 Stefano Ferrari*
Stefano Ferrari* Christian Gentili
Christian Gentili- Institute of Molecular Cancer Research, University of Zurich, Zurich, Switzerland
The implementation of decisions affecting cell viability and proliferation is based on prompt detection of the issue to be addressed, formulation and transmission of a correct set of instructions and fidelity in the execution of orders. While the first and the last are purely mechanical processes relying on the faithful functioning of single proteins or macromolecular complexes (sensors and effectors), information is the real cue, with signal amplitude, duration, and frequency ultimately determining the type of response. The cellular response to DNA damage is no exception to the rule. In this review article we focus on DNA damage responses in G2 and Mitosis. First, we set the stage describing mitosis and the machineries in charge of assembling the apparatus responsible for chromosome alignment and segregation as well as the inputs that control its function (checkpoints). Next, we examine the type of issues that a cell approaching mitosis might face, presenting the impact of post-translational modifications (PTMs) on the correct and timely functioning of pathways correcting errors or damage before chromosome segregation. We conclude this essay with a perspective on the current status of mitotic signaling pathway inhibitors and their potential use in cancer therapy.
Introduction
Signaling pathways have been initially depicted as linear cascades, with elements organized in a hierarchical manner and unidirectional arrows connecting a stimulus to the final response through a defined number of intermediates (Rodbell, 1980). The advent of systems biology, following completion of animal and plant genome sequencing, has changed this view. The amount of information available today allows to more realistically depict signaling pathways as networks, where the arrangement of components (nodes) is such that some are more connected than others in a so-called scale-free topology, and where sets of components are organized in modular fashion, with a clear hierarchy among modules (Barabási and Oltvai, 2004). Such architecture has been shown to ensure fault tolerance (robustness) in response to challenges (Barabási and Oltvai, 2004; Zhu et al., 2007). Corollary to system-level approaches has been the development of mathematical models where the fluctuation of variables as it actually occurs in defined biological systems can be computed, hence realistically representing the dynamic flow of information in signaling networks (Samaga and Klamt, 2013; Gerard et al., 2015).
The descriptive power of systems biology and its ability to predict scenarios do not, however, dwarf the contribution of reductionism when it comes to identification of network components and to dissection of their molecular mechanism of action, including elucidation of the inputs that affect their sub-cellular localization, the interaction with partner proteins and biochemical properties such as stability and enzymatic activity. It is only thanks to the wealth of information provided by reductionist approaches that rational design of small molecule inhibitors able to interfere with the correct functioning of networks could be successfully guided (Asghar et al., 2015). Since the constitutive elements of network modules hierarchically relate to each other, modification of structural or enzymatic traits of one or more elements in a network will necessary affect network properties and result in outputs that are directly observable. Protein post-translational modification (PTM), in form of covalent addition of chemical groups or entire peptidyl moieties to one or more amino acids of a protein target, is the means to rapidly and, in most cases, reversibly affect such traits. The hierarchical, synergistic or antagonistic combination of PTMs defines a code that translates into distinct outputs, hence contributing to shape the emergent properties of complex systems like living organisms (Lorenz et al., 2011).
In this review, we focus on mitosis and examine how DNA damage occurring during transition through mitosis is addressed to avoid genome instability. Special emphasis will be set on the impact of PTMs on mechanisms of genome surveillance. We conclude with an up-to-date perspective on drugs designed for therapeutic purposes and that entered clinical trials.
Mitosis and Checkpoints
Transition through the cell cycle sets the conditions for cell division. This results in the generation of two daughter cells genetically identical to the mother, according to a principle originally formulated by Rudolf Virchow who first made such observation in 1858 and stated that every cell derives from a pre-exiting cell, “omnis cellula e cellula” (Mazzarello, 1999). The major events characterizing transition through the cell cycle are cell growth, by which means cells increase their size and the number of organelles, and duplication of genetic material in S-phase. If not perturbed, upon completion of DNA replication cells enter mitosis, a term that originally described nuclear division (Mazzarello, 1999). Perturbations of this program may be caused by external agents such as ionizing radiation or certain chemotherapeutic drugs as well as by endogenous metabolic processes, leading to the formation of double-strand breaks (DSBs). Inappropriate repair of DSBs may cause gross chromosomal aberrations, the activation of oncogenes or the inactivation of tumor suppressor genes resulting in carcinogenesis. Direct demonstration of the importance of surveillance pathways in the maintenance of genome stability (Hanahan and Weinberg, 2011) is provided by genetic conditions characterized by dysfunction of the machinery that signals DNA damage and/or addresses its repair, which are associated with a predisposition to the development of cancer (Curtin, 2012).
Mitosis
Mitosis is probably the most spectacular event a cell undergoes to during its lifetime and it is essentially the process by which the duplicated genetic information is equally distributed to the daughter cells. Morphological changes that are easily observable with a microscope allow distinguishing sub-phases of mitosis consisting of prophase, metaphase, anaphase and telophase. These are followed by cytokinesis, ultimately causing physical separation of the daughter cells. The use of suitable model organisms and the support provided by modern technology has led us to a deep understanding of mechanistic aspects and regulatory pathways controlling the onset, execution and completion of mitosis. Briefly, in S-phase newly synthesized DNA emerging behind replication complexes that processively move on template DNA is maintained catenated throughout its length by ring-shaped cohesins and sister chromatids are held together at the centromeric region where kinetochores have been assembled (Kenney and Heald, 2006; Walczak et al., 2010). As cells move to prophase, chromatin condensation takes place, leading to the formation of visible rod-shaped structures, with a reduction of the length of DNA to an extent compatible with the distance that chromatids cover when moving to the opposite poles of the mitotic spindle (Walczak et al., 2010). Chromatin condensation results from the action of a multi-subunit protein complex called condensin, whose recruitment and activity are positively controlled by phosphorylation through CDK1, Aurora-B and PLKs and opposed by phosphorylation through CK2 (Hirano, 2012). Topoisomerase II, which undergoes phosphorylation and sumoylation in mitosis (Dephoure et al., 2008; Hendriks et al., 2014), ensures decatenation of sister chromatids prior to condensation (Hirano, 2015). Segregation of compacted chromosomes is initially prevented by cohesins (Peters et al., 2008) that are controlled by a combination of PTMs at lysine residues involving acetylation and sumoylation (Rudra and Skibbens, 2013) and are first removed at chromosome arms during prophase through PLK1-mediated phosphorylation (Hauf et al., 2005). At this time centromeric regions are protected by the protein shugoshin that, through recruitment of the phosphatase PP2A, counteracts PLK1 activity (Kitajima et al., 2006; Liu et al., 2013b). Construction of the mitotic spindle is the necessary step for physical separation of chromatids, with different strategies employed in distinct organisms to promote microtubule-to-kinetochore contacts (Boettcher and Barral, 2013). Microtubules forming the cell's cytoskeleton are disassembled in late prophase and highly dynamic microtubules radiate at this point from mature centrosomes or self-organize around chromosomes (Heald et al., 1996, 1997; Karsenti and Vernos, 2001), driving migration of centrosomes to opposite poles of the cell (inter-polar microtubules), anchoring centrosomes to the plasma membrane and positioning the spindle (astral microtubules) and initiating the capture of chromosomes (kinetochore microtubules). All these events are controlled by mitotic kinases (Nigg, 2001; Walczak et al., 2010).
In prophase, more than 100 proteins assemble around each centromeric region forming the kinetochore, while in the cytoplasm pairs of centrioles that have duplicated during S phase remain linked together at the proximal ends by a proteinaceous link containing C-Nap1 and rootletin, which is removed at mitotic entry through NEK2-mediated phosphorylation (Bahe et al., 2005; Hardy et al., 2014). Microtubule-chromosome interactions are characterized by the dynamic process of capture and release of erroneous attachments, as for instance merotelic attachments, which defines the condition of a single kinetochore being attached to microtubules nucleated from opposite spindle poles. Such interactions are principally regulated by Aurora-B-mediated phosphorylation of kinetochore components (Cheeseman, 2014), occur in prometaphase and metaphase, and largely affect the duration of these sub-phases (Pereira and Maiato, 2012). The subsequent chromosome congression to the spindle equator (metaphase plate) is coordinated by the action of motor proteins such as dynein and CENP-E, the latter being controlled by an Aurora-A/PP1-dependent phosphorylation switch (Kim et al., 2010), and is followed by a process called bi-orientation, where kinetochores of sister chromatids attach to microtubule bundles that have nucleated from opposite centrosomes (Tanaka et al., 2005). Upon congression of all sister pairs to the metaphase plate, licensing of a multimeric E3-ligase, the anaphase promoting complex/cyclosome (APC/C), ensues and leads to ubiquitylation and degradation of proteins such as Cyclin B, switching off CDK1 activity, and securin, freeing the enzyme separase that is now able to cleave and remove centromeric cohesins (Sivakumar and Gorbsky, 2015). This point marks the metaphase-to-anaphase transition where mechanical processes, consisting of inter-polar microtubule elongation and kinetochore microtubule shortening, as well as biochemical events mediated by the action of APC/C, determine the movement of chromatids to spindle poles (Castro et al., 2005; Goshima and Scholey, 2010). The process is completed by re-establishment of the nuclear membrane around decondensing chromosomes at telophase and is followed by physical separation of daughter cells, or cytokinesis (Pines and Rieder, 2001), assisted by the action of an acto-myosin contractile ring (D'Avino, 2009).
G2/M Checkpoint
Entry and transition through mitosis is highly controlled by molecular constrains (checkpoints) that have evolved to prevent genomic instability and consist of the G2/M and the spindle assembly checkpoints. The G2/M checkpoint prevents mitotic entry to cells that have suffered DNA damage during G2 or that have progressed into G2 with unrepaired DNA lesions from previous cell cycle phases. Final target of the G2/M DNA damage checkpoint is CDK1, the master regulator of mitosis. The cascade of phosphorylation events impinging on CDK1 is briefly sketched below. Signals from unfinished DNA replication (through ATR/CHK1), damaged DNA (through ATM/CHK2) or DNA resected at sites of damage (through ATR/CHK1), activate the kinases WEE1/MYT1 that, in turn, phosphorylate T14 and Y15 in the Gly-rich P-loop of CDK1, causing inhibition of enzymatic activity (Heald et al., 1993; Figure 1). Phosphorylation at these sites does not impair ATP binding, neither sterically nor by electrostatic repulsion (Gould and Nurse, 1989), but rather hampers catalysis (Atherton-Fessler et al., 1993). Additionally, WEE1 enforces the signal of “NO-entry” into mitosis by inactivating CDC25 (Donzelli and Draetta, 2003), the phosphatase responsible for CDK1 dephosphorylation. Specifically, CHK1-dependent phosphorylation of CDC25A at Ser124/Thr507 and of CDC25C at Ser216 mediates interaction with 14-3-3 proteins that, in turn, displace the phosphatases from the nucleus, a mechanism that appears to be the primary way to inhibit the function of these two phosphatases during G2 and mitosis (Uto et al., 2004). On the other hand, inhibition of CDC25B, the phosphatase mediating activation of CDK1/Cyclin B at centrosomes during prophase, has been extensively studied in relation to its mitotic role (Gabrielli et al., 1996) but is less characterized in the context of the DNA damage response. Factors upstream of CDC25 or Cyclin B/CDK1, such as the Polo-like kinases PLK1 and PLK3 (Nyberg et al., 2002; Bahassi el et al., 2006), Aurora-A (Ferrari et al., 2005; Krystyniak et al., 2006; Bhatia et al., 2010) and protein phosphatase PP2A (Yan et al., 2010) are also part of the G2/M checkpoint signaling network. Maintenance of the G2/M checkpoint activation partly relies on transcriptional regulation by p53 that induces transcription of the cell-cycle inhibitor p21CIP1∕WAF1, and on expressions of 14-3-3s (a scaffold and signaling protein), PUMA (BCL2 binding component 3), BAX (BCL2 partner and apoptotic activator) and GADD45 (growth arrest and DNA-damage-inducible gene) (Nyberg et al., 2002; Riley et al., 2008). Upon completion of DNA synthesis or repair of damage, signals from WEE1 cease, resulting in progressive dephosphorylation and reactivation of CDC25C. The latter, in turn, initiates selective dephosphorylation of the inhibiting sites in CDK1 (Izumi and Maller, 1993), creating an auto-catalytic loop in which CDK1- (Hoffmann et al., 1993; Strausfeld et al., 1994) and Polo-like kinase 1 (PLK1)-dependent CDC25C phosphorylation (Strausfeld et al., 1994; Toyoshima-Morimoto et al., 2002) increase phosphatase activity leading to full dephosphorylation and activation of CDK1. As a result, the checkpoint is silenced and cell cycle progression ensues.
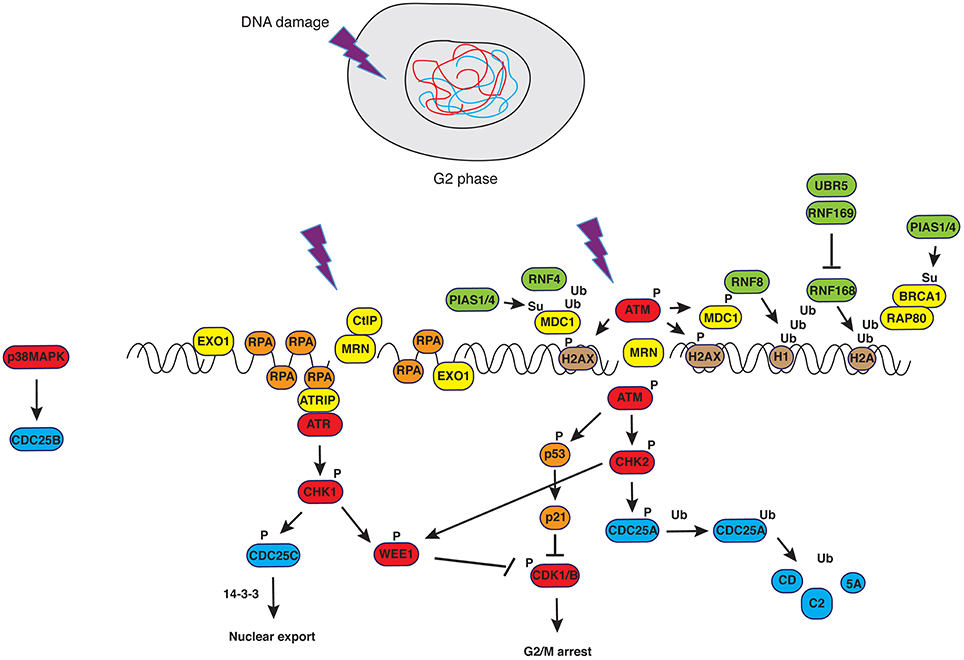
Figure 1. DNA damage response in G2. Upon generation of double strand breaks (DSBs), ATM is recruited to DNA ends in a MRN-dependent manner. Phosphorylation of H2AX creates epitopes facilitating the recruitment of DNA damage signaling and repair factors in a manner that depends on PTMs such as ubiquitylation and sumoylation (see text for details and Bologna and Ferrari, 2013). Successful activation of ATM-dependent signals causes controlled resection of DNA ends that, in turn, trigger ATR-dependent pathways. The latter converge with the former on the master regulator of mitosis, CDK1, blocking its activity.
Spindle Assembly Checkpoint
The spindle assembly checkpoint (SAC) ensures that chromosomes are properly bi-oriented, preventing missegregation that would otherwise result in aneuploidy (Musacchio and Salmon, 2007). Target of the SAC is the APC/C, an E3 ubiquitin ligase composed of approximately 15 subunits, which binds its substrates by recognizing so-called degron sequences (Pines, 2011). APC/C is activated in mitosis by its co-activators CDC20 and CDH1 in a Cyclin B/CDK1-dependent manner (Wieser and Pines, 2015) and works in tandem with two distinct E2 conjugating enzymes: UBCH5 or UBCH10 that add the first ubiquitin moiety to APC/C substrates, and UBE2S that extends the chain (Rodrigo-Brenni and Morgan, 2007; Garnett et al., 2009) mediating preferentially the formation of K11-linked ubiquitin chains (Wu et al., 2010; Bremm and Komander, 2012). K11-chains show a distinct fold with respect to K48- or K63-linked ubiquitin (Matsumoto et al., 2010). A phosphorylation-dependent switch controls timely activation of the E2 UBE2S by the APC/C complex, whereby phosphorylation of Ser92 in CDC20 prevents delivery of UBE2S to the APC/C, and its dephosphorylation by PP2AB56 allows UBE2S to bind the APC/C, catalyzing ubiquitin chain elongation (Craney et al., 2016).
Major players of the SAC are Mad1, Mad2, Bub1, BubR1/Mad3, Bub3, and Mps1, proteins that essentially monitor kinetochore—microtubule attachments and convert this to signals that inhibit metaphase-to-anaphase transition (Cheeseman, 2014). The main trigger of signals from SAC is Mad2, a protein that can assume an “open” (inactive) or a “close” (active) conformation. Mechanistically, the closed conformation into which Mad2 folds once bound to kinetochores that are improperly attached to spindle microtubules is induced in further neighboring Mad2 molecules that diffuse away from kinetochores and associate with BubR1 and Bub3 forming the so-called mitotic checkpoint complex (MCC). The latter binds and sequesters the first co-activator of APC/C, Cdc20, in an MPS1-dependent manner (Wieser and Pines, 2015), blocking degradation of securin and effectively arresting cells in metaphase (Cheeseman, 2014; Sivakumar and Gorbsky, 2015). The kinases Aurora-B, CDK1 and PLK1 participate in regulating kinetochore function, with Aurora-B-dependent phosphorylation of Ndc80 N-terminus reducing the microtubule-binding affinity of the Ndc80 complex and eliminating incorrect kinetochore-microtubule attachments (Cheeseman et al., 2006). PLK1 associates and regulates several kinetochore proteins, including those localized in the inner centromere like CENP-U, phosphorylation of which facilitates PLK1 recruitment to the kinetochore (Kang et al., 2006), and PLK1-interacting checkpoint helicase (PICH) that binds the kinase through its Polo-box domain (Baumann et al., 2007). Once appropriate attachment is established (i.e., bi-orientation) such that sufficient tension is created and the kinase is spatially separated from its substrates (Liu et al., 2009), PP1 dephosphorylates Aurora-B targets (Cheeseman, 2014), with additional support from PP2A (Foley et al., 2011). Satisfaction of the checkpoint upon appropriate bi-orientation of chromosomes triggers the metaphase to anaphase transition.
Challenges to the Genome and Responses in Mitosis
In order to preserve the integrity of information contained in the genome, DNA is continuously monitored by proteins that recognize distinct types of damage. Such proteins or protein complexes—so called sensors—inform signal transducers that, in turn, prompt effectors to orchestrate repair of the damage (Bologna and Ferrari, 2013; Jackson and Durocher, 2013; Dantuma and van Attikum, 2016). In parallel, transducers trigger checkpoint pathways impinging on key cell cycle controllers (see above) that ultimately slow down or arrests transition through the cell cycle (Kastan and Bartek, 2004). Inappropriate detection or untimely repair of DNA damage before the onset of mitosis may lead to chromosome breaks, rearrangements or fusions—comprehensively know as “structural abnormalities”—that facilitate the development of cancer (Branzei and Foiani, 2010; Curtin, 2012) and have been focus of intense research in the last decades (Aguilera and García-Muse, 2013).
DNA repair involves chromatin remodeling that, in turn, facilitates binding of repair factors to the region(s) where the lesion occurred (Aydin et al., 2014). This sequence of events has been observed during transition through the cell cycle, when the DNA repair machinery called to action faces simple or more challenging tasks, depending on whether damage is in euchromatin or in heterochromatin (Lemaître and Soutoglou, 2014). It appears, however, that DNA damage responses operative till completion of G2 and mediated through checkpoint kinases converging on WEE1 and CDK1 (Boddy et al., 1998; Smith et al., 2010; Figure 1), must be blocked at the time of chromosome condensation and segregation. In cells carrying a wild type complement of checkpoint genes, entry into prometaphase with ensuing chromosome condensation and nuclear envelope breakdown defines a point of non-return and puts an end to the checkpoint that was operative in G2.
Termination of activities on DNA in mitosis is exemplified by the repression of transcriptional activity (Martínez-Bálbas et al., 1995) that occurs through a passive process, consisting in limited access of transcription machinery to compacted chromatin, and an active mechanism, entailing CDK1-dependent phosphorylation of its components (Gottesfeld and Forbes, 1997). Similar mechanisms control DNA repair proteins, to avoid that active DSB repair during mitosis may result in telomere fusions, aneuploidy (Cesare, 2014; Orthwein et al., 2014) and whole chromosome missegregation through collateral stabilization of kinetochore-microtubules interactions (Bakhoum et al., 2014). Indeed, it was observed that in the absence of genotoxic stress, DNA repair proteins are phosphorylated in mitosis in a CDK- or PLK1-dependent manner to exclude them from chromatin (Figure 2). This is the case for BRCA2 (Lee et al., 2004), RAP80 (Cho et al., 2013), 53BP1 (Orthwein et al., 2014; Benada et al., 2015), RNF8 (Orthwein et al., 2014) and XRCC4 (Terasawa et al., 2014), to mention just few examples. RNF8, a well-characterized E3 ubiquitin ligase recruited to sites of damage through interaction of its N-terminal FHA domain with phosphorylated MDC1 and HERC2 (Bologna and Ferrari, 2013), the latter acting as coordinator of ubiquitin-dependent assembly of DNA repair factors (Bekker-Jensen et al., 2010), is phosphorylated by mitotic kinases to suppress its interaction with MDC1 (Orthwein et al., 2014). In the case of 53BP1, phosphorylation of two residues within the ubiquitylation-dependent recruitment (UDR) motif of 53BP1 in mitosis blocks binding to K15-ubiquitylated histone H2A, thus impairing its recruitment to foci (Benada et al., 2015). On the other hand, PP4C/R3β-mediated dephosphorylation of these sites in G1 re-establishes 53BP1 binding to chromatin (Lee et al., 2014). In the case of BRCA2, PLK1-dependent phosphorylation at S193, 205, 206 and T203, 207 causes dissociation from the histone acetyltransferase protein p300/CBP-associated factor (P/CAF) (Lin et al., 2003), and CDK-dependent S3291 phosphorylation at the onset of mitosis inhibits BRCA2-mediated stabilization of RAD51 nucleofilaments that are normally generated at sites of recombination (Esashi et al., 2005).
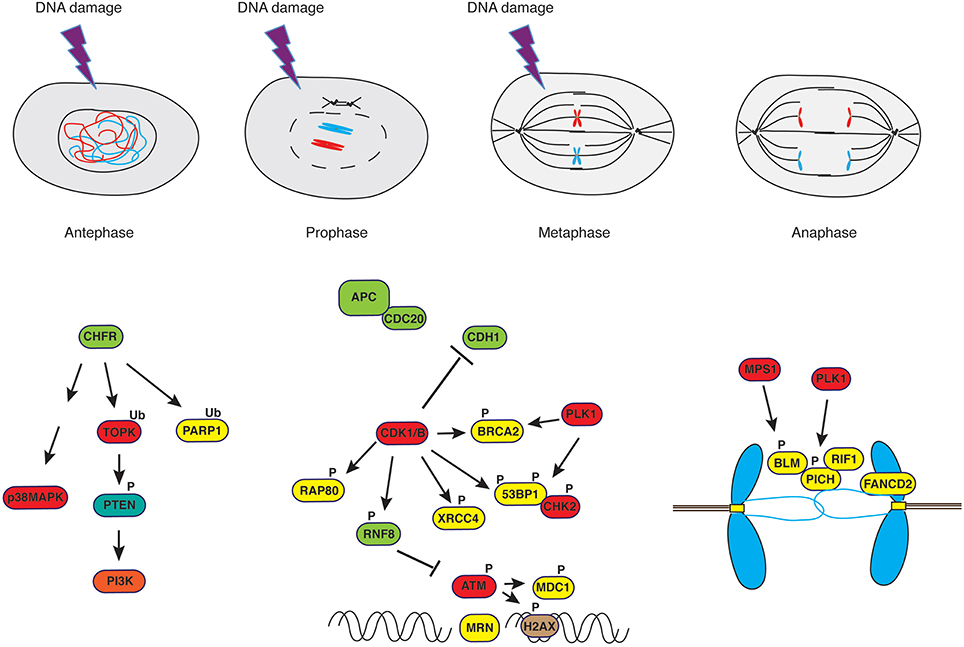
Figure 2. DNA damage response in mitosis. Irradiation of cells in antephase or in early prophase triggers a response that is independent of PI-3K-like kinases such as ATM but rather depends on the E3-ubiquitin ligase CHFR and the stress-response kinase p38MAPK. On the other hand, irradiation of cells in late prophase or in metaphase leads to a curtailed DNA damage response. Ultrafine anaphase bridges, caused by improper resolution of replication or recombination intermediates, are addressed by the coordinated action of the helicases PICH and BLM supported by RIF1 (see text for details).
Shutting off repair in mitosis, however, does not imply that DNA damage is ignored if it occurs in this phase of the cell cycle. Evidence obtained in early studies conducted in vertebrate somatic cells showed that chromosome fragmentation caused by irradiation at the beginning of mitosis persisted till anaphase (Zirkle and Bloom, 1953), possibly indicating that repair pathways were not activated in this period of time. On the other hand, recent studies on the outcome of laser irradiation of mitotic chromosomes indicated that DNA damage response is triggered within 30 s from the treatment (Gomez-Godinez et al., 2010). Studies in the budding yeast S. cerevisiae, where activation of a dicentric chromosome was used to introduce a double strand DNA break into a chromosome at mitosis, showed that cells paused in mid-anaphase, triggering RAD9-dependent events that were reminiscent of a DNA damage response (Yang et al., 1997). Subsequent work conducted in yeast, where cells were irradiated in mitosis, showed that stabilization of Pds1, an anaphase inhibitor and APC/Cdc20 target, led to delay of anaphase and mitotic exit, facilitating repair of damage (Tinker-Kulberg and Morgan, 1999). Delay of the metaphase-to-anaphase transition was also reported for Drosophila embryos undergoing irradiation or being treated with methly metane sulfonate (MMS) and it was shown to depend on the stabilization of Cyclin A (Su and Jaklevic, 2001). Mitotic DNA damage in X. laevis and DT40 cells was shown to prevent spindle assembly in an ATM/ATR-dependent manner (Smith et al., 2009), and proposed to be an additional means to monitor chromosome breaks that have escaped the G2/M checkpoint.
It has been reported that eukaryotic cells are able to delay the execution of mitosis or, in some instances, reverse progression through mitosis, in response to DNA damage (Rieder and Cole, 1998) or microtubule poisons (Rieder and Cole, 2000) administered in antephase, a time when microscopic changes in the cell are not yet detectable and that physically spans from the conclusion of G2 to the initiation of chromosome condensation (Chin and Yeong, 2010). The same response was observed upon damage caused in early prophase (Rieder and Cole, 1998). In antephase, cells activate a checkpoint that is not mediated by PI-3K-like kinases such as ATM but rather depends on two proteins, the CHFR E3-ubiquitin ligase (Matsusaka and Pines, 2004; Shinde et al., 2013) that principally catalyzes poly-ubiquitylation of its substrates via K48 and K63 (Kang et al., 2002; Bothos et al., 2003; Oh et al., 2009) and is involved in the first wave of ubiquitylation at DNA damage sites (Liu et al., 2013a), and the Pro-directed p38 MAPK (Mikhailov et al., 2004; Figure 2). Cells containing a wild-type antephase checkpoint undergo chromosome decondensation and revert to a G2-like state (Rieder and Cole, 1998; Matsusaka and Pines, 2004), whereas cells lacking a functional CHFR progress into mitosis (Scolnick and Halazonetis, 2000). Extensive damage occurring upon completion of antephase does not normally cause reversion to an early stage of the cell cycle but rather triggers mitotic arrest through activation of SAC (Mikhailov et al., 2002, 2004; Choi and Lee, 2008), the only mechanism left in the arsenal of cells at this point of the cell cycle. SAC, however, does not orchestrate repair of damaged DNA but monitors that distribution of chromosomes to daughter cells occurs equally, hence avoiding aneuploidy. As mentioned above, SAC is active at kinetochores where the state of microtubule attachment is monitored, and signaling pathways preventing anaphase remain active as long as mono-oriented or incorrectly attached kinetochores are detected (Mikhailov et al., 2002; Cheeseman and Desai, 2008). Observations made in yeast (Pangilinan and Spencer, 1996) and in mammalian cells (Mikhailov et al., 2002) indicate that altering the topology of chromatin, particularly at regions that affect kinetochore structure, prevents satisfaction of SAC and delays transition through mitosis. These studies showed that ATM-dependent pathways (Mikhailov et al., 2002) or DDR genes (Pangilinan and Spencer, 1996) are not involved in the response to chromosome damage and that the metaphase block can be rapidly overridden by dominant-negative Mad2 (Mikhailov et al., 2002). However, a study addressing the effect of decatenation inhibitors (topoisomerase-II inhibitors) on nocodazole-arrested cells described a number of ATM-dependent events in response to these drugs, including H2AX phosphorylation, CDK1 inactivation, histone H3 dephosphorylation and chromosome decondensation, paralleled by stabilization of Cyclins A and B1, with cells apparently unable to exit mitosis (Chow et al., 2003). Studies conducted in our laboratory on cells that were synchronized in mitosis without disturbing microtubule dynamics, hence without “pre-sensitizing” cells by activation of stress responses that are known to increase levels of γH2AX (Giunta et al., 2010), and that we treated with ionizing radiation at metaphase, showed CHK2 activation, rapid inhibition of CDK1 and Aurora-A activities accompanied by reactivation of PP1, increased APC/CDH1 E3-ubiquitin ligase activity and chromosome decondensation (Bhatia et al., 2010). Our data clearly showed that metaphase-irradiated cells completed mitosis at the expenses of genome stability, displaying increased chromosome segregation defects and the formation of micronuclei (Bhatia et al., 2010).
A comprehensive study that assessed entity and amplitude of the DDR in mitosis by scoring formation of IR-induced foci (IRIF) and comparing mitotic to interphase cells concluded that only a subset of IRIF could form in mitosis, namely those comprising γ-H2AX, NBS1 and MDC1, but not RNF8, RNF168, BRCA1 or 53BP1 (Giunta et al., 2010) as also confirmed by others (Nelson et al., 2009). Exclusion of RNF8 and 53BP1 from chromatin was shown to be the consequence of phosphorylation by mitotic kinases (see above) (Orthwein et al., 2014) and association of 53BP1 to IRIF was observed only upon nuclear envelope reformation around decompacting chromosomes in telophase (Giunta et al., 2010; Figure 2).
Specifically regarding ATM, its activation has been examined in mammalian cells both during undisturbed transition through mitosis or upon stress. In the absence of DNA damage, the kinase Aurora-B phosphorylates ATM on S1403 in mitosis, and abrogation of this event was shown to impair signaling through the spindle assembly checkpoint (Yang et al., 2011). Administration of taxol, a drug suppressing microtubule dynamics and causing mitotic stress, was reported to trigger ATM activity, though none of the known ATM targets in DDR such as SMC-1, NBS-1 or CHK-2 was phosphorylated under these conditions (Shen et al., 2006). DNA damage response-related roles for ATM in mitosis were inferred from early observations made in lymphoblastoid cells derived from A-T patients, which displayed a defective SAC upon treatment with radiation (Takagi et al., 1998; Shigeta et al., 1999). Another report described the activation of ATM in response to chromosomal breaks generated during mitotic catastrophe (Imreh et al., 2011). ATM activation was also examined upon irradiation of cells synchronized in mitosis with drugs that interfere with microtubule polymerization. Under these conditions, ionizing radiation triggered ATM activity, though CHK2 failed to fire and cells remained in mitosis with elevated phosphorylation at MPM-2 epitopes, indicative of high CDK1 activity. Mechanistically, the absence of a productive DDR signal following ATM activation was proposed to result from PLK1-dependent phosphorylation of CHK2, with 53BP1 acting as platform to bring PLK1 and CHK2 in close proximity (van Vugt et al., 2010; Figure 2).
As a whole, these studies confirm that ATM can fire when the minimal requirement for its activation is satisfied, namely the presence of exposed double-stranded ends (You et al., 2007), independently on the cell cycle position, though a productive DDR downstream of ATM seems not to be triggered in early mitosis.
In addition to DNA damage occurring during transition through mitosis, cells reaching mitosis are confronted with other problems: these are the structures resulting from incomplete DNA replication, improper resolution of replication intermediates or unresolved intermediates of homology-directed repair carried over from S-phase (Liu et al., 2014). Such structures become a threat at the time of chromosome segregation since they can cause sister chromatid entanglement and non-disjunction (Gelot et al., 2015). Incomplete DNA replication occurs at regions encompassing so-called “replication barriers.” Predominant among those are common-fragile sites (CFSs) (Durkin and Glover, 2007), cytologically defined as segments in metaphase chromosomes displaying brakes at runs of flexible AT-rich repeats (Aguilera and García-Muse, 2013). CFSs constitute up to 80% of the breakpoints that lead to the gross chromosomal rearrangements (GCRs) observed in precancerous cells (Bartkova et al., 2006). Part of under-replicated CFSs observed in cells at anaphase remain connected through thin threads of DNA called ultrafine bridges (UFBs) (Liu et al., 2014). To avoid DNA breaks resulting from segregation of incompletely replicated chromosomes, these structures are addressed before cell division. It has been observed that BLM, along with topoisomerase IIIα, RMI1, RMI2 (BTRR complex) and PICH (PLK1-Interacting Checkpoint Helicase), coat anaphase UFBs (Baumann et al., 2007; Chan and Hickson, 2009; Chan et al., 2009; Naim and Rosselli, 2009; Figure 2). An earlier report on BLM phosphorylation by MPS1, facilitating accurate chromosome segregation (Leng et al., 2006), anticipated the important role played by this DNA helicase in mitosis. BLM recruitment to UBFs is facilitated by FANCD2, a key component of the Fanconi Anemia pathway, which was shown to form sister foci in mitosis (Naim and Rosselli, 2009; Harrigan et al., 2011; Lukas et al., 2011; Figure 2) and be necessary to prevent the generation of micronuclei (Naim and Rosselli, 2009). The SNF2 ATPase family member PICH plays an essential role at kinetochores and the inner centromere, as demonstrated by studies in which PICH depletion caused loss of Mad2 from kinetochores and abrogated the spindle checkpoint, events that were followed by chromosome missegregation (Baumann et al., 2007). Also PICH was proposed to help recruiting the BTRR complex at UFBs, cooperating to the resolution of DNA bridges by the end of anaphase (Liu et al., 2014; Figure 2). A recent addition to the pool of proteins present at UFBs is RIF1, ortholog of a yeast telomeric protein. RIF1 is recruited to UFBs in a PICH-dependent manner but independently of 53BP1, ATM or BLM, and phosphorylation by CDK1 restricts its ability to bind DNA at anaphase (Hengeveld et al., 2015). In addition to the BTRR complex, the Holliday Junction resolvases SLX1–SLX4–MUS81–EME1 (SLX–MUS complex) and GEN1 (Wyatt et al., 2013; Chan and West, 2014) contribute to process structures caused by under-replication at CFSs (Naim et al., 2013; Ying et al., 2013). The SLX–MUS complex cooperates with TopBP1, a scaffold protein composed of nine BRCT domains and recruited at sites of DNA damage in a 9-1-1-dependent manner (Delacroix et al., 2007; Lee et al., 2007; Wardlaw et al., 2014). TopBP1 is necessary for ATR activation (Kumagai et al., 2006), colocalizes with RPA and FANCD2 (Pedersen et al., 2015) forming foci on condensing chromatin through its BRCT5 domain, and recruits TOP2A to help resolving DNA entanglements between sister chromatids (Broderick et al., 2015). CDK1-dependent phosphorylation of EME1 in the MUS81-EME1 structure-specific endonuclease, promoting interaction with SLX1-SLX4, controls the resolution of DNA recombination intermediates in mitosis (Matos et al., 2011; Matos and West, 2014). Proteome-wide studies have identified a number of ubiquitylation sites in GEN1, MUS81, EME1, TopBP1 (Kim et al., 2011; Wagner et al., 2011; Mertins et al., 2013), though the biological function of such PTM and its eventual connection with mitotic functions of these proteins has not been addressed to date. Finally, human GEN1 acts as back up to the above-mentioned machinery at anaphase, moving in place and gaining access to DNA after nuclear envelope breakdown (NEB) (Wechsler et al., 2011; Chan and West, 2014; Sarbajna et al., 2014). For the yeast homolog of GEN1, Yen1, it was shown that activity and access to the nucleus depend on a reversible CDK1/Cdc14 phosphorylation switch (Eissler et al., 2014; Matos and West, 2014).
In addition to the role of the above mentioned scaffold proteins in tethering nucleases to UFBs to the end of resolving DNA bridges in anaphase, unscheduled DNA synthesis at UFBs marked by TopBP1 (Pedersen et al., 2015) or SLX4 (Minocherhomji et al., 2015) has been reported and interpreted as an attempt to fill-in unreplicated regions, hence restoring genome integrity before cell division.
In case lesions generated by replication stress remain unrepaired, they are passed to daughter cells in a manner that shelters them from further damage through sequestration in 53BP1 nuclear bodies, thus allowing repair in the next cell cycle (Lukas et al., 2011). In the presence of extensive damage that remains unaddressed, cells experience sudden mitotic death also known as mitotic catastrophe (Morrison and Rieder, 2004; Vitale et al., 2011).
We have mentioned above that the DNA damage checkpoint is in place to facilitate DNA repair by blocking transition from G2 to M (Figure 1). A non-trivial consequence of prolonged arrest before mitosis is centrosome amplification, an event that is observed with high incidence in cancer cells carrying mutations of DNA repair genes. This event, which is alleviated upon bypass of the checkpoint in a manner that is only partially dependent on ATM, was postulated to be a mechanism ensuring death of cells that manage to evade the G2/M checkpoint or the SAC (Dodson et al., 2004). The metaphase-to-anaphase transition is a critical cell cycle stage during which chromosome missegregation may occur. Loss or gain of entire chromosomes—known as “numerical abnormalities”—resulting from chromosome missegregation during mitosis, is a characteristic of tumors known from more than a century and described as “aneuploidy” (Pellman, 2007). Mechanisms leading to aneuploidy have been amply reviewed elsewhere (Holland and Cleveland, 2009) and comprise (i) defective attachment of sister chromatids to spindle microtubules (merotelic attachment), often linked to centrosome amplification, (ii) malfunction of the spindle assembly checkpoint and (iii) defects in chromosome cohesion.
Key to a fully-fledged response to DNA damage is the network of signals that orchestrate assembly of DNA repair proteins at sites of damage and informs the cell cycle machinery. Ultimate target of G2/M checkpoint pathways is CDK1, the master regulator of mitosis (see “Mitosis and Checkpoints”) that is maintained in an “OFF” status by direct negative inputs (WEE1) and inactivation of its positive regulators (CDC25), in conjunction with modulation of other enzymatic activities such as those of the kinases PLK1 (Smits et al., 2000), Aurora-A (Krystyniak et al., 2006) and protein phosphatase PP2A (Yan et al., 2010; Figure 1). The budding yeast S. cerevisiae represents a notable exception in this respect. Whereas in high eukaryotes CDKs have acquired specific functions throughout evolution, with CDK1 being the master controller of mitosis and undergoing immediate inhibition in an ATM/ATR-CHK1/CHK2-dependent manner upon DNA damage, S. cerevisiae possesses only one Cyclin-dependent kinase, Cdc28, controlling pathways and transitions in all phases of the cell cycle and whose activity depends on interaction with different Cyclins (Enserink and Kolodner, 2010). As opposed to CDK1, budding yeast Cdc28 is not inhibited by DNA damage response pathways, since the status of Tyr19 phosphorylation in the P-loop of Cdc28 is not a determinant for entry into mitosis (Amon et al., 1992). The key control of budding yeast mitosis is operative at the metaphase-to-anaphase transition, where degradation of the Esp1 (separase) inhibitor Pds1 (securin) allows cleavage of the Scc1 subunit in the cohesin complex and separation of the sisters (Ciosk et al., 1998; Sanchez et al., 1999). Hence, in yeast, mitotic arrest in response to DNA damage occurs in metaphase and depends on the abundance of Pds1 (Sanchez et al., 1999). This mechanism liberates Cdc28 of the control that CDK1 undergoes to in higher eukaryotes. Contrary to rapid inhibition upon DNA damage, Cdc28 is absolutely required in DDR and participates to the control of genome stability (Enserink et al., 2009). Cdc28 triggers homologous directed repair of DSBs through phosphorylation of Sae2 (Huertas et al., 2008), prompting initial resection of DNA ends (Ira et al., 2004), and other components of error-free repair pathways such as Dna2 (Ubersax et al., 2003) and Srs2 (Chiolo et al., 2005; Saponaro et al., 2010). Interestingly, Cdc28 targets such as Sae2 are also phosphorylated by classic DDR kinases, whereby mutation of phosphorylation sites for either set of kinases hampers repair and recombination functions of the protein (Baroni et al., 2004).
Hence, the rapid inactivation of vertebrate CDK1 in response to damage is difficult to reconcile with claims on its involvement in DNA damage responses at G2/M. Although it has been suggested that the gap between checkpoint triggering and CDK1 shutoff in vertebrate cells may be sufficient for CDK1 to orchestrate initial phases of repair, a much wiser interpretation of the experimental evidence is that repair of DNA damage in checkpoint-arrested cells depends on CDK2 (Wohlbold and Fisher, 2009) and other Proline-directed kinases involved in stress responses (Bulavin et al., 2001). As a matter of fact, high CDK1 activity, along with the activity of other mitotic kinases (Benada et al., 2015), is sufficient to suppress responses to DNA damage occurring during transition through mitosis in mammalian cells (Zhang et al., 2011). A further layer of regulation is imposed by phosphatases such as WIP1, a CDK1 target that undergoes ubiquitin-mediated degradation in mitosis, which sets the threshold for DDR signaling in mitosis by controlling the phosphorylation state of DDR proteins (Macurek et al., 2013).
Mitotic PTMs and Cancer Therapy
Mitosis is the cell cycle phase that is most vulnerable to injury, regardless on whether damage is caused by radiation, heat-shock or chemicals (Westra and Dewey, 1971; Stobbe et al., 2002; Chan et al., 2012). Based on this indication, targeting mitotic cells has been largely exploited in the clinic as means to contain tumor growth (Doménech and Malumbres, 2013; Marzo and Naval, 2013). Molecular studies have highlighted the role of PTMs, and the enzymes that mediate them, in mechanisms controlling the mitotic responses to stress (Pearce and Humphrey, 2001). This comes as no surprise, considering that essentially all mechanistic aspects of normal transition through mitosis are controlled by PTMs of mitotic machinery components, with reversible PTMs allowing a certain degree of flexibility in the decisions implemented and irreversible PTMs conferring directionality to the process (Nigg, 2001; Ma and Poon, 2011; Teixeira and Reed, 2013). Hence, mitotic protein kinases and E3-ubiquitin ligases with established role in cancer have become the focus of interest for chemists and pharmacologists designing and testing novel therapeutics that target cells in mitosis (Dominguez-Brauer et al., 2015). Such interest was also motivated by considerations on the side effects of classic anti-mitotic drugs like taxanes and vinca alkaloids that are currently deployed to the treatment of a variety of solid tumors such as breast, ovarian and lung cancer. Anti-mitotic drugs, due to their mode of action that alters microtubules' dynamic instability, result in neurotoxicity and neutropenia (Marzo and Naval, 2013). Furthermore, their lack of efficiency when used as single agents has evidenced another important limitation of these anti-mitotics (Doménech and Malumbres, 2013; Marzo and Naval, 2013). Shifting the focus to the discovery of drugs that target mitotic kinases or E3-ubiquitin ligases, however, did not solve the major caveat for cell cycle—and mitotic—inhibitors, namely the fact that the efficacy of a drug depends on the tumor proliferative rate: fast proliferation makes leukemia and myeloma relatively favorable conditions to treat, whereas a mitotic index (i.e., the percentage of mitotic cells in the whole populations) as little as 1% and doubling time of more than 1 year, as observed in some solid tumors, are negative factors to be taken into account when planning a treatment and its length.
Here below we provide a report on the current status of drug discovery and clinical trials for compounds targeting mitotic kinases and phosphatases as well as ubiquitin-proteasome system components (Table 1 and Figure 3).
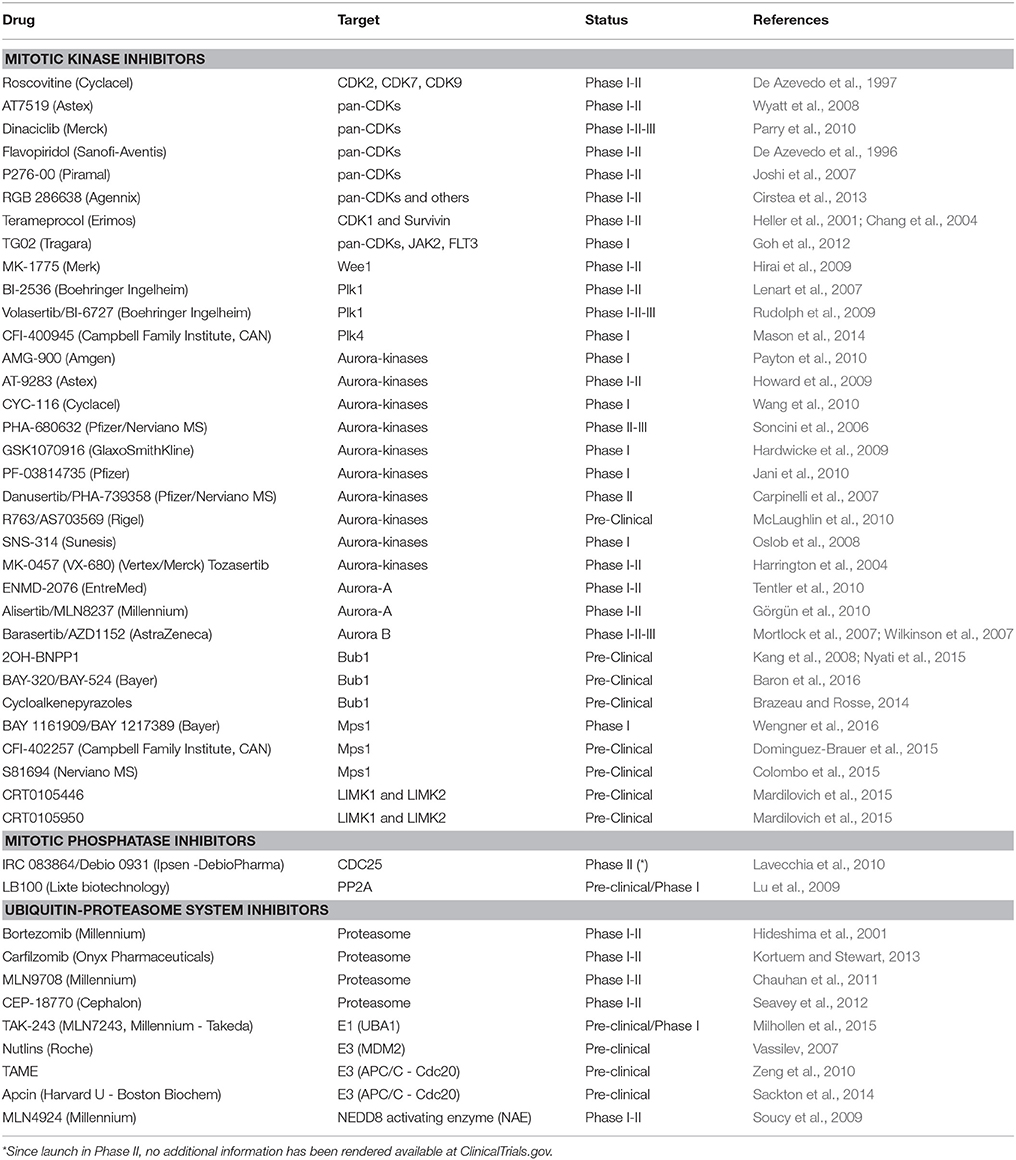
Table 1. List of drugs, their mitotic targets and current clinical trial phase.
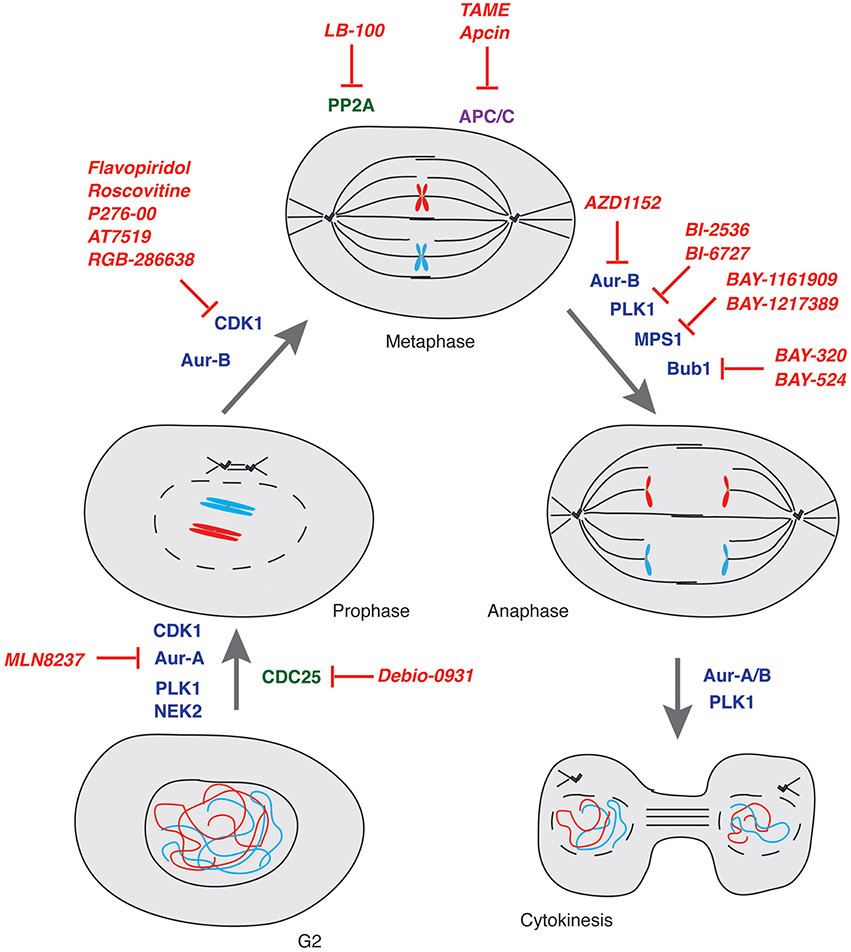
Figure 3. Mitosis and its control by kinases, phosphatases and E3-ubiquitin ligases. Schematic representation of key controllers of the onset, transition and exit from mitosis with indication of the major drugs inhibiting their function. Kinases: blue; Phosphatases: green; E3-ubiquitin ligases: purple; Drugs: red.
Inhibition of Mitotic Kinases
CDK1 Inhibitors
CDK1 is the master regulator of mitosis (Nigg, 2001). Flavopiridol is the first CDK1 inhibitor that underwent >60 clinical trials to date (www.clinicaltrials.gov). The poor efficacy of the compound, however, prevented its approval as anti-tumor drug (Shapiro, 2006; Stone et al., 2012; Galons et al., 2013). Other CDK1 inhibitors displaying high potency on cancer cell lines are currently in Phase I or II (with none of them being yet available for patients, see Table 1 and references therein). So far, only the CDK1 inhibitor Dinaciclib was tested in a phase III study that was concluded in 2015 and aimed at treating refractory chronic lymphocytic leukemia patients (NCT01580228). In general, however, inhibition of CDK1 in healthy cells and the poor selectivity of CDK1 inhibitors, possibly due to the high degree of sequence conservation in the catalytic domain of CDK members, often result in major side effects when deployed in the clinic. For such reason, CDK1 inhibitors are currently used in combination therapies with other mitotic inhibitors (see below).
Aurora Kinases Inhibitors
Aurora kinase family members, Aurora-A, -B and -C exert different roles in the cell. Early studies showed that Aurora-A controls centrosomes maturation and separation, bipolar spindle formation and chromosomes segregation, while Aurora-B, as member of the Chromosome Passenger Complex, participates in the control of chromosome condensation and orientation on the mitotic spindle, ensuring correct kinetochore-microtubule attachments (Nigg, 2001). Both Aurora-A and -B were shown to stabilize midzone microtubules and regulate cytokinesis (Carmena et al., 2009). Aurora-C has a role in gametogenesis, it is expressed in testis, thyroid, and placenta and its contribution to cancer development was shown in mouse models (Khan et al., 2011). Several inhibitors against Aurora-A and Aurora-B have been developed during the last decade (Doménech and Malumbres, 2013; Bavetsias and Linardopoulos, 2015; D'Assoro et al., 2015; Falchook et al., 2015). As for CDKs, most Aurora kinases inhibitors target all family members and a major effort has been done to develop drugs that are more selective for individual Aurora kinases (Table 1 and Figure 3). Among them, two reversible ATP competitive inhibitors, MLN8054 (Manfredi et al., 2007) and its derivative MLN8237 (Manfredi et al., 2011) have shown to be potent and selective Aurora-A inhibitors (Sells et al., 2015). Both were deployed in several studies and since MLN8237 has shown to be safer, it is currently under evaluation in Phase III clinical trials (NCT01482962).
Polo-Like Kinases Inhibitors
Polo-like family members constitute another class of Serine/Threonine (Ser/Thr) kinases with key roles in mitosis (Nigg, 2001). Five PLKs are expressed in human cells, PLK1-5, with PLK1 and PLK4 being the major representatives of this family (Zitouni et al., 2014). Distinguishing feature of PLKs is the polo-box domain that flanks the catalytic domain and allows docking to substrates primed by CDKs to carry on their phosphorylation (Zitouni et al., 2014).
Mechanistically, PLK1 is activated by Aurora-A (Ferrari et al., 2005; Macurek et al., 2008) at the onset of mitosis and functions to promote centrosome maturation and separation, assembly and elongation of the mitotic spindle as well as cytokinesis (Barr et al., 2004; Degenhardt and Lampkin, 2010). PLK1 is overexpressed in various malignancies (Holtrich et al., 1994; Eckerdt et al., 2005; Mito et al., 2005; Takai et al., 2005; Strebhardt and Ullrich, 2006; Renner et al., 2009; Weiß and Efferth, 2012). PLK1 inhibition in cancer patients has been pursued with some success using two ATP-competitive kinase inhibitors: BI-2536 and BI-6727 (see Table 1). The potency, pharmacokinetic and pharmacodynamic properties of BI-6727 as well as its antitumor activity in a number of cancer models (Rudolph et al., 2009) has promoted the drug to a phase III trial for acute myeloid leukemia patients where BI-6727 was tested in combination with the DNA-synthesis blocking agent cytarabine (NCT01721876).
PLK4 has a fundamental role in centriole duplication (Bettencourt-Dias et al., 2005; Habedanck et al., 2005). PLK4 overexpression leads to the formation of extra centrosomes resulting in aberrant mitotic spindles and aneuploid daughter cells (Basto et al., 2008; Ganem et al., 2009; Holland et al., 2010). Evidence on PLK4 overexpression in tumors (Macmillan et al., 2001; van de Vijver et al., 2002; Miller et al., 2005; Hu et al., 2006; Salvatore et al., 2007; Chng et al., 2008) raised the interest to develop small molecule inhibitors of this kinase. CFI-400945 was shown to be a potent and selective PLK4 inhibitor exerting a dose-dependent effect on centriole biogenesis (Mason et al., 2014). At high concentration, CFI-400945 inhibits centriole duplication, while at low concentration it causes the generation of supernumerary centrosomes. Interestingly, in both cases, cells arrest or die (Mason et al., 2014). In the same study, the anti-cancer potential of CFI-400945 was also shown in mice and the drug is currently under evaluation in advanced cancer patients (NCT01954316).
Supernumerary centrosomes occur at high frequency in cancer cells but not in non-transformed cells and were originally proposed by Theodor Boveri to be linked to cancer development (Brinkley and Goepfert, 1998; Brinkley, 2001). Supernumerary centrosomes tend to cluster at mitosis forming pseudo-bipolar spindles to avoid multipolar mitoses that would result in the generation of unviable progeny (Ganem et al., 2009). Formation of pseudo-bipolar spindles where merotelic chromosome attachments is frequent, is among the major causes of aneuploidy (Ganem et al., 2009). The anti-fungal drug griseofulvin was shown to freeze the process of centrosome clustering (Raab et al., 2012) and since then a number of small molecule inhibitors of this process have been synthesized and examined (Kawamura et al., 2013; Ogden et al., 2014; Bhakta-Guha et al., 2015). Centrosome declustering drugs are, however, still in pre-clinical studies (Krämer et al., 2011; Pannu et al., 2014) given two main considerations: The first is that eliminating the subpopulation of cancer cells carrying centrosome amplifications in the heterogeneous collection of cells making up a tumor is yet to be proven beneficial in anticancer therapy. The second is that identification of individuals suitable to treatment with centrosome declustering drugs still awaits routine screening methods to define the genetic makeup of patients with centrosome amplification who would benefit of such treatment (Godinho and Pellman, 2014).
Mitotic Phosphatase Inhibitors
Members of the CDC25 family of protein phosphatases act as positive regulators of CDKs that are their unique targets (see above). The only report on CDC25 targeting drugs is for phase II clinical trials initiated in 2010 with IRC 083864 under the name Debio-0931 (Lavecchia et al., 2010), a drug that has previously shown activity against pancreatic and cervical cancer xenografts (Brezak et al., 2009). To date, LB-100 is the only know drug targeting the Ser/Thr phosphatase PP2A to have entered phase I trials in combination with cytotoxic drugs or irradiation for the indication “solid tumors” (NCT01837667) (Hong et al., 2015). Inhibition of enzymes with multiple functions such as PP2A, by many considered unfeasible due to the associated high toxicity of such treatments, was shown to be well-tolerated if the drug is administered intermittently (http://www.lixte.com/Product_Development.php). LB-100 has been granted licensing in Asia for treatment of Hepatocellular Carcinoma in December 2015 (http://adisinsight.springer.com/drugs/800037966).
Ubiquitin-Proteasome Inhibitors
The established role of ubiquitin-dependent pathways in the degradation of mitotic apparatus components has made them an ideal site of intervention in cancer therapy and possible applications of proteasome inhibitors to the treatment of cancer, their mode of action and mechanisms of resistance have been amply reviewed (Crawford et al., 2011; Zhang et al., 2013). Approval of Bortezomid over a decade ago for the indications multiple myeloma and multiple cell lymphoma paved the way to the discovery of candidates with reduced side effects and improved efficacy that are currently in clinical trial (Zhang et al., 2013). Specifically to mitosis, a new perspective was provided in a report describing the use of spindle poisons in conjunction with inhibition of the ATPase activity of components of the proteasome to increase apoptosis in cancer cells (Yamada and Gorbsky, 2006), offering further possibilities of intervention.
The majority of drugs that we discussed above halt cells before mitosis or in early mitosis. Prolonged treatment with drugs interfering with microtubules dynamics has been described to lead to mitotic exit—operationally defined mitotic slippage—(Brito and Rieder, 2006), a condition that leads to the acquisition of further aneuploidy and aggressiveness (Kuukasjarvi et al., 1997; McClelland et al., 2009). Hence, significant effort has been devoted in recent years to block mitotic exit. Inhibiting the interaction of CDC20 with APC/C by TAME (tosyl-L-arginine methyl ester) has shown to effectively halt cells in mitosis and channel them to death (Zeng et al., 2010; Zeng and King, 2012). The more recently developed APC/C inhibitor Apcin, showing the ability to bind CDC20 and to prevent ubiquitylation of D-box containing APC/C targets, has provided an additional means to block mitotic exit (Sackton et al., 2014). The combined use of Apcin and TAME was reported to synergistically halt mitotic exit, hence opening new therapeutic perspectives (Sackton et al., 2014).
In a similar fashion Nutlins were described to impair physical interaction between p53 and the E3 ubiquitin ligase MDM2, promoting p53 stabilization and enhancing its tumor suppressor activity (Vassilev et al., 2004; Vassilev, 2007). Enthusiasm for these drugs, however, was mitigated by two major drawbacks: first the observation that MDM2 interacts preferentially with wild-type p53 (Lukashchuk and Vousden, 2007) and, second, the report that Nutlins exert a cytostatic effects in p53-deficient cells, indicating that they do not solely inhibit the p53/MDM2 interaction (VanderBorght et al., 2006).
In conclusion, it is foreseeable that the development of novel and specific drugs targeting components of pathways that control mitosis and/or interfere with signals that fine-tune their function, in conjunction with stratification of patients based on their genetic background, will allow to better determine combination therapies for each individual patient, taking us a step closer to personalized medicine.
Author Contributions
SF conceived the review topic and wrote the manuscript. CG contributed to write the manuscript.
Funding
This work was supported by grants of the Promedica-Stiftung, the Stiftung zur Krebsbekämpfung and the Stiftung für wissenschaftliche Forschung.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Pavel Janscak and members of SF laboratory for critical reading of the manuscript as well as to Doriano Fabbro for helpful suggestions. We apologize to those colleagues whose work has not been cited due to space constraints.
References
Aguilera, A., and García-Muse, T. (2013). Causes of genome instability. Annu. Rev. Genet. 47, 1–32. doi: 10.1146/annurev-genet-111212-133232
Amon, A., Surana, U., Muroff, I., and Nasmyth, K. (1992). Regulation of p34CDC28 tyrosine phosphorylation is not required for entry into mitosis in S. cerevisiae. Nature 355, 368–371. doi: 10.1038/355368a0
Asghar, U., Witkiewicz, A. K., Turner, N. C., and Knudsen, E. S. (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146. doi: 10.1038/nrd4504
Atherton-Fessler, S., Parker, L. L., Geahlen, R. L., and Piwnica-Worms, H. (1993). Mechanisms of p34cdc2 regulation. Mol. Cell. Biol. 13, 1675–1685. doi: 10.1128/MCB.13.3.1675
Aydin, Ö. Z., Vermeulen, W., and Lans, H. (2014). ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle 13, 3016–3025. doi: 10.4161/15384101.2014.956551
Bahassi el, M., Myer, D. L., McKenney, R. J., Hennigan, R. F., and Stambrook, P. J. (2006). Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage. Mutat. Res. 596, 166–176. doi: 10.1016/j.mrfmmm.2005.12.002
Bahe, S., Stierhof, Y. D., Wilkinson, C. J., Leiss, F., and Nigg, E. A. (2005). Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 171, 27–33. doi: 10.1083/jcb.200504107
Bakhoum, S. F., Kabeche, L., Murnane, J. P., Zaki, B. I., and Compton, D. A. (2014). DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 4, 1281–1289. doi: 10.1158/2159-8290.CD-14-0403
Barabási, A. L., and Oltvai, Z. N. (2004). Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113. doi: 10.1038/nrg1272
Baron, A. P., von Schubert, C., Cubizolles, F., Siemeister, G., Hitchcock, M., Mengel, A., et al. (2016). Probing the catalytic functions of Bub1 kinase using the small molecule inhibitors BAY-320 and BAY-524. Elife 5:e12187. doi: 10.7554/eLife.12187
Baroni, E., Viscardi, V., Cartagena-Lirola, H., Lucchini, G., and Longhese, M. P. (2004). The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol. Cell. Biol. 24, 4151–4165. doi: 10.1128/MCB.24.10.4151-4165.2004
Barr, F. A., Silljé, H. H., and Nigg, E. A. (2004). Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5, 429–440. doi: 10.1038/nrm1401
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. doi: 10.1038/nature05268
Basto, R., Brunk, K., Vinadogrova, T., Peel, N., Franz, A., Khodjakov, A., et al. (2008). Centrosome amplification can initiate tumorigenesis in flies. Cell 133, 1032–1042. doi: 10.1016/j.cell.2008.05.039
Baumann, C., Körner, R., Hofmann, K., and Nigg, E. A. (2007). PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 128, 101–114. doi: 10.1016/j.cell.2006.11.041
Bavetsias, V., and Linardopoulos, S. (2015). Aurora kinase inhibitors: current status and outlook. Front. Oncol. 5:278. doi: 10.3389/fonc.2015.00278
Bekker-Jensen, S., Rendtlew Danielsen, J., Fugger, K., Gromova, I., Nerstedt, A., Lukas, C., et al. (2010). HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 12, 80–86; sup pp 1–12. doi: 10.1038/ncb2008
Benada, J., Burdová, K., Lidak, T., von Morgen, P., and Macurek, L. (2015). Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 14, 219–231. doi: 10.4161/15384101.2014.977067
Bettencourt-Dias, M., Rodrigues-Martins, A., Carpenter, L., Riparbelli, M., Lehmann, L., Gatt, M. K., et al. (2005). SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 15, 2199–2207. doi: 10.1016/j.cub.2005.11.042
Bhakta-Guha, D., Saeed, M. E., Greten, H. J., and Efferth, T. (2015). Dis-organizing centrosomal clusters: specific cancer therapy for a generic spread? Curr. Med. Chem. 22, 685–694. doi: 10.2174/0929867322666141212114529
Bhatia, P., Menigatti, M., Brocard, M., Morley, S. J., and Ferrari, S. (2010). Mitotic DNA damage targets the Aurora A/TPX2 complex. Cell Cycle 9, 4592–4599. doi: 10.4161/cc.9.22.13665
Boddy, M. N., Furnari, B., Mondesert, O., and Russell, P. (1998). Replication checkpoint enforced by kinases Cds1 and Chk1. Science 280, 909–912. doi: 10.1126/science.280.5365.909
Boettcher, B., and Barral, Y. (2013). The cell biology of open and closed mitosis. Nucleus 4, 160–165. doi: 10.4161/nucl.24676
Bologna, S., and Ferrari, S. (2013). It takes two to tango: Ubiquitin and SUMO in the DNA damage response. Front. Genet. 4:106. doi: 10.3389/fgene.2013.00106
Bothos, J., Summers, M. K., Venere, M., Scolnick, D. M., and Halazonetis, T. D. (2003). The Chfr mitotic checkpoint protein functions with Ubc13-Mms2 to form Lys63-linked polyubiquitin chains. Oncogene 22, 7101–7107. doi: 10.1038/sj.onc.1206831
Branzei, D., and Foiani, M. (2010). Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219. doi: 10.1038/nrm2852
Brazeau, J. F., and Rosse, G. (2014). Novel cycloalkenepyrazoles as inhibitors of bub1 kinase. ACS Med. Chem. Lett. 5, 280–281. doi: 10.1021/ml5000492
Bremm, A., and Komander, D. (2012). Synthesis and analysis of K11-linked ubiquitin chains. Methods Mol. Biol. 832, 219–228. doi: 10.1007/978-1-61779-474-2_15
Brezak, M. C., Valette, A., Quaranta, M., Contour-Galcera, M. O., Jullien, D., Lavergne, O., et al. (2009). IRC-083864, a novel bis quinone inhibitor of CDC25 phosphatases active against human cancer cells. Int. J. Cancer 124, 1449–1456. doi: 10.1002/ijc.24080
Brinkley, B. R. (2001). Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 11, 18–21. doi: 10.1016/S0962-8924(00)01872-9
Brinkley, B. R., and Goepfert, T. M. (1998). Supernumerary centrosomes and cancer: Boveri's hypothesis resurrected. Cell Motil. Cytoskeleton 41, 281–288.
Brito, D. A., and Rieder, C. L. (2006). Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 16, 1194–1200. doi: 10.1016/j.cub.2006.04.043
Broderick, R., Nieminuszczy, J., Blackford, A. N., Winczura, A., and Niedzwiedz, W. (2015). TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat. Commun. 6, 6572. doi: 10.1038/ncomms7572
Bulavin, D. V., Higashimoto, Y., Popoff, I. J., Gaarde, W. A., Basrur, V., Potapova, O., et al. (2001). Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107. doi: 10.1038/35075107
Carmena, M., Ruchaud, S., and Earnshaw, W. C. (2009). Making the Auroras glow: regulation of Aurora, A., and B kinase function by interacting proteins. Curr. Opin. Cell Biol. 21, 796–805. doi: 10.1016/j.ceb.2009.09.008
Carpinelli, P., Ceruti, R., Giorgini, M. L., Cappella, P., Gianellini, L., Croci, V., et al. (2007). PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol. Cancer Ther. 6, 3158–3168. doi: 10.1158/1535-7163.MCT-07-0444
Castro, A., Bernis, C., Vigneron, S., Labbé, J. C., and Lorca, T. (2005). The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene 24, 314–325. doi: 10.1038/sj.onc.1207973
Cesare, A. J. (2014). Mitosis, double strand break repair, and telomeres: a view from the end: how telomeres and the DNA damage response cooperate during mitosis to maintain genome stability. Bioessays 36, 1054–1061. doi: 10.1002/bies.201400104
Chan, K. L., and Hickson, I. D. (2009). On the origins of ultra-fine anaphase bridges. Cell Cycle 8, 3065–3066. doi: 10.4161/cc.8.19.9513
Chan, K. L., Palmai-Pallag, T., Ying, S., and Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760. doi: 10.1038/ncb1882
Chan, K. S., Koh, C. G., and Li, H. Y. (2012). Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 3, e411. doi: 10.1038/cddis.2012.148
Chan, Y. W., and West, S. C. (2014). Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat. Commun. 5, 4844. doi: 10.1038/ncomms5844
Chang, Y. C., Chou, F. P., Huang, H. P., Hsu, J. D., and Wang, C. J. (2004). Inhibition of cell cycle progression by penta-acetyl geniposide in rat C6 glioma cells. Toxicol. Appl. Pharmacol. 198, 11–20. doi: 10.1016/j.taap.2004.03.004
Chauhan, D., Tian, Z., Zhou, B., Kuhn, D., Orlowski, R., Raje, N., et al. (2011). In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 17, 5311–5321. doi: 10.1158/1078-0432.CCR-11-0476
Cheeseman, I. M. (2014). The kinetochore. Cold Spring Harb. Perspect. Biol. 6:a015826. doi: 10.1101/cshperspect.a015826
Cheeseman, I. M., Chappie, J. S., Wilson-Kubalek, E. M., and Desai, A. (2006). The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997. doi: 10.1016/j.cell.2006.09.039
Cheeseman, I. M., and Desai, A. (2008). Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33–46. doi: 10.1038/nrm2310
Chin, C. F., and Yeong, F. M. (2010). Safeguarding entry into mitosis: the antephase checkpoint. Mol. Cell Biol. 30, 22–32. doi: 10.1128/MCB.00687-09
Chiolo, I., Carotenuto, W., Maffioletti, G., Petrini, J. H., Foiani, M., and Liberi, G. (2005). Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol. Cell. Biol. 25, 5738–5751. doi: 10.1128/MCB.25.13.5738-5751.2005
Chng, W. J., Braggio, E., Mulligan, G., Bryant, B., Remstein, E., Valdez, R., et al. (2008). The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood 111, 1603–1609. doi: 10.1182/blood-2007-06-097774
Cho, H. J., Oh, Y. J., Han, S. H., Chung, H. J., Kim, C. H., Lee, N. S., et al. (2013). Cdk1 protein-mediated phosphorylation of receptor-associated protein 80 (RAP80) serine 677 modulates DNA damage-induced G2/M checkpoint and cell survival. J. Biol. Chem. 288, 3768–3776. doi: 10.1074/jbc.M112.401299
Choi, E., and Lee, H. (2008). Chromosome damage in mitosis induces BubR1 activation and prometaphase arrest. FEBS Lett. 582, 1700–1706. doi: 10.1016/j.febslet.2008.04.028
Chow, J. P., Siu, W. Y., Fung, T. K., Chan, W. M., Lau, A., Arooz, T., et al. (2003). DNA damage during the spindle-assembly checkpoint degrades CDC25A, inhibits cyclin-CDC2 complexes, and reverses cells to interphase. Mol. Biol. Cell 14, 3989–4002. doi: 10.1091/mbc.E03-03-0168
Ciosk, R., Zachariae, W., Michaelis, C., Shevchenko, A., Mann, M., and Nasmyth, K. (1998). An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 93, 1067–1076. doi: 10.1016/S0092-8674(00)81211-8
Cirstea, D., Hideshima, T., Santo, L., Eda, H., Mishima, Y., Nemani, N., et al. (2013). Small-molecule multi-targeted kinase inhibitor RGB-286638 triggers P53-dependent and -independent anti-multiple myeloma activity through inhibition of transcriptional CDKs. Leukemia 27, 2366–2375. doi: 10.1038/leu.2013.194
Colombo, R., Burbridge, M., Rodriguez, M., Cantero, F., Caldarelli, M., Giorgini, M. L., et al. (2015). Abstract 1638: Preclinical characterization of the novel TTK kinase inhibitor S81694 for the treatment of triple negative breast cancer. Cancer Res. 75:1638. doi: 10.1158/1538-7445.AM2015-1638
Craney, A., Kelly, A., Jia, L., Fedrigo, I., Yu, H., and Rape, M. (2016). Control of APC/C-dependent ubiquitin chain elongation by reversible phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 113, 1540–1545. doi: 10.1073/pnas.1522423113
Crawford, L. J., Walker, B., and Irvine, A. E. (2011). Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 5, 101–110. doi: 10.1007/s12079-011-0121-7
Curtin, N. J. (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 12, 801–817. doi: 10.1038/nrc3399
Dantuma, N. P., and van Attikum, H. (2016). Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 35, 6–23. doi: 10.15252/embj.201592595
D'Assoro, A. B., Haddad, T., and Galanis, E. (2015). Aurora-A kinase as a promising therapeutic target in cancer. Front. Oncol. 5:295. doi: 10.3389/fonc.2015.00295
D'Avino, P. P. (2009). How to scaffold the contractile ring for a safe cytokinesis - lessons from Anillin-related proteins. J. Cell Sci. 122, 1071–1079. doi: 10.1242/jcs.034785
De Azevedo, W. F., Leclerc, S., Meijer, L., Havlicek, L., Strnad, M., and Kim, S. H. (1997). Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 243, 518–526. doi: 10.1111/j.1432-1033.1997.0518a.x
De Azevedo, W. F. Jr., Mueller-Dieckmann, H. J., Schulze-Gahmen, U., Worland, P. J., Sausville, E., and Kim, S. H. (1996). Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. U.S.A. 93, 2735–2740. doi: 10.1073/pnas.93.7.2735
Degenhardt, Y., and Lampkin, T. (2010). Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. 16, 384–389. doi: 10.1158/1078-0432.CCR-09-1380
Delacroix, S., Wagner, J. M., Kobayashi, M., Yamamoto, K., and Karnitz, L. M. (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21, 1472–1477. doi: 10.1101/gad.1547007
Dephoure, N., Zhou, C., Villén, J., Beausoleil, S. A., Bakalarski, C. E., Elledge, S. J., et al. (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767. doi: 10.1073/pnas.0805139105
Dodson, H., Bourke, E., Jeffers, L. J., Vagnarelli, P., Sonoda, E., Takeda, S., et al. (2004). Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 23, 3864–3873. doi: 10.1038/sj.emboj.7600393
Doménech, E., and Malumbres, M. (2013). Mitosis-targeting therapies: a troubleshooting guide. Curr. Opin. Pharmacol. 13, 519–528. doi: 10.1016/j.coph.2013.03.011
Dominguez-Brauer, C., Thu, K. L., Mason, J. M., Blaser, H., Bray, M. R., and Mak, T. W. (2015). Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 60, 524–536. doi: 10.1016/j.molcel.2015.11.006
Donzelli, M., and Draetta, G. F. (2003). Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 4, 671–677. doi: 10.1038/sj.embor.embor887
Durkin, S. G., and Glover, T. W. (2007). Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192. doi: 10.1146/annurev.genet.41.042007.165900
Eckerdt, F., Yuan, J., and Strebhardt, K. (2005). Polo-like kinases and oncogenesis. Oncogene 24, 267–276. doi: 10.1038/sj.onc.1208273
Eissler, C. L., Mazón, G., Powers, B. L., Savinov, S. N., Symington, L. S., and Hall, M. C. (2014). The Cdk/cDc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol. Cell 54, 80–93. doi: 10.1016/j.molcel.2014.02.012
Enserink, J. M., Hombauer, H., Huang, M. E., and Kolodner, R. D. (2009). Cdc28/Cdk1 positively and negatively affects genome stability in S. cerevisiae. J Cell Biol 185, 423–437. doi: 10.1083/jcb.200811083
Enserink, J. M., and Kolodner, R. D. (2010). An overview of Cdk1-controlled targets and processes. Cell Div. 5:11. doi: 10.1186/1747-1028-5-11
Esashi, F., Christ, N., Gannon, J., Liu, Y., Hunt, T., Jasin, M., et al. (2005). CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434, 598–604. doi: 10.1038/nature03404
Falchook, G. S., Bastida, C. C., and Kurzrock, R. (2015). Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin. Oncol. 42, 832–848. doi: 10.1053/j.seminoncol.2015.09.022
Ferrari, S., Marin, O., Pagano, M. A., Meggio, F., Hess, D., El-Shemerly, M., et al. (2005). Aurora-A site specificity: a study with synthetic peptide substrates. Biochem. J. 390, 293–302. doi: 10.1042/BJ20050343
Foley, E. A., Maldonado, M., and Kapoor, T. M. (2011). Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat. Cell Biol. 13, 1265–1271. doi: 10.1038/ncb2327
Gabrielli, B. G., De Souza, C. P., Tonks, I. D., Clark, J. M., Hayward, N. K., and Ellem, K. A. (1996). Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 109 (Pt 5), 1081–1093.
Galons, H., Oumata, N., Gloulou, O., and Meijer, L. (2013). Cyclin-dependent kinase inhibitors closer to market launch? Expert Opin. Ther. Pat. 23, 945–963. doi: 10.1517/13543776.2013.789861
Ganem, N. J., Godinho, S. A., and Pellman, D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282. doi: 10.1038/nature08136
Garnett, M. J., Mansfeld, J., Godwin, C., Matsusaka, T., Wu, J., Russell, P., et al. (2009). UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat. Cell Biol. 11, 1363–1369. doi: 10.1038/ncb1983
Gelot, C., Magdalou, I., and Lopez, B. S. (2015). Replication stress in Mammalian cells and its consequences for mitosis. Genes (Basel). 6, 267–298. doi: 10.3390/genes6020267
Gerard, C., Tyson, J. J., Coudreuse, D., and Novak, B. (2015). Cell cycle control by a minimal Cdk network. PLoS Comput. Biol. 11:e1004056. doi: 10.1371/journal.pcbi.1004056
Giunta, S., Belotserkovskaya, R., and Jackson, S. P. (2010). DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 190, 197–207. doi: 10.1083/jcb.200911156
Godinho, S. A., and Pellman, D. (2014). Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369:20130467. doi: 10.1098/rstb.2013.0467
Goh, K. C., Novotny-Diermayr, V., Hart, S., Ong, L. C., Loh, Y. K., Cheong, A., et al. (2012). TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 26, 236–243. doi: 10.1038/leu.2011.218
Gomez-Godinez, V., Wu, T., Sherman, A. J., Lee, C. S., Liaw, L. H., Zhongsheng, Y., et al. (2010). Analysis of DNA double-strand break response and chromatin structure in mitosis using laser microirradiation. Nucleic Acids Res. 38, e202. doi: 10.1093/nar/gkq836
Görgün, G., Calabrese, E., Hideshima, T., Ecsedy, J., Perrone, G., Mani, M., et al. (2010). A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 115, 5202–5213. doi: 10.1182/blood-2009-12-259523
Goshima, G., and Scholey, J. M. (2010). Control of mitotic spindle length. Annu. Rev. Cell Dev. Biol. 26, 21–57. doi: 10.1146/annurev-cellbio-100109-104006
Gottesfeld, J. M., and Forbes, D. J. (1997). Mitotic repression of the transcriptional machinery. Trends Biochem. Sci. 22, 197–202. doi: 10.1016/S0968-0004(97)01045-1
Gould, K. L., and Nurse, P. (1989). Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature 342, 39–45. doi: 10.1038/342039a0
Habedanck, R., Stierhof, Y. D., Wilkinson, C. J., and Nigg, E. A. (2005). The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 7, 1140–1146. doi: 10.1038/ncb1320
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hardwicke, M. A., Oleykowski, C. A., Plant, R., Wang, J., Liao, Q., Moss, K., et al. (2009). GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol. Cancer Ther. 8, 1808–1817. doi: 10.1158/1535-7163.MCT-09-0041
Hardy, T., Lee, M., Hames, R. S., Prosser, S. L., Cheary, D. M., Samant, M. D., et al. (2014). Multisite phosphorylation of C-Nap1 releases it from Cep135 to trigger centrosome disjunction. J. Cell Sci. 127, 2493–2506. doi: 10.1242/jcs.142331
Harrigan, J. A., Belotserkovskaya, R., Coates, J., Dimitrova, D. S., Polo, S. E., Bradshaw, C. R., et al. (2011). Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108. doi: 10.1083/jcb.201011083
Harrington, E. A., Bebbington, D., Moore, J., Rasmussen, R. K., Ajose-Adeogun, A. O., Nakayama, T., et al. (2004). VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 10, 262–267. doi: 10.1038/nm1003
Hauf, S., Roitinger, E., Koch, B., Dittrich, C. M., Mechtler, K., and Peters, J. M. (2005). Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 3:e69. doi: 10.1371/journal.pbio.0030069
Heald, R., McLoughlin, M., and McKeon, F. (1993). Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74, 463–474. doi: 10.1016/0092-8674(93)80048-J
Heald, R., Tournebize, R., Blank, T., Sandaltzopoulos, R., Becker, P., Hyman, A., et al. (1996). Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature 382, 420–425. doi: 10.1038/382420a0
Heald, R., Tournebize, R., Habermann, A., Karsenti, E., and Hyman, A. (1997). Spindle assembly in Xenopus egg extracts: respective roles of centrosomes and microtubule self-organization. J. Cell Biol. 138, 615–628. doi: 10.1083/jcb.138.3.615
Heller, J. D., Kuo, J., Wu, T. C., Kast, W. M., and Huang, R. C. (2001). Tetra-O-methyl nordihydroguaiaretic acid induces G2 arrest in mammalian cells and exhibits tumoricidal activity in vivo. Cancer Res. 61, 5499–5504.
Hendriks, I. A., D'Souza, R. C., Yang, B., Verlaan-de Vries, M., Mann, M., and Vertegaal, A. C. (2014). Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 21, 927–936. doi: 10.1038/nsmb.2890
Hengeveld, R. C., de Boer, H. R., Schoonen, P. M., de Vries, E. G., Lens, S. M., and van Vugt, M. A. (2015). Rif1 is required for resolution of ultrafine DNA bridges in anaphase to ensure genomic stability. Dev. Cell 34, 466–474. doi: 10.1016/j.devcel.2015.06.014
Hideshima, T., Richardson, P., Chauhan, D., Palombella, V. J., Elliott, P. J., Adams, J., et al. (2001). The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 61, 3071–3076.
Hirai, H., Iwasawa, Y., Okada, M., Arai, T., Nishibata, T., Kobayashi, M., et al. (2009). Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 8, 2992–3000. doi: 10.1158/1535-7163.MCT-09-0463
Hirano, T. (2012). Condensins: universal organizers of chromosomes with diverse functions. Genes Dev. 26, 1659–1678. doi: 10.1101/gad.194746.112
Hirano, T. (2015). Chromosome Dynamics during Mitosis. Cold Spring Harb. Perspect. Biol. 7:a015792. doi: 10.1101/cshperspect.a015792
Hoffmann, I., Clarke, P. R., Marcote, M. J., Karsenti, E., and Draetta, G. (1993). Phosphorylation and activation of human cdc25-C by cdc2–cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 12, 53–63.
Holland, A. J., and Cleveland, D. W. (2009). Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487. doi: 10.1038/nrm2718
Holland, A. J., Lan, W., Niessen, S., Hoover, H., and Cleveland, D. W. (2010). Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J. Cell Biol. 188, 191–198. doi: 10.1083/jcb.200911102
Holtrich, U., Wolf, G., Bräuninger, A., Karn, T., Böhme, B., Rübsamen-Waigmann, H., et al. (1994). Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc. Natl. Acad. Sci. U.S.A. 91, 1736–1740. doi: 10.1073/pnas.91.5.1736
Hong, C. S., Ho, W., Zhang, C., Yang, C., Elder, J. B., and Zhuang, Z. (2015). LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 16, 821–833. doi: 10.1080/15384047.2015.1040961
Howard, S., Berdini, V., Boulstridge, J. A., Carr, M. G., Cross, D. M., Curry, J., et al. (2009). Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J. Med. Chem. 52, 379–388. doi: 10.1021/jm800984v
Hu, Z., Fan, C., Oh, D. S., Marron, J. S., He, X., Qaqish, B. F., et al. (2006). The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 7:96. doi: 10.1186/1471-2164-7-96
Huertas, P., Cortés-Ledesma, F., Sartori, A. A., Aguilera, A., and Jackson, S. P. (2008). CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692. doi: 10.1038/nature07215
Imreh, G., Norberg, H. V., Imreh, S., and Zhivotovsky, B. (2011). Chromosomal breaks during mitotic catastrophe trigger gammaH2AX-ATM-p53-mediated apoptosis. J. Cell Sci. 124, 2951–2963. doi: 10.1242/jcs.081612
Ira, G., Pellicioli, A., Balijja, A., Wang, X., Fiorani, S., Carotenuto, W., et al. (2004). DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017. doi: 10.1038/nature02964
Izumi, T., and Maller, J. L. (1993). Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol. Biol. Cell 4, 1337–1350. doi: 10.1091/mbc.4.12.1337
Jackson, S. P., and Durocher, D. (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807. doi: 10.1016/j.molcel.2013.01.017
Jani, J. P., Arcari, J., Bernardo, V., Bhattacharya, S. K., Briere, D., Cohen, B. D., et al. (2010). PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol. Cancer Ther. 9, 883–894. doi: 10.1158/1535-7163.MCT-09-0915
Joshi, K. S., Rathos, M. J., Joshi, R. D., Sivakumar, M., Mascarenhas, M., Kamble, S., et al. (2007). In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276-00. Mol. Cancer Ther. 6, 918–925. doi: 10.1158/1535-7163.MCT-06-0613
Kang, D., Chen, J., Wong, J., and Fang, G. (2002). The checkpoint protein Chfr is a ligase that ubiquitinates Plk1 and inhibits Cdc2 at the G2 to M transition. J. Cell Biol. 156, 249–259. doi: 10.1083/jcb.200108016
Kang, J., Yang, M., Li, B., Qi, W., Zhang, C., Shokat, K. M., et al. (2008). Structure and substrate recruitment of the human spindle checkpoint kinase Bub1. Mol. Cell 32, 394–405. doi: 10.1016/j.molcel.2008.09.017
Kang, Y. H., Park, J. E., Yu, L. R., Soung, N. K., Yun, S. M., Bang, J. K., et al. (2006). Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell 24, 409–422. doi: 10.1016/j.molcel.2006.10.016
Karsenti, E., and Vernos, I. (2001). The mitotic spindle: a self-made machine. Science 294, 543–547. doi: 10.1126/science.1063488
Kastan, M. B., and Bartek, J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. doi: 10.1038/nature03097
Kawamura, E., Fielding, A. B., Kannan, N., Balgi, A., Eaves, C. J., Roberge, M., et al. (2013). Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 4, 1763–1776. doi: 10.18632/oncotarget.1198
Kenney, R. D., and Heald, R. (2006). Essential roles for cohesin in kinetochore and spindle function in Xenopus egg extracts. J. Cell Sci. 119, 5057–5066. doi: 10.1242/jcs.03277
Khan, J., Ezan, F., Crémet, J. Y., Fautrel, A., Gilot, D., Lambert, M., et al. (2011). Overexpression of active Aurora-C kinase results in cell transformation and tumour formation. PLoS ONE 6:e26512. doi: 10.1371/journal.pone.0026512
Kim, W., Bennett, E. J., Huttlin, E. L., Guo, A., Li, J., Possemato, A., et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340. doi: 10.1016/j.molcel.2011.08.025
Kim, Y., Holland, A. J., Lan, W., and Cleveland, D. W. (2010). Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 142, 444–455. doi: 10.1016/j.cell.2010.06.039
Kitajima, T. S., Sakuno, T., Ishiguro, K., Iemura, S., Natsume, T., Kawashima, S. A., et al. (2006). Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 441, 46–52. doi: 10.1038/nature04663
Kortuem, K. M., and Stewart, A. K. (2013). Carfilzomib. Blood 121, 893–897. doi: 10.1182/blood-2012-10-459883
Krämer, A., Maier, B., and Bartek, J. (2011). Centrosome clustering and chromosomal (in)stability: a matter of life and death. Mol. Oncol. 5, 324–335. doi: 10.1016/j.molonc.2011.05.003
Krystyniak, A., Garcia-Echeverria, C., Prigent, C., and Ferrari, S. (2006). Inhibition of Aurora A in response to DNA damage. Oncogene 25, 338–348.
Kumagai, A., Lee, J., Yoo, H. Y., and Dunphy, W. G. (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. doi: 10.1016/j.cell.2005.12.041
Kuukasjärvi, T., Karhu, R., Tanner, M., Kähkönen, M., Schäffer, A., Nupponen, N., et al. (1997). Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 57, 1597–1604.
Lavecchia, A., Di Giovanni, C., and Novellino, E. (2010). Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert Opin. Ther. Pat. 20, 405–425. doi: 10.1517/13543771003623232
Lee, D. H., Acharya, S. S., Kwon, M., Drane, P., Guan, Y., Adelmant, G., et al. (2014). Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 54, 512–525. doi: 10.1016/j.molcel.2014.03.020
Lee, J., Kumagai, A., and Dunphy, W. G. (2007). The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 282, 28036–28044. doi: 10.1074/jbc.M704635200
Lee, M., Daniels, M. J., and Venkitaraman, A. R. (2004). Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 23, 865–872. doi: 10.1038/sj.onc.1207223
Lemaître, C., and Soutoglou, E. (2014). Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair (Amst). 19, 163–168. doi: 10.1016/j.dnarep.2014.03.015
Lénárt, P., Petronczki, M., Steegmaier, M., Di Fiore, B., Lipp, J. J., Hoffmann, M., et al. (2007). The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17, 304–315. doi: 10.1016/j.cub.2006.12.046
Leng, M., Chan, D. W., Luo, H., Zhu, C., Qin, J., and Wang, Y. (2006). MPS1-dependent mitotic BLM phosphorylation is important for chromosome stability. Proc. Natl. Acad. Sci. U.S.A. 103, 11485–11490. doi: 10.1073/pnas.0601828103
Lin, H. R., Ting, N. S., Qin, J., and Lee, W. H. (2003). M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex. J. Biol. Chem. 278, 35979–35987. doi: 10.1074/jbc.M210659200
Liu, C., Wu, J., Paudyal, S. C., You, Z., and Yu, X. (2013a). CHFR is important for the first wave of ubiquitination at DNA damage sites. Nucleic Acids Res. 41, 1698–1710. doi: 10.1093/nar/gks1278
Liu, D., Vader, G., Vromans, M. J., Lampson, M. A., and Lens, S. M. (2009). Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 323, 1350–1353. doi: 10.1126/science.1167000
Liu, H., Rankin, S., and Yu, H. (2013b). Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat. Cell Biol. 15, 40–49. doi: 10.1038/ncb2637
Liu, Y., Nielsen, C. F., Yao, Q., and Hickson, I. D. (2014). The origins and processing of ultra fine anaphase DNA bridges. Curr. Opin. Genet. Dev. 26, 1–5. doi: 10.1016/j.gde.2014.03.003
Lorenz, D. M., Jeng, A., and Deem, M. W. (2011). The emergence of modularity in biological systems. Phys. Life Rev. 8, 129–160. doi: 10.1016/j.plrev.2011.02.003
Lu, J., Kovach, J. S., Johnson, F., Chiang, J., Hodes, R., Lonser, R., et al. (2009). Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl. Acad. Sci. U.S.A. 106, 11697–11702. doi: 10.1073/pnas.0905930106
Lukas, C., Savic, V., Bekker-Jensen, S., Doil, C., Neumann, B., Pedersen, R. S., et al. (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253. doi: 10.1038/ncb2201
Lukashchuk, N., and Vousden, K. H. (2007). Ubiquitination and degradation of mutant p53. Mol. Cell Biol. 27, 8284–8295. doi: 10.1128/MCB.00050-07
Ma, H. T., and Poon, R. Y. (2011). How protein kinases co-ordinate mitosis in animal cells. Biochem. J. 435, 17–31. doi: 10.1042/BJ20100284
Macmillan, J. C., Hudson, J. W., Bull, S., Dennis, J. W., and Swallow, C. J. (2001). Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann. Surg. Oncol. 8, 729–740. doi: 10.1007/s10434-001-0729-6
Macurek, L., Benada, J., Müllers, E., Halim, V. A., Krejcíková, K., Burdová, K., et al. (2013). Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle 12, 251–262. doi: 10.4161/cc.23057
Macurek, L., Lindqvist, A., Lim, D., Lampson, M. A., Klompmaker, R., Freire, R., et al. (2008). Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 455, 119–123. doi: 10.1038/nature07185
Manfredi, M. G., Ecsedy, J. A., Chakravarty, A., Silverman, L., Zhang, M., Hoar, K. M., et al. (2011). Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 17, 7614–7624. doi: 10.1158/1078-0432.CCR-11-1536
Manfredi, M. G., Ecsedy, J. A., Meetze, K. A., Balani, S. K., Burenkova, O., Chen, W., et al. (2007). Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc. Natl. Acad. Sci. U.S.A. 104, 4106–4111. doi: 10.1073/pnas.0608798104
Mardilovich, K., Baugh, M., Crighton, D., Kowalczyk, D., Gabrielsen, M., Munro, J., et al. (2015). LIM kinase inhibitors disrupt mitotic microtubule organization and impair tumor cell proliferation. Oncotarget 6, 38469–38486. doi: 10.18632/oncotarget.6288
Martínez-Bálbas, M. A., Dey, A., Rabindran, S. K., Ozato, K., and Wu, C. (1995). Displacement of sequence-specific transcription factors from mitotic chromatin. Cell 83, 29–38. doi: 10.1016/0092-8674(95)90231-7
Marzo, I., and Naval, J. (2013). Antimitotic drugs in cancer chemotherapy: promises and pitfalls. Biochem. Pharmacol. 86, 703–710. doi: 10.1016/j.bcp.2013.07.010
Mason, J. M., Lin, D. C., Wei, X., Che, Y., Yao, Y., Kiarash, R., et al. (2014). Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 26, 163–176. doi: 10.1016/j.ccr.2014.05.006
Matos, J., Blanco, M. G., Maslen, S., Skehel, J. M., and West, S. C. (2011). Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147, 158–172. doi: 10.1016/j.cell.2011.08.032
Matos, J., and West, S. C. (2014). Holliday junction resolution: regulation in space and time. DNA Repair (Amst). 19, 176–181. doi: 10.1016/j.dnarep.2014.03.013
Matsumoto, M. L., Wickliffe, K. E., Dong, K. C., Yu, C., Bosanac, I., Bustos, D., et al. (2010). K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell 39, 477–484. doi: 10.1016/j.molcel.2010.07.001
Matsusaka, T., and Pines, J. (2004). Chfr acts with the p38 stress kinases to block entry to mitosis in mammalian cells. J. Cell Biol. 166, 507–516. doi: 10.1083/jcb.200401139
Mazzarello, P. (1999). A unifying concept: the history of cell theory. Nat. Cell Biol. 1, E13–E15. doi: 10.1038/8964
McClelland, S. E., Burrell, R. A., and Swanton, C. (2009). Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle 8, 3262–3266. doi: 10.4161/cc.8.20.9690
McLaughlin, J., Markovtsov, V., Li, H., Wong, S., Gelman, M., Zhu, Y., et al. (2010). Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. J. Cancer Res. Clin. Oncol. 136, 99–113. doi: 10.1007/s00432-009-0641-1
Mertins, P., Qiao, J. W., Patel, J., Udeshi, N. D., Clauser, K. R., Mani, D. R., et al. (2013). Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637. doi: 10.1038/nmeth.2518
Mikhailov, A., Cole, R. W., and Rieder, C. L. (2002). DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 12, 1797–1806. doi: 10.1016/S0960-9822(02)01226-5
Mikhailov, A., Shinohara, M., and Rieder, C. L. (2004). Topoisomerase, I. I., and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 166, 517–526. doi: 10.1083/jcb.200405167
Milhollen, M. A., Shi, J., Traore, T., Huck, J., Sappal, D., Duffy, J., et al. (2015). Abstract A164: The small molecule UAE inhibitor TAK-243 (MLN7243) prevents DNA damage repair and reduces cell viability/tumor growth when combined with radiation, carboplatin and docetaxel. Mol. Cancer Ther. 14:A164. doi: 10.1158/1535-7163.TARG-15-A164
Miller, L. D., Smeds, J., George, J., Vega, V. B., Vergara, L., Ploner, A., et al. (2005). An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. U.S.A. 102, 13550–13555. doi: 10.1073/pnas.0506230102
Minocherhomji, S., Ying, S., Bjerregaard, V. A., Bursomanno, S., Aleliunaite, A., Wu, W., et al. (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. doi: 10.1038/nature16139
Mito, K., Kashima, K., Kikuchi, H., Daa, T., Nakayama, I., and Yokoyama, S. (2005). Expression of Polo-Like Kinase (PLK1) in non-Hodgkin's lymphomas. Leuk. Lymphoma 46, 225–231. doi: 10.1080/10428190400015709
Morrison, C., and Rieder, C. L. (2004). Chromosome damage and progression into and through mitosis in vertebrates. DNA Repair (Amst). 3, 1133–1139. doi: 10.1016/j.dnarep.2004.03.005
Mortlock, A. A., Foote, K. M., Heron, N. M., Jung, F. H., Pasquet, G., Lohmann, J. J., et al. (2007). Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 50, 2213–2224. doi: 10.1021/jm061335f
Musacchio, A., and Salmon, E. D. (2007). The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393. doi: 10.1038/nrm2163
Naim, V., and Rosselli, F. (2009). The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 11, 761–768. doi: 10.1038/ncb1883
Naim, V., Wilhelm, T., Debatisse, M., and Rosselli, F. (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015. doi: 10.1038/ncb2793
Nelson, G., Buhmann, M., and von Zglinicki, T. (2009). DNA damage foci in mitosis are devoid of 53BP1. Cell Cycle 8, 3379–3383. doi: 10.4161/cc.8.20.9857
Nigg, E. A. (2001). Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2, 21–32. doi: 10.1038/35048096
Nyati, S., Schinske-Sebolt, K., Pitchiaya, S., Chekhovskiy, K., Chator, A., Chaudhry, N., et al. (2015). The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-beta signaling. Sci. Signal. 8, ra1. doi: 10.1126/scisignal.2005379
Nyberg, K. A., Michelson, R. J., Putnam, C. W., and Weinert, T. A. (2002). Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656. doi: 10.1146/annurev.genet.36.060402.113540
Ogden, A., Cheng, A., Rida, P. C., Pannu, V., Osan, R., Clewley, R., et al. (2014). Quantitative multi-parametric evaluation of centrosome declustering drugs: centrosome amplification, mitotic phenotype, cell cycle and death. Cell Death Dis. 5, e1204. doi: 10.1038/cddis.2014.164
Oh, Y. M., Kwon, Y. E., Kim, J. M., Bae, S. J., Lee, B. K., Yoo, S. J., et al. (2009). Chfr is linked to tumour metastasis through the downregulation of HDAC1. Nat. Cell Biol. 11, 295–302. doi: 10.1038/ncb1837
Orthwein, A., Fradet-Turcotte, A., Noordermeer, S. M., Canny, M. D., Brun, C. M., Strecker, J., et al. (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. doi: 10.1126/science.1248024
Oslob, J. D., Romanowski, M. J., Allen, D. A., Baskaran, S., Bui, M., Elling, R. A., et al. (2008). Discovery of a potent and selective aurora kinase inhibitor. Bioorg. Med. Chem. Lett. 18, 4880–4884. doi: 10.1016/j.bmcl.2008.07.073
Pangilinan, F., and Spencer, F. (1996). Abnormal kinetochore structure activates the spindle assembly checkpoint in budding yeast. Mol. Biol. Cell 7, 1195–1208. doi: 10.1091/mbc.7.8.1195
Pannu, V., Rida, P. C., Celik, B., Turaga, R. C., Ogden, A., Cantuaria, G., et al. (2014). Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis. 5, e1538. doi: 10.1038/cddis.2014.505
Parry, D., Guzi, T., Shanahan, F., Davis, N., Prabhavalkar, D., Wiswell, D., et al. (2010). Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 9, 2344–2353. doi: 10.1158/1535-7163.MCT-10-0324
Payton, M., Bush, T. L., Chung, G., Ziegler, B., Eden, P., McElroy, P., et al. (2010). Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 70, 9846–9854. doi: 10.1158/0008-5472.CAN-10-3001
Pearce, A. K., and Humphrey, T. C. (2001). Integrating stress-response and cell-cycle checkpoint pathways. Trends Cell Biol. 11, 426–433. doi: 10.1016/S0962-8924(01)02119-5
Pedersen, R. T., Kruse, T., Nilsson, J., Oestergaard, V. H., and Lisby, M. (2015). TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 210, 565–582. doi: 10.1083/jcb.201502107
Pereira, A. J., and Maiato, H. (2012). Maturation of the kinetochore-microtubule interface and the meaning of metaphase. Chromosome Res. 20, 563–577. doi: 10.1007/s10577-012-9298-8
Peters, J. M., Tedeschi, A., and Schmitz, J. (2008). The cohesin complex and its roles in chromosome biology. Genes Dev. 22, 3089–3114. doi: 10.1101/gad.1724308
Pines, J. (2011). Cubism and the cell cycle: the many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 12, 427–438. doi: 10.1038/nrm3132
Pines, J., and Rieder, C. L. (2001). Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3, E3–E6. doi: 10.1038/35050676
Raab, M. S., Breitkreutz, I., Anderhub, S., Rønnest, M. H., Leber, B., Larsen, T. O., et al. (2012). GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res. 72, 5374–5385. doi: 10.1158/0008-5472.CAN-12-2026
Renner, A. G., Dos Santos, C., Recher, C., Bailly, C., Créancier, L., Kruczynski, A., et al. (2009). Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 114, 659–662. doi: 10.1182/blood-2008-12-195867
Rieder, C. L., and Cole, R. (2000). Microtubule disassembly delays the G2-M transition in vertebrates. Curr. Biol. 10, 1067–1070. doi: 10.1016/S0960-9822(00)00678-3
Rieder, C. L., and Cole, R. W. (1998). Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. J. Cell Biol. 142, 1013–1022. doi: 10.1083/jcb.142.4.1013
Riley, T., Sontag, E., Chen, P., and Levine, A. (2008). Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412. doi: 10.1038/nrm2395
Rodbell, M. (1980). The role of hormone receptors and GTP-regulatory proteins in membrane transduction. Nature 284, 17–22. doi: 10.1038/284017a0
Rodrigo-Brenni, M. C., and Morgan, D. O. (2007). Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell 130, 127–139. doi: 10.1016/j.cell.2007.05.027
Rudolph, D., Steegmaier, M., Hoffmann, M., Grauert, M., Baum, A., Quant, J., et al. (2009). BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 15, 3094–3102. doi: 10.1158/1078-0432.CCR-08-2445
Rudra, S., and Skibbens, R. V. (2013). Cohesin codes - interpreting chromatin architecture and the many facets of cohesin function. J. Cell Sci. 126, 31–41. doi: 10.1242/jcs.116566
Sackton, K. L., Dimova, N., Zeng, X., Tian, W., Zhang, M., Sackton, T. B., et al. (2014). Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 514, 646–649. doi: 10.1038/nature13660
Salvatore, G., Nappi, T. C., Salerno, P., Jiang, Y., Garbi, C., Ugolini, C., et al. (2007). A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 67, 10148–10158. doi: 10.1158/0008-5472.CAN-07-1887
Samaga, R., and Klamt, S. (2013). Modeling approaches for qualitative and semi-quantitative analysis of cellular signaling networks. Cell Commun. Signal. 11:43. doi: 10.1186/1478-811X-11-43
Sanchez, Y., Bachant, J., Wang, H., Hu, F., Liu, D., Tetzlaff, M., et al. (1999). Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286, 1166–1171. doi: 10.1126/science.286.5442.1166
Saponaro, M., Callahan, D., Zheng, X., Krejci, L., Haber, J. E., Klein, H. L., et al. (2010). Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet. 6:e1000858. doi: 10.1371/journal.pgen.1000858
Sarbajna, S., Davies, D., and West, S. C. (2014). Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 28, 1124–1136. doi: 10.1101/gad.238303.114
Scolnick, D. M., and Halazonetis, T. D. (2000). Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 406, 430–435. doi: 10.1038/35019108
Seavey, M. M., Lu, L. D., Stump, K. L., Wallace, N. H., and Ruggeri, B. A. (2012). Novel, orally active, proteasome inhibitor, delanzomib (CEP-18770), ameliorates disease symptoms and glomerulonephritis in two preclinical mouse models of SLE. Int. Immunopharmacol. 12, 257–270. doi: 10.1016/j.intimp.2011.11.019
Sells, T. B., Chau, R., Ecsedy, J. A., Gershman, R. E., Hoar, K., Huck, J., et al. (2015). MLN8054 and Alisertib (MLN8237): discovery of selective oral Aurora A inhibitors. ACS Med. Chem. Lett. 6, 630–634. doi: 10.1021/ml500409n
Shapiro, G. I. (2006). Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 24, 1770–1783. doi: 10.1200/JCO.2005.03.7689
Shen, K., Wang, Y., Brooks, S. C., Raz, A., and Wang, Y. A. (2006). ATM is activated by mitotic stress and suppresses centrosome amplification in primary but not in tumor cells. J. Cell. Biochem. 99, 1267–1274. doi: 10.1002/jcb.20848
Shigeta, T., Takagi, M., Delia, D., Chessa, L., Iwata, S., Kanke, Y., et al. (1999). Defective control of apoptosis and mitotic spindle checkpoint in heterozygous carriers of ATM mutations. Cancer Res. 59, 2602–2607.
Shinde, S. R., Gangula, N. R., Kavela, S., Pandey, V., and Maddika, S. (2013). TOPK and PTEN participate in CHFR mediated mitotic checkpoint. Cell Signal. 25, 2511–2517. doi: 10.1016/j.cellsig.2013.08.013
Sivakumar, S., and Gorbsky, G. J. (2015). Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 16, 82–94. doi: 10.1038/nrm3934
Smith, E., Dejsuphong, D., Balestrini, A., Hampel, M., Lenz, C., Takeda, S., et al. (2009). An ATM- and ATR-dependent checkpoint inactivates spindle assembly by targeting CEP63. Nat. Cell Biol. 11, 278–285. doi: 10.1038/ncb1835
Smith, J., Tho, L. M., Xu, N., and Gillespie, D. A. (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112. doi: 10.1016/B978-0-12-380888-2.00003-0
Smits, V. A., Klompmaker, R., Arnaud, L., Rijksen, G., Nigg, E. A., and Medema, R. H. (2000). Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2, 672–676. doi: 10.1038/35023629
Soncini, C., Carpinelli, P., Gianellini, L., Fancelli, D., Vianello, P., Rusconi, L., et al. (2006). PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin. Cancer Res. 12, 4080–4089. doi: 10.1158/1078-0432.CCR-05-1964
Soucy, T. A., Smith, P. G., and Rolfe, M. (2009). Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin. Cancer Res. 15, 3912–3916. doi: 10.1158/1078-0432.CCR-09-0343
Stobbe, C. C., Park, S. J., and Chapman, J. D. (2002). The radiation hypersensitivity of cells at mitosis. Int. J. Radiat. Biol. 78, 1149–1157. doi: 10.1080/09553000210166570
Stone, A., Sutherland, R. L., and Musgrove, E. A. (2012). Inhibitors of cell cycle kinases: recent advances and future prospects as cancer therapeutics. Crit. Rev. Oncog. 17, 175–198. doi: 10.1615/CritRevOncog.v17.i2.40
Strausfeld, U., Fernandez, A., Capony, J. P., Girard, F., Lautredou, N., Derancourt, J., et al. (1994). Activation of p34cdc2 protein kinase by microinjection of human cdc25C into mammalian cells. Requirement for prior phosphorylation of cdc25C by p34cdc2 on sites phosphorylated at mitosis. J. Biol. Chem. 269, 5989–6000.
Strebhardt, K., and Ullrich, A. (2006). Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 6, 321–330. doi: 10.1038/nrc1841
Su, T. T., and Jaklevic, B. (2001). DNA damage leads to a Cyclin A-dependent delay in metaphase-anaphase transition in the Drosophila gastrula. Curr. Biol. 11, 8–17. doi: 10.1016/S0960-9822(00)00042-7
Takagi, M., Delia, D., Chessa, L., Iwata, S., Shigeta, T., Kanke, Y., et al. (1998). Defective control of apoptosis, radiosensitivity, and spindle checkpoint in ataxia telangiectasia. Cancer Res. 58, 4923–4929.
Takai, N., Hamanaka, R., Yoshimatsu, J., and Miyakawa, I. (2005). Polo-like kinases (Plks) and cancer. Oncogene 24, 287–291. doi: 10.1038/sj.onc.1208272
Tanaka, T. U., Stark, M. J., and Tanaka, K. (2005). Kinetochore capture and bi-orientation on the mitotic spindle. Nat. Rev. Mol. Cell Biol. 6, 929–942. doi: 10.1038/nrm1764
Teixeira, L. K., and Reed, S. I. (2013). Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 82, 387–414. doi: 10.1146/annurev-biochem-060410-105307
Tentler, J. J., Bradshaw-Pierce, E. L., Serkova, N. J., Hasebroock, K. M., Pitts, T. M., Diamond, J. R., et al. (2010). Assessment of the in vivo antitumor effects of ENMD-2076, a novel multitargeted kinase inhibitor, against primary and cell line-derived human colorectal cancer xenograft models. Clin. Cancer Res. 16, 2989–2998. doi: 10.1158/1078-0432.CCR-10-0325
Terasawa, M., Shinohara, A., and Shinohara, M. (2014). Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 10:e1004563. doi: 10.1371/journal.pgen.1004563
Tinker-Kulberg, R. L., and Morgan, D. O. (1999). Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev. 13:1936–1949. doi: 10.1101/gad.13.15.1936
Toyoshima-Morimoto, F., Taniguchi, E., and Nishida, E. (2002). Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 3, 341–348. doi: 10.1093/embo-reports/kvf069
Ubersax, J. A., Woodbury, E. L., Quang, P. N., Paraz, M., Blethrow, J. D., Shah, K., et al. (2003). Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864. doi: 10.1038/nature02062
Uto, K., Inoue, D., Shimuta, K., Nakajo, N., and Sagata, N. (2004). Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. EMBO J. 23, 3386–3396. doi: 10.1038/sj.emboj.7600328
VanderBorght, A., Valckx, A., Van Dun, J., Grand-Perret, T., De Schepper, S., Vialard, J., et al. (2006). Effect of an hdm-2 antagonist peptide inhibitor on cell cycle progression in p53-deficient H1299 human lung carcinoma cells. Oncogene 25, 6672–6677. doi: 10.1038/sj.onc.1209667
van de Vijver, M. J., He, Y. D., van't Veer, L. J., Dai, H., Hart, A. A., Voskuil, D. W., et al. (2002). A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347, 1999–2009. doi: 10.1056/NEJMoa021967
van Vugt, M. A., Gardino, A. K., Linding, R., Ostheimer, G. J., Reinhardt, H. C., Ong, S. E., et al. (2010). A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 8:e1000287. doi: 10.1371/journal.pbio.1000287
Vassilev, L. T. (2007). MDM2 inhibitors for cancer therapy. Trends Mol. Med. 13, 23–31. doi: 10.1016/j.molmed.2006.11.002
Vassilev, L. T., Vu, B. T., Graves, B., Carvajal, D., Podlaski, F., Filipovic, Z., et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. doi: 10.1126/science.1092472
Vitale, I., Galluzzi, L., Castedo, M., and Kroemer, G. (2011). Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 12, 385–392. doi: 10.1038/nrm3115
Wagner, S. A., Beli, P., Weinert, B. T., Nielsen, M. L., Cox, J., Mann, M., et al. (2011). A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10:M111 013284. doi: 10.1074/mcp.M111.013284
Walczak, C. E., Cai, S., and Khodjakov, A. (2010). Mechanisms of chromosome behaviour during mitosis. Nat. Rev. Mol. Cell Biol. 11, 91–102. doi: 10.1038/nrm2832
Wang, S., Midgley, C. A., Scaërou, F., Grabarek, J. B., Griffiths, G., Jackson, W., et al. (2010). Discovery of N-phenyl-4-(thiazol-5-yl)pyrimidin-2-amine aurora kinase inhibitors. J. Med. Chem. 53, 4367–4378. doi: 10.1021/jm901913s
Wardlaw, C. P., Carr, A. M., and Oliver, A. W. (2014). TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst). 22, 165–174. doi: 10.1016/j.dnarep.2014.06.004
Wechsler, T., Newman, S., and West, S. C. (2011). Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471, 642–646. doi: 10.1038/nature09790
Weiß, L., and Efferth, T. (2012). Polo-like kinase 1 as target for cancer therapy. Exp. Hematol. Oncol. 1:38. doi: 10.1186/2162-3619-1-38
Wengner, A. M., Siemeister, G., Koppitz, M., Schulze, V., Kosemund, D., Klar, U., et al. (2016). Novel Mps1 Kinase Inhibitors with Potent Antitumor Activity. Mol. Cancer Ther. 15, 583–592. doi: 10.1158/1535-7163.MCT-15-0500
Westra, A., and Dewey, W. C. (1971). Variation in sensitivity to heat shock during the cell-cycle of Chinese hamster cells in vitro. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 19, 467–477. doi: 10.1080/09553007114550601
Wieser, S., and Pines, J. (2015). The biochemistry of mitosis. Cold Spring Harb. Perspect. Biol. 7:a015776. doi: 10.1101/cshperspect.a015776
Wilkinson, R. W., Odedra, R., Heaton, S. P., Wedge, S. R., Keen, N. J., Crafter, C., et al. (2007). AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res. 13, 3682–3688. doi: 10.1158/1078-0432.CCR-06-2979
Wohlbold, L., and Fisher, R. P. (2009). Behind the wheel and under the hood: functions of cyclin-dependent kinases in response to DNA damage. DNA Repair (Amst). 8, 1018–1024. doi: 10.1016/j.dnarep.2009.04.009
Wu, T., Merbl, Y., Huo, Y., Gallop, J. L., Tzur, A., and Kirschner, M. W. (2010). UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc. Natl. Acad. Sci. U.S.A. 107, 1355–1360. doi: 10.1073/pnas.0912802107
Wyatt, H. D., Sarbajna, S., Matos, J., and West, S. C. (2013). Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247. doi: 10.1016/j.molcel.2013.08.035
Wyatt, P. G., Woodhead, A. J., Berdini, V., Boulstridge, J. A., Carr, M. G., Cross, D. M., et al. (2008). Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. J. Med. Chem. 51, 4986–4999. doi: 10.1021/jm800382h
Yamada, H. Y., and Gorbsky, G. J. (2006). Inhibition of TRIP1/S8/hSug1, a component of the human 19S proteasome, enhances mitotic apoptosis induced by spindle poisons. Mol. Cancer Ther. 5, 29–38. doi: 10.1158/1535-7163.MCT-05-0126
Yan, Y., Cao, P. T., Greer, P. M., Nagengast, E. S., Kolb, R. H., Mumby, M. C., et al. (2010). Protein phosphatase 2A has an essential role in the activation of gamma-irradiation-induced G2/M checkpoint response. Oncogene 29, 4317–4329. doi: 10.1038/onc.2010.187
Yang, C., Tang, X., Guo, X., Niikura, Y., Kitagawa, K., Cui, K., et al. (2011). Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol. Cell 44, 597–608. doi: 10.1016/j.molcel.2011.09.016
Yang, S. S., Yeh, E., Salmon, E. D., and Bloom, K. (1997). Identification of a mid-anaphase checkpoint in budding yeast. J. Cell Biol. 136, 345–354. doi: 10.1083/jcb.136.2.345
Ying, S., Minocherhomji, S., Chan, K. L., Palmai-Pallag, T., Chu, W. K., Wass, T., et al. (2013). MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007. doi: 10.1038/ncb2773
You, Z., Bailis, J. M., Johnson, S. A., Dilworth, S. M., and Hunter, T. (2007). Rapid activation of ATM on DNA flanking double-strand breaks. Nat. Cell Biol. 9, 1311–1318. doi: 10.1038/ncb1651
Zeng, X., and King, R. W. (2012). An APC/C inhibitor stabilizes cyclin B1 by prematurely terminating ubiquitination. Nat. Chem. Biol. 8, 383–392. doi: 10.1038/nchembio.801
Zeng, X., Sigoillot, F., Gaur, S., Choi, S., Pfaff, K. L., Oh, D. C., et al. (2010). Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18, 382–395. doi: 10.1016/j.ccr.2010.08.010
Zhang, J., Wu, P., and Hu, Y. (2013). Clinical and marketed proteasome inhibitors for cancer treatment. Curr. Med. Chem. 20, 2537–2551. doi: 10.2174/09298673113209990122
Zhang, W., Peng, G., Lin, S. Y., and Zhang, P. (2011). DNA damage response is suppressed by the high cyclin-dependent kinase 1 activity in mitotic mammalian cells. J. Biol. Chem. 286, 35899–35905. doi: 10.1074/jbc.M111.267690
Zhu, X., Gerstein, M., and Snyder, M. (2007). Getting connected: analysis and principles of biological networks. Genes Dev. 21, 1010–1024. doi: 10.1101/gad.1528707
Zirkle, R. E., and Bloom, W. (1953). Irradiation of parts of individual cells. Science 117, 487–493. doi: 10.1126/science.117.3045.487
Keywords: cancer therapy, checkpoint, DNA damage, mitosis, phosphorylation, ubiquitylation
Citation: Ferrari S and Gentili C (2016) Maintaining Genome Stability in Defiance of Mitotic DNA Damage. Front. Genet. 7:128. doi: 10.3389/fgene.2016.00128
Received: 24 May 2016; Accepted: 06 July 2016;
Published: 21 July 2016.
Edited by:
Kristijan Ramadan, University of Oxford, UKReviewed by:
Howard Donninger, University of Louisville, USAClaude Prigent, Centre National de la Recherche Scientifique, France
Copyright © 2016 Ferrari and Gentili. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Ferrari, c2ZlcnJhcmlAaW1jci51emguY2g=