 Xia Hua
Xia Hua Lindell Bromham
Lindell Bromham- Centre for Macroevolution and Macroecology, Research School of Biology, Australian National University, Canberra, ACT, Australia
A growing body of evidence suggests that rates of diversification of biological lineages are correlated with differences in genome-wide mutation rate. Given that most research into differential patterns of diversification rate have focused on species traits or ecological parameters, a connection to the biochemical processes of genome change is an unexpected observation. While the empirical evidence for a significant association between mutation rate and diversification rate is mounting, there has been less effort in explaining the factors that mediate this connection between genetic change and species richness. Here we draw together empirical studies and theoretical concepts that may help to build links in the explanatory chain that connects mutation to diversification. First we consider the way that mutation rates vary between species. We then explore how differences in mutation rates have flow-through effects to the rate at which populations acquire substitutions, which in turn influences the speed at which populations become reproductively isolated from each other due to the acquisition of genomic incompatibilities. Since diversification rate is commonly measured from phylogenetic analyses, we propose a conceptual approach for relating events of reproductive isolation to bifurcations on molecular phylogenies. As we examine each of these relationships, we consider theoretical models that might shine a light on the observed association between rate of molecular evolution and diversification rate, and critically evaluate the empirical evidence for these links, focusing on phylogenetic comparative studies. Finally, we ask whether we are getting closer to a real understanding of the way that the processes of molecular evolution connect to the observable patterns of diversification.
Introduction
Darwinism unites genetics (heritable characteristics) with population biology (change in frequency of heritable traits through differential reproduction) and biodiversity (formation of new lineages through speciation). The neo-Darwinian synthesis codifies the belief that these processes are all parts of a single evolutionary process, such that the generation of genetic variation that makes individuals differ from each other feeds the change in the characteristics of populations by selection and drift which drives speciation and the diversification of lineages. Thus the neo-Darwinian synthesis “puts an equals sign between microevolution and macroevolution” (Dobzhanksy, 1937). Most biologists accept this unified view of evolutionary process. Yet evolution is still predominantly studied in a remarkably partite fashion, with most researchers concentrating on one particular aspect of this process. While discipline specialization is a necessary part of modern science, there are some evolutionary phenomena that require an appreciation of many different levels of biological organization.
One such phenomenon is the observed correlation between differences in the mutation rate, estimated by comparing DNA sequences of different species, with net diversification rate, estimated from the number of extant species in different lineages (Barraclough and Savolainen, 2001; Pagel et al., 2006; Eo and DeWoody, 2010; Lanfear et al., 2010; Duchene and Bromham, 2013; Bromham et al., 2015). This relationship might initially seem surprising: how could the rate at which mistakes or damage are repaired in individual genomes lead to differences in species richness between lineages? The correlates of diversity are usually examined at the level of lineages, regions and biological communities. For example, diversification rate has been found to be associated with life history, dispersal rate, generalized feeding strategies, geographic range size, environmental energy and latitude (Cardillo, 1999; Cardillo et al., 2003; Davies et al., 2004; Phillimore et al., 2006). But evolutionary change must ultimately start with heritable changes to genetic information, so the supply of variation is a critical first step in the formation of new evolutionary lineages.
Our aim with this Hypothesis and Theory paper is to examine the possible links in the chain of causation that connects the average mutation rate, estimated by comparing nucleotide sequences, to differences in lineage net diversification rate, as measured from molecular phylogenies. In order to investigate how mutation rate could be linked to diversification rate, we need to consider how an increase in the supply of genetic variation could speed the rate of formation of new species. To do this we will first consider why mutation rate varies between species. Then we will examine the factors that shape the number and type of mutations that become fixed features of the genome within a population (substitutions). Then we consider how the accumulation of these substitutions can cause population to diverge from each other, until the genetic differences between their genomes prevent populations from exchanging genetic material. At this point, the populations can no longer interbreed, and we consider them separate species. Lastly, we propose a conceptual approach to relating the formation of new species to bifurcations on molecular phylogenies.
We explore both the theoretical foundations and empirical evidence for the links in the causal chain between mutation rates and macroevolution. These connections bring together topics typically considered in different biological disciplines, namely molecular evolution, population genetics and macroevolution, but we illustrate some of the approaches that allow us to effectively view all of these processes simultaneously by comparing DNA sequences between individuals, populations, species and lineages. Because the relationship has been noted by comparing gene sequences between lineages, we confine our discussion to mutations that change nucleotide sequences. However, there are many other kinds of genetic change that may also be important drivers of diversification, such as chromosomal rearrangement, regulatory changes or epigenetic modification.
Mutation Rates Evolve
To explain why variation in mutation rate is associated with differences in diversification rate, the first topic we have to address is why species differ in their average mutation rate. Mutation rate is often in the background of molecular evolutionary studies. It is frequently assigned an arbitrary constant value, providing a steady drip of genetic variation into populations. But mutation rate is a dynamic and highly variable phenomenon, changing across the genome and over time, varying among individuals and lineages. We should not be surprised that mutation rate is highly responsive to evolutionary pressures: it is, after all, the fundamental process at the heart of all evolutionary change.
Because we wish to examine the links between diversification rate and the rate of molecular evolution as measured from DNA sequence comparisons, we will consider only point mutations that change single nucleotides in gene sequences. Point mutations arise when damage to DNA is imperfectly repaired or errors in DNA replication are not fully corrected, resulting in permanent change to the base sequence such that the changed sequence will be included in any subsequent copies. DNA is a large, complex molecule: it is inevitable that it will occasionally suffer damage that affects the genetic information it encodes. In addition, the genome must be replicated every time a cell divides, so even a phenomenally low replication error rate (typically only one error per billion bases copied) will result in frequent changes to the DNA sequence. To protect the integrity of the genetic information needed to make essential components of the organism, there is a diverse and sophisticated set of cellular machinery that is directed at detecting and correcting any damage to the DNA or mistakes made in copying the genome. The costs of DNA repair to the cell are not well known, but it seems fair to say that investment in repair apparatus must come at cost of other cellular functions. Therefore, while we expect organisms to invest resources in reducing the risk of harmful mutation, there might come a point where the payoffs of further reduction in mutation rate are outweighed by the increasing costs of DNA fidelity. All species must walk a tightrope, finding a balance between fidelity and error, but they may find different points of balance between repair and mutation (Denamur and Matic, 2006; Bromham, 2009; Lynch, 2010).
One aspect of this balancing act, initially proposed on the basis of information theory, is the need to maintain mutation rates below an “error threshold,” the copy error rate above which a replicating sequence effectively goes extinct by failing to produce sufficiently faithful copies of itself (Eigen, 1971). In the earliest life forms, this may have led to a trade-off between genome size and mutation rate: for a given per-base error rate, a small genome is more likely to be copied without error, but a small genome may also have limited capacity for evolving mechanisms that reduce the mutation rate (Maynard Smith and Szathmáry, 1995). The error threshold model appears to place real-world constraints on the evolution of RNA viruses (Holmes, 2003). The reliance of RNA viruses on the error-prone reverse transcriptase to complete their life cycle endows them with a mutation rate more than an order of magnitude greater than DNA viruses (Sanjuán et al., 2010). While the high mutation rate in RNA viruses has been considered to contribute to their rapid evolvability, it may also place restrictions on adaptation by limiting genome size. Small genomes may speed replication, but it could also place constraints on coding capacity. Therefore it has been suggested that the high mutation rate of RNA viruses may, by limiting genome size, constrain the capacity for evolving new genetic features (Holmes, 2003). Increase in mutation rate beyond the error threshold has been reported as causing extinction of yeast lines (Herr et al., 2011a).
It has been suggested that many species settle on a similar point of balance between the per-genome copy error rate and genome size that represents a chance of error roughly once in every three hundred genome copies (Drake et al., 1998), at least relative to the “non-frivolous fraction” of the genome in which mutations can influence fitness (Drake, 1991). A larger genome with more functional sequences provides more targets for deleterious mutations, so a lower per-base mutation rate is required to have the same per-genome-replication risk of fitness-damaging mutations. The balancing point for the per-genome mutation rates has been most frequently examined for single-celled organisms. It is less clear whether the same relationship between genome size and mutation rate applies to large, complex multicellular organisms. There is surprisingly sparse data on the association between mutation rate and genome size in large multicellular organisms (Bromham et al., 2015), but observed patterns have been considered consistent with an overarching relationship between the effective genome size and the per-generation mutation rate (Lynch, 2010).
The balancing point between the costs and benefits of mutation and repair can be altered by circumstance. Both theory and experiment have suggested that populations of microbes subject to rapidly changing conditions can show a transient increase in mutation rate because “mutator” alleles that increase the rate of mutation can hitchhike to fixation through their association with advantageous mutations (Chao and Cox, 1983; Haraguchi and Sasaki, 1996; Sniegowski et al., 1997; Taddei, 1997; Giraud, 2001). These theoretical models and laboratory experiments have found additional support in observations from medical settings. For example, bacteria infecting the lungs of patients with cystic fibrosis can only persist if they can adapt to ongoing antibiotic treatment and a changing environment as the patient’s lung condition deteriorates, resulting in a higher frequency of bacteria with raised mutation rates (Oliver et al., 2000; Oliver and Mena, 2010).
Both models and experiments suggest that it is possible for mutation rates to be shaped by adaptation to circumstance. However, most of these experiments and simulations produce only transient increases in mutation rate, as the association between the mutator allele and beneficial mutations may be uncoupled by recombination, and the burden of deleterious mutations will result in selection against the mutator allele (McDonald et al., 2012). Can selection produce long-term differences in mutation rate between species through balancing the costs and benefits of mutation and repair? There are several observations that suggest it can. Firstly, in many populations there is naturally occurring heritable variation in mutation rates. For example, individuals can vary in the proofreading efficiency of their polymerase enzymes or their mismatch repair systems (Oliver et al., 2002; Sundin and Weigand, 2007; Reha-Krantz, 2010). Mutations that disable essential DNA repair genes can lead to disease phenotypes with dramatically increased mortality for affected individuals (Bradford et al., 2011), but slight variations in DNA repair genes that cause relatively mild reductions in DNA repair efficiency and fidelity can also influence fitness (e.g., Mohrenweiser et al., 2003; Gokkusu et al., 2013; Kabzinski et al., 2016). Although most studied “antimutator” alleles are changes that can compensate for deficiencies in mutator strains (Herr et al., 2011b), there is some evidence of naturally occurring alleles that can increase DNA repair efficiency or copy fidelity (Schaaper, 1998; Foury and Szczepanowska, 2011). The existence of spatial variation in DNA repair enzymes provides further evidence of the evolvability of mutation rate in natural populations (Miner et al., 2015; Svetec et al., 2016). So the raw material for selection to act on mutation rates does not seem to be lacking in natural populations.
Secondly, it seems fair to suppose that DNA repair is costly, such that organisms must find a level of investment that maximizes survival yet minimizes costs. Higher DNA repair efficiency may come at cost of other key cellular functions. For example, costs of repair may explain why the induction of resource-intensive stress response pathways in bacteria can lead to an increased mutation rate (Torres-Barceló et al., 2013). The potential trade-off between investment in DNA repair and other cellular functions is supported by the observation that some DNA repair systems are inducible under mutagenic conditions. For example, plants can have repair systems that are turned on or increased under high UV conditions, to prevent an upsurge in DNA damage (Ries et al., 2000). This suggests that maximum DNA repair efficiency is not maintained at all times, instead the repair effort is scaled to a level that allows the maintenance of cellular processes and reproductive potential. The metabolic cost of DNA repair is supported by observations that individuals in poor condition or subjected to mild stress can have elevated mutation rates, presumably because they are unable to invest as much in DNA repair (Agrawal and Wang, 2008; Goho and Bell, 2000). For example, it has been reported that male birds with lower levels of antioxidant carotenoids have higher rates of DNA damage and also reduced survivorship and lower mating success (Freeman-Gallant et al., 2011).
Thirdly, species can adapt to different levels of risk of mutation through altered investment in DNA repair. Species living in highly mutagenic environments might require greater investment in DNA repair in order to be able to maintain a persistent population against mutational meltdown. Microbes living in high-altitude lakes, exposed to high salinity, high levels of UV radiation and high concentrations of heavy metals, have been shown to have enhanced levels of DNA repair (Albarracin et al., 2012). Bacteria adapted to survive long periods of desiccation, which results in accumulation of DNA double-strand breaks, can have enhanced DNA repair that incidentally allows them to survive other mutagens such as ionizing radiation (Mattimore and Battista, 1996). Escherichia coli selected to survive ionizing radiation can evolve DNA repair proteins that are more efficient and less prone to inhibition by perturbations of normal metabolism, potentially making these repair proteins able to function under a broader range of environmental conditions (Piechura et al., 2015).
Conversely, species living in low mutagen environments may need to invest less in DNA repair. For example, species living in low UV environments may have lost some of their photolyase repair genes (Lucas-Lledó and Lynch, 2009). Populations can have consistently different levels of efficiency of UV-induced DNA repair, apparently reflecting different points of adaptation of DNA repair efficiency to match local environmental conditions. For example, water fleas from natural ponds with higher levels of UV exposure have more efficient DNA repair of light-induced mutation (Miner et al., 2015) and fruit fly populations from different latitudes show different levels of UV repair efficiency (Svetec et al., 2016). The adjustment of DNA repair to the level of mutation risk might also explain the puzzling lack of evidence for the prediction that basal metabolic rate should have a direct influence on the mutation rate, due to the production of free oxygen radicals that can damage DNA (Gillooly et al., 2007). Although this relationship has been modeled based on body size and temperature, comparative studies have found no significant variation of rate of molecular evolution with metabolic rate, above and beyond the association with other life history traits such as body size or generation time (Bromham et al., 1996; Lanfear et al., 2007; Galtier et al., 2009b). One possible explanation for the lack of a significant association between metabolic rate and mutation rate is that DNA repair efficiency may be adjusted to ameliorate any additional damage, so that species with high metabolic rates also evolve greater levels of protection against damage from free-oxygen radicals (Lanfear et al., 2007).
DNA repair proteins thus can be considered as a “highly adaptable scaffold readily tailored by evolution to the requirements for genome maintenance in each particular organism” (Piechura et al., 2015). Evolution shapes mutation rates just as it shapes other species traits, balancing costs and benefits to suit the species form and lifestyle. In the next section, we will consider how differences in mutation rates between species can vary predictably with species traits.
Species Traits Influence Mutation Rate Evolution
Given that DNA repair is likely to impose a non-trivial cost on individuals, that higher rates of mutation can lead to lower survival and reproduction, and that individuals show genetic variation in DNA repair efficiency, we should expect selection to find a balance between the cost of repair and the cost of mutation. But the balance between these competing costs might vary with other species characteristics (Sniegowski et al., 2000).
In multicellular organisms, the costs of mutation are expected to increase with increasing body size (Bromham, 2011). The larger the body, the more cell generations it takes to build it. Every cell division requires the genome to be copied in its entirety, and every genome replication brings the risk of copy errors that might ruin important DNA sequences. The influence of number of genome replications on the mutation rate is supported by the observation of higher per-generation mutation rates in males than in females. Due to the large number of cell generations required for sperm production, the male germline is copied many times more per generation that the female germline, and so the majority of de novo mutations arise in males (Wilson Sayres and Makova, 2011). Furthermore, germline mutations accumulate with male age, suggesting that the increasing number of germline divisions per gamete results in more mutations (Kong et al., 2012; Venn et al., 2014). Species with stronger potential for sperm competition, and therefore selective pressure to increase sperm production, have been found in some cases to have higher rates of molecular evolution (Bartosch-Harlid et al., 2003), though this pattern is not supported in all studies (Wilson Sayres et al., 2011). The influence of number of DNA replications on mutation rate has been proposed as the explanation for the generation time effect on rates of molecular evolution, on the assumption that species with faster generation turnovers copy their germline DNA more often per unit time (Gaut et al., 1992; Bromham et al., 1996; Welch et al., 2008; Thomas et al., 2010; Lehtonen and Lanfear, 2014).
However, the copy error effect on its own does not seem sufficient to explain the observed life history patterns in mutation rate (Nabholz et al., 2008; Welch et al., 2008; Gaut et al., 2011; Hua et al., 2015). The difference in mutation rates between species is not commensurate with the difference in number of genome copies per unit time. For example, mice can go through 50 generations for every human generation, yet the rate of molecular evolution in mice is only a few times faster than that in humans. This suggests that the influence of copy number on mutation rate is modulated by other factors. Other life history characteristics have a significant relationship with mutation rate above and beyond their covariation with generation time, particularly fecundity and longevity (Nabholz et al., 2008; Welch et al., 2008). The significant association between generation time and molecular evolution rates may be partly due to it covarying with other causal traits, but being measured more reliably. Most studies of longevity are based on maximum recorded lifespan, which is strongly influenced by number of observations made (that is, it can only go up as more data points are included), so it is possible that for species with relatively little data on longevity, generation time is a better proxy for life history differences.
What effect will selection for longer life spans have on the evolution of a species’ mutation rate? Larger-bodied species tend to have longer lives, requiring the maintenance of more genome copies from incidental damage over a longer time period. Unrepaired damage to any cell’s genome can result in life-shortening damage, such as somatic mutations causing cancer. In particular, the accumulation of mutations in mitochondrial genomes has been proposed as a driver of aging (Fridovich, 2004; Kujoth et al., 2005; Loeb et al., 2005; Larsson, 2010; Yang et al., 2013). Reactive oxygen species produced in the mitochondria as a byproduct of metabolism can cause damage to DNA and other biomolecules. As damage accumulates over time, mitochondrial function may be impeded. So evolving a longer life span may require increased investment in cellular mechanisms that reduce the overall mutation rate in order to keep lifetime mutation risk to a tolerable level. For example, the mutation rate in long-lived species might be reduced by greater investment in mechanisms that prevent oxidative damage (Pamplona and Barja, 2011). This prediction is borne out by observations that long-lived species of mammals, birds and fish tend to have lower per-base mutation rates (as measured by the synonymous substitution rate), above and beyond the association between longevity and other aspects of life history (Nabholz et al., 2008; Welch et al., 2008; Galtier et al., 2009a,b; Hua et al., 2015).
Yet, molecular phylogenetic studies measure the germline mutation rate, not the somatic mutation rate. Although there must be evolutionary pressure on the mutation rate in both somatic and germline cells, the somatic mutation rate need not be the same as the mutation rate in the germline. Indeed, one of the possible evolutionary advantages of multicellularity is the ability to set aside a quiescent germ line in which DNA (particularly organelle DNA) is relatively protected from the harmful byproducts of metabolism, and for which expression levels are kept at a minimum (Bendich, 2010). Keeping germline copies in a quiescent state might be particularly valuable for mitochondrial genomes, hence the evolution of separate sexes: the mitochondrial genomes of motile and metabolically active male gametes can be discarded at fertilization, leaving only the quiescent, relatively unimpaired mitochondria from the immobile female gamete (Allen, 1995; de Paula et al., 2013).
While there is good reason to believe that selection acts on mutation rates, the power of natural selection to shape DNA repair might in itself by limited by species traits, specifically by traits that influence the mutation rate (affecting the supply of variation) and population size (affecting the power of selection). In small populations, fewer mutations that improve DNA repair will arise (Baer et al., 2007), and selection will be less effective at promoting slight improvements to DNA repair or removing mutations that increase the mutation rate, potentially limiting the effectiveness of selection in optimizing mutation rate (Knight et al., 2005; Lynch, 2007; Hodgkinson and Eyre-Walker, 2011).
Whatever the mechanistic or evolutionary forces that shape the differences in mutation rate between species – whether an incidental effect of copy frequency, an adaptive compromise between competing costs, or some other phenomena – the practical upshot is that even closely related species can differ in their average rate of mutation. So the rate of supply of new genetic variation to a species’ gene pool is at least partly dependent on a variety of species traits that influence the mutation rate. What effect will differences in the supply of variation have on the evolutionary process? The role of mutation rate in governing the rate of evolution has been given relatively little attention for non-microbial taxa, perhaps due to a general conviction that adaptation in complex multicellular creatures is typically not mutation-limited. But we should not be quick to dismiss the rate of mutation as playing an important role in population diversification. Although there is debate about the relationship between mutation rate, standing variation, and rate of adaptation (Barrett and Schluter, 2008), empirical studies suggest that adaptation to rapid environmental change or strong selection pressures can come from both standing variation (existing alleles) and de novo mutations (Hartley et al., 2006; Durand et al., 2010; Jerome et al., 2011). Even if adaptation is not mutation limited, population divergence can also be driven by non-adaptive genome evolution that may be influenced by the mutation rate.
Species-specific differences in mutation rate will only influence the evolutionary rate of lineage divergence if the supply of variation influences the rate of fixation of genetic differences: that is, if the mutation rate at least partly determines the substitution rate. So now that we have explored the evolutionary factors that shape the supply of variation to populations at any point in time, we need to consider how the supply of variation through mutation contributes to rates of genome evolution.
Mutation Rate Influences Substitution Rate
Mutations occur in individual DNA molecules. For a mutation to be detected as a consistent difference in the DNA sequences sampled from different populations or species, it must clear several hurdles. First it must be copied from the genome it occurred in and passed on to that individual’s offspring, in order to enter a new generation. If the mutation is included in one or more offspring in the next generation, then each of those individuals has a chance to reproduce and pass the mutation to their offspring. While the mutation is at low frequency in the population, there is a high chance of being lost by chance if those few individuals fail to reproduce. If it increases in representation in the population, the mutation becomes established as a polymorphism, carried by some but not all members of the population. Eventually it may rise in frequency until it replaces all other variants in the population, so now all new individuals born in that population will carry a copy of that mutation (barring a new mutation occurring at the same site). At this point, we call the mutation a substitution and say that it has been fixed in the population. In this section, we will consider the factors that influence the rate at which new mutations become substitutions, thereby contribute to population divergence.
For each neutral mutation that has no effect on fitness, such as most synonymous substitutions that change the DNA sequence of a gene but not the amino acid sequence it codes for, the probability of being passed to future generations is entirely due to chance, and so its frequency in the population is determined only by the mutation rate and the effective population size (Ne). However, the observed level of standing genetic variation within populations is often much lower than would be expected (Lande, 1976; Turelli, 1984; Lynch and Hill, 1986) if the amount of variation was shaped only by the processes of mutation and genetic drift (Frankham, 2012; Hodgins-Davis et al., 2015). This suggests that that there must be additional factors limiting the accumulation of genetic variation in populations (Eyre-Walker and Keightley, 2007).
For non-neutral mutations, such as most non-synonymous mutations that change the amino acid sequence of a protein, their frequency in the population will be determined not only by the mutation rate and the effective population size, but also the selection coefficient of the mutation (s). The influence of chance events on allele frequencies is potentially much greater in small populations. For example, the lucky survival and reproduction of an individual with a mutation that slightly lowers fitness will have a proportionally greater effect on allele frequencies in a small population than in a large population. In a large population, random fluctuations in allele frequencies are less likely to result in chance fixation, because they have proportionally less effect on average frequencies from one generation to the next. The larger the population, the less influence of chance events on allele frequencies. Therefore a beneficial mutation (s > 0) is more likely to increase in frequency in larger population than in a smaller population. Mutations with a substantial fitness costs (s << 0) will not become substitutions, as they will be removed from the population by purifying selection. But slightly deleterious changes (s < 0) will be nearly neutral and are more likely to go to fixation by chance in smaller population (Ohta, 1992).
We can describe the probability that a new mutation will becomes a substitution, found in all genomes in the population. We call this probability the fixation probability Pfix. Theoretical studies provide an analytical approximation of the fixation probability (Waxman, 2011):
This approximation accounts for a general situation where the values of effective population size and selection coefficient can change over T amount of time. So T = 0 means the population has constant effective population size and selection coefficient. According to the equation, the fixation probability depends on the product of the effective population size and the selection coefficient of the mutation, and on the frequency of the mutation at time T [X(T)].
Over evolutionary time periods, the overall substitution rate in the population approximates the product of the mutation rate in the population and the fixation probability of each mutation, that is, 2NeuPfix, where u is the mutation rate. When most mutations are neutral (s = 0) and when effective population size stays constant, Pfix depends only on the initial frequency of a mutation. For a new mutation occurring in a population of diploid individuals, its initial frequency is 1/2N, where N is the number of individuals in the population. Under these conditions, substitution rate equals uNe/N. To investigate the impact of changing effective population size over time on the substitution rate, we simulated changes in the frequency of a mutation when the effective population size increases, decreases, or stays constant. The resulting frequencies were used to calculate the expectation value of e-4NesX(T) in the equation for Pfix. Results suggest that changing the effective population size does not have a significant effect on the neutral substitution rate (Figure 1). Therefore, the rate of neutral substitutions always approximates uNe/N, regardless of the demographic history of the population.
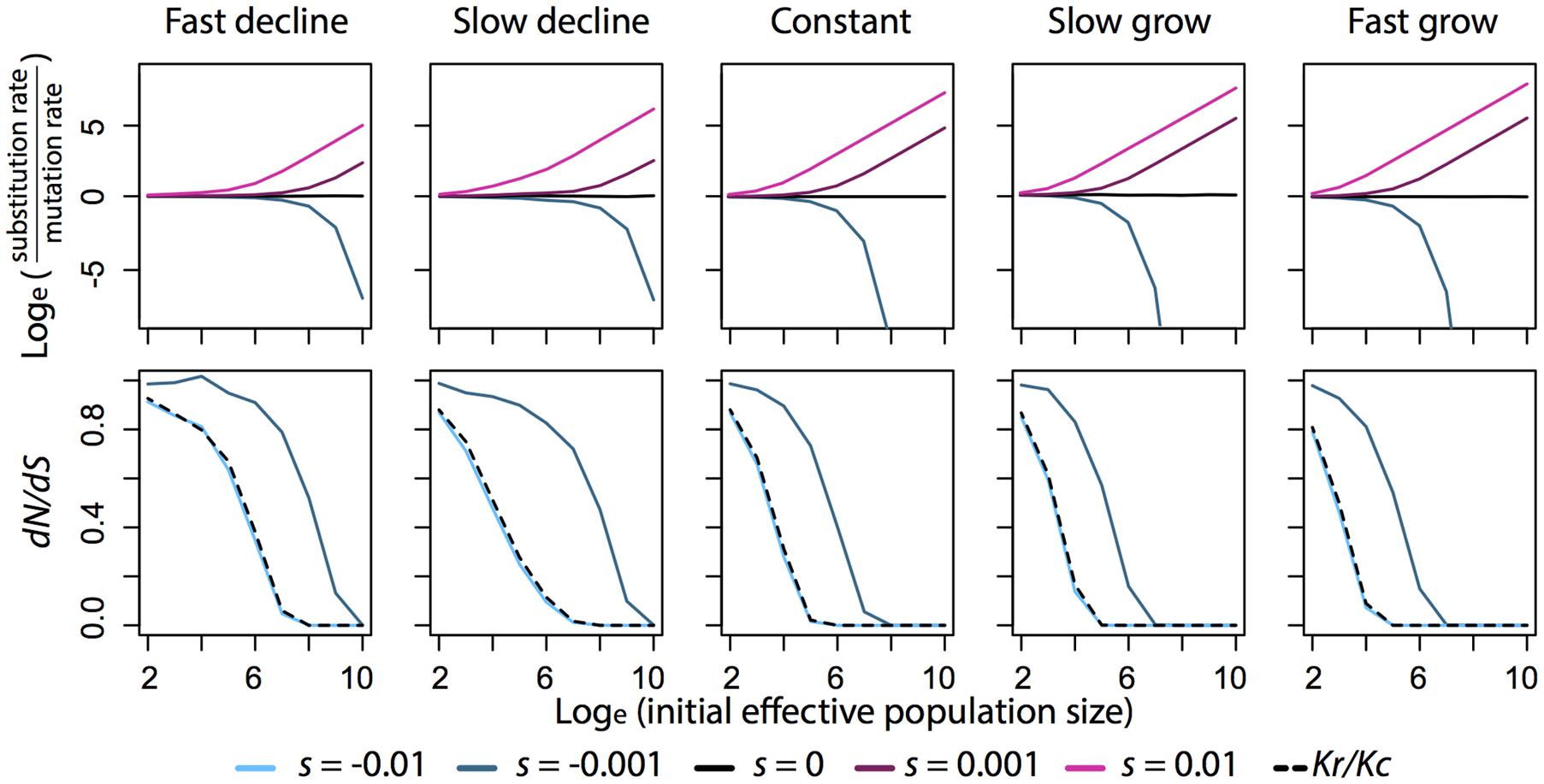
FIGURE 1. Relationship between substitution rate, dN/dS, Kr/Kc, and effective population size (N𝜀) under different scenarios of population growth and selection coefficient (s). Fixation probability is calculated by the general formula described in the text. To approximate the expectation value in the formula, 500000 replicates of an allele are simulated under a Wright-Fisher model described in Waxman (2011). In each simulation, selection coefficient stays constant and population size changes deterministically over generations. When population grows, the final population size is 1.9 times the initial population size. When population declines, the final population size is 0.1 times the initial population size. Fast population change takes 20 generations to reach the final population size. Slow population change takes 200 generations. Substitution rate is then calculated from the fixation probability and is plotted on natural log scale with respect to mutation rate, so when substitution rate equals mutation rate, the value is 0 in the plots. dN/dS is calculated as the ratio between the fixation probability of a slightly deleterious mutation (s = -0.001) to that of a neutral mutation (s = 0). Kr/Kc is calculated as the ratio between the fixation probability of a deleterious mutation (s = -0.01) to that of a slightly deleterious mutation (s = -0.001).
Theory predicts the rate of fixation of neutral mutations will be unaffected by population size. The substitution of beneficial mutations will be faster in large population and the effect is greater in an expanding population (Figure 1). The substitution of deleterious mutations will be faster in small populations and the effect is greater in a declining population (Figure 1). Given that advantageous mutations are relatively rare compared to deleterious mutations (Eyre-Walker and Keightley, 2007), we expect smaller populations to have the greater overall substitution rate for a given mutation rate.
We can test these predictions by comparing different classes of substitutions in DNA sequence comparisons. Since all types of substitution rates are related to mutation rate, taking the ratio between any two of them will cancel out the influence of mutation rate. Therefore, a common test for the effect of population size on substitution rate is to correlate population size to the ratio between non-synonymous to synonymous substitution rates (dN/dS). Similarly, radical substitutions (from one class of amino acid residue to another) are more likely to be deleterious than conservative substitutions (within the same functional category) because radical substitutions cause more changes in the physiochemical properties of the protien, so the ratio between them (Kr/Kc) is expected to show the same pattern with population size as dN/dS (Figure 1; Zhang, 2000; Smith, 2003).
The predicted negative relationship between dN/dS and population size has been supported by observed correlations between dN/dS and life history traits that scale with population size (Nikolaev et al., 2007; Popadin et al., 2007; Lartillot and Delsuc, 2012; Romiguier et al., 2013; Figuet et al., 2014). It has also been supported by comparing sister lineages that differ in effective population size, such as domesticated populations to wild populations (Björnerfeldt et al., 2006; Wang et al., 2011) and island lineages to their mainland close relatives (Johnson and Seger, 2001; Woolfit and Bromham, 2005), although island lineages do not necessarily have smaller effective population size than mainland lineages (Wright et al., 2009; James et al., 2016). A puzzling exception is that dN/dS in birds shows no correlation to body size (which is often related to population size), but the correlation between Kr/Kc and body size is as predicted (Nabholz et al., 2013; Weber et al., 2014; Figuet et al., 2016). While there are many possible causes of the lack of relationship in birds, such as a lack of correlation between life history traits and historical population size or unreliable estimates of dN/dS (Lartillot, 2013), there is thusfar a puzzling lack of evidence that they provide an explanation for the lack of correlation between dN/dS and body size in birds (Figuet et al., 2016).
Interestingly, the formula of fixation probability never suggests a linear relationship between dN/dS and population size. In fact, we show in Figure 1 that in the range of effective population size where it has strong negative relationship with Kr/Kc, dN/dS may show weak or even no dependence on population size particularly when population declines. So, a simple correlation test between dN/dS and population size may not be a robust way to test our theoretical understanding of substitutions against empirical data. We suggest that the formula of fixation probability should be explicitly accounted in future analyses on the relationship between population size and substitution rates. In particular, the formula can be written into a form similar to logistic regression, where the selection coefficient is the regression coefficient and the expectation term becomes a parameter in the link function to estimate.
We have considered the way that increases in mutation rate should flow through to increases in substitution rate, in many classes of substitution (neutral, advantageous, slightly deleterious). Now we can look at the way the accumulation of substitutions makes populations genetically distinct from each other. In the next section, we will consider how isolation between populations is achieved either by spatial separation or through the acquisition of genetic traits that reduce the chances of interbreeding. Once populations are isolated, they will accumulate unique sets of substitutions.
Isolated Populations Acquire Different Substitutions
The simplest way for populations to become isolated is through the imposition of a physical barrier to the movement of alleles from one population to another. When a once-continuous population is divided by unsuitable habitat, there are three possible outcomes. One is that the barrier continues to prevent gene flow between the divided populations. The second is that the barrier is removed, allowing individuals or gametes to move between populations once more. The third is that one or both of the daughter populations acquires adaptations that allow them to overcome the barrier and re-establish contact with the other population. The first two processes are entirely determined by the frequency at which isolating mechanisms are created or removed. But the third possibility is a biological process and its frequency will depend not only on the nature of the isolating mechanism but also on the ability of either daughter population to adapt to the novel intervening habitat and overcome the barrier. Adaptation is found to occur on both new mutations and standing genetic variation (Olson-Manning et al., 2012). Standing genetic variation is also positively correlated with direct estimates of mutation rate (Vigouroux et al., 2002; Phillips et al., 2009). So, higher mutation rate could facilitate local adaptation, potentially allowing secondary contact between divided populations (Barton, 2001; Sexton et al., 2009).
It’s possible for substitutions to cause genetic isolation between populations in the absence of a physical barrier to interbreeding. Divergent selection for different substitutions within the population may contribute to non-random mating (Servedio et al., 2011). For example, divergent selection on flowering time has been detected in two genetically distinct populations of Mimulus guttatus, causing temporal isolation between the two populations (Lowry et al., 2008), and divergent selection on color pattern mimicry has been found to cause assortative mating in sister species Heliconius melpomene (Jiggins et al., 2001). Such substitutions can happen rapidly under local adaptation, generating genetically isolating populations (Servedio et al., 2011), so the effectiveness of this process will depend on how common these kinds of disruptive traits are, and on the rate of generation of relevant alleles by mutation. Therefore, the major limit on the evolution of non-random mating is the initial frequency of alleles associated with these disruptive traits in the populations, which is related to the mutation rate and the rarity of the traits. In theory, any substitutions that make individuals more likely to mate with individuals of the same population will facilitate genetic isolation between populations. The most general case is when substitutions make immigrants much less viable and/or fertile than local individuals (Servedio et al., 2011).
If divergent selection is removed, then non-random mating may breakdown. For example, light gradient has been found to drive divergent evolution of female sensory bias in some cichlid fish, such that females at different water depths prefer different male colors (Seehausen et al., 2008). But the same species living in turbid water does not show the same preference, presumably because light of the certain wavelengths is absorbed so fast that most water depths have monochromatic light, under which females cannot discriminate color (Seehausen and van Alphen, 1998).
We have now considered how sub-populations can acquire different sets of substitutions, either because physical barriers prevent movement of substitutions between them, or because divergent selection fixes different traits in different parts of a species’ range which directly or indirectly engender genetic isolation between populations. Now we will consider how the acquisition of these different sets of substitutions can contribute to genetic incompatibility between populations, paving the way for the formation of new species.
Substitutions Lead to Incompatibility
Given some level of population isolation, genetic differences between populations will inevitably accumulate over time. If these genetic differences reduce the chance of successful reproduction between members of different populations, they contribute to the reproductive isolation (RI), preventing or reducing gene flow between populations. Broadly speaking, there are three ways that substitutions can contribute to reproductive isolation: by local adaptation of populations making hybrids unfit in either of the parent environment (e.g., Via et al., 2000), by influencing the chance of successful mating or fertilization (pre-zygotic isolation: e.g., Quinn et al., 2000), or by generating sets of alleles that reduce hybrid viability and/or fertility when mixed with alleles from other populations (post-zygotic isolation, or genomic incompatibility: e.g., Yamamoto et al., 2010).
However, there may be no clear line between the different ways that alleles contribute to RI. For example, local adaptation that involves changes to metabolic genes might involve co-ordinated changes to both mitochondrial and nuclear genes, potentially causing cytonuclear conflicts if mitochondrial alleles from one population are combined with nuclear alleles from another population (Johnson, 2010). Furthermore, if mutations conferring local adaptation are physically linked on the same chromosome to mutations causing genomic incompatibility, locally incompatible regions of the genomes (“speciation islands”) can reduce interbreeding between populations and accelerate their genetic differentiation (Via, 2009, 2012). It is possible for RI to evolve within a population, for example through polyploidy or chromosomal rearrangement, as long as the mutant individuals can reproduce by selfing or mating with other mutants, but have greatly reduced chance of successfully combining chromosomes with wildtype members of the population (Ptacek et al., 1994; Guelbeogo et al., 2005; Wood et al., 2009; Twyford and Friedman, 2015).
Following Dobzhansky’s pioneering experiments in the early 1900s that involved introgressing small regions of the genome from one species to another (Dobzhansky, 1936), “speciation genes” have been identified which confer low fitness in hybrid genetic backgrounds but not in their original genetic backgrounds. The genomic incompatibility caused by these genes stems from antagonistic interactions between parental genomes. Empirical studies on speciation genes have agreed on four general patterns (Coyne and Orr, 2004; Welch, 2004; Presgraves, 2010). First, the completion of RI can involve multiple incompatibilities. Second, the evolution of each incompatibility can be driven by multiple substitutions. Third, these substitutions are often found in uniparentally inherited genes. Fourth, these substitutions cause significant decrease in individual fitness only when they are in particular combinations.
Most of the models developed to account for these patterns are derivatives of the Dobzhansky–Muller-Incompatibility (DMI) model (Dobzhanksy, 1937; Muller, 1942). The basic argument of a DMI model is that combinations of alleles found in high frequencies in a given population must be compatible with each other; otherwise they would be removed from the population by purifying selection. Given that any individual unfortunate enough to inherit a deleterious combination of alleles has lower fitness, so will pass on fewer copies of those alleles to the next generation, we expect incompatible alleles to reduce in frequency within an interbreeding population. But alleles in one isolated population are not tested against alleles in other populations, so combinations of alleles that can only be formed by hybridization between populations may have low overall fitness. The DMI model is particularly good at explaining the four general patterns of “speciation genes,” because it predicts that incompatibility is a feature of particular allele combinations, and it assumes that each incompatibility involves multiple substitutions as it requires different substitutions in different populations (Welch, 2004). Note that, although the DMI model was originally proposed to describe the evolution of genomic incompatibility, it can be generalized to model the other two ways that substitutions can contribute to reproductive isolation through prezygotic incompatibility or local adaptation. We can use the DMI model in any cases by where substitutions have positive or neutral effects on fitness in their own populations, but hybrids may have deleterious combinations of alleles from both parent populations and so contribute to RI.
When a population is divided, each subpopulation will acquire substitutions, and every substitution has some chance of creating incompatibility between the subpopulations. Orr (1995) formulated the DMI model by assuming that each derived allele in a population has equal probability of being incompatible with each of the derived alleles in the other population. As a result, the number of incompatibilities is the number of all possible combinations of derived alleles, which increases with the square of the number of substitutions fixed between the two populations. This prediction that the rate of acquisition of RI increases quadratically with genetic differences between populations is called the snowball effect (Orr, 1995). While empirical studies have given some support for the snowball effect (Matute et al., 2010; Moyle and Nakazato, 2010; Wang et al., 2015), it has been suggested that these results suffer from inaccurate estimates of genetic divergence between species (Stadler et al., 2012) or overestimates in the frequency of hybrid inviability (Barbash, 2011).
The assumption that every derived allele in a population has equal probability of being incompatible with each of the derived alleles in the other population does not seem realistic, as we might expect alleles associated with the same function to have a greater risk of being incompatible than substitutions affecting distinct functions that do not interact. For example, if different copies of a duplicated gene are silenced by mutations in different populations, then a hybrid may inherit both silenced copies and so lose the gene function (Masly et al., 2006; Bikard et al., 2009). Similarly, if one population fixes two alleles in succession, one of which compensates for the deleterious effect of the other allele, then a hybrid may suffer reduction in fitness if it inherits only one of the alleles without the compensating effect of the other allele. Some speciation genes are associated with the suppression of molecular drive by cytoplasmic genomes, pathogens or selfish genetic elements, which favor their own transmission at the expense of fitness of gametes not carrying them (Johnson, 2010; Presgraves, 2010). A hybrid between two separate populations might have incompatible sets of alleles, having the elements from one population but the suppression mechanisms of another. Situations such as these that involve pairs of compatible alleles could drive a linear relationship between the number of incompatibilities and genetic divergence between populations. So if paired substitutions affecting related functions are the primary cause of genomic incompatibility, then we might expect the rate of increase in RI to be less than quadratic.
But we should not expect all incompatible alleles to be paired up. Indeed, observation suggests that multiple substitutions are often needed to confer an incompatibility (Coyne and Orr, 2004; Welch, 2004). Given the complexity in the way derived alleles interact to cause incompatibilities, we might not expect either a strict linear or quadratic increase in RI with the number of substitutions fixed between two populations. If incompatibilities can be identified from genetic mapping data, then the number of incompatibilities can be regressed against the number of substitutions fixed between species (Matute et al., 2010; Moyle and Nakazato, 2010). Then we could use the degree of greater-than-linear increase as a continuous measure of how many alleles in one population can be incompatible with each allele in another population. We can also explicitly model the accumulation of incompatibilities along phylogenies under different models and compare the goodness-of-fit between models against the observed number of incompatibilities among species (Wang et al., 2013).
We have seen that species differ in the rate of supply of new mutations, and that this should influence the rate of acquisition of substitutions. Isolation between populations – whether by physical, behavioral or genetic barriers to interbreeding – will cause different sets of substitutions to accumulate in sister populations. These substitutions must be compatible with other alleles in the population, but may be incompatible with substitutions accumulated independently in the sister populations. Now we will consider how the accumulation of incompatible substitutions leads to the formation of new species.
Incompatibility Leads to Speciation
Both spatial isolation and genetic isolation may drive speciation if the isolating conditions can be sustained for a sufficiently long time. The DMI model suggests that isolation makes speciation inevitable, as divided populations will eventually accumulate sufficient differences to prevent the formation of successful hybrids during secondary contact. Even if RI is not complete during secondary contact, as long as there is some form of selection against hybridization and the selection is strong enough, traits that facilitate premating isolation can be selected for to complete RI (Servedio and Noor, 2003; Otto et al., 2008; Bank et al., 2012). The more substitutions contribute to RI, the stronger is the selection against hybridization. So in order to link mutation, substitution and speciation, we need to ask how the rate of accumulation of substitutions is linked to the rate of formation of reproductive isolation.
To answer this question, Gavrilets and Gravner (1997) extended the DMI model to the holey landscape model, which considers an adaptive landscape in which the “holes” are unfit genotypes. The holey landscape model demonstrates the conditions under which incompatible substitutions can be fixed in different populations, and provides quantitative predictions concerning the number of substitutions and the amount of time required to reach RI. Because strongly deleterious mutations are unlikely to be fixed, we need to look for the conditions under which incompatible substitutions can be fixed in different populations without incurring large fitness costs. In other words, we want to find evolutionary paths that move along the adaptive landscape without falling into a hole. Given a DMI prediction on the relationship between incompatibilities and substitutions, the holey landscape model is able to predict the number of evolutionary paths that a genotype can move along (Gavrilets, 2004).
When all the incompatible substitutions are paired, the number of incompatibilities is a linear function of the number of substitutions 𝜀d, where 𝜀 is the probability that each substitution causes reduction in fitness and d is the number of substitutions that differ from a starting genotype. If the starting genotype has L mutational targets, there are L number of steps the genotype can take with one substitution and the probability that any step will not lead to a hole is the chance that the next substitution is compatible with any previous substitutions and so causes no reduction in fitness: 1–𝜀. Then the expected number of paths for the starting genotype is L(1–𝜀). So the conditions where the starting genotype can move along the fitness landscape without falling into a hole is when L(1–𝜀) > 1, which is almost always true. Following similar arguments, the conditions for a snowball effect (quadratic increase in incompatibility) is 𝜀 < lnL/L (Gavrilets, 2004). In this case, having more mutational targets (L) should have lower values of 𝜀 to fulfill the condition of a snowball effect. In principle, 𝜀 can be estimated from a regression model between the number of incompatibilities and the number of substitutions fixed between species.
Assuming the simplest case where any one pair of incompatible substitutions causes complete RI, and all substitutions have the same chance of being incompatible, we can consider the probability that two populations differ in d substitutions can still interbreed. Two populations can interbreed if the path of d substitutions between them along the landscape does not fall in a hole, the probability of which is (1 - 𝜀)d. The probability that the k-th substitution causes RI is (1 - 𝜀)k-1𝜀. So the expectation of the number of substitutions required to complete RI is 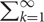 k(1 - 𝜀)k-1𝜀 = 1/𝜀. The expected amount of time to complete RI is then the ratio between the number of substitutions to reach RI and the substitution rate (Gavrilets, 2004). This model allows us to make predictions concerning the relationship between mutation rates, substitution rates and time to speciation. If each step along the evolutionary paths of the populations is neutral or nearly neutral, the overall substitution rate in a population should be primarily determined by the mutation rate (see Mutation Rate Influences Substitution Rate). If the incompatible substitutions are the result of adaptation, then this will reduce the time to reach complete RI, proportional to the product between effective population size and selection coefficient (Gavrilets, 2004). Because this calculation is based solely on new mutations and neglects the role of standing genetic variation within populations, it may overestimate time to achieve RI (Gavrilets, 2004). To account for standing genetic variation, one can numerically model changes in genetic variation both within and between populations (Gavrilets, 1999).
k(1 - 𝜀)k-1𝜀 = 1/𝜀. The expected amount of time to complete RI is then the ratio between the number of substitutions to reach RI and the substitution rate (Gavrilets, 2004). This model allows us to make predictions concerning the relationship between mutation rates, substitution rates and time to speciation. If each step along the evolutionary paths of the populations is neutral or nearly neutral, the overall substitution rate in a population should be primarily determined by the mutation rate (see Mutation Rate Influences Substitution Rate). If the incompatible substitutions are the result of adaptation, then this will reduce the time to reach complete RI, proportional to the product between effective population size and selection coefficient (Gavrilets, 2004). Because this calculation is based solely on new mutations and neglects the role of standing genetic variation within populations, it may overestimate time to achieve RI (Gavrilets, 2004). To account for standing genetic variation, one can numerically model changes in genetic variation both within and between populations (Gavrilets, 1999).
Clearly, the waiting time to speciation will depend on many interacting factors, including the rate of supply of genetic variation, the level of gene flow between isolated populations, the nature of the genetic changes underlying reproductive isolation, population size and nature of selective pressures. The holey landscape model only provides general predictions on the relationship between mutation rate, substitution rate, and speciation rate. But general predictions are useful for comparative studies, in which the influences of confounding factors may be treated as random effects when sample size is large.
Now that we have seen how species differ in mutation rates, and how these will influence the rate at which populations acquire substitutions that will cause them to become genetically incompatible, we can explore the relationship between the formation of genetically isolated populations and the rate of diversification. To do this, we need to consider the possible evolutionary fates of newly isolated populations once they have formed.
Speciation Drives Diversification
Diversification is the net result of the processes that change the number of independent evolving lineages. The possible component processes of diversification are speciation adding lineages, extinction removing lineages, and merger of existing lineages through hybridization. So to understand how the processes that lead to genetically isolated populations contribute to diversification, we need to consider the factors that influence whether these new isolated populations will persist. If they persist, they may potentially divide again. Or, one or more of them may go extinct, resulting in the loss of any unique substitutions that had accumulated in that species. Or, if RI is not complete, isolated populations may reconnect and merge genetically with each other during secondary contact, losing their separate identity and becoming a single intermixed lineage.
While the process of speciation is typically studied by comparing closely related populations, diversification is usually studied by comparing the diversity of lineages over time and space. Because we are focusing only on evolutionary analysis of DNA sequences, we will not attempt to consider the ways that palaeontological, taxonomic and biogeographic data are used to shed light on the process of diversification. Instead we will consider only how diversification rate is measured from molecular phylogenies. Rather than considering the existence of RI between populations, phylogenetic studies of diversification rate typically rely on counting the number of recognized taxa within genetically distinct lineages. Speciation rate is typically estimated from the distribution of branching events in a phylogeny (e.g., Pagel et al., 2006), and differences in the net diversification rate estimated by comparing the number of extant species per lineage (e.g., Bromham et al., 2015).
These phylogenetic measures of diversification rate do not map exactly to the process being considered in speciation models. Taxonomic counts of species in a lineage are typically based on the number of physically or biogeographically distinct forms rather than direct measures of reproductive isolation. Phylogenetic identification of species recognizes populations that show deep genetic divergence (Simpson, 1962; de Queiroz, 1998), but not all the genetic differences between populations cause RI, and in some cases previously isolated populations can rejoin and fuse (Coyne and Orr, 2004), and physically or spatially distinct forms may be connected by interbreeding populations (Irwin et al., 2001). So phylogenetic estimates of diversification are not direct measures of time to reach RI as predicted by studies on speciation (Wiens, 2004).
To understand the relationship between phylogenetic patterns and speciation processes, we need to consider the interplay between population isolation, secondary contact, and reproductive isolation, and the relative amounts of evolutionary time between them. First we can define the expected waiting time between isolating events, which we will call TI (time to isolation). If isolating events are physical barriers preventing interbreeding, then TI may depend on chance environmental factors that divide populations. If isolating events are caused by substitutions that cause non-random mating, then TI depends not only on the occurrence of environmental factors that trigger divergent selection on the subpopulations, but also the amount of time the populations take to evolve non-random mating.
Similarly, we can consider the expected waiting time for two previously divided populations to come back into contact, reestablishing the potential for gene flow (TS, time to secondary contact). In the case of physical barriers, TS could depend, like TI, on chance environmental factors that remove isolating barriers (e.g., a river changing course), or TS may be due to ecological succession (e.g., burnt forest re-growing). Alternatively, TS might be governed by genetic change within the population, if one or both of the divided populations is able to adapt to the intervening unsuitable habitat. So when populations are isolated by unsuitable habitat, TS equals the length of time the intervening habitat remains unsuitable or the amount of time it takes for populations to adapt to the unsuitable habitat, whichever is shorter. When isolation is due to non-random mating, TS may be rapid (e.g., if a sudden change in environment removes divergent selection) or more gradual (e.g., if the breakdown in divergent selection is due to selection for the same new adaptive trait in both populations).
Finally, we can consider the waiting time to complete RI, TR, which describes the amount of time needed to accumulate sufficient incompatibilities to become irreversibly genetically incompatible, even if secondary contact between populations is restored. At TR, members of the population can no longer interbreed even if brought back into contact.
We can consider the pattern of lineage evolution under different relative values of TI, TS and TR (Figure 2). When the waiting time to secondary contact tends to be greater than the time needed to complete RI (TS > TR), previously isolated populations won’t be able to interbreed if they come into secondary contact. In these circumstances, isolation leads to distinct species, and the internodal branch lengths of an accurately reconstructed phylogeny should equal TI (Figures 2A–B). In this situation, the diversification rate estimated from branching events reflects the frequency of the isolating events.
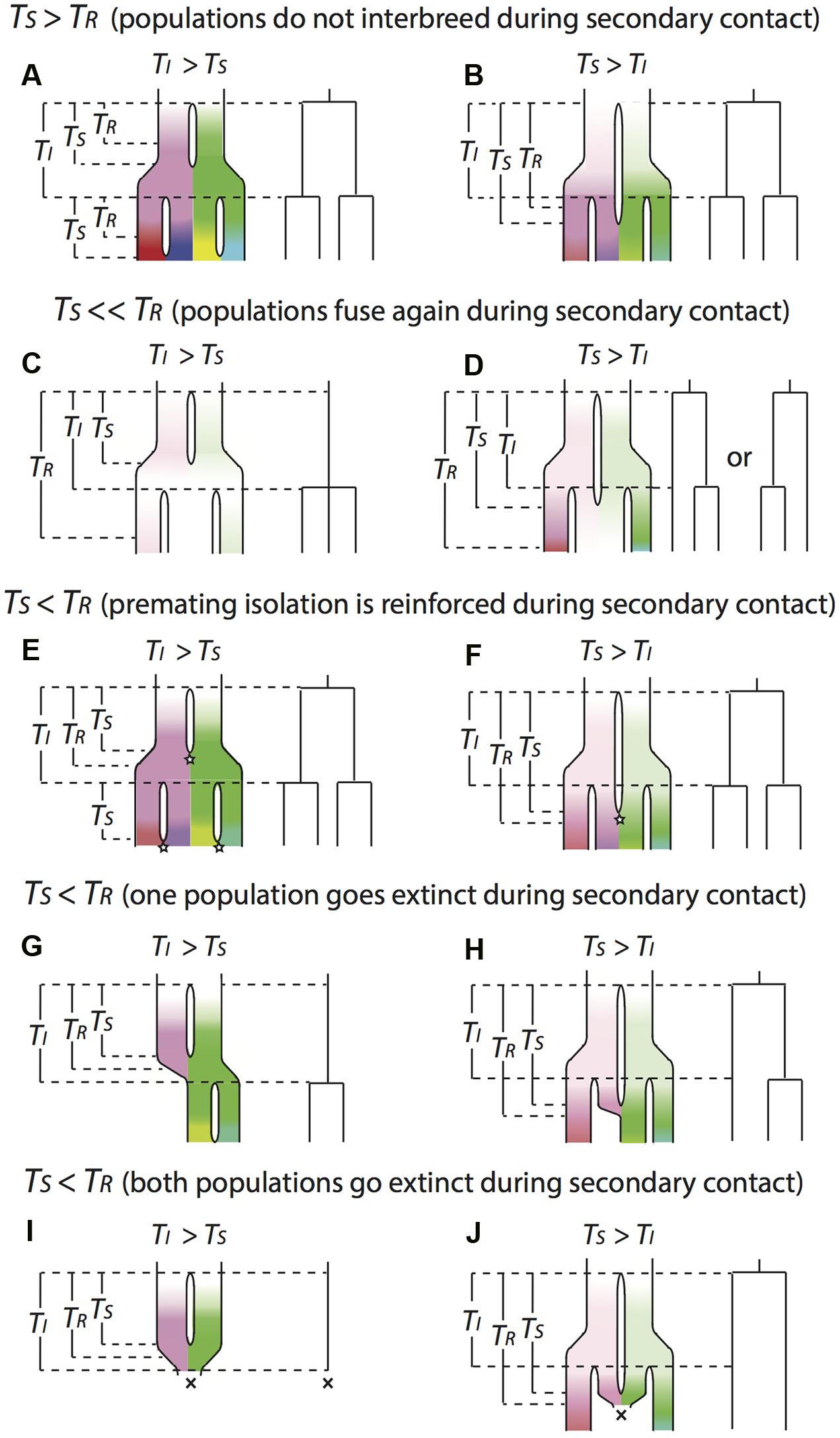
FIGURE 2. Illustration for the conceptual approach to relating events of reproductive isolation to bifurcations on molecular phylogenies. Each scenario includes the true history of diversification on the left and the reconstructed molecular phylogeny on the right, assuming the phylogeny is accurately reconstructed. Gaps between lineages in the true history indicate population isolation. The color indicates the genetic distinctiveness of each lineage (in terms of how many different substitutions the populations have acquired). The brighter the color is, the more distinctive the lineage is to the others. Diversification starts with a white lineage that bifurcates into a pink and a green lineage. During the process, populations are expanding and encounter new isolation events. Then pink lineage bifurcates into a red and a purple lineage, and green lineage bifurcates into a yellow and a blue lineage. TI is the expected waiting time between isolating events, which are kept the same under all the scenarios in order to make the internodal branch lengths of the reconstructed phylogenies under different scenarios comparable to each other. TS is the amount of time that populations remain isolated. TR is the amount of time that populations accumulate enough incompatibilities to reach complete RI. Stars represent reinforcement events and crosses represent extinction events. (A–J) describe different evolutionary scenarios: see text for details.
When waiting time to secondary contact is much less than the time needed to develop complete reproductive isolation (TS ≪ TR), previously isolated populations can interbreed if they come into secondary contact. If waiting time to secondary contact is shorter than the typical period between events that separate populations (TI > TS), then separated populations are able to fuse again before the next isolation event happens. If we were to reconstruct this history on a phylogeny, we would not be able to detect the period of isolation, instead we would detect only the most recent isolating event that causes a split (Figure 2C). If waiting time to secondary contact is longer than the period between isolating events (TI < TS), the next isolation event happens before populations have a chance to merge, keeping the most spatially distant populations isolated (Figure 2D). If we were to reconstruct this history on a phylogeny, we would observe the most spatially distant populations having deepest genetic divergence (even though they may not be the oldest isolating events), and the phylogenetic relationships between these populations and the population that is spatially distributed in the middle of them might not be fully resolved.
When waiting time to secondary contact is slightly less than the time needed to develop complete reproductive isolation (TS < TR), some degree of reproductive isolation has evolved between populations, so hybrids produced by secondary contact will have reduced fitness, in which case there are two possible outcomes. First, traits that facilitate premating isolation may be selected for to prevent production of low-fitness hybrids, and this selection will favor mutations that bring about complete RI. Under this scenario, we would observe similar phylogenetic patterns as the case of TS > TR (Figures 2E,F). Second, the loss of reproductive output on unsuccessful hybrid mating could be severe enough to result in extinction of one or both populations (Todesco et al., 2016). If one population goes extinct, we would observe similar phylogenetic patterns as the case of TS ≪ TR (Figures 2G,H). If isolation events are less frequent than secondary contact events (TI > TS; Figure 2I), then both populations may go extinct on merger due to loss of reproductive output. However, an isolation event may save a population from extinction by hybridization, allowing persistence of the lineage, leading to a pattern of species with discrete spatial distributions. In this case each internode on the phylogeny would represent multiple isolating and merging events (Figure 2J).
In our illustration, we assumed that the values of TI, TS, and TR were constant over the phylogeny, but it might be more realistic to model them as random variables. If the values can change over time and between lineages, then all the different scenarios illustrated in Figure 2 may be found in a single reconstructed phylogeny reconstructed from the sequence data of a species group.
Using our conceptual approach to relating events of reproductive isolation to bifurcations on molecular phylogenies, we can make some predictions about the role of mutation rate in shaping average speciation rates. As outlined in the previous section, we expect mutation rate to have a role in determining the rate of formation of reproductive isolation between populations, so higher mutation rate should result in shorter TR. When the rate of secondary contact is controlled primarily by environmental change, populations with shorter TR are more likely to evolve reproductive isolation before secondary contact, so these populations will show the phylogenetic patterns under the scenario of TS > TR (Figures 2A,B), where internodal branch lengths are shorter and speciation rate is higher than other scenarios. Higher mutation rate may also result in faster evolution of non-random mating under divergent selection and so shorter TI. Populations with shorter TI are more likely to show the phylogenetic patterns under scenarios of T1 < TS (Figures 2B,D,F,H,J), where speciation rate is also higher than the corresponding scenarios of T1 > TS (Figures 2A,C,E,G,I). As a result, greater mutation rates may lead to faster speciation due to more rapid accumulation of reproductive isolation and/or faster evolution of non-random mating. But when secondary contact is driven by one or both of the divided populations evolving to occupy the intervening habitat, we would expect populations with faster mutation rate to have both shorter TS and TR, because greater genetic variation in populations may speed both adaptation to novel environments and the evolution of reproductive isolation. Under this situation, whether faster mutation can accelerate speciation depends on the relative magnitude of TS versus TS, which seems unlikely to have a general pattern.
It is also possible that mutation rate could influence diversification rate through relative extinction rates. Several theoretical studies have suggested that populations with faster mutation rate should adapt to novel environments faster (Lynch, 1996; Barton and Partridge, 2000; Stockwell et al., 2003; Orr and Unckless, 2008). This may provide some reduction in extinction rates if extinction is largely driven by failing to adapt to environmental changes. However, some population genetic models have predicted an opposite effect, where the accumulation of deleterious mutations in extremely small populations causes extinction by “mutational meltdown” (Lynch et al., 1995; Lande, 1998). Due to difficulties in measuring the extinction rate for most lineages, it is difficult to test these hypotheses directly. In terms of the effect on diversification rates measured from phylogenies, extinction influences phylogenetic branch lengths because in most cases we expect to have direct evidence for only a relatively small proportion of all of the extinct species. Missing data from the extinct lineages increases internodal branch lengths in the phylogeny and decreases estimates of diversification rate.
Linking Mutation Rates to Diversification Rates
We have discussed how the rate of supply of new genetic variation is determined by the mutation rate that varies between species. We have also seen how patterns and rates of fixation of mutations in populations are influenced by factors that are in themselves shaped by species traits. Therefore we expect that patterns and rates of accumulation of genetic differences will differ between lineages. The acquisition of substitutions causes populations to diverge from each other, until they become so different that they cannot freely interbreed. These links – more mutations, more substitutions, more incompatibilities, a higher rate of speciation, and a higher rate of diversification – connect biochemical events in single cells to the generation of biodiversity. This is not to say that the production of variation by itself explains the process of diversification. Clearly, individual speciation events will be driven by particular local circumstances and the biology of the organism in question. But given that a link between mutation and diversification has been detected in comparative studies, we conclude that the production of variation makes some degree of contribution to the rate of evolutionary change at both the microevolutionary and macroevolutionary levels.
Empirical support for this connection between mutation and diversification comes primarily from molecular phylogenetic studies that show a correlation between estimates of rate of molecular evolution (estimated from phylogenies either from the tips, all branches, or root-to-tip paths) and measures of net diversification rate (either species richness or number of nodes in a tree). Association between rates of molecular evolution and species richness has been noted for a wide range of plants and animals (Barraclough and Savolainen, 2001; Davies et al., 2004; Pagel et al., 2006; Eo and DeWoody, 2010; Lancaster, 2010; Duchene and Bromham, 2013; Ezard et al., 2013; Bromham et al., 2015; Dugo-Cota et al., 2015). While individual studies may be subject to measurement biases or analytical artifacts, the diversity of approaches taken and the wide variety of data analyzed supports the contention that these studies reveal a widespread phenomenon.
While comparative studies demonstrate statistically significant and consistent patterns across many taxa, they don’t reveal the underlying cause of the relationship between rates of molecular evolution and diversification. We have hypothesized how mutation rate could promote diversification, either by providing more raw materials for adaptation, or by contributing to the evolution of reproductive isolation, or both. Conversely, it has been suggested that the process of speciation could cause acceleration in rates of molecular evolution, resulting in longer phylogenetic branch lengths in more species-rich lineages (Pagel et al., 2006). However, this hypothesis is difficult to reconcile with studies that have identified a correlation between the synonymous substitution rate and species richness (Barraclough and Savolainen, 2001; Lanfear et al., 2007; Duchene and Bromham, 2013; Bromham et al., 2015). Given that variation in synonymous substitution rate is considered to reflect differences in the mutation rate, it is difficult to see how speciation can directly influence mutation rate.
Alternatively, the association between molecular rates and diversification rate may be an incidental consequence of both being associated with some other factor, although it is difficult to think of a convincing indirect link. While body size and other associated life history characteristics correlate with rate of molecular evolution in a wide range of taxa, size is a surprisingly poor predictor of species richness in animals (Owens et al., 1999; Orme et al., 2002; Stuart-Fox and Owens, 2003; Isaac et al., 2005). Population size could offer an indirect link, if greater rates of speciation or extinction were associated with consistent reduction in population size, which could increase the fixation of slightly deleterious substitutions, however, there is currently little empirical evidence to support either a consistently lower population size or increased dN/dS in species-rich lineages (Bromham et al., 2015).
Various attempts have been made to provide a general theory of the tempo and mode of evolution by following the causal chain from biochemical processes to macroevolutionary change, for example by linking available kinetic energy to individual metabolic rate to both mutation rate and generation time, which may then influence the rate of evolutionary change (Gillooly and Allen, 2007). Such theories have been used to explain spatial patterns in biodiversity, on the assumption that higher temperatures drive greater rates of genetic change, either directly through an effect on rate of biochemical reactions, or indirectly through faster life histories reducing generation time (Rohde, 1992; Gillman and Wright, 2013). If rate of phenotypic evolution or niche change is increased by higher mutation rate or faster generation turnover, then lineages in warmer environments might diversify more rapidly (Smith and Beaulieu, 2009). However, theories linking energy availability to molecular change to diversity have been challenged on both empirical (noting exceptions to the rule) and theoretical grounds (questioning the validity of the underlying assumptions) (Duncan et al., 2007; Price et al., 2012). The proposed links in the causal chains might often be overwhelmed by other evolutionary forces operating on particular species (Dowle et al., 2013; Bromham et al., 2015; Glazier, 2015).
In particular, we might expect some of the evolutionary feedback loops discussed in this paper to have an impact on the knock-on effects of environmental temperature on rate of molecular evolution. Species are not passive in the face of environmental variation in mutagens such as UV or temperature, instead there is evidence that they adapt to their local conditions (Albarracin et al., 2012; Miner et al., 2015; Svetec et al., 2016), which may iron out some of the predicted environmental variation in mutation rates. Similarly, while variation in growth rates and generation turnover may influence the rate of accumulation of DNA replication errors, it seems that copy frequency effects are modulated by selection on copy fidelity mechanisms in order to produce acceptable levels of per generation error rates (Drake et al., 1998; Bromham, 2011; Sung et al., 2012). Life history and mutation rate must be matched to the environment and optimized to each other if a lineage is to persist through evolutionary time.
Conclusion
The search for simple unifying theories in macroevolution and macroecology seems unlikely to succeed given the vast number of factors that can influence a particular lineage’s evolutionary trajectory, including rare events and the weight of history. Patterns in biodiversity are shaped by a great many factors, both intrinsic and extrinsic to organisms. Both evidence and theory suggests that one such factor is variation in the mutation rate between species. But the explanatory power of the observed relationship between molecular rates and biodiversity is relatively modest, so it does not provide anything like the predictive power that might be hoped for in a unifying theory. However, we feel that the evidence is growing that, in addition to the many and varied influences on the generation of diversity, the differential rate supply of variation through species-specific differences in mutation rate has some role to play in generating different rates of diversification.
Consideration of the forces shaping molecular evolution provides one piece of an intricate macroevolutionary puzzle. Molecular phylogenetic analysis has given us the ability to be able to consider both molecular processes and diversification rates simultaneously, giving us a new tool with which to explore the connections between the supply of variation and the production of biodiversity. We can’t help but think that Darwin would be pleased with these new views of the evolutionary process that molecular analyses afford us, as it offers the potential to demonstrate the links in the Darwinian chain that connects variation between individuals to divergence between populations to the generation of biodiversity:
“It cannot be asserted that organic beings in a state of nature are subject to no variation; it cannot be proved that the amount of variation in the course of long ages is a limited quantity; no clear distinction has been, or can be, drawn between species and well-marked varieties. It cannot be maintained that species when intercrossed are invariably sterile, and varieties invariably fertile; or that sterility is a special endowment and sign of creation.... But the chief cause of our natural unwillingness to admit that one species has given birth to other and distinct species, is that we are always slow in admitting any great change of which we do not see the intermediate steps.” (Darwin, 1859)
Author Contributions
XH and LB designed the work and wrote the article. XH developed models and conducted analyses.
Funding
We thank the Australian Research Council for funding this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David Waxman for allowing us use his simulation code.
References
Agrawal, A. F., and Wang, A. D. (2008). Increased transmission of mutations by low-condition females: evidence for condition-dependent DNA repair. PLoS Biol. 6:e30. doi: 10.1371/journal.pbio.0060030
Albarracin, V., Pathak, G., Douki, T., Cadet, J., Borsarelli, C., Gurtner, W., et al. (2012). Extremophilic Acinetobacter strains from high-altitude lakes in Argentinean Puna: remarkable UV-B resistance and efficient DNA damage repair. Orig. Life Evol. Biosph. 42, 201–221. doi: 10.1007/s11084-012-9276-3
Allen, J. (1995). Separate sexes and the mitochondrial theory of ageing. J. Theor. Biol. 180, 135–140. doi: 10.1006/jtbi.1996.0089
Baer, C., Miyamoto, M. M., and Denver, D. R. (2007). Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat. Rev. Genet. 8, 619–631. doi: 10.1038/nrg2158
Bank, C., Hermisson, J., and Kirkpatrick, M. (2012). Can reinforcement complete speciation? Evolution 66, 229–239. doi: 10.1111/j.1558-5646.2011.01423.x
Barbash, D. A. (2011). Comment on “a test of the snowball theory for the rate of evolution of hybrid incompatibilities”. Science 333:1576. doi: 10.1126/science.1202876
Barraclough, T. G., and Savolainen, V. (2001). Evolutionary rates and species diversity in flowering plants. Evolution 55, 677–683. doi: 10.1111/j.0014-3820.2001.tb00803.x
Barrett, R. D., and Schluter, D. (2008). Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44. doi: 10.1016/j.tree.2007.09.008
Barton, N. (2001). “Adaptation at the edge of a species’ range,” in Integrating Ecology and Evolution in a Spatial Context, eds J. Silvertown and J. Antonovics (London: Blackwell), 365–392.
Barton, N., and Partridge, L. (2000). Limits to natural selection. Bioessays 22, 1075–1084. doi: 10.1002/1521-1878(200012)22:12<1075::AID-BIES5<3.0.CO;2-M
Bartosch-Harlid, A., Berlin, S., Smith, N. G. C., Ller, A. P., and Ellegren, H. (2003). Life history and the male mutation bias. Evolution 57, 2398–2406. doi: 10.1111/j.0014-3820.2003.tb00251.x
Bendich, A. J. (2010). Hypothesis mitochondrial DNA, chloroplast DNA and the origins of development in eukaryotic organisms. Biol. Direct 5:42. doi: 10.1186/1745-6150-5-42
Bikard, D., Patel, D., Le Mette, C., Giorgi, V., Camilleri, C., Bennett, M. J., et al. (2009). Divergent evolution of duplicate genes leads to genetic incompatibilities within A. thaliana. Science 323, 623–626. doi: 10.1126/science.1165917
Björnerfeldt, S., Webster, M. T., and Vilà, C. (2006). Relaxation of selective constraint on dog mitochondrial DNA following domestication. Genome Res. 16, 990–994. doi: 10.1101/gr.5117706
Bradford, P. T., Goldstein, A. M., Tamura, D., Khan, S. G., Ueda, T., Boyle, J., et al. (2011). Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterizes the role of DNA repair. J. Med. Genet. 48, 168–176. doi: 10.1136/jmg.2010.083022
Bromham, L. (2009). Why do species vary in their rate of molecular evolution? Biol. Lett. 5, 401–404. doi: 10.1098/rsbl.2009.0136
Bromham, L. (2011). The genome as a life-history character: why rate of molecular evolution varies between mammal species. Philos. Trans. Roy. Soc. B 366, 2503–2513. doi: 10.1098/rstb.2011.0014
Bromham, L., Hua, X., Lanfear, R., and Cowman, P. F. (2015). Exploring the relationships between mutation rates, life history, genome size, environment, and species richness in flowering plants. Am. Nat. 185, 507–524. doi: 10.1086/680052
Bromham, L., Rambaut, A., and Harvey, P. H. (1996). Determinants of rate variation in mammalian DNA sequence evolution. J. Mol. Evol. 43, 610–621. doi: 10.1007/BF02202109
Cardillo, M. (1999). Latitude and rates of diversification in birds and butterflies. Proc. R. Soc. B 266, 1221–1225. doi: 10.1098/rspb.1999.0766
Cardillo, M., Huxtable, J., and Bromham, L. (2003). Geographic range size, life history and rates of diversification in Australian mammals. J. Evol. Biol. 16, 282–288. doi: 10.1046/j.1420-9101.2003.00513.x
Chao, L., and Cox, E. C. (1983). Competition between high and low mutating strains of Escherichia coli. Evolution 37, 125–134. doi: 10.2307/2408181
Coyne, J. A., and Orr, H. A. (2004). Speciation. Sunderland, MA: Sinauer Associates, Inc Publishers.
Darwin, C. (1859). The Origin of Species by Means of Natural Selection: or the Preservation of Favoured Races in the Struggle for Life. London: John Murray.
Davies, T. J., Savolainen, V., Chase, M. W., Moat, J., and Barraclough, T. G. (2004). Environmental energy and evolutionary rates in flowering plants. Proc. R. Soc. B 271, 2195–2200. doi: 10.1098/rspb.2004.2849
de Paula, W. B. M., Lucas, C. H., Agip, A. N., Vizcay-Barrena, G., and Allen, J. F. (2013). Energy, ageing, fidelity and sex: oocyte mitochondrial DNA as a protected genetic template. Philos. Trans. R. Soc. B 368, 20120263. doi: 10.1098/rstb.2012.0263
de Queiroz, K. (1998). “The general lineage concept of species, species criteria, and the process of speciation: a conceptual unification and terminological recommendations,” in Endless Forms: Species and Speciation, eds D. J. Howard and S. H. Berlocher (New York, NY: Oxford University Press), 57–75.
Denamur, E., and Matic, I. (2006). Evolution of mutation rates in bacteria. Mol. Microbiol. 60, 820–827. doi: 10.1111/j.1365-2958.2006.05150.x
Dobzhansky, T. (1936). Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics 21, 113–135.
Dowle, E., Morgan-Richards, M., and Trewick, S. (2013). Molecular evolution and the latitudinal biodiversity gradient. Heredity 110, 501–510. doi: 10.1038/hdy.2013
Drake, J. W. (1991). A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 88, 7160–7164. doi: 10.1073/pnas.88.16.7160
Drake, J. W., Charlesworth, B., Charlesworth, D., and Crow, J. F. (1998). Rates of spontaneous mutation. Genetics 148, 1667–1686.
Duchene, D., and Bromham, L. (2013). Rates of molecular evolution and diversification in plants: chloroplast substitution rates correlate with species-richness in the Proteaceae. BMC Evol. Biol. 13:1. doi: 10.1186/1471-2148-13-65
Dugo-Cota,Á., Castroviejo-Fisher, S., Vilà, C., and Gonzalez-Voyer, A. (2015). A test of the integrated evolutionary speed hypothesis in a Neotropical amphibian radiation. Global Ecol. Biogeogr. 24, 804–813. doi: 10.1111/geb.12318
Duncan, R. P., Forsyth, D. M., and Hone, J. (2007). Testing the metabolic theory of ecology: allometric scaling exponents in mammals. Ecology 88, 324–333. doi: 10.1890/0012-9658200788[324:TTMTOE]2.0.CO;2
Durand, E., Tenaillon, M. I., Ridel, C., Coubriche, D., Jamin, P., Jouanne, S., et al. (2010). Standing variation and new mutations both contribute to a fast response to selection for flowering time in maize inbreds. BMC Evol. Biol. 10:2. doi: 10.1186/1471-2148-10-2
Eigen, M. (1971). Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 58, 465–523. doi: 10.1007/bf00623322
Eo, S. H., and DeWoody, J. A. (2010). Evolutionary rates of mitochondrial genomes correspond to diversification rates and to contemporary species richness in birds and reptiles. Proc. R. Soc. B 277, 3587–3592. doi: 10.1098/rspb.2010.0965
Eyre-Walker, A., and Keightley, P. D. (2007). The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618. doi: 10.1038/nrg2146
Ezard, T. H., Thomas, G. H., and Purvis, A. (2013). Inclusion of a near-complete fossil record reveals speciation-related molecular evolution. Methods Ecol. Evol. 4, 745–753. doi: 10.1111/2041-210X.12089
Figuet, E., Nabholz, B., Bonneau, M., Carrio, E. M., Nadachowska-Brzyska, K., Ellegren, H., et al. (2016). Life-history traits, protein evolution, and the nearly neutral theory in amniotes. Mol. Biol. Evol. 33, 1517–1527. doi: 10.1093/molbev/msw033
Figuet, E., Romiguier, J., Dutheil, J. Y., and Galtier, N. (2014). Mitochondrial DNA as a tool for reconstructing past life-history traits in mammals. J. Evol. Biol. 27, 899–910. doi: 10.1111/jeb.12361
Foury, F., and Szczepanowska, K. (2011). Antimutator alleles of yeast DNA polymerase gamma modulate the balance between DNA synthesis and excision. PLoS ONE 6:e27847. doi: 10.1371/journal.pone.0027847
Frankham, R. (2012). How closely does genetic diversity in finite populations conform to predictions of neutral theory? Large deficits in regions of low recombination. Heredity 108, 167–178. doi: 10.1038/hdy.2011.66
Freeman-Gallant, C. R., Amidon, J., Berdy, B., Wein, S., Taff, C. C., and Haussmann, M. F. (2011). Oxidative damage to DNA related to survivorship and carotenoid-based sexual ornamentation in the common yellowthroat. Biol. Lett. 7, 429–432. doi: 10.1098/rsbl.2010.1186
Fridovich, I. (2004). Mitochondria: are they the seat of senescence? Aging Cell 3, 13–16. doi: 10.1046/j.1474-9728.2003.00075.x
Galtier, N., Blier, P. U., and Nabholz, B. (2009a). Inverse relationship between longevity and evolutionary rate of mitochondrial proteins in mammals and birds. Mitochondrion 9, 51–57. doi: 10.1016/j.mito.2008.11.006
Galtier, N., Jobson, R. W., Nabholz, B., Glemin, S., and Blier, P. U. (2009b). Mitochondrial whims: metabolic rate, longevity and the rate of molecular evolution. Biol. Lett. 5, 413–416. doi: 10.1098/rsbl.2008.0662
Gaut, B., Yang, L., Takuno, S., and Eguiarte, L. E. (2011). The patterns and causes of variation in plant nucleotide substitution rates. Annu. Rev. Ecol. Evol. Syst. 42, 245–266. doi: 10.1146/annurev-ecolsys-102710-145119
Gaut, B. S., Muse, S. V., Clark, W. D., and Clegg, M. T. (1992). Relative rates of nucleotide subsitution at the rbcL locus of monocotyledonous plants. J. Mol. Evol. 35, 292–303. doi: 10.1007/BF00161167
Gavrilets, S. (1999). A dynamical theory of speciation on holey adaptive landscapes. Am. Nat. 154, 1–22. doi: 10.1016/S0169-5347(97)01098-7
Gavrilets, S. (2004). Fitness Landscapes and the Origin of Species. Princeton, NJ: Princeton University Press.
Gavrilets, S., and Gravner, J. (1997). Percolation on the fitness hypercube and the evolution of reproductive isolation. J. Theor. Biol. 184, 51–64. doi: 10.1006/jtbi.1996.0242
Gillman, L. N., and Wright, S. D. (2013). Patterns of evolutionary speed: in search of a causal mechanism. Diversity 5, 811–823. doi: 10.3390/d5040811
Gillooly, J. F., and Allen, A. P. (2007). Linking global patterns in biodiversity to evolutionary dynamics using metabolic theory. Ecology 88, 1890–1894. doi: 10.1890/06-1935.1
Gillooly, J. F., Mccoy, M. W., and Allen, A. P. (2007). Effects of metabolic rate on protein evolution. Biol. Lett. 3, 655–660. doi: 10.1098/rsbl.2007.0403
Giraud, A. (2001). Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291, 2606–2608. doi: 10.1126/science.1056421
Glazier, D. S. (2015). Is metabolic rate a universal ‘pacemaker’ for biological processes? Biol. Rev. 90, 377–407. doi: 10.1111/brv.12115
Goho, S., and Bell, G. (2000). Mild environmental stress elicits mutations affecting fitness in Chlamydomonas. Proc. R. Soc. B 267, 123–129. doi: 10.1098/rspb.2000.0976
Gokkusu, C., Cakmakoglu, B., Dasdemir, S., Tulubas, F., Elitok, A., Tamer, S., et al. (2013). Association between genetic variants of DNA repair genes and coronary artery disease. Genet. Test. Mol. Biomarkers 17, 307–313. doi: 10.1089/gtmb.2012.0383
Guelbeogo, W. M., Grushko, O., Boccolini, D., Ouedraogo, P. A., Besansky, N. J., Sagnon, N. F., et al. (2005). Chromosomal evidence of incipient speciation in the Afrotropical malaria mosquito Anopheles funestus. Med. Vet. Entomol. 19, 458–469. doi: 10.1111/j.1365-2915.2005.00595.x
Haraguchi, Y., and Sasaki, A. (1996). Host-parasite arms-race in mutation modifications-indefinate escalation despite a heavy load? J. Theor. Biol. 183, 121–137. doi: 10.1006/jtbi.1996.9999
Hartley, C., Newcomb, R., Russell, R., Yong, C., Stevens, J., Yeates, D., et al. (2006). Amplification of DNA from preserved specimens shows blowflies were preadapted for the rapid evolution of insecticide resistance. Proc. Natl. Acad. Sci. U.S.A. 103, 8757–8762. doi: 10.1073/pnas.0509590103
Herr, A. J., Ogawa, M., Lawrence, N. A., Williams, L. N., Eggington, J. M., Singh, M., et al. (2011a). Mutator suppression and escape from replication error?induced extinction in yeast. PLoS Genet. 7:e1002282. doi: 10.1371/journal.pgen.1002282
Herr, A. J., Williams, L. N., and Preston, B. D. (2011b). Antimutator variants of DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 46, 548–570. doi: 10.3109/10409238.2011.620941
Hodgins-Davis, A., Rice, D. P., and Townsend, J. P. (2015). Gene expression evolves under a house-of-cards model of stabilizing selection. Mol. Biol. Evol. 32, 2130–2140. doi: 10.1093/molbev/msv094
Hodgkinson, A., and Eyre-Walker, A. (2011). Variation in the mutation rate across mammalian genomes. Nature Rev. Genet. 12, 756–766. doi: 10.1038/nrg3098
Holmes, E. C. (2003). Error thresholds and the constraints to RNA virus evolution. Trends Microbiol. 11, 543–546. doi: 10.1016/j.tim.2003.10.006
Hua, X., Cowman, P., Warren, D., and Bromham, L. (2015). Longevity is linked to mitochondrial mutation rates in rockfish: a test using poisson regression. Mol. Biol. Evol. 32, 2633–2645. doi: 10.1093/molbev/msv137
Irwin, D. E., Irwin, J. H., and Price, T. D. (2001). Ring species as bridges between microevolution and speciation. Genetica 112, 223–243. doi: 10.1023/A:1013319217703
Isaac, N. J., Jones, K. E., Gittleman, J. L., and Purvis, A. (2005). Correlates of species richness in mammals: body size, life history, and ecology. Am. Nat. 165, 600–607. doi: 10.1086/429148
James, J. E., Lanfear, R., and Eyre-Walker, A. (2016). Molecular evolutionary consequences of island colonization. Genome Biol. Evol. 8, 1876–1888. doi: 10.1093/gbe/evw120
Jerome, J. P., Bell, J. A., Plovanich-Jones, A. E., Barrick, J. E., Brown, C. T., and Mansfield, L. S. (2011). Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel nost. PLoS ONE 6:e16399. doi: 10.1371/journal.pone.0016399
Jiggins, C. D., Naisbit, R. E., Coe, R. L., and Mallet, J. (2001). Reproductive isolation caused by colour pattern mimicry. Nature 411, 302–305. doi: 10.1038/35077075
Johnson, K. P., and Seger, J. (2001). Elevated rates of nonsynonymous substitution in island birds. Mol. Biol. Evol. 18, 874–881. doi: 10.1093/oxfordjournals.molbev.a003869
Johnson, N. A. (2010). Hybrid incompatibility genes: remnants of a genomic battlefield? Trends Genet. 26, 317–325. doi: 10.1016/j.tig.2010.04.005
Kabzinski, J., Mucha, B., Cuchra, M., Markiewicz, L., Przybylowska, I. K., Dziki, A., et al. (2016). Efficiency of base excision repair of oxidative DNA damage and its impact on the risk of colorectal cancer in the polish population. Oxid. Med. Cell. Longev 2016:9. doi: 10.1155/2016/3125989
Knight, C. A., Molinari, N. A., and Petrov, D. A. (2005). The large genome constraint hypothesis: evolution, ecology and phenotype. Ann. Bot. 95, 177–190. doi: 10.1093/aob/mci011
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations, father’s age, and disease risk. Nature 488, 471–475. doi: 10.1038/nature11396
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. doi: 10.1126/science.1112125
Lancaster, L. T. (2010). Molecular evolutionary rates predict both extinction and speciation in temperate angiosperm lineages. BMC Evol. Biol. 10:1. doi: 10.1186/1471-2148-10-162
Lande, R. (1976). Natural selection and random genetic drift in phenotypic evolution. Evolution 30, 314–334. doi: 10.2307/2407703
Lande, R. (1998). Risk of population extinction from fixation of deleterious and reverse mutations. Genetica 102, 21–27. doi: 10.1023/a:1017018405648
Lanfear, R., Ho, S. Y. W., Love, D., and Bromham, L. (2010). Mutation rate influences diversification rate in birds. Proc. Natl Acad. Sci. U.S.A. 107, 20423–20428. doi: 10.1073/pnas.1007888107
Lanfear, R., Thomas, J. A., Welch, J. J., and Bromham, L. (2007). Metabolic rate does not calibrate the molecular clock. Proc. Natl. Acad. Sci. U.S.A. 104, 15388–15393. doi: 10.1073/pnas.0703359104
Larsson, N.-G. R. (2010). Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79, 683–706. doi: 10.1146/annurev-biochem-060408-093701
Lartillot, N. (2013). Interaction between selection and biased gene conversion in mammalian protein-coding sequence evolution revealed by a phylogenetic covariance analysis. Mol. Biol. Evol. 30, 356–368. doi: 10.1093/molbev/mss231
Lartillot, N., and Delsuc, F. (2012). Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model. Evolution 66, 1773–1787. doi: 10.1111/j.1558-5646.2011.01558.x
Lehtonen, J., and Lanfear, R. (2014). Generation time, life history and the substitution rate of neutral mutations. Biol. Lett. 10:20140801. doi: 10.1098/rsbl.2014.0801
Loeb, L. A., Wallace, D. C., and Martin, G. M. (2005). The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci.U.S.A. 102, 18769–18770. doi: 10.1073/pnas.0509776102
Lowry, D. B., Modliszewski, J. L., Wright, K. M., Wu, C. A., and Willis, J. H. (2008). The strength and genetic basis of reproductive isolating barriers in flowering plants. Philos. Trans. R. Soc. B 363, 3009–3021. doi: 10.1098/rstb.2008.0064
Lucas-Lledó, J. I., and Lynch, M. (2009). Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family. Mol. Biol. Evol. 26, 1143–1153. doi: 10.1093/molbev/msp029
Lynch, M. (1996). “A quantitative-genetic perspective on conversation issues,” in Conservation Genetics: Case Studies From Nature, eds J. C. Avise and J. L. Hamrick (London: Chapman & Hall), 471–501.
Lynch, M. (2010). Evolution of the mutation rate. Trends Genet. 26, 345–352. doi: 10.1016/j.tig.2010.05.003
Lynch, M., Conery, J., and Burger, R. (1995). Mutation accumulation and the extinction of small populations. Am. Nat. 146, 489–518. doi: 10.1086/285812
Lynch, M., and Hill, W. G. (1986). Phenotypic evolution by neutral mutation. Evolution 40, 915–935. doi: 10.2307/2408753
Masly, J. P., Jones, C. D., Noor, M. A. F., Locke, J., and Orr, H. A. (2006). Gene transposition as a cause of hybrid sterility in Drosophila. Science 313, 1448–1450. doi: 10.1126/science.1128721
Mattimore, V., and Battista, J. R. (1996). Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J. Bacteriol. 178, 633–637. doi: 10.1128/jb.178.3.633-637.1996
Matute, D. R., Butler, I. A., Turissini, D. A., and Coyne, J. A. (2010). A test of the snowball theory for the rate of evolution of hybrid incompatibilities. Science 329, 1518–1521. doi: 10.1126/science.1193440
Maynard Smith, J., and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford: W. H. Freeman.
McDonald, M. J., Hsieh, Y. Y., Yu, Y. H., Chang, S. L., and Leu, J. Y. (2012). The evolution of low mutation rates in experimental mutator populations of Saccharomyces cerevisiae. Curr. Biol. 22, 1235–1240. doi: 10.1016/j.cub.2012.04.056
Miner, B. E., Kulling, P. M., Beer, K. D., and Kerr, B. (2015). Divergence in DNA photorepair efficiency among genotypes from contrasting UV radiation environments in nature. Mol. Ecol. 24, 6177–6187. doi: 10.1111/mec.13460
Mohrenweiser, H. W., Wilson, D. M., and Jones, I. M. (2003). Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat. Res. 526, 93–125. doi: 10.1016/S0027-5107(03)00049-6
Moyle, L. C., and Nakazato, T. (2010). Hybrid incompatibility ”snowballs” between Solanum Species. Science 329, 1521–1523. doi: 10.1126/science.1193063
Nabholz, B., Glemin, S., and Galtier, N. (2008). Strong variations of mitochondrial mutation rate across mammals - the longevity hypothesis. Mol. Biol. Evol. 25, 120–130. doi: 10.1093/molbev/msm248
Nabholz, B., Uwimana, N., and Lartillot, N. (2013). Reconstructing the phylogenetic history of long-term effective population size and life-history traits using patterns of amino acid replacement in mitochondrial genomes of mammals and birds. Genome Biol. Evol. 5, 1273–1290. doi: 10.1093/gbe/evt083
Nikolaev, S. I., Montoya-Burgos, J. I., Popadin, K., Parand, L., Margulies, E. H., Antonarakis, S. E., et al. (2007). Life-history traits drive the evolutionary rates of mammalian coding and noncoding genomic elements. Proc. Natl. Acad. Sci. U.S.A. 104, 20443–20448. doi: 10.1073/pnas.0705658104
Ohta, T. (1992). The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23, 263–286. doi: 10.1146/annurev.es.23.110192.001403
Oliver, A., Baquero, F., and Blázquez, J. (2002). The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol. Microbiol. 43, 1641–1650. doi: 10.1046/j.1365-2958.2002.02855.x
Oliver, A., Cantoìn, R., Campo, P., Baquero, F., and Blaìzquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1253. doi: 10.1126/science.288.5469.1251
Oliver, A., and Mena, A. (2010). Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infect. 16, 798–808. doi: 10.1111/j.1469-0691.2010.03250.x
Olson-Manning, C. F., Wagner, M. R., and Mitchell-Olds, T. (2012). Adaptive evolution: evaluating empirical support for theoretical predictions. Nat. Rev. Genet. 13, 867–877. doi: 10.1038/nrg3322
Orme, C., Quicke, D., Cook, J., and Purvis, A. (2002). Body size does not predict species richness among the metazoan phyla. J. Evol. Biol. 15, 235–247. doi: 10.1046/j.1420-9101.2002.00379.x
Orr, H. A. (1995). The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139, 1805–1813.
Orr, H. A., and Unckless, R. L. (2008). Population extinction and the genetics of adaptation. Am. Nat. 172, 160–169. doi: 10.1086/589460
Otto, S. P., Servedio, M. R., and Nuismer, S. L. (2008). Frequency-dependent selection and the evolution of assortative mating. Genetics 179, 2091–2112. doi: 10.1534/genetics.107.084418
Owens, I. P., Bennett, P. M., and Harvey, P. H. (1999). Species richness among birds: body size, life history, sexual selection or ecology? Proc. R. Soc. B 266, 933–939. doi: 10.1098/rspb.1999.0726
Pagel, M., Venditti, C., and Meade, A. (2006). Large punctuational contribution of speciation to evolutionary divergence at the molecular level. Science 314, 119–121. doi: 10.1126/science.1129647
Pamplona, R., and Barja, G. (2011). An evolutionary comparative scan for longevity-related oxidative stress resistance mechanisms in homeotherms. Biogerontology 12, 409–435. doi: 10.1007/s10522-011-9348-1
Phillimore, A. B., Freckleton, R. P., Orme, C. D., and Owens, I. P. (2006). Ecology predicts large-scale patterns of phylogenetic diversification in birds. Am. Nat. 168, 220–229. doi: 10.1086/505763
Phillips, N., Salomon, M., Custer, A., Ostrow, D., and Baer, C. F. (2009). Spontaneous mutational and standing genetic (co)variation at dinucleotide microsatellites in Caenorhabditis briggsae and Caenorhabditis elegans. Mol. Biol. Evol. 26, 659–699. doi: 10.1093/molbev/msn287
Piechura, J. R., Tseng, T.-L., Hsu, H.-F., Byrne, R. T., Windgassen, T. A., Chitteni-Pattu, S., et al. (2015). Biochemical characterization of RecA variants that contribute to extreme resistance to ionizing radiation. DNA Repair 26, 30–43. doi: 10.1016/j.dnarep.2014.12.001
Popadin, K., Polishchuk, L. V., Mamirova, L., Knorre, D., and Gunbin, K. (2007). Accumulation of slightly deleterious mutations in mitochondrial protein-coding genes of large versus small mammals. Proc. Natl. Acad. Sci. U.S.A. 104, 13390–13395. doi: 10.1073/pnas.0701256104
Presgraves, D. C. (2010). The molecular evolutionary basis of species formation. Nat. Rev. Genet. 11, 175–180. doi: 10.1038/nrg2718
Price, C. A., Weitz, J. S., Savage, V. M., Stegen, J., Clarke, A., Coomes, D. A., et al. (2012). Testing the metabolic theory of ecology. Ecol. Lett. 15, 1465–1474. doi: 10.1111/j.1461-0248.2012.01860.x
Ptacek, M. B., Gerhardt, H. C., and Sage, R. D. (1994). Speciation by polyploidy in treefrogs: multiple origins of the tetraploid, Hyla versicolor. Evolution 48, 898–908. doi: 10.2307/2410495
Quinn, T. P., Unwin, M. J., and Kinnison, M. T. (2000). Evolution of temporal isolation in the wild: genetic divergence in timing of migration and breeding by introduced chinook salmon populations. Evolution 54, 1372–1385. doi: 10.1111/j.0014-3820.2000.tb00569.x
Reha-Krantz, L. J. (2010). DNA polymerase proofreading: multiple roles maintain genome stability. Biochim. Biophys. Acta 1804, 1049–1063. doi: 10.1016/j.bbapap.2009.06.012
Ries, G., Heller, W., Puchta, H., Sandermann, H., Seidlitz, H. K., and Hohn, B. (2000). Elevated UV-B radiation reduces genome stability in plants. Nature 406, 98–101. doi: 10.1038/35017595
Rohde, K. (1992). Latitudinal gradients in species diversity: the search for the primary cause. Oikos 65, 514–527. doi: 10.2307/3545569
Romiguier, J., Ranwez, V., Douzery, E. J. P., and Galtier, N. (2013). Genomic evidence for large, long-lived ancestors to placental mammals. Mol. Biol. Evol. 30, 5–13. doi: 10.1093/molbev/mss211
Sanjuán, R., Nebot, M. R., Chirico, N., Mansky, L. M., and Belshaw, R. (2010). Viral mutation rates. J. Virol. 84, 9733–9748. doi: 10.1128/jvi.00694-10
Schaaper, R. M. (1998). Antimutator mutants in bacteriophage T4 and Escherichia coli. Genetics 148, 1579–1585.
Seehausen, O., Terai, Y., Magalhaes, I. S., Carleton, K. L., Mrosso, H. D. J., Miyagi, R., et al. (2008). Speciation through sensory drive in cichlid fish. Nature 455, 620–623. doi: 10.1038/nature07285
Seehausen, O., and van Alphen, J. J. M. (1998). The effect of male coloration on female mate choice in closely related Lake Victoria cichlids (Haplochromis nyererei complex). Behav. Ecol. Sociobiol. 42, 1–8. doi: 10.1007/s002650050405
Servedio, M. R., and Noor, M. A. F. (2003). The role of reinforcement in speciation: theory and data. Annu. Rev. Ecol. Evol. Syst. 34, 339–364. doi: 10.1146/annurev.ecolsys.34.011802.132412
Servedio, M. R., Van Doorn, G. S., Kopp, M., Frame, A. M., and Nosil, P. (2011). Magic traits in speciation: ’magic’ but not rare? Trends Ecol. Evol. 26, 389–397. doi: 10.1016/j.tree.2011.04.005
Sexton, J. P., McIntyre, P. J., Angert, A. L., and Rice, K. J. (2009). Evolution and ccology of species range limits. Annu. Rev. Ecol. Evol. Syst. 40, 415–436. doi: 10.1146/annurev.ecolsys.110308.120317
Smith, N. G. C. (2003). Are radical and conservative substitution rates useful statistics in molecular evolution? J. Mol. Evol. 57, 467–478. doi: 10.1007/s00239-003-2500-z
Smith, S. A., and Beaulieu, J. M. (2009). Life history influences rates of climatic niche evolution in flowering plants. Proc. R. Soc. B 276, 4345–4352. doi: 10.1098/rspb.2009.1176
Sniegowski, P. D., Gerrish, P. J., Johnson, T., and Shaver, A. (2000). The evolution of mutation rates: separating causes from consequences. Bioessays 22, 1057–1066. doi: 10.1002/1521-1878(200012)22:12<1057::AID-BIES3<3.0.CO;2-W
Sniegowski, P. D., Gerrish, P. J., and Lenski, R. E. (1997). Evolution of high mutation rates in experimental populations of E. coli. Nature 387, 703–705. doi: 10.1038/42701
Stadler, T., Florez-Rueda, A. M., and Paris, M. (2012). Testing for ”snowballing” hybrid incompatibilities in Solanum: impact of ancestral polymorphism and divergence estimates. Mol. Biol. Evol. 29, 31–34. doi: 10.1093/molbev/msr218
Stockwell, C. A., Hendry, A. P., and Kinnison, M. T. (2003). Contemporary evolution meets conservation biology. Trends Ecol. Evol. 18, 94–101. doi: 10.1016/S0169-5347(02)00044-7
Stuart-Fox, D., and Owens, I. P. F. (2003). Species richness in agamid lizards: chance, body size, sexual selection or ecology? J. Evol. Biol. 16, 659–669. doi: 10.1046/j.1420-9101.2003.00573.x
Sundin, G. W., and Weigand, M. R. (2007). The microbiology of mutability. FEMS Microbiol. Lett. 277, 11–20. doi: 10.1111/j.1574-6968.2007.00901.x
Sung, W., Tucker, A. E., Doak, T. G., Choi, E., Thomas, W. K., and Lynch, M. (2012). Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proc. Natl. Acad. Sci. U.S.A. 109, 19339–19344. doi: 10.1073/pnas.1210663109
Svetec, N., Cridland, J. M., Zhao, L., and Begun, D. J. (2016). The adaptive significance of natural genetic variation in the DNA damage response of Drosophila melanogaster. PLoS Genet. 12:e1005869. doi: 10.1371/journal.pgen.1005869
Taddei, F. (1997). Role of mutator alleles in adaptive evolution. Nature 387, 700–702. doi: 10.1038/42696
Thomas, J. A., Welch, J. J., Lanfear, R., and Bromham, L. (2010). A generation time effect on the rate of molecular evolution in invertebrates. Mol. Biol. Evol. 27, 1173–1180. doi: 10.1093/molbev/msq009
Todesco, M., Pascual, M. A., Owens, G. L., Ostevik, K. L., Moyers, B. T., Hübner, S., et al. (2016). Hybridization and extinction. Evol. Appl. 9, 892–908. doi: 10.1111/eva.12367
Torres-Barceló, C., Cabot, G., Oliver, A., Buckling, A., and MacLean, R. C. (2013). A trade-off between oxidative stress resistance and DNA repair plays a role in the evolution of elevated mutation rates in bacteria. Proc. R. Soc. B 280:20130007. doi: 10.1098/rspb.2013.0007
Turelli, M. (1984). Heritable genetic variation via mutation selection balance: Lerch’s Zeta meets the abdominal bristle. Theor. Popul. Biol. 25, 138–193. doi: 10.1016/0040-5809(84)90017-0
Twyford, A. D., and Friedman, J. (2015). Adaptive divergence in the monkey flower Mimulus guttatus is maintained by a chromosomal inversion. Evolution 69, 1476–1486. doi: 10.1111/evo.12663
Venn, O., Turner, I., Mathieson, I., de Groot, N., Bontrop, R., and McVean, G. (2014). Strong male bias drives germline mutation in chimpanzees. Science 344, 1272–1275. doi: 10.1126/science.344.6189.1272
Via, S. (2009). Natural selection in action during speciation. Proc. Natl. Acad. Sci. U.S.A. 106, 9939–9946. doi: 10.1073/pnas.0901397106
Via, S. (2012). Divergence hitchhiking and the spread of genomic isolation during ecological speciation-with-gene-flow. Philos. Trans. R. Soc. B 367, 451–460. doi: 10.1098/rstb.2011.0260
Via, S., Bouck, A. C., and Skillman, S. (2000). Reproductive isolation between divergent races of pea aphids on two hosts. II. Selection against migrants and hybrids in the parental environments. Evolution 54, 1626–1637. doi: 10.1111/j.0014-3820.2000.tb00707.x
Vigouroux, Y., Jaqueth, J. S., Matsuoka, Y., Smith, O. S., Beavis, W. F., Smith, J. S. C., et al. (2002). Rate and pattern of mutation at microsatellite loci in maize. Mol. Biol. Evol. 19, 1251–1260. doi: 10.1093/oxfordjournals.molbev.a004186
Wang, R. J., Ane, C., and Payseur, B. A. (2013). The evolution of hybrid incompatibilities along a phylogeny. Evolution 67, 2905–2922. doi: 10.1111/evo.12173
Wang, R. J., White, M. A., and Payseur, B. A. (2015). The pace of hybrid incompatibility evolution in house mice. Genetics 201, 229–242. doi: 10.1534/genetics.115.179499
Wang, Z., Yonezawa, T., Liu, B., Ma, T., Shen, X., Su, J., et al. (2011). Domestication relaxed selective constraints on the yak mitochondrial genome. Mol. Biol. Evol. 28, 1553–1556. doi: 10.1093/molbev/msq336
Waxman, D. (2011). A unified treatment of the probability of fixation when population size and the strength of selection change over time. Genetics 188, 907–913. doi: 10.1534/genetics.111.129288
Weber, C. C., Nabholz, B., Romiguier, J., and Ellegren, H. (2014). Kr/Kc but not dN/dS correlates positively with body mass in birds, raising implications for inferring lineage-specific selection. Genome Biol. 15, 542. doi: 10.1186/s13059-014-0542-8
Welch, J. J. (2004). Accumulating Dobzhansky-Muller incompatibilities: reconciling theory and data. Evolution 58, 1145–1156. doi: 10.1554/03-502
Welch, J. J., Bininda-Emonds, O. R. P., and Bromham, L. (2008). Correlates of substitution rate variation in mammalian protein-coding sequences. BMC Evol. Biol. 8:53. doi: 10.1186/1471-2148-8-53
Wiens, J. J. (2004). What is speciation and how should we study it? Am. Nat. 163, 914–923. doi: 10.1086/386552
Wilson Sayres, M. A., and Makova, K. D. (2011). Genome analyses substantiate male mutation bias in many species. Bioessays 33, 938–945. doi: 10.1002/bies.201100091
Wilson Sayres, M. A., Venditti, C., Pagel, M., and Makova, K. D. (2011). Do variations in substitution rates and male mutation bias correlate with life-history traits? A study of 32 mammalian genomes. Evolution 65, 2800–2815. doi: 10.1111/j.1558-5646.2011.01337.x
Wood, T. E., Takebayashi, N., Barker, M. S., Mayrose, I., Greenspoon, P. B., and Rieseberg, L. H. (2009). The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. U.S.A. 106, 13875–13879. doi: 10.1073/pnas.0811575106
Woolfit, M., and Bromham, L. (2005). Population size and molecular evolution on islands. Proc. R. Soc. B 272, 2277–2282. doi: 10.1098/rspb.2005.3217
Wright, S. D., Gillman, L. N., Ross, H. A., and Keeling, D. J. (2009). Slower tempo of microevolution in island birds: implications for conservation biology. Evolution 63, 2275–2287. doi: 10.1111/j.1558-5646.2009.00717.x
Yamamoto, E., Takashi, T., Morinaka, Y., Lin, S., Wu, J., Matsumoto, T., et al. (2010). Gain of deleterious function causes an autoimmune response and Bateson–Dobzhansky–Muller incompatibility in rice. Mol. Genet. Genomics 283, 305–315. doi: 10.1007/s00438-010-0514-y
Yang, J. N., Seluanov, A., and Gorbunova, V. (2013). Mitochondrial inverted repeats strongly correlate with lifespan: mtDNA inversions and aging. PLoS ONE 8:e73318. doi: 10.1371/journal.pone.0073318
Keywords: molecular evolution, macroevolution, phylogeny, comparative studies, reproductive isolation
Citation: Hua X and Bromham L (2017) Darwinism for the Genomic Age: Connecting Mutation to Diversification. Front. Genet. 8:12. doi: 10.3389/fgene.2017.00012
Received: 09 August 2016; Accepted: 19 January 2017;
Published: 07 February 2017.
Edited by:
Scott V. Edwards, Harvard University, USAReviewed by:
Brian C. Verrelli, Virginia Commonwealth University, USAAmy Litt, University of California, Riverside, USA
Copyright © 2017 Hua and Bromham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Hua, eGlhLmh1YUBhbnUuZWR1LmF1