 Darine Villela
Darine Villela Claudia K. Suemoto
Claudia K. Suemoto Carlos A. Pasqualucci3,4
Carlos A. Pasqualucci3,4 Lea T. Grinberg
Lea T. Grinberg- 1Human Genome and Stem-Cell Research Center, Department of Genetics and Evolutionary Biology, Institute of Biosciences, University of São Paulo, São Paulo, Brazil
- 2Discipline of Geriatrics, Department of Clinical Medicine, School of Medicine, University of São Paulo, São Paulo, Brazil
- 3Brazilian Aging Brain Study Group – LIM22, Department of Pathology, School of Medicine, University of São Paulo, São Paulo, Brazil
- 4Department of Pathology, School of Medicine, University of São Paulo, São Paulo, Brazil
- 5Memory and Aging Center, Department of Neurology, University of California, San Francisco, San Francisco, CA, USA
Dysregulation of calcium (Ca2+) homeostasis is now being recognized to be a key step in the pathogenesis of Alzheimer’s disease (AD). Data from the literature, in particular the association between AD and polymorphism that interfere with Ca2+ homeostasis indicates the presence of genetic factors in this process; further, presenilins mutations, which are known to cause the familial form of AD, are involved in the regulation of intracellular Ca2+ stores. Here, we wish to draw attention to rare DNA copy number variations identified in two subjects with late-onset AD that led to partial or full duplication of genes that encode different subunits of the same type of voltage-gated Ca2+ channels; these duplications of voltage-gated Ca2+ channel genes is consistent with the critical role of calcium signaling in molecular processes underlying memory as has been demonstrated by several studies.
It’s widely known that Ca2+ represents one of the most important second messengers in the brain and plays an essential role in neuronal development, synaptic transmission and plasticity, besides regulating various metabolic pathways (Striessnig et al., 2006). The regulation of Ca2+ homeostasis in the central nervous system is a very complex process involving proteins localized in the plasma membrane, endoplasmic reticulum (ER), mitochondria, and cytoplasm that together are responsible for maintaining a higher concentration of Ca2+ in the extracellular space (Berridge et al., 2003). Notably, as demonstrated by several studies, a dysregulation in Ca2+ homeostasis is associated with many pathological mechanisms, especially those related with neurodegenerative disorders (Berridge, 2013; Sulzer and Surmeier, 2013).
Based on previous observations that intracellular Ca2+ levels are increased in aging neurons, it has been suggested that a dysregulation of Ca2+ homeostasis could be a key step in the pathogenesis of Alzheimer’s disease (AD; Small, 2009). In fact, this is the basis of the calcium hypothesis of AD, which offers a potential correlation between β-amyloid (Aβ) toxicity and a progressive decline in memory and neuronal cell death (Fedrizzi and Carafoli, 2011). The central idea is that activation of the amyloidogenic pathway leads to a remodeling of the neuronal Ca2+ signaling, and this remodeling usually appears as upregulated in AD, triggered by either an enhance in the entry of external Ca2+ or a release from internal stores (Berridge, 2010).
The Aβ peptide, present in the extracellular senile plaques, is one of the hallmarks of the disease and its aggregation in oligomers has been found to induce Ca2+ influx into neurons functioning as channels and/or by activating channels in the plasma membrane (Demuro et al., 2010). The Aβ oligomers could form non-selective pores with high conductance for Ca2+ but they could also alter the fluidity of the membrane affecting the activity of several Ca2+ permeable channels, including acetylcholine receptors, glutamate receptors, dopamine receptors, and serotonin receptors (Eckert et al., 2005). On the other hand, within the neurons, Aβ interacts with two types of Ca2+ channels in the ER membrane that regulate calcium release from internal stores: (i) ryanodine receptors (RyRs) and (ii) inositol trisphosphate receptors (IP3Rs); this leads to an increase of their opening and the amount of Ca2+ being released by the ER (Chakroborty et al., 2009). It is relevant to note, though, that data from literature demonstrate that genetic factors may be crucial in the dysregulation of Ca2+ homeostasis in AD. Presenilins mutations, which are known to cause the familial form of AD, have been found to increase the release of intracellular Ca2+ from internal stores (Honarnejad and Herms, 2012; Bezprozvanny, 2013). The activity of the SERCA pump, which is responsible for maintain the Ca2+ concentration gradient across the ER membrane (higher inside) is enhanced by the presenilins (Green et al., 2008). Additionally, there is increasing evidence that presenilins may be leak channel. Tu et al. (2006) reported that the mutated form of presenilin 1 (PS1) reduces the passive leak in neurons resulting in an increase of cytoplasmatic Ca2+.
Of particular interest, it was demonstrated that a polymorphism in CALHM1 (Ca2+ homeostasis modulator-1) was significantly associated with the sporadic form of the disease in a case-control study (Dreses-Werringloer et al., 2008). Based on its sequence similarity to the ionotropic glutamate receptor NMDA, it has been proposed that this gene encodes a glycoprotein that is a cerebral Ca2+ channel component, which is localized both in the plasma membrane and ER, and may also function as a leak channel (Dreses-Werringloer et al., 2008). The expression of CALHM1 polymorphic variant reduces Ca2+ entry and this reduced Ca2+ permeability would increase the level of stored Ca2+ by reducing this putative leak pathyway; consequently, a decreased intracelullar Ca2+ levels is observed (Dreses-Werringloer et al., 2008). The authors also suggest a direct impact of CALHM1 on Aβ production. Although some studies argue against the latter observation, it seems that the problem is the different methods used for analysis of the data (Bertram et al., 2008; Rubio-Moscardo et al., 2013). Besides, even though it seems controversial that in the familial form of AD an increase of intracellular Ca2+ is observed while in the sporadic form studies report a reduce in intracellular Ca2+ levels, dysregulation of Ca2+ homeostasis in the two forms of AD, as described above, arise by different mechanisms, but what is pretty clear in the literature is that dysregulation of Ca2+ homeostasis is a major feature in AD.
More recently, a multicenter study reported a robust and consistently significant enrichment for genes constituting the calcium signaling pathway, especially those related to the elevation of cytosolic calcium, in independent cohorts of young and elderly participants (Heck et al., 2015). The authors also showed that the same gene set identified in those participants was significantly enriched in a very large case-control study of sporadic AD, making evident that calcium signaling is crucial in human memory processes, both in cognitive health and disease (Heck et al., 2015).
In an investigation about the frequency of DNA copy number variations (CNVs) in a cohort of 46 individual’s diagnosed post-mortem with late-onset AD provided by the Brain Bank of the Brazilian Aging Brain Study Group (BBBABSG; Grinberg et al., 2007). We found two cases with rare microduplications partially or fully affecting genes that encode different subunits of the same type of voltage-gated Ca2+ channels, the L-type (L-VGCC). One individual presented a genomic imbalance of 665 kb at 3p21.1 that incudes, among other genes, CACNA1D and CACNA2D3 (Figure 1A). CACNA1D encodes the subunit α1 (subtype 1D), which form the ion-conducting pore of the L-VGCC, and CACNA2D3 is responsible for encoding the subunit α-2δ, that influences the channels’ biophysical properties by increasing the current amplitude and regulating the activation and inactivation kinetics of the L-VGCC. On the other hand, the other subject showed a genomic imbalance of 300 kb at 3p21.31 that encompass the CACNA2D2 gene, which acts similarly to CACNA2D3, i.e., also encodes the subunit α-2δ of the L-VGCC (Figure 1B). Both duplications have intragenic breakpoints; depending on the resulting structure of the rearrangement, the partial gene duplications can result in loss of function of the genes involved.
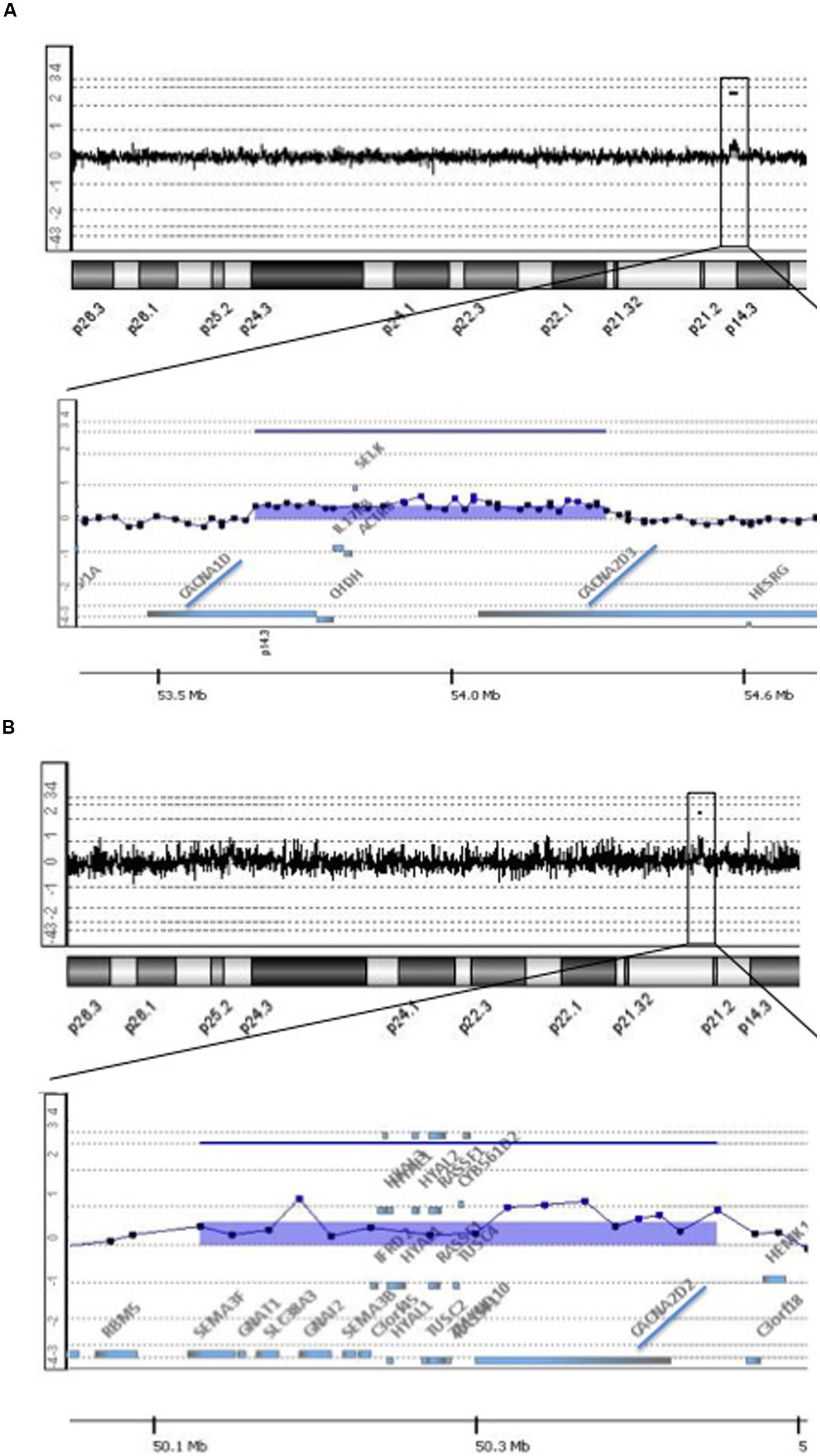
FIGURE 1. Rare copy number variations (CNVs) implicated in voltage gate calcium channels detected in two individuals with late-onset Alzheimer’s disease. The figure shows array-CGH results where each dot represents a probe. A genomic segment was considered duplicated or deleted when the log2 ratio of the Test/Reference fluorescent intensities of a given region encompassing at least three probes was above 0.3 or below -0.3, respectively, (Images were extracted from Genomic Workbench software). (A) Top shows array-CGH profile of chromosome 3 of Individual 1; the black box indicates the location of the microduplication on the short arm of the chromosome; underneath a detailed view of the area comprised in the black box at 3p21.1; the image shows a 665 kb duplication containing among other genes, CACNA1D and CACNA2D3, highlighted by a blue trace. (B) Top shows array-CGH profile of chromosome 3 of Individual 2; the black box indicates the location of the microduplication on the short arm of the chromosome; underneath a detailed view of the area comprised in the black box at 3p21.31; the image shows a 300 kb duplication containing among other genes, CACNA2D2, highlighted by a blue trace.
To exclude that these rare CNVs (rare defined as frequency <0.1% of population, based on the Database of Genomic Variants – DGV) represent common variants in the Brazilian population, we compile CNV data from more than 1,300 individuals studied by array-CGH in our laboratory for reasons other than dementia, such as cancer predisposition, deafness, congenital abnormalities, and intellectual disability. The rare CNVs documented in this study were never detected in any of these subjects. Additionally, in contrary to the control sample in this study, which consisted in individuals with no anatomophatological signs of AD, this set of more than 1,300 individuals is supposedly similar to the population regarding AD predisposition factors. In particular, despite some of the other individuals investigated in our casuistic also presented rare CNVs, these two cases are the only ones who showed copy number changes in genes that constitute the calcium signaling pathway. However, we cannot exclude that the remaining 44 individuals may have point mutations in relevant genes of calcium signaling pathway that could not be observed due to a limited resolution of array-CGH.
CNVs are recognized to be a prevalent form of common genetic variation and represent a substantial proportion of total genetic variability in human population. The functional impact of CNVs has been demonstrated at all biological levels, from cellular effects on gene expression to their association with several types of complex traits and genetic diseases (Stankiewicz and Lupski, 2010). To date, copy number changes in genes encoding calcium-signaling molecules have been associated especially with neuronal development disorders, such as autism (Rosenfeld et al., 2010), psychiatric diseases (Lee et al., 2012), and amyotrophic lateral sclerosis.
The present evidence is the first to highlight genomic imbalances in genes that are known to encode important calcium-signaling molecules, and link those CNVs to late-onset AD. Of note, in the manuscript from Heck et al. (2015) it was demonstrated that the same gene family of CACNA1D, CACNA2D2, and CACNA2D3 was found to be significantly enriched in patients with AD, which reinforces the significance of our findings. The present data also contribute adding new mutations in genes of calcium signaling pathway and associate these mutations with AD since CACNA1D, CACNA1D, CACNA2D2, and CACNA2D3 are not in the list of genes studied by Heck et al. (2015). Thus, considering the importance of L-VGCC in the central nervous system and Ca2+ being such an important regulator of synaptic plasticity, we could suppose that a disruption in CACNA1D, CACNA2D3, and CACNA2D3 caused by CNVs may lead to a dysregulation in Ca2+ homeostasis and contribute to the pathogenesis of AD. Also, using the Variant Effect Predictor (VEP) tool from Ensemble1 to predict the effects of these two microduplications on genes, transcripts, and protein sequence, as well as regulatory regions, we found that the most likely consequence (34%) is amplification of the amount of transcript. Therefore, this prediction suggests a possible upregulation of Ca2+ signaling, which meets the central idea of the calcium hypothesis of AD (Berridge, 2010). Yet, the crucial question of whether the microduplications showed in this study constitutes a predisposing risk factor for AD can only be answered by similar investigations in large cohorts, which we hope will be stimulated by this report. Also, we believe that the present evidence may stimulate further functional studies and eventually confirm the impact of these copy number changes in the pathogenesis of AD.
In summary, we wish to draw attention to rare DNA CNVs identified in two subjects with late-onset AD that led to partial or full duplication of genes that encode different subunits of the same type of voltage-gated Ca2+ channels and point out that the present finding is consistent with the critical role of calcium signaling in molecular processes underlying memory as has been demonstrated by several studies.
Author Contributions
This study was coordinated by CR and DV, as the principal investigator, provided conceptual and technical guidance for all aspects of this study. CS, LG, and CP coordinate the Brain Bank of the Brazilian Aging Study Group and provided all the samples. This commentary was written by DV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by FAPESP grants CR and CEPID-FAPESP (2009/00898-1; 2013/08028-1), and DV was supported by FAPESP PhD fellowship (2010/15503-0). We thank the Brazilian Aging Brain Study Group for providing DNA samples.
Footnotes
References
Berridge, M. J. (2010). Calcium hypothesis of Alzheimer’s disease. Pflugers. Arch. 459, 441–449. doi: 10.1007/s00424-009-0736-1
Berridge, M. J. (2013). Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 7, 2–13. doi: 10.4161/pri.21767
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Bertram, L., Schjeide, B. M., Hooli, B., Mullin, K., Hiltunen, M., Soininen, H., et al. (2008). No association between CALHM1 and Alzheimer’s disease risk. Cell 135, 993–994; author reply 994–996. doi: 10.1016/j.cell.2008.11.030
Bezprozvanny, I. (2013). Presenilins and calcium signaling-systems biology to the rescue. Sci. Signal. 6, e24. doi: 10.1126/scisignal.2004296
Chakroborty, S., Goussakov, I., Miller, M. B., and Stutzmann, G. E. (2009). Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 29, 9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009
Demuro, A., Parker, I., and Stutzmann, G. E. (2010). Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285, 12463–12468. doi: 10.1074/jbc.R109.080895
Dreses-Werringloer, U., Lambert, J. C., Vingtdeux, V., Zhao, H., Vais, H., Siebert, A., et al. (2008). A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 133, 1149–1161. doi: 10.1016/j.cell.2008.05.048
Eckert, G. P., Wood, W. G., and Muller, W. E. (2005). Membrane disordering effects of beta-amyloid peptides. Subcell Biochem. 38, 319–337. doi: 10.1007/0-387-23226-5_16
Fedrizzi, L., and Carafoli, E. (2011). Ca2+ dysfunction in neurodegenerative disorders: Alzheimer’s disease. Biofactors. 37, 189–196. doi: 10.1002/biof.157
Green, K. N., Demuro, A., Akbari, Y., Hitt, B. D., Smith, I. F., Parker, I., et al. (2008). SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Gen. Physiol. 181, 1107–1116. doi: 10.1085/JGP1322OIA1
Grinberg, L. T., Ferretti, R. E., Farfel, J. M., Leite, R., Pasqualucci, C. A., Rosemberg, S., et al. (2007). Brain bank of the Brazilian aging brain study group - a milestone reached and more than 1,600 collected brains. Cell Tissue Bank. 8, 151–162. doi: 10.1007/s10561-006-9022-z
Heck, A., Fastenrath, M., Coynel, D., Auschra, B., Bickel, H., Freytag, V., et al. (2015). Genetic analysis of association between calcium signaling and hippocampal activation, memory performance in the young and old, and risk for sporadic alzheimer disease. JAMA Psychiatry 72, 1029–1036. doi: 10.1001/jamapsychiatry.2015.1309
Honarnejad, K., and Herms, J. (2012). Presenilins: role in calcium homeostasis. Int. J. Biochem. Cell Biol. 44, 1983–1986. doi: 10.1016/j.biocel.2012.07.019
Lee, K. W., Woon, P. S., Teo, Y. Y., and Sim, K. (2012). Genome wide association studies (GWAS) and copy number variation (CNV) studies of the major psychoses: what have we learnt? Neurosci. Biobehav. Rev. 36, 556–571. doi: 10.1016/j.neubiorev.2011.09.001
Rosenfeld, J. A., Ballif, B. C., Torchia, B. S., Sahoo, T., Ravnan, J. B., Schultz, R., et al. (2010). Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet. Med. 12, 694–702. doi: 10.1097/GIM.0b013e3181f0c5f3
Rubio-Moscardo, F., Seto-Salvia, N., Pera, M., Bosch-Morato, M., Plata, C., Belbin, O., et al. (2013). Rare variants in calcium homeostasis modulator 1 (CALHM1) found in early onset Alzheimer’s disease patients alter calcium homeostasis. PLoS ONE 8:e74203. doi: 10.1371/journal.pone.0074203
Small, D. H. (2009). Dysregulation of calcium homeostasis in Alzheimer’s disease. Neurochem. Res. 34, 1824–1829. doi: 10.1007/s11064-009-9960-5
Stankiewicz, P., and Lupski, J. R. (2010). Structural variation in the human genome and its role in disease. Annu. Rev. Med. 61, 437–455. doi: 10.1146/annurev-med-100708-204735
Striessnig, J., Koschak, A., Sinnegger-Brauns, M. J., Hetzenauer, A., Nguyen, N. K., Busquet, P., et al. (2006). Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem. Soc. Trans. 34, 903–909. doi: 10.1042/BST0340903
Sulzer, D., and Surmeier, D. J. (2013). Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 28, 715–724. doi: 10.1002/mds.25187
Keywords: Alzheimer’s disease, copy number variations, CNVs, array-CGH, Ca2+
Citation: Villela D, Suemoto CK, Pasqualucci CA, Grinberg LT and Rosenberg C (2016) Do Copy Number Changes in CACNA2D2, CACNA2D3, and CACNA1D Constitute a Predisposing Risk Factor for Alzheimer’s Disease? Front. Genet. 7:107. doi: 10.3389/fgene.2016.00107
Received: 23 February 2016; Accepted: 27 May 2016;
Published: 14 June 2016.
Edited by:
Shin Murakami, Touro University California, USAReviewed by:
Ancha Baranova, George Mason University, USASangkyu Kim, Tulane University Health Sciences Center, USA
Anatoliy I. Yashin, Duke University, USA
Copyright © 2016 Villela, Suemoto, Pasqualucci, Grinberg and Rosenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carla Rosenberg, Y2FybGFyb3NlbmJlcmdAdW9sLmNvbS5icg==