 Jennifer Larimore1*
Jennifer Larimore1* Stephanie A. Zlatic2†
Stephanie A. Zlatic2† Miranda Arnold1†
Miranda Arnold1† Kaela S. Singleton1
Kaela S. Singleton1 Rebecca Cross1
Rebecca Cross1 Hannah Rudolph1
Hannah Rudolph1 Martha V. Bruegge1
Martha V. Bruegge1 Andrea Sweetman1
Andrea Sweetman1 Cecilia Garza1Eli Whisnant1
Cecilia Garza1Eli Whisnant1 Victor Faundez2*
Victor Faundez2*
- 1Department of Biology, Agnes-Scott College, Decatur, GA, USA
- 2Department of Cell Biology, Emory University, Atlanta, GA, USA
The neurodevelopmental factor dysbindin is required for synapse function and GABA interneuron development. Dysbindin protein levels are reduced in the hippocampus of schizophrenia patients. Mouse dysbindin genetic defects and other mouse models of neurodevelopmental disorders share defective GABAergic neurotransmission and, in several instances, a loss of parvalbumin-positive interneuron phenotypes. This suggests that mechanisms downstream of dysbindin deficiency, such as those affecting GABA interneurons, could inform pathways contributing to or ameliorating diverse neurodevelopmental disorders. Here we define the transcriptome of developing wild type and dysbindin null Bloc1s8sdy/sdy mouse hippocampus in order to identify mechanisms downstream dysbindin defects. The dysbindin mutant transcriptome revealed previously reported GABA parvalbumin interneuron defects. However, the Bloc1s8sdy/sdy transcriptome additionally uncovered changes in the expression of molecules controlling cellular excitability such as the cation-chloride cotransporters NKCC1, KCC2, and NCKX2 as well as the potassium channel subunits Kcne2 and Kcnj13. Our results suggest that dysbindin deficiency phenotypes, such as GABAergic defects, are modulated by the expression of molecules controlling the magnitude and cadence of neuronal excitability.
Introduction
Dysbindin is a neurodevelopmental gene product encoded by DTNBP1, a gene whose polymorphisms influence cognitive and neuroanatomical traits in non-disease individuals. (Straub et al., 2002; Van Den Bogaert et al., 2003; Bray et al., 2005; Luciano et al., 2009; Markov et al., 2009, 2010; Mechelli et al., 2010; Cerasa et al., 2011; Tognin et al., 2011; Wolf et al., 2011; Ayalew et al., 2012; Trost et al., 2013). DTNBP1 polymorphisms have been considered risk factors for schizophrenia onset (Straub et al., 2002; Van Den Bogaert et al., 2003), yet this is not a consensus view (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Farrell et al., 2015). Dysbindin polypeptide expression is reduced in the brain of schizophrenia affected individuals, in particular synaptic fields of the hippocampal formation suggesting a dysbindin requirement for synapse function (Talbot et al., 2004, 2006, 2011; Tang et al., 2009). Dysbindin’s necessity for normal synapse architecture and function has been best documented in dysbindin mutant organisms. The sandy mouse, a dysbindin null mutation (Bloc1s8sdy/sdy), affects the biogenesis of synaptic vesicles and impairs glutamatergic and GABAergic neurotransmission (Jentsch et al., 2009; Ji et al., 2009; Carlson et al., 2011; Ghiani and Dell’angelica, 2011; Karlsgodt et al., 2011; Larimore et al., 2011; Mullin et al., 2011; Yuan et al., 2016). Similarly, Drosophila dysbindin mutants are characterized by impaired neurotransmission, abrogated synaptic homeostasis, pre- and post-synaptic morphological alterations, and defects in short term memory (Dickman and Davis, 2009; Cheli et al., 2010; Shao et al., 2011; Dickman et al., 2012; Gokhale et al., 2015a, 2016; Mullin et al., 2015). The reduction of dysbindin in the hippocampus of patients with schizophrenia, alterations in excitatory/inhibitory signaling in the mouse, and changes in neurotransmission impacting short term memory in Drosophila demonstrate that dysbindin-dependent pathways provide insight into mechanisms of schizophrenia and other neurodevelopmental disorders. The focus of this work is the characterization of these dysbindin-dependent mechanisms and pathways in the developing mouse brain.
Dysbindin Bloc1s8sdy/sdy mutant possesses impaired GABAergic neurotransmission, a consequence of decreased parvalbumin positive interneurons (Ji et al., 2009; Carlson et al., 2011; Larimore et al., 2014; Yuan et al., 2016). Similarly, GABA neurotransmission dysfunction has been implicated in multiple neurodevelopmental disorders including autism and schizophrenia (Akbarian et al., 1995; Guidotti et al., 2000; Hashimoto et al., 2003, 2005, 2008a,b; Tabuchi et al., 2007; Gogolla et al., 2009; Sohal et al., 2009; Chao et al., 2010; Han et al., 2012; Marin, 2012; Del Pino et al., 2013; Gonzalez-Burgos et al., 2015; Mariani et al., 2015; Wohr et al., 2015). The convergence on GABA interneuron defects among multiple models of neurodevelopmental disorders, including dysbindin mutants, prompted us to interrogate transcriptional responses of developing hippocampal neurons bearing null mutations in dysbindin. We reasoned mechanisms sensitive to dysbindin mutations would inform us about GABA response pathways implicated in diverse neurodevelopmental disorders. Here we describe transcript modifications in the developing Bloc1s8sdy/sdy hippocampus. The dysbindin deficiency transcriptome not only captured the previously described GABA interneuron phenotype but, in addition, revealed changes in the expression of molecules controlling cellular excitability such as cation-chloride cotransporters and potassium channel subunits. Our results suggest that GABAergic phenotypes in dysbindin deficiency are developmentally modulated by complex changes in the expression of channels and transporters controlling the magnitude and tempo of neuronal excitability.
Results
Null Alleles of the BLOC-1 Subunits Dysbindin, Muted, and Pallid Differentially Affect GABAergic Interneurons
Dysbindin (Bloc1s8) is a subunit of the cytosolic hetero-octamer known as the biogenesis of lysosome-related organelles complex 1 (BLOC-1). This complex consists of Bloc1s1–8 subunits (Li et al., 2003; Wei, 2006; Ghiani et al., 2009; Ghiani and Dell’angelica, 2011). Dysbindin-null Bloc1s8sdy/sdy mouse hippocampi have reduced numbers of GABAergic interneurons and diverse interneuron markers resulting in impaired inhibitory neurotransmission in the hippocampus (Carlson et al., 2011; Larimore et al., 2014). Here, we explored the ontological and anatomical penetrance of interneuron phenotypes in mouse mutants affecting three subunits of the dysbindin-BLOC-1 complex: dysbindin, muted, and pallid; which are encoded by the genes Bloc1s8, Bloc1s5, and Bloc1s6, respectively, in mouse (Larimore et al., 2014). As a first step, we sought to identify dysbindin-BLOC-1 complex subunit mutant mouse age and anatomical region with the most prominent interneuron phenotypes. We then used this information to identify transcriptional responses to dysbindin-BLOC-1 dependent impaired inhibitory neurotransmission.
We performed quantitative immunofluorescence microscopy of parvalbumin-positive interneurons in the hippocampal formation of adult (P50) wild type and dysbindin-BLOC-1 null mice: Bloc1s8sdy/sdy, Bloc1s5mu/mu, and Bloc1s6pa/pa (Figure 1A). We focused on parvalbumin positive cells because of their abundance in hippocampus as compared to other interneuron types (Celio, 1986; Tamamaki et al., 2003; Whissell et al., 2015). In addition, parvalbumin positive interneurons phenotypes in adult P50 Bloc1s8sdy/sdy hippocampus are shared with other interneurons, which we previously scored with diverse GABAergic transcript markers (Larimore et al., 2014). We determined the numbers of parvalbumin-positive cells in the dentate gyrus, CA1, and CA3 regions of the hippocampus and confirmed previously described decreases in CA1 and CA3 parvalbumin-positive cells in Bloc1s8sdy/sdy (Figure 1B) (Carlson et al., 2011). These results are also in agreement with reduced levels of diverse GABAergic interneuron mRNAs in adult Bloc1s8sdy/sdy hippocampus (Larimore et al., 2014). In contrast, parvalbumin cell count phenotypes were less pronounced or absent from the CA1 and CA3 regions of Bloc1s5mu/mu and Bloc1s6pa/pa (Figures 1A,B). We did not detect changes in parvalbumin cell counts in the dentate gyrus in any of the dysbindin-BLOC-1 mutant genotypes analyzed (Figure 1B). These results indicate that the most severe and anatomically penetrant GABAergic phenotypes are observed in Bloc1s8sdy/sdy.
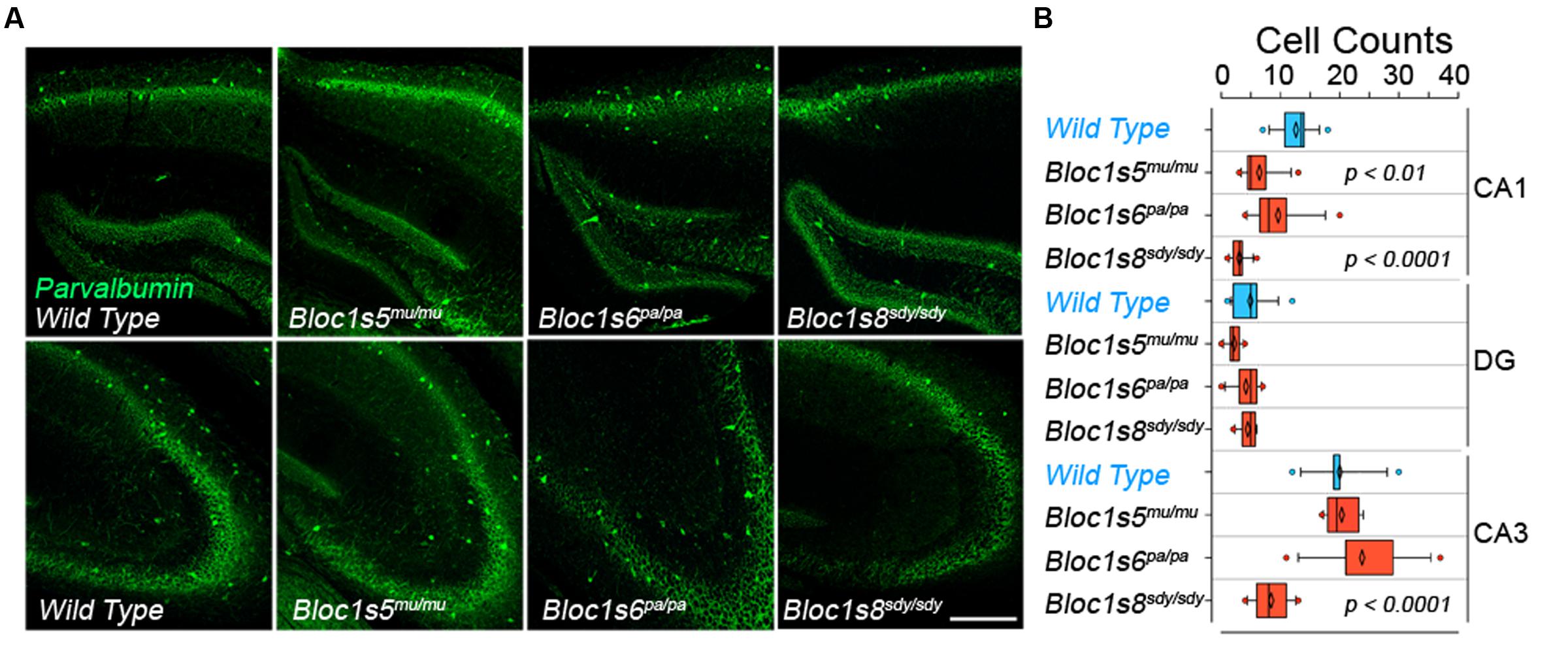
FIGURE 1. Differential Effects of BLOC-1 Complex Null Mutations in the Number of Parvalbumin-Positive Neurons. Hippocampal sections from wild type, Bloc1s8sdy/sdy, Bloc1s5mu/mu, and Bloc1s6pa/pa postnatal day 50 adults were stained with antibodies against parvalbumin and imaged by confocal microscopy. (A) Depicts sections of the dentate gyrus and CA1 (upper panels) and the CA3 area (bottom panels) for four genotypes. (B) Depicts quantitation of the number of parvalbumin positive cells per section in each one of the genotypes. Cell counts were performed blind to the genotype of the animal. Wild type (n = 9), Bloc1s8sdy/sdy (n = 7), Bloc1s5mu/mu (n = 7), and Bloc1s6pa/pa (n = 6) hippocampi analyzed. One Way ANOVA followed by Dunnett’s Multiple Comparison.
In order to define the ontology and anatomical penetrance of the Bloc1s8sdy/sdy parvalbumin phenotype, we used quantitative RT-PCR to measure transcripts encoding GABAergic interneuron markers parvalbumin (Pvalb), the vesicular GABA transporter (VGAT, Slc32a1), the plasma membrane GABA transporter GAT2 (Slc6a13), and glutamate decarboxylase 2 (Gad2), an enzyme catalyzing the production of gamma-aminobutyric acid from L-glutamic acid (Figure 2). mRNA levels of all of these GABA interneurons markers were significantly decreased in the hippocampal formation from embryonic day 18 to 14 days post-natal age (Figure 2A). The selectivity of these mRNA modifications was determined by measuring the transcript of synaptophysin (Syp), a globally expressed synaptic vesicle protein whose transcripts are insensitive to Bloc1s8sdy/sdy mutation (Larimore et al., 2014). We reasoned that reductions in GABAergic interneuron function would be reflected in possible compensatory postsynaptic changes in the transcripts of chloride channels that control the inhibitory or excitatory responses of target neurons. NKCC1 and KCC2 are sodium-potassium and potassium-chloride co-transporters, respectively, whose expression levels determine whether GABA induces depolarization or hyperpolarization of target cells (Kaila et al., 2014). Normally, NKCC1 predominates in immature target neurons whereas KCC2 expression is low in immature cells (Kaila et al., 2014). This NKCC1 and KCC2 pattern of expression was drastically accentuated in embryonic 18 and newborn Bloc1s8sdy/sdy mouse hippocampus (Figure 2A). NKCC1 transcripts were increased two- to sixfold in E18 and newborn Bloc1s8sdy/sdy hippocampus. In contrast, the expression of KCC2 was reduced by several orders of magnitude in Bloc1s8sdy/sdy hippocampus as compared to age matched wild type animals (Figure 2A). These Bloc1s8sdy/sdy NKCC1 and KCC2 expression trends were reverted (NKCC1) or ameliorated (KCC2) by postnatal day 14, a developmental time where the chloride electrochemical equilibrium potential reaches adult levels (Figure 2A) (Berglund et al., 2006; Kaila et al., 2014). We compared these hippocampal GABA neuron marker and chloride transporter mRNA phenotypes with expression of these markers in the prefrontal cortex of embryonic 18 and newborn wild type and Bloc1s8sdy/sdy (Figure 2B). Prefrontal cortex has decreased parvalbumin, VGAT, and KCC2 mRNA levels yet the magnitude of these changes was modest as compared to the mutant hippocampus (Figure 2B). These data argue that the most penetrant GABAergic molecular phenotypes occur in the hippocampus of Bloc1s8sdy/sdy between birth and postnatal age 14. Furthermore, these Bloc1s8sdy/sdy GABAergic defects associate with changes in the expression of postsynaptic chloride channels that determine GABA-dependent excitatory or inhibitory tone during development.
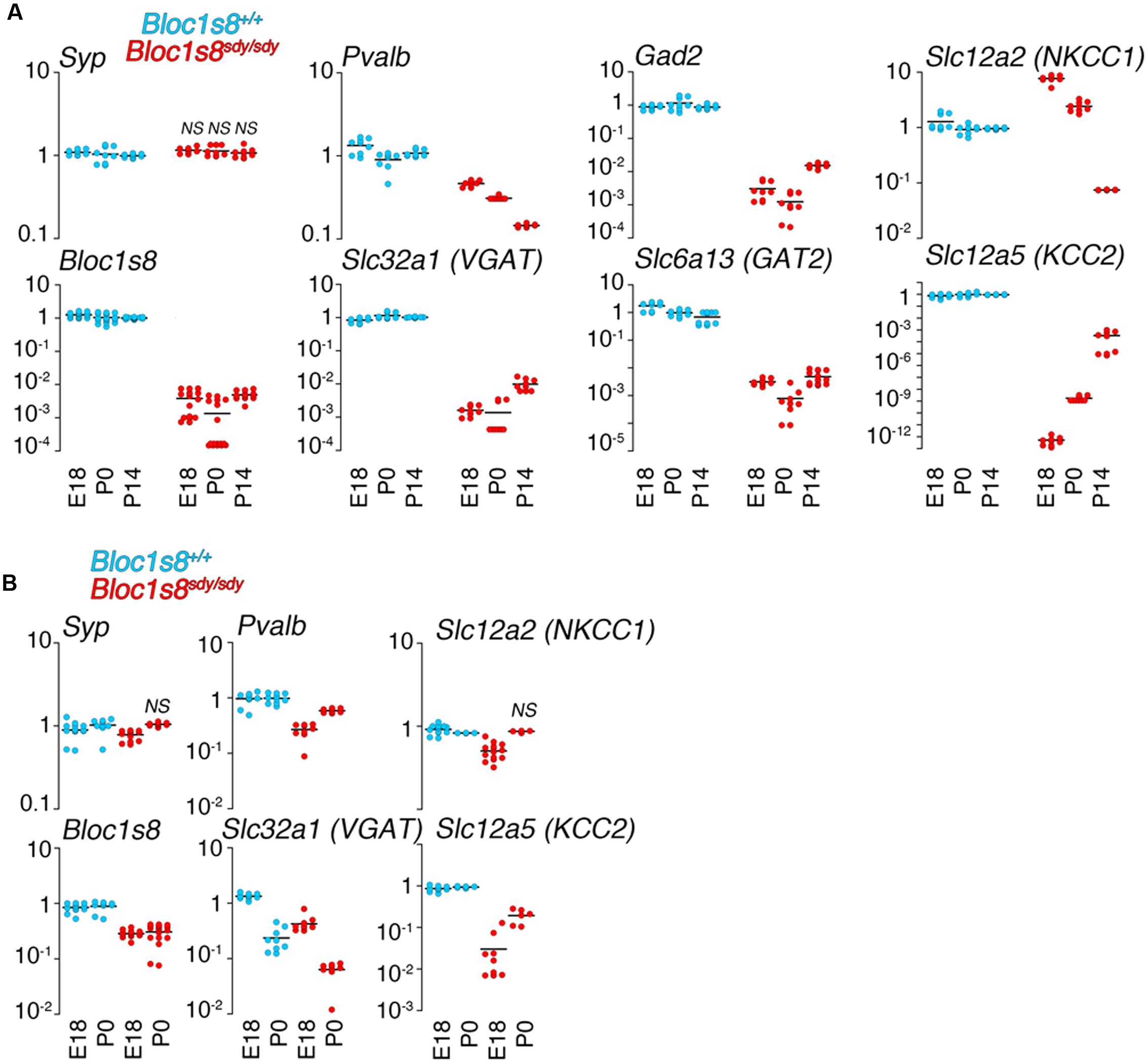
FIGURE 2. Developmental Changes in the Expression of GABA Neuron Markers and Chloride Cotransporters Transcripts in Dysbindin Null Hippocampus. (A) mRNA was isolated from wild type and Bloc1s8sdy/sdy embryonic day 18, newborn (P0), and postnatal day 14 (P14) hippocampi. Transcripts were quantified by real time quantitative PCR for the indicated GABA neuron gene products and the chloride transporters NKCC1 and KCC2. (B) Shows a similar analysis performed in prefrontal cortex. Tissue collections and processing were performed for each age independently, thus qRT-PC comparisons were performed within a specific age. Wild type values for a transcript were defined as 1 at each specified age. All determinations were performed from at least three animals per genotype and two independent cDNA preparations for qRT-PCR. Comparisons between wild type and Bloc1s8sdy/sdy were performed with Wilcoxon–Mann–Whitney Rank Sum Test. All comparisons were significantly different with a p < 0.00041 unless specified otherwise as not significant (NS).
Transcriptome Analysis of Bloc1s8sdy/sdy Hippocampus
We performed RNAseq of the hippocampus of wild type and Bloc1s8sdy/sdy hippocampus at postnatal day seven (Figure 3 and Supplementary Table S1). We selected this age because it is in between the ages with the most pronounced GABAergic transcript levels modifications (Figure 2), and it coincides with peak hippocampal synaptogenesis in mice (Mody et al., 2001). We prepared normalized polyA cDNA libraries of three wild type and three Bloc1s8sdy/sdy hippocampi and determined the abundance of transcripts by Illumina HiSeq2000 sequencing (Mortazavi et al., 2008). Illumina sequencing quantitation identified 111 transcripts whose content was significantly modified in Bloc1s8sdy/sdy hippocampi (q < 0.005, see Supplementary Table S1). Eighty of the transcripts were upregulated and 31 were downregulated (Figures 3A,B and Supplementary Table S1). The transcriptome did not detect the changes in expression of NKCC1 or KCC2 detected by qRT-PCR (compare Figure 2 and Supplementary Table S1). However, the transcriptome identified landmark mRNAs that validated the transcript dataset. First, we found that transcripts encoding dysbindin (DTNBP1, Bloc1s8) and parvalbumin (Pvalb) were reduced in Bloc1s8sdy/sdy hippocampi by 50% (Figures 3B,D,E and Supplementary Table S1). Additionally, transcripts of synaptic vesicle proteins insensitive to the Bloc1s8sdy/sdy mutation and used as controls, such as synaptophysin and VAMP2, were not affected by the Bloc1s8sdy/sdy mutation (Figures 3B,C and Supplementary Table S1) (Larimore et al., 2014). We confirmed mRNA expression of all of these markers by qRT-PCR in wild type and Bloc1s8sdy/sdy seven day postnatal hippocampi (Figure 3F).
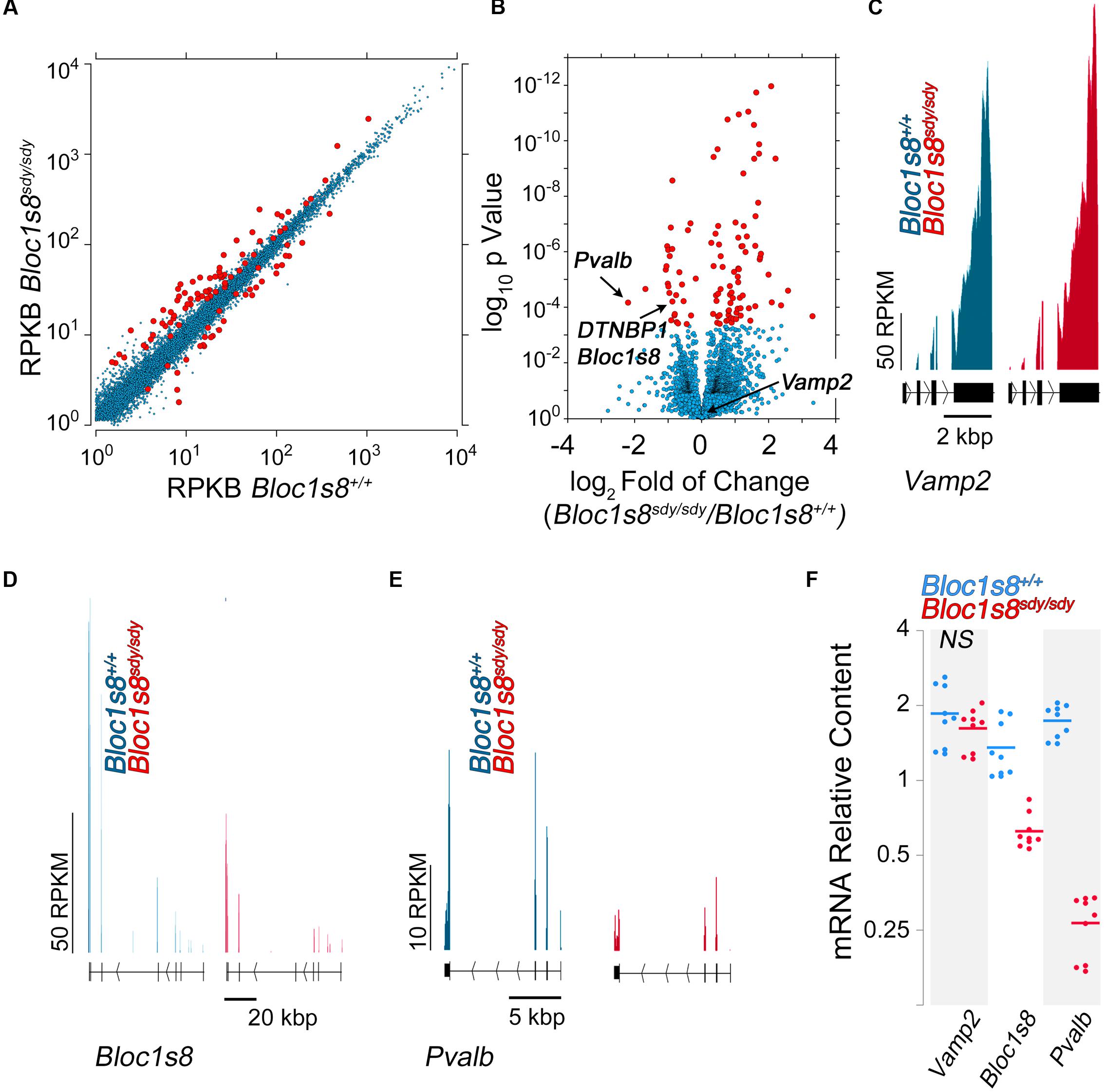
FIGURE 3. Transcriptome of the Dysbindin Null Developing Hippocampus. (A) Scatter plot of normalized read counts per gene calculated by Cuffdiff analysis in postnatal seven hippocampi from wild type and Bloc1s8sdy/sdy animals, n = 3. RKBP = reads per kilobase of transcript per million mapped reads. Red dots represent genes differentially expressed with a q-value <0.05. (B) Volcano plot of Cuffdiff analysis shows (Trapnell et al., 2012) differentially expressed genes in Bloc1s8sdy/sdy hippocampus as in (A). Red dots correspond to genes with >2 log2-fold differential expression or <-1 log2-fold differential expression. (C–E) Illumina sequence reads maps for the listed mouse genes from assembly mm9. VAMP2 is a control gene whose expression is not sensitive to Bloc1s8sdy/sdy. (F) Postnatal day 7 hippocampi were quantified by real time quantitative PCR for the indicated gene products. All determinations were performed from at least three animals per genotype and three independent cDNA preparations for qRT-PCR. Comparisons between wild type and Bloc1s8sdy/sdy were performed with Wilcoxon–Mann–Whitney Rank Sum Test. All comparisons were significantly different with a p < 0.0005 unless specified otherwise as not significant (NS).
We performed gene ontology analysis to prioritize transcripts associated to pathways and mechanisms for independent confirmation and further study (Figure 4). We used two different bioinformatics algorithms, ENRICHR and DAVID, to statistically assess gene product enrichment within gene ontological (GO) terms (Huang da et al., 2009; Chen et al., 2013). The major biological process GO terms associated to the Bloc1s8sdy/sdy-sensitive transcriptome were Wnt signaling planar cell polarity (BP GO:2000095, p < 0.00006), excitatory postsynaptic membrane potential (BP GO:0060079, p < 0.00002) as well as ontological terms associated to solute transport and ion permeation through membranes (all p-values <0.0036, Figure 4A). Similar outcomes were obtained either by analyzing molecular function GO terms where the top term was channel activity (MF GO:0015267, p < 0.0027, Figure 4B) or by using the REACTOME database, where top terms where implicated in solute transport (R-HSA-425366 and R-HSA-425407, p-values <0.000032 and <0.00017, respectively, Figure 4C). Gene ontology analysis using the DAVID algorithm lent the same results as ENRICHR. Ion transport emerged as the top biological process category (BP GO:0006811 p < 4.29E-04). Gene ontology categories identified in the Bloc1s8sdy/sdy-sensitive transcriptome were not recognized within a random brain gene dataset of comparable size (Figure 4D). Similarly, simultaneous ontology analyses of the random brain gene data set and the Bloc1s8sdy/sdy-sensitive transcriptome using the engine GeneCodis showed no ontology overlap (Tabas-Madrid et al., 2012).
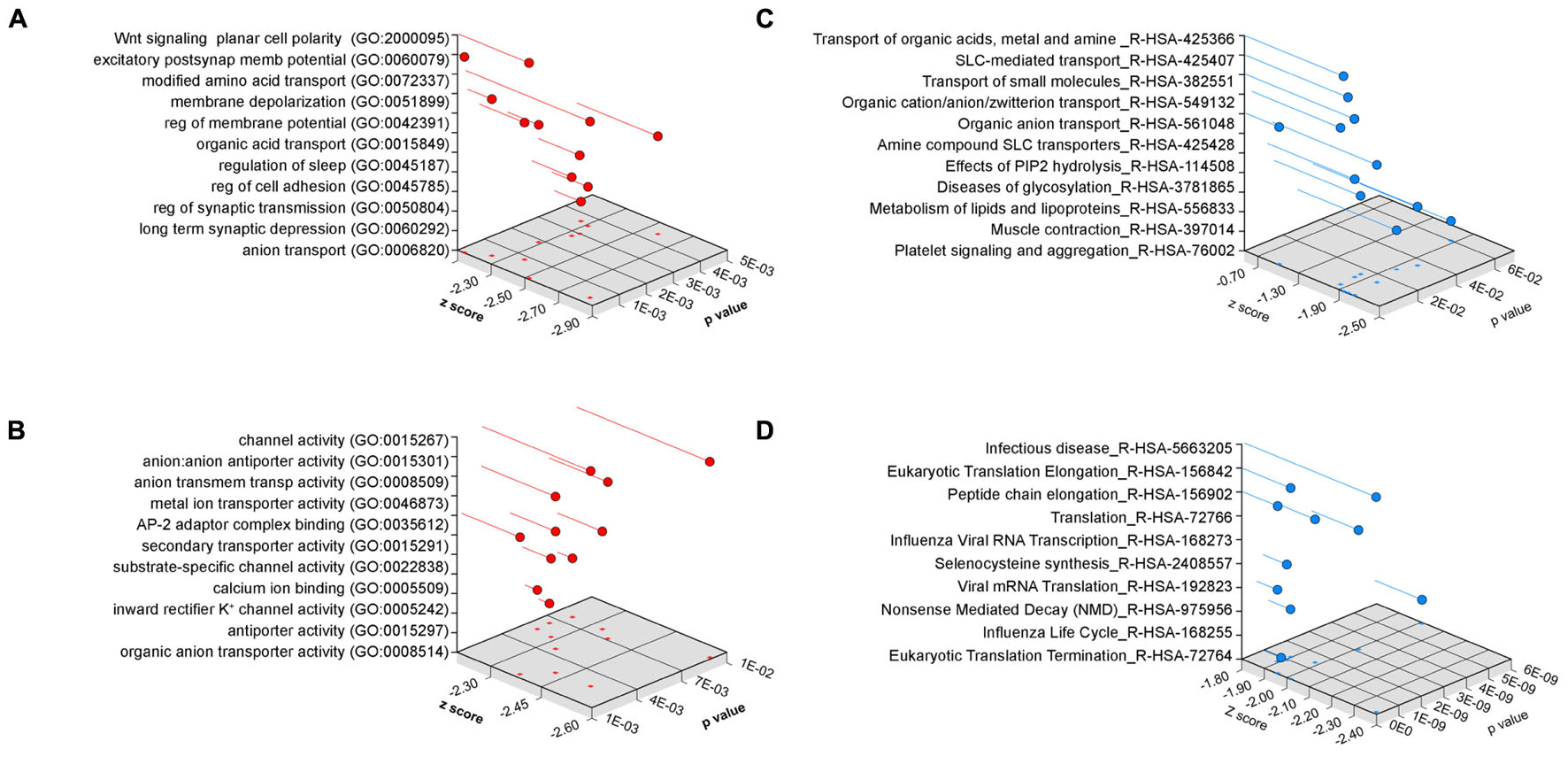
FIGURE 4. Ontology Analysis of the Dysbindin Null Developing Hippocampus. Gene ontology analysis of the Bloc1s8sdy/sdy P7 hippocampus using ENRICHR for (A) biological process, (B) molecular function, and (C) the REACTOME database. (D) Depicts ENRICHR REACTOME analysis for a custom generated random human brain gene data set presented in Supplementary Table S2.
Twelve percent of the 111 transcripts sensitive to Bloc1s8sdy/sdy belonged to ion transport gene ontology terms. Among these gene products are those encoded by Fxyd1, Trpm3, Slc39a10, Slc22a8, Slc24a2, Slc22a6, Slc13a4, Tcn2, Trf, Kcnj13, Kcne2, Camk2a, and Steap1. We focused on these ion transport gene products to confirm by qRT-PCR the Bloc1s8sdy/sdy transcriptome dataset. In total, we chose for confirmation by qRT-PCR 19 of the 111 transcriptome hits. Among these confirmed hits were Fxyd1, Trpm3, Slc39a10, Slc24a2, Trf, Kcnj13, Kcne2, and Camk2a, which belong to the ion transport ontology term (GO:0006811, Figures 5A,B). In addition, we confirmed Cap2, Chgb, Cpne6, Lynx1, and Mbp; which were selected among gene products implicated in schizophrenia and neurological disorders by the DAVID algorithm (p < 2.35E-03, Figure 5C). These results validate the Bloc1s8sdy/sdy hippocampal transcriptome dataset by means of an ontological prioritization of mRNA phenotypes.
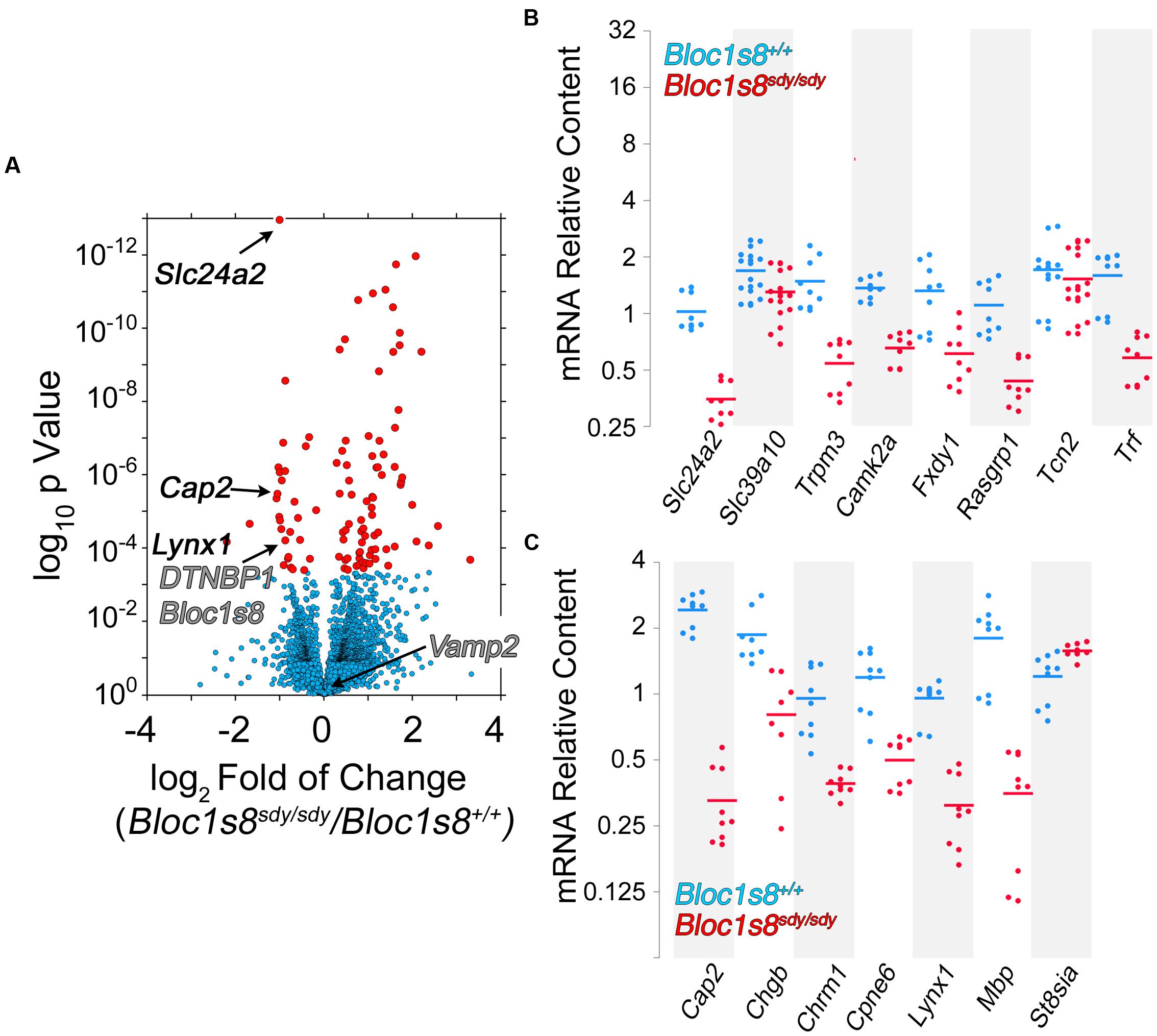
FIGURE 5. Dysbindin null developing hippocampus changes the expression of ion transport gene products. (A) Volcano plot of Cuffdiff analysis (Trapnell et al., 2012) shows differentially expressed genes in Bloc1s8sdy/sdy hippocampus as in Figure 3B. Arrows mark genes belonging the ion transport ontology terms and neurological disorder according to DAVID analysis. (B,C) Postnatal day 7 hippocampi mRNA were quantified by real time quantitative PCR for the indicated gene products belonging to ion transport ontology terms (B) and neurological disorder (C). All determinations were performed from at least three animals per genotype and three independent cDNA preparations for qRT-PCR. Comparisons between wild type and Bloc1s8sdy/sdy were performed with Wilcoxon–Mann–Whitney Rank Sum Test. All comparisons were significantly different with a p < 0.005.
NCKX2, which is encoded by Slc24a2 is the most significantly reduced transcript in Bloc1s8sdy/sdy hippocampus (Figure 5). NCKX2 is a sodium-potassium-calcium exchanger that regulates calcium transients and excitability (Lee et al., 2002; Jeon et al., 2003; Kim et al., 2005; Stephan et al., 2011). Kcnj13 and Kcne2 are the most significantly upregulated transcripts in Bloc1s8sdy/sdy hippocampus (Figure 6A). We focused our studies in these potassium channels as they may represent compensatory mechanisms secondary to impaired GABAergic neurotransmission in Bloc1s8sdy/sdy brain. Kcnj13 is an inward rectifier potassium channel, also known as Kir7.1, whereas Kcne2 or MiRP1 is a voltage gated potassium channel modulatory subunit. Loss of any one of these two channel subunits results in increased membrane excitability both in neurons and muscle cells (Ying et al., 2012; Abbott et al., 2014; McCloskey et al., 2014). Illumina sequencing digital quantitation of these two transcripts showed that Kcnj13 increased threefold while Kcne2 increased 4.5-fold in mutant hippocampus as compared to controls (Figures 6A,B). We confirmed these Bloc1s8sdy/sdy mRNA changes by qRT-PCR and found that Kcnj13 and Kcne2 increased their expression by 9.6- and 10.5- fold, respectively, in postnatal day seven mutant hippocampus (Figure 6C). These changes in mRNA led to increased protein expression. We measured protein levels of Kcne2 polypeptide in postnatal day 50 wild type and mutant hippocampus. We focused on Kcne2 because antibodies recognized bands of the proper molecular weight, which were abolished by preincubating antibodies with the antigenic Kcne2 peptide (Figures 6D,E). We used as loading controls actin and the mitochondrial chaperone mortalin (Hspa9) and as positive controls dysbindin (Bloc1s8) and the GABA vesicular transporter (Vgat) (Figures 6D,E). Protein levels of Kcne3 increased by two- to threefold in mutant hippocampus (Figures 6D,E). Since these immunoblots were performed in young adult hippocampus, postnatal day 50, these results demonstrate that increased expression of a potassium channel subunit that decreases excitability is persistent phenotypes downstream of Bloc1s8sdy/sdy. We propose that changes in the expression of NKCC1, KCC2, NCKX2, Kcne2, and Kcnj13 represent developmental adaptive responses to a reduced GABAergic tone induced by genetic defects in a neurodevelopmental gene, Bloc1s8.
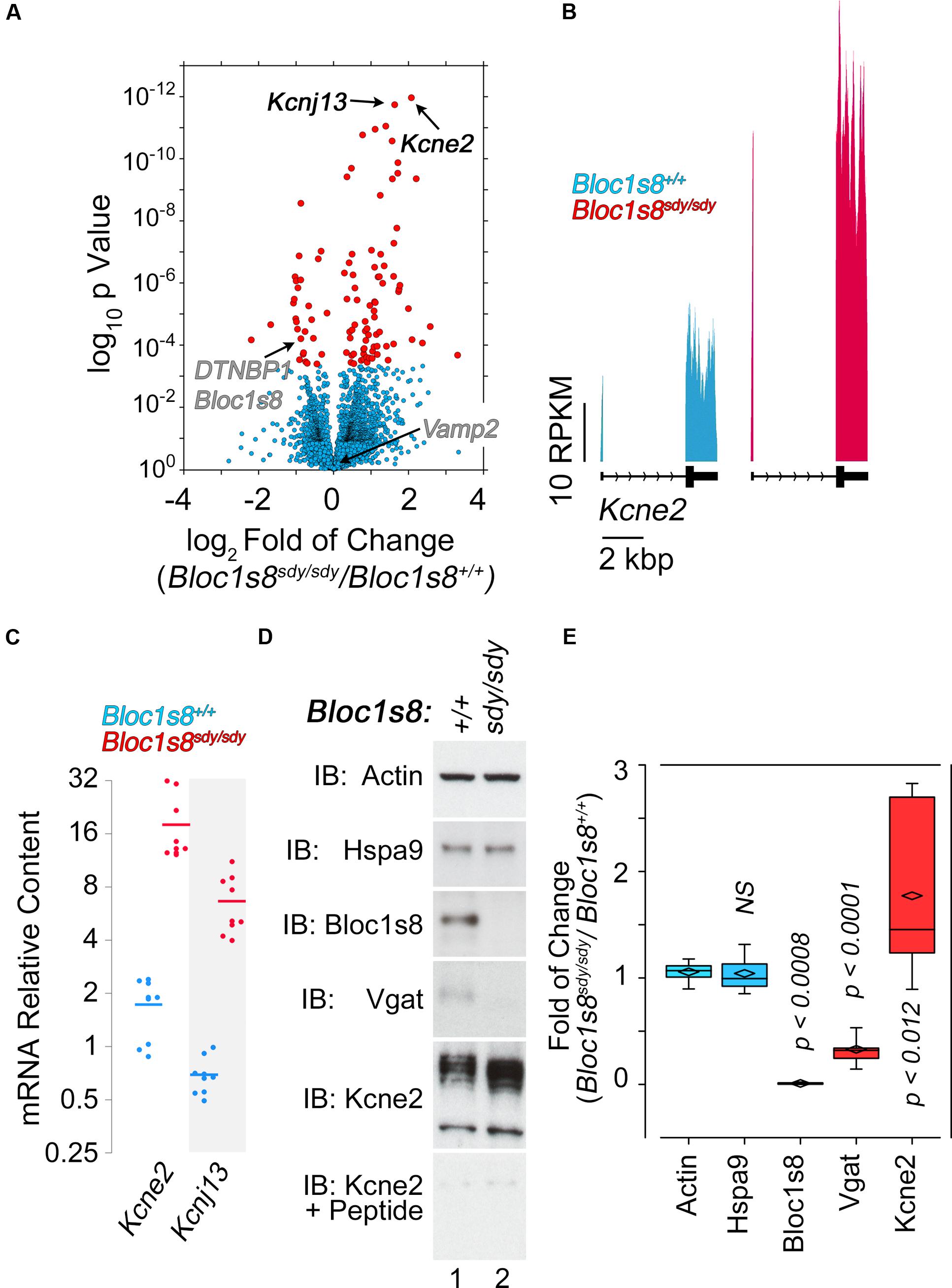
FIGURE 6. Dysbindin null developing hippocampus changes the expression of potassium channel subunits. (A) Volcano plot of Cuffdiff analysis shows (Trapnell et al., 2012) differentially expressed genes in Bloc1s8sdy/sdy hippocampus as in Figure 3B. Arrows mark potassium channel subunit genes for reference. (B) Illumina sequence reads maps for the listed genes from assembly mm9. (C) Postnatal day 7 hippocampi mRNA were quantified by real time quantitative PCR for Kcne2 and Kcnj13. All determinations were performed from at least three animals per genotype and three independent cDNA preparations for qRT-PCR. Comparisons between wild type and Bloc1s8sdy/sdy were performed with Wilcoxon–Mann–Whitney Rank Sum Test. All comparisons were significantly different with a p < 0.0001. (D,E) Immunoblot analysis of Kcne2 expression in wild type and Bloc1s8sdy/sdy hippocampus P50 adult hippocampus in the absence or presence of the Kcne2 antigenic peptide. Control blots with dysbindin antibodies (Bloc1s8), Vgat and loading controls (actin and Hspa9) are presented. (E) Comparisons between actin and other antigens were performed Kruskal–Wallis Rank Sum Test followed by pairwise Wilcoxon–Mann–Whitney Rank Sum Test.
Discussion
We described GABA neuron defects and transcriptional responses downstream of a mutation in a neurodevelopmental gene product, dysbindin, encoded by DTNBP1 and Bloc1s8 in human and mice, respectively. We identified two novel molecular phenotypes in dysbindin deficiency (Figure 7). First, we found drastic changes in the expression of the cation-chloride cotranspoters NKCC1 and KCC2 during hippocampal development. These NKCC1 and KCC2 expression ratio changes are similar to those described in schizophrenia patient brains where the NKCC1 and KCC2 expression ratio is increased (Hyde et al., 2011; Sullivan et al., 2015). Second, the transcriptome of Bloc1s8sdy/sdy postnatal day 7 hippocampus is enriched in transcripts implicated in ionic regulation. In particular, one of the most prominently downregulated messages was SLC24A2 which encodes the sodium-potassium-calcium exchanger 2 or NCKX2. Loss of NCKX2 protein or its transport function increases cytoplasmic calcium levels, thus prolonging the tempo of cellular excitability (Lee et al., 2002; Jeon et al., 2003; Kim et al., 2005; Stephan et al., 2011). In contrast, we found that two of the most upregulated messages in Bloc1s8sdy/sdy hippocampus encode potassium channel subunits Kcne2 and Kcnj13. We documented increased expression of the message and protein expression of Kcne2, an ancillary regulatory potassium channel subunit in the hippocampus of postnatal day 7 and adult Bloc1s8sdy/sdy animals. Loss of Kcne2 or Kcnj13 increase cellular excitability (Ying et al., 2012; Abbott et al., 2014; McCloskey et al., 2014), thus our findings suggest that their overexpression in Bloc1s8sdy/sdy hippocampus may decrease cellular excitability.
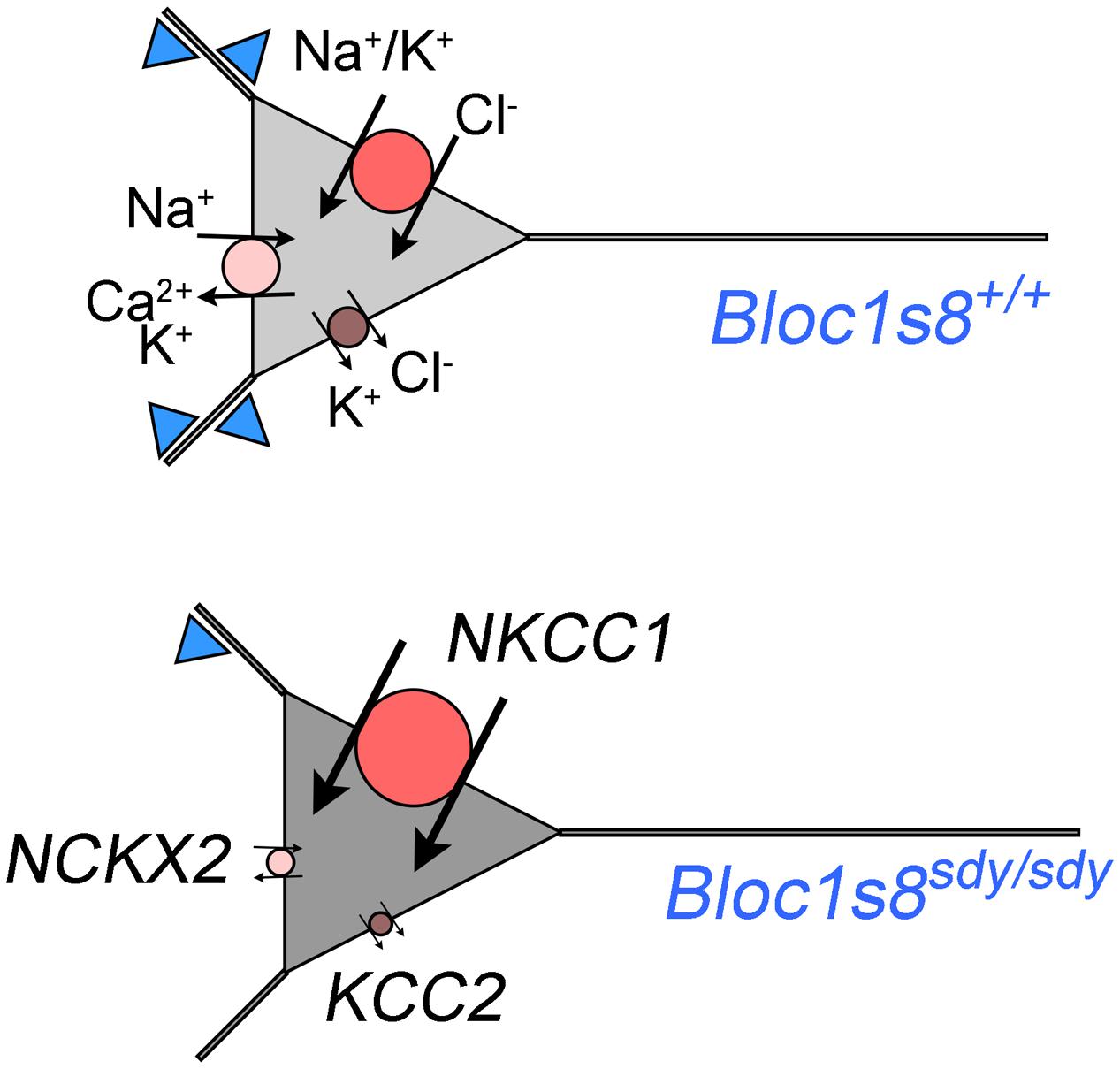
FIGURE 7. A model of phenotypes downstream of GABA interneurons in dysbindin null developing hippocampus. Diagram depicts a model of wild type and Bloc1s8sdy/sdy neuron innervated by parvalbumin-positive GABA terminals (blue triangles) at postnatal day 7. The Bloc1s8sdy/sdy mutation decreases the number of GABA cells and synapses (reduced blue triangles). Changes in the expression of NKCC1, KCC2, and NCKX2 are represented by the size of the circle. Arrows depict direction of ion flows. The shade of gray represents predicted intracellular chloride concentration, note an increased concentration of intracellular chloride in Bloc1s8sdy/sdy neurons due to changes in expression in transporters expression.
The transcriptome of Bloc1s8sdy/sdy hippocampus identified reduced expression of Bloc1s8, Pvalb, and 111 additional transcripts, yet it did not detect modifications in the expression of NKCC1 and KCC2 transcripts documented here by qRT-PCR. Similarly, the transcriptome did not detect changes in the expression of the copper homeostasis molecules Atp7a, Atox1, and Lox; as well as other GABA neuron markers previously documented by us (Larimore et al., 2014; Gokhale et al., 2015b). This argues that our transcriptome either was not sufficiently powered to identify all transcripts sensitive to the Bloc1s8sdy/sdy mutation in the developing hippocampus or the depth of library sequencing was insufficient. However, the transcriptome still illuminated new ion channel and transport mechanisms secondary to the Bloc1s8sdy/sdy mutation. A compromised discovery of transcripts sensitive to the Bloc1s8sdy/sdy allele may be a contributing factor to explain why the transcriptome gene ontology poorly overlaps with the ontology of the Bloc1s8sdy/sdy and the BLOC-1-sensitive proteomes (Gokhale et al., 2016). An additional factor to account for the poor overlap between ontologies of the proteome and transcriptome in Bloc1s8sdy/sdy genetic defects is the relative protein abundance of channels and transporters as compared to other proteins. For example, beta actin is present in 114 million copies per HeLa cell. In contrast, NKCC1 is present in ∼100,000 copies per HeLa cell while the Arp2/3 complex is present in 2 million copies per cell (Itzhak et al., 2016). We found that the Arp2/3 complex is downregulated by proteomics in dysbindin mutations and that the Arp2/3 complex genetically and biochemically interacts with dysbindin (Gokhale et al., 2016). Thus, the Bloc1s8sdy/sdy transcriptome likely contributes hits that escape the partial coverage and comparative sensitivity afforded by proteomic studies. It is interesting that adding the Bloc1s8sdy/sdy transcriptome hits to the BLOC-1 sensitive proteome hits minimally modifies the gene ontology terms associated to the BLOC-1 sensitive proteome (Supplementary Figure S1). In contrast, ionic channel ontology terms discovered with the Bloc1s8sdy/sdy sensitive transcriptome are no longer top priority when added to the BLOC-1 sensitive proteome. Thus, proteome and transcriptome gene ontologies associated to Bloc1s8sdy/sdy are complementary in the identification of mechanisms that require dysbindin or compensate Bloc1s8 genetic defects.
How can we reconcile that loss of dysbindin during development modifies the expression of molecules with apparent opposite roles in excitability? On one hand, the Bloc1s8sdy/sdy mutation would increase neuronal excitability by changes in NKCC1, KCC2, and NCKX2 expression (Figure 7). On the other hand, dysbindin mutation would increase the expression of potassium channel subunits (Kcne2 and Kcnj13), which would decrease neuronal excitability. We favor a model where pro-excitatory changes in the expression of NKCC1, KCC2, and NCKX2 would be confined to a cell type in the developing hippocampus distinct from cells where increased expression Kcne2 and Kcnj13 occurs (Figure 7). Increase expression of the potassium channel ancillary subunit Kcne2 is predicted to decrease neuronal excitability based on the functional consequences of Kcne2 mutation in excitable tissues (Ying et al., 2012; Abbott et al., 2014; McCloskey et al., 2014). Mutations in KCNE2 in human heart causes a long QT-syndrome and ventricular fibrillation by diminishing potassium currents while mouse null mutations in Kcne2 increase the excitability of cortical pyramidal neurons (Abbott et al., 1999, 2014). However, the function of Kcne potassium channel subunits is more nuanced. Overexpression of Kcne2 could influence diverse potassium channels at the level of gating, selectivity, conductivity, and channel traffic such as their movement along the exocytic route, endocytosis, and channel polarized cell distribution. (McCrossan and Abbott, 2004; Kanda et al., 2011a,b,c; Abbott, 2015). Some of these properties associated to Kcne2 family members could occur in the same cells where the expression of NKCC1 and KCC2 is modified as a way to modulate the frequency of GABA excitatory potentials in developing hippocampus.
Increased NKCC1, and decreased KCC2 and NCKX2 expression changes in Bloc1s8sdy/sdy hippocampus are more pronounced between post-natal days 0 to 7, a time where GABA neurotransmission is excitatory due to high intracellular concentration of chloride (Berglund et al., 2006; Ben-Ari et al., 2007; Kaila et al., 2014). We speculate that the increased expression of NKCC1 and the drastically reduced expression of KCC2 would increase intracellular chloride concentrations in developing P7 hippocampal Bloc1s8sdy/sdy neurons (Figure 7). An increased intracellular chloride concentration would in turn compensate the reduction in parvalbumin-positive cells by increasing depolarizing responses to GABA neurotransmission in Bloc1s8sdy/sdy hippocampus (Figure 7). Although this model is speculative, we believe it is of importance. The function of NKCC1 and/or KCC2 can be inhibited by the FDA approved drugs bumetanide and furosemide, respectively (Loscher et al., 2013). Bumetamide has been used to revert persistently increased intracellular chloride concentrations in a mouse model of Down syndrome and improve cognitive outcomes (Deidda et al., 2015). This observation raises the prospect of NKCC1 inhibitors as developmental modulators of GABAergic neurotransmission in Bloc1s8sdy/sdy and other disorders of GABAergic neurotransmission such as schizophrenia. However, if as we postulate here, increased NKCC1 expression in the developing Bloc1s8sdy/sdy hippocampus is a compensatory mechanism for reduced GABAergic innervation, then bumetanide may be ineffective or have deleterious effects in Bloc1s8sdy/sdy and other neurodevelopmental genetic defects affecting GABA neurotransmission such as schizophrenia. In fact, chronic inhibition of NKCC1 with bumetanide during development induces endophenotypes that resemble those in schizophrenia and the Bloc1s8sdy/sdy mutation (Wang and Kriegstein, 2011). Bumetanide is ineffective in treating positive and negative symptoms in schizophrenia patients (Rahmanzadeh et al., 2016). However, inhibition of KCC2 during development may be an effective strategy to ameliorate neurodevelopmental disorders with impaired GABA neurotransmission. Our findings indicate that the GABA neurotransmission defects observed in Bloc1s8sdy/sdy mice offer a model to study the functional and anatomical consequences of modulating GABA neurotransmission and cellular excitability by pharmacological agents that target potassium channels and cation transporters that modulate intracellular chloride and calcium concentration.
Materials and Methods
Antibodies
Antibodies utilized in this study: rabbit anti-parvalbumin (ThermoFisher Scientific PA1-933), mouse anti-synaptophysin (EMD Millipore MAB5258), mouse anti-actin-beta (Sigma A5451), mouse anti-Hspa9/mortalin (NeuroMab N52A/42), rabbit anti-Bloc1s8 (gift from Dr. Talbot), mouse anti-Vgat (Synaptic Systems 131011), and rabbit anti-Kcne2 (Alomone APC-054).
Animals and Tissue Preparation
Mice null for dysbindin (Bloc1s8sdy/sdy), muted (Bloc1s5mu/mu) and pallidin (Bloc1s6pa/pa) were previously described (Larimore et al., 2014). Mice were bred in-house following IUCAC approved protocols. All animals were in in the C57/Black6 background
qRT-PCR
Hippocampal regions were dissected from E18 to P14 and young adult animals between P48-P52 then flash frozen. mRNA was isolated using TRIzol (Invitrogen Life Technologies, Grand Island, NY, USA) extraction and then reverse transcribed into a cDNA using Super Script III First-Strand Synthesis (Invitrogen Life Technologies, Grand Island, NY, USA). Quantitative PCR amplifications were performed on a LightCycler480 Real Time plate reader using Light Cycler 480 SYBR Green reagents (Roche, Indianapolis, IN) at 95°C for 5 min followed by 45 cycles of 95°C for 5 s, 65°C for 10 s, and 72°C for 20 s followed by a 95°C for 5 s, 65°C for 2 min and a 97°C incubation to determine melting curves. Supplementary Primers Table describes the primers used in this study (Supplementary Primers Table S1).
Immunofluorescence Labeling for Confocal Microscopy
Brain slices were prepared from adult mice at P50 as described (Larimore et al., 2011; Larimore et al., 2013). Following ketamine treatment, animals were transcardially perfused with Ringer’s solution and then perfused with fixative (4% paraformaldehyde with 0.1% glutaraldehyde). For 12-18 h, brains were post-fixed in 4% paraformaldehyde. Following post-fixation, brains were cut into 60 μm thick sections and stored in antifreeze (0.1 M sodium phosphate monobasic, 0.1 M sodium phosphate dibasic heptahydrate, 30% ethylene glycol, 30% glycerol) at –20°C. Brain sections containing the hippocampus were incubated in 1% sodium borohydride. For 60 minutes, tissue was pre-incubated (5% NHS and 1% BSA and 0.3% Triton X-100). Tissue was incubated in primary antibody overnight (anti-Parvalbumin 1:200 with anti-Synaptophysin 1:10,000, 1% NHS and 1% BSA) followed by 60 min in a secondary antibody (1% NHS and 1% BSA 1:500 anti-mouse 568 and anti-rabbit 488) (Invitrogen Molecular Probes, Carlsbad, CA, USA). Finally, tissue was incubated for 30 min in cupric sulfate (3.854 W/V Ammonium Acetate, 1.596 W/V Cupric Sulfate, pH 5). Tissue sections were mounted on slides with Vectashield (Vector Laboratories). Confocal microscopy of immunofluorescent samples was performed with an Anxiovert 100M (Carl Zeiss) coupled to an Argon laser, HeNe1 laser, and Titanium Sapphire laser. Z-stacks were acquired using Plan Apochromat 20X/0.5 dry objective. The emission filters used for fluorescence imaging were BP 505–530 and LP 560. Images were acquired with ZEN (Carl Zeiss). Parvalbumin-positive cells were scored by creating a region of interest (ROI) for each region of the hippocampus. Experimenters were blinded to which genotype was analyzed and the intensity of fluorescence in the section was not taken into account for scoring. Cells were considered parvalbumin-positive if the cell body and dendrites were distinguishable from the surrounding fluorescence in the ROI. More than one blind experimenter scored each genotype and each region. For each experiment, a minimal of four control hippocampi and four null hippocampi were processed independently.
Immunoblot Analyses
Brain lysates were separated for SDS–PAGE and transferred to PVDF membranes (BioRad, Hercules, CA, USA). Membranes were probed with primary antibodies followed by HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA and Invitrogen). Secondary antibodies were detected using Supersignal West Dura Extended Duration or Western Lightning substrate (Pierce Chemical, Rockford, IL, USA and Perkin Elmer) and developed on film. Quantitation was performed as follows, multiple film exposures were obtained and those films within a liner range were used for quantitation using Fiji (Schindelin et al., 2012). Antigen expression was compared to actin and Hspa9 as housekeeping gene products.
Transcriptomic and Gene Ontology Analysis
RNAseq services were contracted from Otogenetics Corporation (Atlanta, GA, USA). Briefly, RNA from P7 control and dysbindin mutant hippocampi were extracted with Trizol Reagent and polyA RNA was isolated. The integrity and purity of RNA were determined with Agilent Bioanalyzer. cDNA was prepared with Clontech SmartPCR cDNA kit (Clontech Laboratories). cDNA fragmentation was done with Covaris shearing, profiled with Agilent Bioanalyzer, and subjected to Illumina library preparation with NEB Next reagents (New England Biolabs). RNA content per sample was normalized with ERCC Spike-in (Ambion Thermo Fisher Scientific). The quality, quantity as well as size span of Illumina libraries were determined with an Agilent Bioanalyzer. The libraries were sequenced with Illumina HiSeq2000 sequencing according to standard procedures with a minimum number of 20 million reads per sample. Sequencing data sets from illumina HiSeq2000 were mapped with Tophat (v2.0.5) against reference assembly UCSC mm9. Mapping from Tophat were analyzed by cufflinks.cuffdiff (v.2.0.2), to measure expression level (Trapnell et al., 2012). Expression levels were measured with RPKM (Reads Per Kilobase per Million mapped). q-value less than 0.05 was considered as statistically significant. Data are accessible upon request through DNANexus.
We used three algorithms to perform gene ontology analysis: GeneTerm Linker1 (Fontanillo et al., 2011), ENRICH2 (Chen et al., 2013), and Database Annotation, Visualization and Integrated Discovery3 (DAVID) (Huang da et al., 2009). Briefly gene lists uploaded to DAVID were analyzed by setting annotations based by species (Huang da et al., 2009). Parameters for these functional annotation charts included P-value cutoff (<0.01), a minimum number of three genes for each GO term, fold enrichment, Bonferroni, and/or Benjamini corrected statistical analysis to control for false discovery (Huang da et al., 2009).
The Bloc1s8sdy/sdy-sensitive transcriptome was compared against a brain enriched random gene data with ENRICHR algorithm described above. We selected the top 100 genes expressed in hippocampus, caudate, and cortex from http://www.gtexportal.org/home/ (Supplementary Table S2) (The GTEx Consortium, 2013, 2015). Three hundred top expressed genes were randomized using the Excel function = RANDBETWEEN(1,n) and the first 150 genes were selected in block for gene ontology analyses. None of the ontology terms in GO BP, MF, and the REACTOME database were common between the Bloc1s8sdy/sdy-sensitive transcriptome gene dataset and the brain random dataset. We further confirmed the lack of overlap in gene ontology terms between the random brain gene data set and the Bloc1s8sdy/sdy-sensitive transcriptome with GeneCodis, and engine that allows for the comparison of two datasets simultaneously (Tabas-Madrid et al., 2012).
Statistical Analysis
Experimental conditions were compared using Synergy Kaleida-Graph, version 4.1.3 (Reading, PA) or Aabel NG2 v5 x64 by Gigawiz as specified in each figure. Transcriptomic data were processed by the cufflinks.cuffdiff (v.2.0.2) which corrected for multiple comparisons using Benjamini–Hochberg procedure. These values are expressed as q-values in Supplementary Table S1. Significance was considered in the transcriptome at a q threshold of 0.05 and marked as yes in Supplementary Table S1. qRT-PCR results were subjected to tests specified in each figure legend without multiple comparisons corrections.
Ethics Statement
This study was carried out in accordance with the recommendations of Emory IUCAC. The protocol was approved by the IUCAC.
Author Contributions
JL, SZ: collected data and edited paper. MA, KS, RC, HR, MB, AS, CG, EW: collected data. VF: designed project, analyzed data, designed figures, wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health NS088503 and the Emory School of Medicine Catalyst Grant to VF. We are indebted to the Faundez lab members and Dr. Peter Wenner for their comments.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fgene.2017.00028/full#supplementary-material
FIGURE S1 | Ontology analysis of the BLOC-1 sensitive proteome with or without addition of the Bloc1s8sdy/sdy hippocampus transcriptome. Gene ontology analysis of the BLOC-1 sensitive proteome using the Geneterm Linker algorithm was performed with and without addition of the Bloc1s8sdy/sdy P7 hippocampus transcriptome hits. The numbers in the Z axis represent the gene ontology term groups defined by Geneterm Linker as metagroups (Fontanillo et al., 2011). The internal tightness of each metagroup and its separation from other metagroups is defined by the Silhouette score. Strong tightness between terms within a metagroup is defined by a positive Silhouette score.
TABLE S1 | Cufflinks Analysis of Bloc1s8-null Transcriptome.
TABLE S2 | Brain Enriched Randomized Gene Set.
PRIMERS TABLE S1 | Primer list for qRT-PCR experiments.
Footnotes
- ^ http://gtlinker.cnb.csic.es/gtset/index
- ^ http://amp.pharm.mssm.edu/Enrichr/
- ^ https://david.ncifcrf.gov
References
Abbott, G. W. (2015). The KCNE2 K(+) channel regulatory subunit: ubiquitous influence, complex pathobiology. Gene 569, 162–172. doi: 10.1016/j.gene.2015.06.061
Abbott, G. W., Sesti, F., Splawski, I., Buck, M. E., Lehmann, M. H., Timothy, K. W., et al. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187. doi: 10.1016/S0092-8674(00)80728-X
Abbott, G. W., Tai, K. K., Neverisky, D. L., Hansler, A., Hu, Z., Roepke, T. K., et al. (2014). KCNQ1, KCNE2, and Na+-coupled solute transporters form reciprocally regulating complexes that affect neuronal excitability. Sci. Signal. 7, ra22. doi: 10.1126/scisignal.2005025
Akbarian, S., Kim, J. J., Potkin, S. G., Hagman, J. O., Tafazzoli, A., Bunney, W. E., et al. (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 52, 258–266. doi: 10.1001/archpsyc.1995.03950160008002
Ayalew, M., Le-Niculescu, H., Levey, D. F., Jain, N., Changala, B., Patel, S. D., et al. (2012). Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol. Psychiatry 17, 887–905. doi: 10.1038/mp.2012.37
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Berglund, K., Schleich, W., Krieger, P., Loo, L. S., Wang, D., Cant, N. B., et al. (2006). Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 35, 207–228. doi: 10.1007/s11068-008-9019-6
Bray, N. J., Preece, A., Williams, N. M., Moskvina, V., Buckland, P. R., Owen, M. J., et al. (2005). Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum. Mol. Genet. 14, 1947–1954. doi: 10.1093/hmg/ddi199
Carlson, G. C., Talbot, K., Halene, T. B., Gandal, M. J., Kazi, H. A., Schlosser, L., et al. (2011). Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 108, E962–E970. doi: 10.1073/pnas.1109625108
Celio, M. R. (1986). Parvalbumin in most gamma-aminobutyric acid-containing neurons of the rat cerebral cortex. Science 231, 995–997. doi: 10.1126/science.3945815
Cerasa, A., Quattrone, A., Gioia, M. C., Tarantino, P., Annesi, G., Assogna, F., et al. (2011). Dysbindin C-A-T haplotype is associated with thicker medial orbitofrontal cortex in healthy population. Neuroimage 55, 508–513. doi: 10.1016/j.neuroimage.2010.12.042
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Cheli, V. T., Daniels, R. W., Godoy, R., Hoyle, D. J., Kandachar, V., Starcevic, M., et al. (2010). Genetic modifiers of abnormal organelle biogenesis in a Drosophila model of BLOC-1 deficiency. Hum. Mol. Genet. 19, 861–878. doi: 10.1093/hmg/ddp555
Chen, E. Y., Tan, C. M., Kou, Y., Duan, Q., Wang, Z., Meirelles, G. V., et al. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. doi: 10.1186/1471-2105-14-128
Deidda, G., Parrini, M., Naskar, S., Bozarth, I. F., Contestabile, A., and Cancedda, L. (2015). Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of down syndrome. Nat. Med. 21, 318–326. doi: 10.1038/nm.3827
Del Pino, I., Garcia-Frigola, C., Dehorter, N., Brotons-Mas, J. R., Alvarez-Salvado, E., Martinez de Lagran, M., et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168. doi: 10.1016/j.neuron.2013.07.010
Dickman, D. K., and Davis, G. W. (2009). The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326, 1127–1130. doi: 10.1126/science.1179685
Dickman, D. K., Tong, A., and Davis, G. W. (2012). Snapin is critical for presynaptic homeostatic plasticity. J. Neurosci. 32, 8716–8724. doi: 10.1523/JNEUROSCI.5465-11.2012
Farrell, M. S., Werge, T., Sklar, P., Owen, M. J., Ophoff, R. A., O’Donovan, M. C., et al. (2015). Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 20, 555–562. doi: 10.1038/mp.2015.16
Fontanillo, C., Nogales-Cadenas, R., Pascual-Montano, A., and De las Rivas, J. (2011). Functional analysis beyond enrichment: non-redundant reciprocal linkage of genes and biological terms. PLoS ONE 6:e24289. doi: 10.1371/journal.pone.0024289
Ghiani, C. A., and Dell’angelica, E. C. (2011). Dysbindin-containing complexes and their proposed functions in brain: from zero to (too) many in a decade. ASN Neuro 3, e00058. doi: 10.1042/AN20110010
Ghiani, C. A., Starcevic, M., Rodriguez-Fernandez, I. A., Nazarian, R., Cheli, V. T., Chan, L. N., et al. (2009). The dysbindin-containing complex (BLOC-1) in brain: developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol. Psychiatry 15, 204–215. doi: 10.1038/mp.2009.58
Gogolla, N., Leblanc, J. J., Quast, K. B., Sudhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev. Disord. 1, 172–181. doi: 10.1007/s11689-009-9023-x
Gokhale, A., Hartwig, C., Freeman, A. H., Das, R., Zlatic, S. A., Vistein, R., et al. (2016). The proteome of BLOC-1 genetic defects identifies the Arp2/3 actin polymerization complex to function downstream of the schizophrenia susceptibility factor dysbindin at the synapse. J. Neurosci. 36, 12393–12411. doi: 10.1523/JNEUROSCI.1321-16.2016
Gokhale, A., Mullin, A. P., Zlatic, S., Easley, C. A., Merritt, M. E., Raj, N., et al. (2015a). The N-ethylmaleimide sensitive factor (NSF) and dysbindin interact to modulate synaptic plasticity. J. Neurosci. 35, 7643–7653. doi: 10.1523/JNEUROSCI.4724-14.2015
Gokhale, A., Vrailas-Mortimer, A., Larimore, J., Comstra, H. S., Zlatic, S. A., Werner, E., et al. (2015b). Neuronal copper homeostasis susceptibility by genetic defects in dysbindin, a schizophrenia susceptibility factor. Hum. Mol. Genet. 24, 5512–5523. doi: 10.1093/hmg/ddv282
Gonzalez-Burgos, G., Cho, R. Y., and Lewis, D. A. (2015). Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 77, 1031–1040. doi: 10.1016/j.biopsych.2015.03.010
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069. doi: 10.1001/archpsyc.57.11.1061
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Hashimoto, T., Arion, D., Unger, T., Maldonado-Aviles, J. G., Morris, H. M., Volk, D. W., et al. (2008a). Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 13, 147–161. doi: 10.1038/sj.mp.4002011
Hashimoto, T., Bazmi, H. H., Mirnics, K., Wu, Q., Sampson, A. R., and Lewis, D. A. (2008b). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165, 479–489. doi: 10.1176/appi.ajp.2007.07081223
Hashimoto, T., Bergen, S. E., Nguyen, Q. L., Xu, B., Monteggia, L. M., Pierri, J. N., et al. (2005). Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 25, 372–383. doi: 10.1523/JNEUROSCI.4035-04.2005
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., et al. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326.
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011
Itzhak, D. N., Tyanova, S., Cox, J., and Borner, G. H. (2016). Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5:e16950. doi: 10.7554/eLife.16950
Jentsch, J. D., Trantham-Davidson, H., Jairl, C., Tinsley, M., Cannon, T. D., and Lavin, A. (2009). Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology 34, 2601–2608. doi: 10.1038/npp.2009.90
Jeon, D., Yang, Y. M., Jeong, M. J., Philipson, K. D., Rhim, H., and Shin, H. S. (2003). Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron 38, 965–976. doi: 10.1016/S0896-6273(03)00334-9
Ji, Y., Yang, F., Papaleo, F., Wang, H. X., Gao, W. J., Weinberger, D. R., et al. (2009). Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proc. Natl. Acad. Sci. U.S.A. 106, 19593–19598. doi: 10.1073/pnas.0904289106
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Kanda, V. A., Lewis, A., Xu, X., and Abbott, G. W. (2011a). KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys. J. 101, 1354–1363. doi: 10.1016/j.bpj.2011.08.015
Kanda, V. A., Lewis, A., Xu, X., and Abbott, G. W. (2011b). KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel alpha-subunit composition. Biophys. J. 101, 1364–1375. doi: 10.1016/j.bpj.2011.08.014
Kanda, V. A., Purtell, K., and Abbott, G. W. (2011c). Protein kinase C downregulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm 8, 1641–1647. doi: 10.1016/j.hrthm.2011.04.034
Karlsgodt, K. H., Robleto, K., Trantham-Davidson, H., Jairl, C., Cannon, T. D., Lavin, A., et al. (2011). Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol. Psychiatry 69, 28–34. doi: 10.1016/j.biopsych.2010.09.012
Kim, M. H., Korogod, N., Schneggenburger, R., Ho, W. K., and Lee, S. H. (2005). Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J. Neurosci. 25, 6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005
Larimore, J., Ryder, P. V., Kim, K. Y., Ambrose, L. A., Chapleau, C., Calfa, G., et al. (2013). MeCP2 regulates the synaptic expression of a Dysbindin-BLOC-1 network component in mouse brain and human induced pluripotent stem cell-derived neurons. PLoS ONE 8:e65069. doi: 10.1371/journal.pone.0065069
Larimore, J., Tornieri, K., Ryder, P. V., Gokhale, A., Zlatic, S. A., Craige, B., et al. (2011). The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol. Biol. Cell 22, 4854–4867. doi: 10.1091/mbc.E11-07-0592
Larimore, J., Zlatic, S. A., Gokhale, A., Tornieri, K., Singleton, K. S., Mullin, A. P., et al. (2014). Mutations in the BLOC-1 subunits dysbindin and muted generate divergent and dosage-dependent phenotypes. J. Biol. Chem. 289, 14291–14300. doi: 10.1074/jbc.M114.553750
Lee, S. H., Kim, M. H., Park, K. H., Earm, Y. E., and Ho, W. K. (2002). K+-dependent Na+/Ca2+ exchange is a major Ca2+ clearance mechanism in axon terminals of rat neurohypophysis. J. Neurosci. 22, 6891–6899.
Li, W., Zhang, Q., Oiso, N., Novak, E. K., Gautam, R., O’Brien, E. P., et al. (2003). Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat. Genet. 35, 84–89. doi: 10.1038/ng1229
Loscher, W., Puskarjov, M., and Kaila, K. (2013). Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74. doi: 10.1016/j.neuropharm.2012.05.045
Luciano, M., Miyajima, F., Lind, P. A., Bates, T. C., Horan, M., Harris, S. E., et al. (2009). Variation in the dysbindin gene and normal cognitive function in three independent population samples. Genes Brain Behav. 8, 218–227. doi: 10.1111/j.1601-183X.2008.00462.x
Mariani, J., Coppola, G., Zhang, P., Abyzov, A., Provini, L., Tomasini, L., et al. (2015). FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162, 375–390. doi: 10.1016/j.cell.2015.06.034
Marin, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Markov, V., Krug, A., Krach, S., Jansen, A., Eggermann, T., Zerres, K., et al. (2010). Impact of schizophrenia-risk gene dysbindin 1 on brain activation in bilateral middle frontal gyrus during a working memory task in healthy individuals. Hum. Brain Mapp. 31, 266–275. doi: 10.1002/hbm.20862
Markov, V., Krug, A., Krach, S., Whitney, C., Eggermann, T., Zerres, K., et al. (2009). Genetic variation in schizophrenia-risk-gene dysbindin 1 modulates brain activation in anterior cingulate cortex and right temporal gyrus during language production in healthy individuals. Neuroimage 47, 2016–2022. doi: 10.1016/j.neuroimage.2009.05.067
McCloskey, C., Rada, C., Bailey, E., McCavera, S., van den Berg, H. A., Atia, J., et al. (2014). The inwardly rectifying K+ channel KIR7.1 controls uterine excitability throughout pregnancy. EMBO Mol. Med. 6, 1161–1174. doi: 10.15252/emmm.201403944
McCrossan, Z. A., and Abbott, G. W. (2004). The MinK-related peptides. Neuropharmacology 47, 787–821. doi: 10.1016/j.neuropharm.2004.06.018
Mechelli, A., Viding, E., Kumar, A., Pettersson-Yeo, W., Fusar-Poli, P., Tognin, S., et al. (2010). Dysbindin modulates brain function during visual processing in children. Neuroimage 49, 817–822. doi: 10.1016/j.neuroimage.2009.07.030
Mody, M., Cao, Y., Cui, Z., Tay, K. Y., Shyong, A., Shimizu, E., et al. (2001). Genome-wide gene expression profiles of the developing mouse hippocampus. Proc. Natl. Acad. Sci. U.S.A. 98, 8862–8867. doi: 10.1073/pnas.141244998
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Mullin, A. P., Gokhale, A., Larimore, J., and Faundez, V. (2011). Cell biology of the BLOC-1 complex subunit dysbindin, a schizophrenia susceptibility gene. Mol. Neurobiol. 44, 53–64. doi: 10.1007/s12035-011-8183-3
Mullin, A. P., Sadanandappa, M. K., Ma, W., Dickman, D. K., VijayRaghavan, K., Ramaswami, M., et al. (2015). Gene dosage in the dysbindin schizophrenia susceptibility network differentially affect synaptic function and plasticity. J. Neurosci. 35, 325–338. doi: 10.1523/JNEUROSCI.3542-14.2015
Rahmanzadeh, R., Shahbazi, A., Ardakani, M. K., Mehrabi, S., Rahmanzade, R., and Joghataei, M. T. (2016). Lack of the effect of bumetanide, a selective NKCC1 inhibitor, in patients with schizophrenia: a double-blind randomized trial. Psychiatry Clin. Neurosci. 71, 72–73. doi: 10.1111/pcn.12475
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Shao, L., Shuai, Y., Wang, J., Feng, S., Lu, B., Li, Z., et al. (2011). Schizophrenia susceptibility gene dysbindin regulates glutamatergic and dopaminergic functions via distinctive mechanisms in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 108, 18831–18836. doi: 10.1073/pnas.1114569108
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. doi: 10.1038/nature07991
Stephan, A. B., Tobochnik, S., Dibattista, M., Wall, C. M., Reisert, J., and Zhao, H. (2011). The Na(+)/Ca(2+) exchanger NCKX4 governs termination and adaptation of the mammalian olfactory response. Nat. Neurosci. 15, 131–137. doi: 10.1038/nn.2943
Straub, R. E., Jiang, Y., MacLean, C. J., Ma, Y., Webb, B. T., Myakishev, M. V., et al. (2002). Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 71, 337–348. doi: 10.1086/341750
Sullivan, C. R., Funk, A. J., Shan, D., Haroutunian, V., and McCullumsmith, R. E. (2015). Decreased chloride channel expression in the dorsolateral prefrontal cortex in schizophrenia. PLoS ONE 10:e0123158. doi: 10.1371/journal.pone.0123158
Tabas-Madrid, D., Nogales-Cadenas, R., and Pascual-Montano, A. (2012). GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 40, W478–W483. doi: 10.1093/nar/gks402
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Talbot, K., Cho, D. S., Ong, W. Y., Benson, M. A., Han, L. Y., Kazi, H. A., et al. (2006). Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum. Mol. Genet. 15, 3041–3054. doi: 10.1093/hmg/ddl246
Talbot, K., Eidem, W. L., Tinsley, C. L., Benson, M. A., Thompson, E. W., Smith, R. J., et al. (2004). Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Invest. 113, 1353–1363. doi: 10.1172/JCI200420425
Talbot, K., Louneva, N., Cohen, J. W., Kazi, H., Blake, D. J., and Arnold, S. E. (2011). Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS ONE 6:e16886. doi: 10.1371/journal.pone.0016886
Tamamaki, N., Yanagawa, Y., Tomioka, R., Miyazaki, J., Obata, K., and Kaneko, T. (2003). Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 467, 60–79. doi: 10.1002/cne.10905
Tang, J., LeGros, R. P., Louneva, N., Yeh, L., Cohen, J. W., Hahn, C. G., et al. (2009). Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 mRNA expression. Hum. Mol. Genet. 18, 3851–3863. doi: 10.1093/hmg/ddp329
The GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585. doi: 10.1038/ng.2653
The GTEx Consortium (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. doi: 10.1126/science.1262110
Tognin, S., Viding, E., McCrory, E. J., Taylor, L., O’Donovan, M. C., McGuire, P., et al. (2011). Effects of DTNBP1 genotype on brain development in children. J. Child Psychol. Psychiatry 52, 1287–1294. doi: 10.1111/j.1469-7610.2011.02427.x
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Trost, S., Platz, B., Usher, J., Scherk, H., Wobrock, T., Ekawardhani, S., et al. (2013). The DTNBP1 (dysbindin-1) gene variant rs2619522 is associated with variation of hippocampal and prefrontal grey matter volumes in humans. Eur. Arch. Psychiatry Clin. Neurosci. 263, 53–63. doi: 10.1007/s00406-012-0320-0
Van Den Bogaert, A., Schumacher, J., Schulze, T. G., Otte, A. C., Ohlraun, S., Kovalenko, S., et al. (2003). The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am. J. Hum. Genet. 73, 1438–1443. doi: 10.1086/379928
Wang, D. D., and Kriegstein, A. R. (2011). Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb. Cortex 21, 574–587. doi: 10.1093/cercor/bhq124
Wei, M. L. (2006). Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 19, 19–42. doi: 10.1111/j.1600-0749.2005.00289.x
Whissell, P. D., Cajanding, J. D., Fogel, N., and Kim, J. C. (2015). Comparative density of CCK- and PV-GABA cells within the cortex and hippocampus. Front. Neuroanat. 9:124. doi: 10.3389/fnana.2015.00124
Wohr, M., Orduz, D., Gregory, P., Moreno, H., Khan, U., Vorckel, K. J., et al. (2015). Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 5, e525. doi: 10.1038/tp.2015.19
Wolf, C., Jackson, M. C., Kissling, C., Thome, J., and Linden, D. E. (2011). Dysbindin-1 genotype effects on emotional working memory. Mol. Psychiatry 16, 145–155. doi: 10.1038/mp.2009.129
Ying, S. W., Kanda, V. A., Hu, Z., Purtell, K., King, E. C., Abbott, G. W., et al. (2012). Targeted deletion of Kcne2 impairs HCN channel function in mouse thalamocortical circuits. PLoS ONE 7:e42756. doi: 10.1371/journal.pone.0042756
Yuan, Q., Yang, F., Xiao, Y., Tan, S., Husain, N., Ren, M., et al. (2016). Regulation of brain-derived neurotrophic factor exocytosis and gamma-aminobutyric acidergic interneuron synapse by the schizophrenia susceptibility gene dysbindin-1. Biol. Psychiatry 80, 312–322. doi: 10.1016/j.biopsych.2015.08.019
Keywords: dysbindin, GABA, parvalbumin, BLOC-1, Neurodevelopmental disorders
Citation: Larimore J, Zlatic SA, Arnold M, Singleton KS, Cross R, Rudolph H, Bruegge MV, Sweetman A, Garza C, Whisnant E and Faundez V (2017) Dysbindin Deficiency Modifies the Expression of GABA Neuron and Ion Permeation Transcripts in the Developing Hippocampus. Front. Genet. 8:28. doi: 10.3389/fgene.2017.00028
Received: 09 January 2017; Accepted: 20 February 2017;
Published: 10 March 2017.
Edited by:
William Davies, Cardiff University, UKReviewed by:
Deepak Prakash Srivastava, King’s College London, UKHyunsoo Shawn JE, Duke NUS Graduate Medical School, Singapore
Antonieta Lavin, Medical University of South Carolina, USA
Copyright © 2017 Larimore, Zlatic, Arnold, Singleton, Cross, Rudolph, Bruegge, Sweetman, Garza, Whisnant and Faundez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor Faundez, dmZhdW5kZUBlbW9yeS5lZHU= Jennifer Larimore, amxhcmltb3JlQGFnbmVzc2NvdHQuZWR1
†These authors have contributed equally to this work.