, must cope with intermittent UV damage. In addition to overall UV dose and the pattern of UV injury (acute vs. chronic), it is likely that UV mutagenesis (and ultimately melanoma incidence) will be heavily influenced by the efficiency by which melanocytes repair nuclear UV photodamage. MC1R-defective individuals, because of their tendency to be undermelanized (which allows more UV to penetrate into the basal layer of the epidermis) and because they lack the DNA repair “boost” that an effective MSH-MC1R axis yields, should accumulate more UV mutagenesis over time and would therefore be at higher risk for melanoma as a result. Pharmacologic “rescue” of cAMP signaling would protect such individuals by enhancing pigment production and by promoting genomic stability. Intriguingly, MC1R function might also predict therapeutic response to immune-based anti-melanoma therapies. Though this has yet to be formally tested, it is possible that melanomas with higher somatic mutational burdens (as might occur in the MC1R-defective state) might respond better to immune checkpoint blockade because they would express more mutated proteins, any one of which might be recognized as a tumor-associated antigen for cytotoxic lymphocytes. Because of its central regulatory role for a host of melanocyte physiological responses, the MC1R signaling pathway is emerging as an ever-increasingly important pharmacologic target in preventing or treating melanoma.
Statements
Author contributions
EMW and MCB wrote the review article along with JAD who helped write and edit the manuscript and figures.
Funding
We are grateful for support from the National Cancer Institute (R01 CA131075), the Melanoma Research Alliance (MRA) and the Regina Drury Endowment for Pediatric Research as well as T32CA165990 which supported EMW.
Acknowledgments
We acknowledge Dr. Stuart Jarrett for insights and key experimental discoveries seminal to our understanding of the molecular links between the melanocortin signaling axis and melanocyte UV resistance and DNA repair. We are grateful for support from the National Cancer Institute (R01 CA131075), the Melanoma Research Alliance (MRA) and the Regina Drury Endowment for Pediatric Research as well as T32CA165990 which supported EMW.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Abdel-MalekZ.ScottM. C.SuzukiI.TadaA.ImS.LamoreuxL.et al (2000). The melanocortin-1 receptor is a key regulator of human cutaneous pigmentation.Pigment Cell Res.13(Suppl. 8), 156–162. 10.1034/j.1600-0749.13.s8.28.x
2
Abdel-MalekZ. A.KadekaroA. L.KavanaghR. J.TodorovicA.KoikovL. N.McNultyJ. C.et al (2006). Melanoma prevention strategy based on using tetrapeptide alpha-MSH analogs that protect human melanocytes from UV-induced DNA damage and cytotoxicity.FASEB J.201561–1563. 10.1096/fj.05-5655fje
3
Abdel-MalekZ. A.RuweA.Kavanagh-StarnerR.KadekaroA. L.SwopeV.Haskell-LuevanoC.et al (2009). alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes.Pigment Cell Melanoma Res.22635–644. 10.1111/j.1755-148X.2009.00598.x
4
Abdel-MalekZ. A.ScottM. C.FurumuraM.LamoreuxM. L.OllmannM.BarshG. S.et al (2001). The melanocortin 1 receptor is the principal mediator of the effects of agouti signaling protein on mammalian melanocytes.J. Cell Sci.1141019–1024.
5
Abdel-MalekZ. A.SwopeV. B.StarnerR. J.KoikovL.CassidyP.LeachmanS. (2014). Melanocortins and the melanocortin 1 receptor, moving translationally towards melanoma prevention.Arch. Biochem. Biophys.5634–12. 10.1016/j.abb.2014.07.002
6
AberdamE.BertolottoC.SviderskayaE. V.de ThillotV.HemesathT. J.FisherD. E.et al (1998). Involvement of microphthalmia in the inhibition of melanocyte lineage differentiation and of melanogenesis by agouti signal protein.J. Biol. Chem.27319560–19565. 10.1074/jbc.273.31.19560
7
AbrisquetaM.HerraizC.Perez OlivaA. B.Sanchez-LaordenB. L.OlivaresC.Jimenez-CervantesC.et al (2013). Differential and competitive regulation of human melanocortin 1 receptor signaling by beta-arrestin isoforms.J. Cell Sci.1263724–3737. 10.1242/jcs.128322
8
AncansJ.TobinD. J.HoogduijnM. J.SmitN. P.WakamatsuK.ThodyA. J. (2001). Melanosomal pH controls rate of melanogenesis, eumelanin/phaeomelanin ratio and melanosome maturation in melanocytes and melanoma cells.Exp. Cell Res.26826–35. 10.1006/excr.2001.5251
9
ArnettE.SeveauS. (2011). The multifaceted activities of mammalian defensins.Curr. Pharm. Des.174254–4269. 10.2174/138161211798999348
10
AttramadalH.ArrizaJ. L.AokiC.DawsonT. M.CodinaJ.KwatraM. M.et al (1992). Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family.J. Biol. Chem.26717882–17890.
11
BaigA. H.SwordsF. M.NoonL. A.KingP. J.HunyadyL.ClarkA. J. (2001). Desensitization of the Y1 cell adrenocorticotropin receptor: evidence for a restricted heterologous mechanism implying a role for receptor-effector complexes.J. Biol. Chem.27644792–44797. 10.1074/jbc.M108572200
12
BaxterL. L.PavanW. J. (2013). The etiology and molecular genetics of human pigmentation disorders.Wiley Interdiscip. Rev. Dev. Biol.2379–392. 10.1002/wdev.72
13
BeaumontK. A.ShekarS. N.NewtonR. A.JamesM. R.StowJ. L.DuffyD. L.et al (2007). Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles.Hum. Mol. Genet.162249–2260. 10.1093/hmg/ddm177
14
BeaumontK. A.SmitD. J.LiuY. Y.ChaiE.PatelM. P.MillhauserG. L.et al (2012). Melanocortin-1 receptor-mediated signalling pathways activated by NDP-MSH and HBD3 ligands.Pigment Cell Melanoma Res.25370–374. 10.1111/j.1755-148X.2012.00990.x
15
BenjannetS.RondeauN.DayR.ChretienM.SeidahN. G. (1991). PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues.Proc. Natl. Acad. Sci. U.S.A.883564–3568. 10.1073/pnas.88.9.3564
16
BennettD. C.LamoreuxM. L. (2003). The color loci of mice–a genetic century.Pigment Cell Res.16333–344. 10.1034/j.1600-0749.2003.00067.x
17
BenovicJ. L.PikeL. J.CerioneR. A.StaniszewskiC.YoshimasaT.CodinaJ.et al (1985). Phosphorylation of the mammalian beta-adrenergic receptor by cyclic AMP-dependent protein kinase. Regulation of the rate of receptor phosphorylation and dephosphorylation by agonist occupancy and effects on coupling of the receptor to the stimulatory guanine nucleotide regulatory protein.J. Biol. Chem.2607094–7101.
18
BenschK. W.RaidaM.MagertH. J.Schulz-KnappeP.ForssmannW. G. (1995). hBD-1: a novel beta-defensin from human plasma.FEBS Lett.368331–335. 10.1016/0014-5793(95)00687-5
19
BiebermannH.KrudeH.ElsnerA.ChubanovV.GudermannT.GrutersA. (2003). Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization.Diabetes Metab. Res. Rev522984–2988.
20
BlanchardS. G.HarrisC. O.IttoopO. R.NicholsJ. S.ParksD. J.TruesdaleA. T.et al (1995). Agouti antagonism of melanocortin binding and action in the B16F10 murine melanoma cell line.Biochemistry3410406–10411. 10.1021/bi00033a012
21
BomanH. G. (1995). Peptide antibiotics and their role in innate immunity.Annu. Rev. Immunol.1361–92. 10.1146/annurev.iy.13.040195.000425
22
BoxN. F.WyethJ. R.O’GormanL. E.MartinN. G.SturmR. A. (1997). Characterization of melanocyte stimulating hormone receptor variant alleles in twins with red hair.Hum. Mol. Genet.61891–1897. 10.1093/hmg/6.11.1891
23
BreitwieserG. E. (2004). G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling.Circ. Res.9417–27. 10.1161/01.RES.0000110420.68526.19
24
BultmanS. J.MichaudE. J.WoychikR. P. (1992). Molecular characterization of the mouse agouti locus.Cell711195–1204. 10.1016/S0092-8674(05)80067-4
25
BurchillS. A.ThodyA. J. (1986). Melanocyte-stimulating hormone and the regulation of tyrosinase activity in hair follicular melanocytes of the mouse.J. Endocrinol.111225–232. 10.1677/joe.0.1110225
26
CandilleS. I.KaelinC. B.CattanachB. M.YuB.ThompsonD. A.NixM. A.et al (2007). A -defensin mutation causes black coat color in domestic dogs.Science3181418–1423. 10.1126/science.1147880
27
ChakrabortyA. K.FunasakaY.SlominskiA.ErmakG.HwangJ.PawelekJ. M.et al (1996). Production and release of proopiomelanocortin (POMC) derived peptides by human melanocytes and keratinocytes in culture: regulation by ultraviolet B.Biochim. Biophys. Acta1313130–138. 10.1016/0167-4889(96)00063-8
28
ChalmersD. T.BehanD. P. (2002). The use of constitutively active GPCRs in drug discovery and functional genomics.Nat. Rev. Drug Discov.1599–608. 10.1038/nrd872
29
ChhajlaniV.WikbergJ. E. (1992). Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA.FEBS Lett.309417–420. 10.1016/0014-5793(92)80820-7
30
ChhajlaniV.XuX.BlauwJ.SudarshiS. (1996). Identification of ligand binding residues in extracellular loops of the melanocortin 1 receptor.Biochem. Biophys. Res. Commun.219521–525. 10.1006/bbrc.1996.0266
31
CleaverJ. E. (2005). Cancer in xeroderma pigmentosum and related disorders of DNA repair.Nat. Rev. Cancer5564–573. 10.1038/nrc1652
32
CuiR.WidlundH. R.FeigeE.LinJ. Y.WilenskyD. L.IgrasV. E.et al (2007). Central role of p53 in the suntan response and pathologic hyperpigmentation.Cell128853–864. 10.1016/j.cell.2006.12.045
33
De LucaM.SiegristW.BondanzaS.MathorM.CanceddaR.EberleA. N. (1993). Alpha melanocyte stimulating hormone (alpha MSH) stimulates normal human melanocyte growth by binding to high-affinity receptors.J. Cell Sci.105(Pt 4), 1079–1084.
34
De LuccaA. J.WalshT. J. (1999). Antifungal peptides: novel therapeutic compounds against emerging pathogens.Antimicrob. Agents Chemother.431–11.
35
DesarnaudF.LabbeO.EggerickxD.VassartG.ParmentierM. (1994). Molecular cloning, functional expression and pharmacological characterization of a mouse melanocortin receptor gene.Biochem. J.299(Pt 2), 367–373. 10.1042/bj2990367
36
DonahueB. A.YinS.TaylorJ. S.ReinesD.HanawaltP. C. (1994). Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template.Proc. Natl. Acad. Sci. U.S.A.918502–8506. 10.1073/pnas.91.18.8502
37
DonatienP. D.HuntG.PieronC.LunecJ.TaiebA.ThodyA. J. (1992). The expression of functional MSH receptors on cultured human melanocytes.Arch. Dermatol. Res.284424–426. 10.1007/BF00372074
38
D’OrazioJ. A.NobuhisaT.CuiR.AryaM.SpryM.WakamatsuK.et al (2006). Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning.Nature443340–344. 10.1038/nature05098
39
DoresR. M. (2009). Adrenocorticotropic hormone, melanocyte-stimulating hormone, and the melanocortin receptors: revisiting the work of Robert Schwyzer: a thirty-year retrospective.Ann. N. Y. Acad. Sci.116393–100. 10.1111/j.1749-6632.2009.04434.x
40
DorsamR. T.GutkindJ. S. (2007). G-protein-coupled receptors and cancer.Nat. Rev. Cancer779–94. 10.1038/nrc2069
41
DuhlD. M.StevensM. E.VrielingH.SaxonP. J.MillerM. W.EpsteinC. J.et al (1994a). Pleiotropic effects of the mouse lethal yellow (Ay) mutation explained by deletion of a maternally expressed gene and the simultaneous production of agouti fusion RNAs.Development1201695–1708.
42
DuhlD. M.VrielingH.MillerK. A.WolffG. L.BarshG. S. (1994b). Neomorphic agouti mutations in obese yellow mice.Nat. Genet.859–65. 10.1038/ng0994-59
43
FellG. L.RobinsonK. C.MaoJ.WoolfC. J.FisherD. E. (2014). Skin beta-endorphin mediates addiction to UV light.Cell1571527–1534. 10.1016/j.cell.2014.04.032
44
FrandbergP. A.DoufexisM.KapasS.ChhajlaniV. (2001). Cysteine residues are involved in structure and function of melanocortin 1 receptor: substitution of a cysteine residue in transmembrane segment two converts an agonist to antagonist.Biochem. Biophys. Res. Commun.281851–857. 10.1006/bbrc.2001.4429
45
GantzI.MiwaH.KondaY.ShimotoY.TashiroT.WatsonS. J.et al (1993). Molecular cloning, expression, and gene localization of a fourth melanocortin receptor.J. Biol. Chem.26815174–15179.
46
GantzI.ShimotoY.KondaY.MiwaH.DickinsonC. J.YamadaT. (1994a). Molecular cloning, expression, and characterization of a fifth melanocortin receptor.Biochem. Biophys. Res. Commun.2001214–1220. 10.1006/bbrc.1994.1580
47
GantzI.YamadaT.TashiroT.KondaY.ShimotoY.MiwaH.et al (1994b). Mapping of the gene encoding the melanocortin-1 (alpha-melanocyte stimulating hormone) receptor (MC1R) to human chromosome 16q24.3 by Fluorescence in situ hybridization.Genomics19394–395. 10.1006/geno.1994.1080
48
GarciaJ. R.JaumannF.SchulzS.KrauseA.Rodriguez-JimenezJ.ForssmannU.et al (2001). Identification of a novel, multifunctional beta-defensin (human beta-defensin 3) with specific antimicrobial activity. Its interaction with plasma membranes of Xenopus oocytes and the induction of macrophage chemoattraction.Cell Tissue Res.306257–264. 10.1007/s004410100433
49
Garcia-BorronJ. C.Sanchez-LaordenB. L.Jimenez-CervantesC. (2005). Melanocortin-1 receptor structure and functional regulation.Pigment Cell Res.18393–410. 10.1111/j.1600-0749.2005.00278.x
50
GerardM.FischerL.MoncollinV.ChipouletJ. M.ChambonP.EglyJ. M. (1991). Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2.J. Biol. Chem.26620940–20945.
51
GetherU. (2000). Uncovering molecular mechanisms involved in activation of G protein-coupled receptors.Endocr. Rev.2190–113. 10.1210/edrv.21.1.0390
52
GhanemG. E.ComunaleG.LibertA.Vercammen-GrandjeanA.LejeuneF. J. (1988). Evidence for alpha-melanocyte-stimulating hormone (alpha-MSH) receptors on human malignant melanoma cells.Int. J. Cancer41248–255. 10.1002/ijc.2910410216
53
GlaserR.NavidF.SchullerW.JantschitschC.HarderJ.SchroderJ. M.et al (2009). UV-B radiation induces the expression of antimicrobial peptides in human keratinocytes in vitro and in vivo.J. Allergy Clin. Immunol.1231117–1123. 10.1016/j.jaci.2009.01.043
54
GrahamA.WakamatsuK.HuntG.ItoS.ThodyA. J. (1997). Agouti protein inhibits the production of eumelanin and phaeomelanin in the presence and absence of alpha-melanocyte stimulating hormone.Pigment Cell Res.10298–303. 10.1111/j.1600-0749.1997.tb00689.x
55
GunnT. M.MillerK. A.HeL.HymanR. W.DavisR. W.AzaraniA.et al (1999). The mouse mahogany locus encodes a transmembrane form of human attractin.Nature398152–156. 10.1038/18217
56
HarderJ.BartelsJ.ChristophersE.SchroderJ. M. (2001). Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic.J. Biol. Chem.2765707–5713. 10.1074/jbc.M008557200
57
HausdorffW. P.CaronM. G.LefkowitzR. J. (1990). Turning off the signal: desensitization of beta-adrenergic receptor function.FASEB J.42881–2889.
58
HauserJ. E.KadekaroA. L.KavanaghR. J.WakamatsuK.TerzievaS.SchwembergerS.et al (2006). Melanin content and MC1R function independently affect UVR-induced DNA damage in cultured human melanocytes.Pigment Cell Res.19303–314. 10.1111/j.1600-0749.2006.00315.x
59
HeL.EldridgeA. G.JacksonP. K.GunnT. M.BarshG. S. (2003a). Accessory proteins for melanocortin signaling: attractin and mahogunin.Ann. N. Y. Acad. Sci.994288–298. 10.1111/j.1749-6632.2003.tb03192.x
60
HeL.GunnT. M.BouleyD. M.LuX. Y.WatsonS. J.SchlossmanS. F.et al (2001). A biochemical function for attractin in agouti-induced pigmentation and obesity.Nat. Genet.2740–47. 10.1038/83741
61
HeL.LuX. Y.JollyA. F.EldridgeA. G.WatsonS. J.JacksonP. K.et al (2003b). Spongiform degeneration in mahoganoid mutant mice.Science299710–712. 10.1126/science.1079694
62
HidaT.WakamatsuK.SviderskayaE. V.DonkinA. J.MontoliuL.Lynn LamoreuxM.et al (2009). Agouti protein, mahogunin, and attractin in pheomelanogenesis and melanoblast-like alteration of melanocytes: a cAMP-independent pathway.Pigment Cell Melanoma Res.22623–634. 10.1111/j.1755-148X.2009.00582.x
63
HodisE.WatsonI. R.KryukovG. V.AroldS. T.ImielinskiM.TheurillatJ. P.et al (2012). A landscape of driver mutations in melanoma.Cell150251–263. 10.1016/j.cell.2012.06.024
64
HolstB.SchwartzT. W. (2003). Molecular mechanism of agonism and inverse agonism in the melanocortin receptors: Zn(2+) as a structural and functional probe.Ann. N. Y. Acad. Sci.9941–11. 10.1111/j.1749-6632.2003.tb03156.x
65
HoogduijnM. J.CemeliE.RossK.AndersonD.ThodyA. J.WoodJ. M. (2004). Melanin protects melanocytes and keratinocytes against H2O2-induced DNA strand breaks through its ability to bind Ca2+.Exp. Cell Res.29460–67. 10.1016/j.yexcr.2003.11.007
66
HoutsmullerA. B.RademakersS.NiggA. L.HoogstratenD.HoeijmakersJ. H.VermeulenW. (1999). Action of DNA repair endonuclease ERCC1/XPF in living cells.Science284958–961. 10.1126/science.284.5416.958
67
HuntG.KyneS.WakamatsuK.ItoS.ThodyA. J. (1995). Nle4DPhe7 alpha-melanocyte-stimulating hormone increases the eumelanin:phaeomelanin ratio in cultured human melanocytes.J. Invest. Dermatol.10483–85. 10.1111/1523-1747.ep12613565
68
HuntG.ThodyA. J. (1995). Agouti protein can act independently of melanocyte-stimulating hormone to inhibit melanogenesis.J. Endocrinol.147R1–R4. 10.1677/joe.0.147R001
69
HuttnerK. M.BevinsC. L. (1999). Antimicrobial peptides as mediators of epithelial host defense.Pediatr. Res.45785–794. 10.1203/00006450-199906000-00001
70
ItoS. (2003). The IFPCS presidential lecture: a chemist’s view of melanogenesis.Pigment Cell Res.16230–236. 10.1034/j.1600-0749.2003.00037.x
71
JacksonI. J.BuddP. S.KeighrenM.McKieL. (2007). Humanized MC1R transgenic mice reveal human specific receptor function.Hum. Mol. Genet.162341–2348. 10.1093/hmg/ddm191
72
JagirdarK.YinK.HarrisonM.LimW.MuscatG. E.SturmR. A.et al (2013). The NR4A2 nuclear receptor is recruited to novel nuclear foci in response to UV irradiation and participates in nucleotide excision repair.PLoS ONE8:e78075. 10.1371/journal.pone.0078075
73
JarrettS. G.HorrellE. M.ChristianP. A.VanoverJ. C.BoulangerM. C.ZouY.et al (2014). PKA-Mediated phosphorylation of ATR promotes recruitment of XPA to UV-Induced DNA damage.Mol. Cell54999–1011. 10.1016/j.molcel.2014.05.030
74
JarrettS. G.Wolf HorrellE. M.BoulangerM. C.D’OrazioJ. A. (2015). Defining the contribution of MC1R physiological ligands to ATR phosphorylation at Ser435, a predictor of DNA repair in melanocytes.J. Invest. Dermatol.1353086–3095. 10.1038/jid.2015.280
75
KadekaroA. L.ChenJ.YangJ.ChenS.JamesonJ.SwopeV. B.et al (2012). Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes.Mol. Cancer Res.10778–786. 10.1158/1541-7786.MCR-11-0436
76
KadekaroA. L.KantoH.KavanaghR.Abdel-MalekZ. (2003). Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival.Ann. N. Y. Acad. Sci.994359–365. 10.1111/j.1749-6632.2003.tb03200.x
77
KadekaroA. L.KavanaghR.KantoH.TerzievaS.HauserJ.KobayashiN.et al (2005). alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes.Cancer Res.654292–4299.
78
KadekaroA. L.LeachmanS.KavanaghR. J.SwopeV.CassidyP.SuppD.et al (2010). Melanocortin 1 receptor genotype: an important determinant of the damage response of melanocytes to ultraviolet radiation.FASEB J.243850–3860. 10.1096/fj.10-158485
79
KaidbeyK. H.GroveK. H.KligmanA. M. (1979). The influence of longwave ultraviolet radiation on sunburn cell production by UVB.J. Invest. Dermatol.73743–745. 10.1111/1523-1747.ep12514324
80
KamiuchiS.SaijoM.CitterioE.de JagerM.HoeijmakersJ. H.TanakaK. (2002). Translocation of cockayne syndrome group A protein to the nuclear matrix: possible relevance to transcription-coupled DNA repair.Proc. Natl. Acad. Sci. U.S.A.99201–206. 10.1073/pnas.012473199
81
KennedyC.ter HuurneJ.BerkhoutM.GruisN.BastiaensM.BergmanW.et al (2001). Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color.J. Invest. Dermatol.117294–300. 10.1046/j.0022-202x.2001.01421.x
82
KestingM. R.StoeckelhuberM.HolzleF.MuckeT.NeumannK.WoermannK.et al (2010). Expression of antimicrobial peptides in cutaneous infections after skin surgery.Br. J. Dermatol.163121–127. 10.1111/j.1365-2133.2010.09781.x
83
KhaledM.LevyC.FisherD. E. (2010). Control of melanocyte differentiation by a MITF-PDE4D3 homeostatic circuit.Genes Dev.242276–2281. 10.1101/gad.1937710
84
KokotA.MetzeD.MouchetN.GalibertM. D.SchillerM.LugerT. A.et al (2009). Alpha-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin.Endocrinology1503197–3206. 10.1210/en.2008-1315
85
KwonH. Y.BultmanS. J.LofflerC.ChenW. J.FurdonP. J.PowellJ. G.et al (1994). Molecular structure and chromosomal mapping of the human homolog of the agouti gene.Proc. Natl. Acad. Sci. U.S.A.919760–9764. 10.1073/pnas.91.21.9760
86
LandiM. T.KanetskyP. A.TsangS.GoldB.MunroeD.RebbeckT.et al (2005). MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population.J. Natl. Cancer Inst.97998–1007. 10.1093/jnci/dji176
87
LaneP. W.GreenM. C. (1960). Mahogany, a recessive color mutation in linkage group V of the mouse.J. Hered.51228–230.
88
LawrenceM. S.StojanovP.MermelC. H.RobinsonJ. T.GarrawayL. A.GolubT. R.et al (2014). Discovery and saturation analysis of cancer genes across 21 tumour types.Nature505495–501. 10.1038/nature12912
89
Le PapeE.PasseronT.GiubellinoA.ValenciaJ. C.WolberR.HearingV. J. (2009). Microarray analysis sheds light on the dedifferentiating role of agouti signal protein in murine melanocytes via the Mc1r.Proc. Natl. Acad. Sci. U.S.A.1061802–1807. 10.1073/pnas.0806753106
90
LernerA. B.LeeT. H. (1955). Isolation of homogenous melanocyte stimulating hormone from hog pituitary gland.J. Am. Chem. Soc.771066–1067. 10.1021/ja01609a098
91
LevyC.KhaledM.FisherD. E. (2006). MITF: master regulator of melanocyte development and melanoma oncogene.Trends Mol Med12406–414. 10.1016/j.molmed.2006.07.008
92
LittleC. (1957). The Inheritance of Coat Color in Dogs.Ithacy, NY: Comstock.
93
LovettM.ChengZ. Y.LamelaE. M.YokoiT.EpsteinC. J. (1987). Molecular markers for the agouti coat color locus of the mouse.Genetics115747–754.
94
LuD.WillardD.PatelI. R.KadwellS.OvertonL.KostT.et al (1994). Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor.Nature371799–802. 10.1038/371799a0
95
LuttrellL. M.LefkowitzR. J. (2002). The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals.J. Cell Sci.115455–465.
96
MandrikaI.PetrovskaR.WikbergJ. (2005). Melanocortin receptors form constitutive homo- and heterodimers.Biochem. Biophys. Res. Commun.326349–354. 10.1016/j.bbrc.2004.11.036
97
MasJ. S.GerritsenI.HahmannC.Jimenez-CervantesC.Garcia-BorronJ. C. (2003). Rate limiting factors in melanocortin 1 receptor signalling through the cAMP pathway.Pigment Cell Res.16540–547. 10.1034/j.1600-0749.2003.00073.x
98
McCrayP. B.Jr.BentleyL. (1997). Human airway epithelia express a beta-defensin.Am. J. Respir. Cell Mol. Biol.16343–349. 10.1165/ajrcmb.16.3.9070620
99
McNultyJ. C.JacksonP. J.ThompsonD. A.ChaiB.GantzI.BarshG. S.et al (2005). Structures of the agouti signaling protein.J. Mol. Biol.3461059–1070. 10.1016/j.jmb.2004.12.030
100
MellonI.SpivakG.HanawaltP. C. (1987). Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene.Cell51241–249. 10.1016/0092-8674(87)90151-6
101
MichaudE. J.BultmanS. J.KlebigM. L.van VugtM. J.StubbsL. J.RussellL. B.et al (1994). A molecular model for the genetic and phenotypic characteristics of the mouse lethal yellow (Ay) mutation.Proc. Natl. Acad. Sci. U.S.A.912562–2566. 10.1073/pnas.91.7.2562
102
MillarS. E.MillerM. W.StevensM. E.BarshG. S. (1995). Expression and transgenic studies of the mouse agouti gene provide insight into the mechanisms by which mammalian coat color patterns are generated.Development1213223–3232.
103
MillerK. A.GunnT. M.CarrasquilloM. M.LamoreuxM. L.GalbraithD. B.BarshG. S. (1997). Genetic studies of the mouse mutations mahogany and mahoganoid.Genetics1461407–1415.
104
MillerM. W.DuhlD. M.VrielingH.CordesS. P.OllmannM. M.WinkesB. M.et al (1993). Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation.Genes Dev.7454–467. 10.1101/gad.7.3.454
105
MilliganG. (2003). Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective.Mol. Pharmacol.641271–1276. 10.1124/mol.64.6.1271
106
MillingtonG. W. (2006). Proopiomelanocortin (POMC): the cutaneous roles of its melanocortin products and receptors.Clin. Exp. Dermatol.31407–412. 10.1111/j.1365-2230.2006.02128.x
107
MitraD.LuoX.MorganA.WangJ.HoangM. P.LoJ.et al (2012). An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background.Nature491449–453. 10.1038/nature11624
108
Montero-MelendezT. (2015). ACTH: the forgotten therapy.Semin. Immunol.27216–226. 10.1016/j.smim.2015.02.003
109
MountjoyK. G.RobbinsL. S.MortrudM. T.ConeR. D. (1992). The cloning of a family of genes that encode the melanocortin receptors.Science2571248–1251. 10.1126/science.1325670
110
MuD.SancarA. (1997). Model for XPC-independent transcription-coupled repair of pyrimidine dimers in humans.J. Biol. Chem.2727570–7573. 10.1074/jbc.272.12.7570
111
NagleD. L.McGrailS. H.VitaleJ.WoolfE. A.DussaultB. J.Jr.DiRoccoL.et al (1999). The mahogany protein is a receptor involved in suppression of obesity.Nature398148–152. 10.1038/18210
112
NevesS. R.RamP. T.IyengarR. (2002). G protein pathways.Science2961636–1639. 10.1126/science.1071550
113
NewtonJ. M.WilkieA. L.HeL.JordanS. A.MetallinosD. L.HolmesN. G.et al (2000). Melanocortin 1 receptor variation in the domestic dog.Mamm. Genome1124–30. 10.1007/s003350010005
114
NijenhuisW. A.OosteromJ.AdanR. A. (2001). AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor.Mol. Endocrinol.15164–171. 10.1210/mend.15.1.0578
115
NixM. A.KaelinC. B.TaT.WeisA.MortonG. J.BarshG. S.et al (2013). Molecular and functional analysis of human beta-defensin 3 action at melanocortin receptors.Chem. Biol.20784–795. 10.1016/j.chembiol.2013.04.015
116
OllmannM. M.BarshG. S. (1999). Down-regulation of melanocortin receptor signaling mediated by the amino terminus of Agouti protein in Xenopus melanophores.J. Biol. Chem.27415837–15846. 10.1074/jbc.274.22.15837
117
OllmannM. M.LamoreuxM. L.WilsonB. D.BarshG. S. (1998). Interaction of agouti protein with the melanocortin 1 receptor in vitro and in vivo.Genes Dev.12316–330. 10.1101/gad.12.3.316
118
PalmerJ. S.DuffyD. L.BoxN. F.AitkenJ. F.O’GormanL. E.GreenA. C.et al (2000). Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype?Am. J. Hum. Genet.66176–186. 10.1086/302711
119
ParkC. J.ChoiB. S. (2006). The protein shuffle. Sequential interactions among components of the human nucleotide excision repair pathway.FEBS J.2731600–1608. 10.1111/j.1742-4658.2006.05189.x
120
PhanL. K.LinF.LeDucC. A.ChungW. K.LeibelR. L. (2002). The mouse mahoganoid coat color mutation disrupts a novel C3HC4 RING domain protein.J. Clin. Invest.1101449–1459. 10.1172/JCI16131
121
PitcherJ. A.FreedmanN. J.LefkowitzR. J. (1998). G protein-coupled receptor kinases.Annu. Rev. Biochem.67653–692. 10.1146/annurev.biochem.67.1.653
122
QanbarR.BouvierM. (2003). Role of palmitoylation/depalmitoylation reactions in G-protein-coupled receptor function.Pharmacol. Ther.971–33. 10.1016/S0163-7258(02)00300-5
123
RobbinsL. S.NadeauJ. H.JohnsonK. R.KellyM. A.Roselli-RehfussL.BaackE.et al (1993). Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function.Cell72827–834. 10.1016/0092-8674(93)90572-8
124
RobertsD. W.NewtonR. A.BeaumontK. A.Helen LeonardJ.SturmR. A. (2006). Quantitative analysis of MC1R gene expression in human skin cell cultures.Pigment Cell Res.1976–89. 10.1111/j.1600-0749.2005.00286.x
125
SakaiC.OllmannM.KobayashiT.Abdel-MalekZ.MullerJ.VieiraW. D.et al (1997). Modulation of murine melanocyte function in vitro by agouti signal protein.EMBO J.163544–3552. 10.1093/emboj/16.12.3544
126
Sanchez-LaordenB. L.Jimenez-CervantesC.Garcia-BorronJ. C. (2007). Regulation of human melanocortin 1 receptor signaling and trafficking by Thr-308 and Ser-316 and its alteration in variant alleles associated with red hair and skin cancer.J. Biol. Chem.2823241–3251. 10.1074/jbc.M606865200
127
Sanchez-LaordenB. L.Sanchez-MasJ.Martinez-AlonsoE.Martinez-MenarguezJ. A.Garcia-BorronJ. C.Jimenez-CervantesC. (2006a). Dimerization of the human melanocortin 1 receptor: functional consequences and dominant-negative effects.J. Invest. Dermatol.126172–181. 10.1038/sj.jid.5700036
128
Sanchez-LaordenB. L.Sanchez-MasJ.TurpinM. C.Garcia-BorronJ. C.Jimenez-CervantesC. (2006b). Variant amino acids in different domains of the human melanocortin 1 receptor impair cell surface expression.Cell Mol. Biol. (Noisy-le-grand)5239–46.
129
Sanchez-MasJ.GuilloL. A.ZannaP.Jimenez-CervantesC.Garcia-BorronJ. C. (2005a). Role of G protein-coupled receptor kinases in the homologous desensitization of the human and mouse melanocortin 1 receptors.Mol. Endocrinol.191035–1048. 10.1210/me.2004-0227
130
Sanchez-MasJ.HahmannC.GerritsenI.Garcia-BorronJ. C.Jimenez-CervantesC. (2004). Agonist-independent, high constitutive activity of the human melanocortin 1 receptor.Pigment Cell Res.17386–395. 10.1111/j.1600-0749.2004.00160.x
131
Sanchez-MasJ.Sanchez-LaordenB. L.GuilloL. A.Jimenez-CervantesC.Garcia-BorronJ. C. (2005b). The melanocortin-1 receptor carboxyl terminal pentapeptide is essential for MC1R function and expression on the cell surface.Peptides261848–1857. 10.1016/j.peptides.2004.11.030
132
SawamuraD.GotoM.ShibakiA.AkiyamaM.McMillanJ. R.AbikoY.et al (2005). Beta defensin-3 engineered epidermis shows highly protective effect for bacterial infection.Gene Ther.12857–861. 10.1038/sj.gt.3302472
133
ScharerO. D. (2013). Nucleotide excision repair in eukaryotes.Cold Spring Harb. Perspect. Biol.5:a012609. 10.1101/cshperspect.a012609
134
SchauerE.TrautingerF.KockA.SchwarzA.BhardwajR.SimonM.et al (1994). Proopiomelanocortin-derived peptides are synthesized and released by human keratinocytes.J. Clin. Invest.932258–2262. 10.1172/JCI117224
135
SchererD.KumarR. (2010). Genetics of pigmentation in skin cancer–a review.Mutat. Res.705141–153. 10.1016/j.mrrev.2010.06.002
136
SchiothH. B.PhillipsS. R.RudzishR.Birch-MachinM. A.WikbergJ. E.ReesJ. L. (1999). Loss of function mutations of the human melanocortin 1 receptor are common and are associated with red hair.Biochem. Biophys. Res. Commun.260488–491. 10.1006/bbrc.1999.0935
137
SchneiderJ. J.UnholzerA.SchallerM.Schafer-KortingM.KortingH. C. (2005). Human defensins.J. Mol. Med. (Berl.)83587–595. 10.1007/s00109-005-0657-1
138
SchroderJ. M.HarderJ. (1999). Human beta-defensin-2.Int. J. Biochem. Cell Biol.31645–651. 10.1016/S1357-2725(99)00013-8
139
SchuleinR.HermosillaR.OkscheA.DeheM.WiesnerB.KrauseG.et al (1998). A dileucine sequence and an upstream glutamate residue in the intracellular carboxyl terminus of the vasopressin V2 receptor are essential for cell surface transport in COS.M6 cells.Mol. Pharmacol.54525–535.
140
SearleA. G. (1968). An extension series in the mouse.J. Hered.59341–342.
141
ShahP.HeY. Y. (2015). Molecular regulation of UV-induced DNA repair.Photochem. Photobiol.91254–264. 10.1111/php.12406
142
ShainA. H.YehI.KovalyshynI.SriharanA.TalevichE.GagnonA.et al (2015). The genetic evolution of melanoma from precursor lesions.N. Engl. J. Med.3731926–1936. 10.1056/NEJMoa1502583
143
ShinyamaH.MasuzakiH.FangH.FlierJ. S. (2003). Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization.Endocrinology1441301–1314. 10.1210/en.2002-220931
144
ShivjiM. K.PodustV. N.HubscherU.WoodR. D. (1995). Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA.Biochemistry345011–5017. 10.1021/bi00015a012
145
SiegristW.SolcaF.StutzS.GiuffreL.CarrelS.GirardJ.et al (1989). Characterization of receptors for alpha-melanocyte-stimulating hormone on human melanoma cells.Cancer Res.496352–6358.
146
SiegristW.StutzS.EberleA. N. (1994). Homologous and heterologous regulation of alpha-melanocyte-stimulating hormone receptors in human and mouse melanoma cell lines.Cancer Res.542604–2610.
147
SiegristW.WillardD. H.WilkisonW. O.EberleA. N. (1996). Agouti protein inhibits growth of B16 melanoma cells in vitro by acting through melanocortin receptors.Biochem. Biophys. Res. Commun.218171–175. 10.1006/bbrc.1996.0030
148
SmithA. G.LukN.NewtonR. A.RobertsD. W.SturmR. A.MuscatG. E. (2008). Melanocortin-1 receptor signaling markedly induces the expression of the NR4A nuclear receptor subgroup in melanocytic cells.J. Biol. Chem.28312564–12570. 10.1074/jbc.M800480200
149
SmithP. E. (1930). Hypophysectomy and a replacement therapy in the rat.Am. J. Anat.45205–273. 10.1002/aja.1000450203
150
SmithR.HealyE.SiddiquiS.FlanaganN.SteijlenP. M.RosdahlI.et al (1998). Melanocortin 1 receptor variants in an Irish population.J. Invest. Dermatol.111119–122. 10.1046/j.1523-1747.1998.00252.x
151
SolcaF.SiegristW.DrozdzR.GirardJ.EberleA. N. (1989). The receptor for alpha-melanotropin of mouse and human melanoma cells. Application of a potent alpha-melanotropin photoaffinity label.J. Biol. Chem.26414277–14281.
152
SongX.MosbyN.YangJ.XuA.Abdel-MalekZ.KadekaroA. L. (2009). alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes.Pigment Cell Melanoma Res.22809–818. 10.1111/j.1755-148X.2009.00615.x
153
StraderC. D.FongT. M.TotaM. R.UnderwoodD.DixonR. A. (1994). Structure and function of G protein-coupled receptors.Annu. Rev. Biochem.63101–132. 10.1146/annurev.bi.63.070194.000533
154
SugasawaK.NgJ. M.MasutaniC.IwaiS.van der SpekP. J.EkerA. P.et al (1998). Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair.Mol. Cell.2223–232. 10.1016/S1097-2765(00)80132-X
155
SugasawaK.OkamotoT.ShimizuY.MasutaniC.IwaiS.HanaokaF. (2001). A multistep damage recognition mechanism for global genomic nucleotide excision repair.Genes Dev.15507–521. 10.1101/gad.866301
156
SuzukiI.TadaA.OllmannM. M.BarshG. S.ImS.LamoreuxM. L.et al (1997). Agouti signaling protein inhibits melanogenesis and the response of human melanocytes to alpha-melanotropin.J. Invest. Dermatol.108838–842. 10.1111/1523-1747.ep12292572
157
SwopeV.AlexanderC.StarnerR.SchwembergerS.BabcockG.Abdel-MalekZ. A. (2014). Significance of the melanocortin 1 receptor in the DNA damage response of human melanocytes to ultraviolet radiation.Pigment Cell Melanoma Res.27601–610. 10.1111/pcmr.12252
158
SwopeV. B.JamesonJ. A.McFarlandK. L.SuppD. M.MillerW. E.McGrawD. W.et al (2012). Defining MC1R regulation in human melanocytes by its agonist alpha-melanocortin and antagonists agouti signaling protein and beta-defensin 3.J. Invest. Dermatol.1322255–2262. 10.1038/jid.2012.135
159
TakeuchiT.KobunaiT.YamamotoH. (1989). Genetic control of signal transduction in mouse melanocytes.J. Invest. Dermatol.92239S–242S. 10.1111/1523-1747.ep13075730
160
TamateH. B.TakeuchiT. (1984). Action of the e locus of mice in the response of phaeomelanic hair follicles to alpha-melanocyte-stimulating hormone in vitro.Science2241241–1242. 10.1126/science.6328651
161
TanC. P.McKeeK. K.WeinbergD. H.MacNeilT.PalyhaO. C.FeighnerS. D.et al (1999). Molecular analysis of a new splice variant of the human melanocortin-1 receptor.FEBS Lett.451137–141. 10.1016/S0014-5793(99)00525-6
162
TapiasA.AuriolJ.ForgetD.EnzlinJ. H.ScharerO. D.CoinF.et al (2004). Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors.J. Biol. Chem.27919074–19083. 10.1074/jbc.M312611200
163
ThodyA. J.HigginsE. M.WakamatsuK.ItoS.BurchillS. A.MarksJ. M. (1991). Pheomelanin as well as eumelanin is present in human epidermis.J. Invest. Dermatol.97340–344. 10.1111/1523-1747.ep12480680
164
TotaM. R.SmithT. S.MaoC.MacNeilT.MosleyR. T.Van der PloegL. H.et al (1999). Molecular interaction of Agouti protein and Agouti-related protein with human melanocortin receptors.Biochemistry38897–904. 10.1021/bi9815602
165
ValverdeP.HealyE.JacksonI.ReesJ. L.ThodyA. J. (1995). Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans.Nat. Genet.11328–330. 10.1038/ng1195-328
166
VenemaJ.van HoffenA.KarcagiV.NatarajanA. T.van ZeelandA. A.MullendersL. H. (1991). Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes.Mol. Cell. Biol.114128–4134. 10.1128/MCB.11.8.4128
167
ViradorV. M.MullerJ.WuX.Abdel-MalekZ. A.YuZ. X.FerransV. J.et al (2002). Influence of alpha-melanocyte-stimulating hormone and ultraviolet radiation on the transfer of melanosomes to keratinocytes.FASEB J.16105–107. 10.1096/fj.01-0518fje
168
WallinE.von HeijneG. (1995). Properties of N-terminal tails in G-protein coupled receptors: a statistical study.Protein Eng.8693–698. 10.1093/protein/8.7.693
169
WangC. Q.AkaluY. T.Suarez-FarinasM.GonzalezJ.MitsuiH.LowesM. A.et al (2013). IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: potential relevance to psoriasis.J. Invest. Dermatol.1332741–2752. 10.1038/jid.2013.237
170
WeinbergA.KrisanaprakornkitS.DaleB. A. (1998). Epithelial antimicrobial peptides: review and significance for oral applications.Crit. Rev. Oral Biol. Med.9399–414. 10.1177/10454411980090040201
171
WilsonB. D.OllmannM. M.KangL.StoffelM.BellG. I.BarshG. S. (1995). Structure and function of ASP, the human homolog of the mouse agouti gene.Hum. Mol. Genet.4223–230. 10.1093/hmg/4.2.223
172
WintzenM.GilchrestB. A. (1996). Proopiomelanocortin, its derived peptides, and the skin.J. Invest. Dermatol.1063–10. 10.1111/1523-1747.ep12326950
173
Wolf HorrellE.D’OrazioJ. (2014). UV-independent induction of beta defensin 3 in neonatal human skin explants.F1000Res3:288. 10.12688/f1000research.5794.2
174
YangY. (2011). Structure, function and regulation of the melanocortin receptors.Eur. J. Pharmacol.660125–130. 10.1016/j.ejphar.2010.12.020
175
YangY.ChenM.LaiY.GantzI.YagmurluA.GeorgesonK. E.et al (2003). Molecular determination of agouti-related protein binding to human melanocortin-4 receptor.Mol. Pharmacol.6494–103. 10.1124/mol.64.1.94
176
YangY.DickinsonC.Haskell-LuevanoC.GantzI. (1997). Molecular basis for the interaction of [Nle4,D-Phe7]melanocyte stimulating hormone with the human melanocortin-1 receptor.J. Biol. Chem.27223000–23010. 10.1074/jbc.272.37.23000
Summary
Keywords
melanocyte, melanoma, MC1R, melanocortin, ASIP, βD3, ATR, DNA repair
Citation
Wolf Horrell EM, Boulanger MC and D’Orazio JA (2016) Melanocortin 1 Receptor: Structure, Function, and Regulation. Front. Genet. 7:95. doi: 10.3389/fgene.2016.00095
Received
25 January 2016
Accepted
13 May 2016
Published
31 May 2016
Volume
7 - 2016
Edited by
Zalfa A. Abdel-Malek, University of Cincinnati, USA
Reviewed by
Massimo Broggini, Istituto di Ricerche Farmacologiche “Mario Negri”, Italy; Salvatore Piscuoglio, University Hospital Basel and University of Basel, Switzerland
Updates
Copyright
© 2016 Wolf Horrell, Boulanger and D’Orazio.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John A. D’Orazio, jdorazio@uky.edu
This article was submitted to Cancer Genetics, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.