 Veronica Bertini1
Veronica Bertini1 Alessia Azzarà1
Alessia Azzarà1 Annalisa Legitimo2Roberta Milone3Roberta Battini3
Annalisa Legitimo2Roberta Milone3Roberta Battini3 Rita Consolini2
Rita Consolini2 Angelo Valetto1*
Angelo Valetto1*- 1Cytogenetics and Molecular Genetics Unit, Department of Laboratory Medicine, Azienda Ospedaliera Univeristaria Pisana, Pisa, Italy
- 2Laboratory of Immunology, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 3Department of Developmental Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico Stella Maris, Pisa, Italy
In humans, the most common genomic disorder is the hemizygous deletion of the chromosome 22q11.2 region, that results in the “22q11.2 deletion syndrome” (22q11.2DS). A peculiarity of 22q11.2DS is its great phenotypic variability that makes this pathology a classic example of a syndrome with variable expressivity and incomplete penetrance. The reasons for this variability have not been elucidated yet, and the molecular substrates underlying the different clinical features of 22q11.2DS are still debated. A cohort of 21 patients has been analyzed by array CGH in order to detect some of the genetic differences that may influence this variability. Two aspects have been investigated: (1) the precise localization of the deletion breakpoints within the low copy repeats (LCRs), (2) the additional Copy Number Variations (CNVs) elsewhere in the genome, by analyzing their gene content. Both protein-coding genes and miRNAs were considered, in order to discover possible epistatic interactions between genes of the 22q11.2 region and the rest of the genome. Eighteen out of twenty-one patients had a deletion of ~3 Mb mediated by LCR22-A and D, whereas 3/21 had a smaller deletion. The breakpoints within the LCR22-A and D do not have a major role in the phenotypic variability since they are rather clustered and the small differences concern genes that are not directly related to clinical signs of 22q11.2DS. A detailed analysis of the gene content of 22q11.2 deleted region indicates that this syndrome could be a bioenergetic disorder or consequence of an altered post-transcriptional gene regulation, due to the presence of DGCR8, a major player of the microRNA (miRNA) biogenesis. Only four genes with mitochondrial function are harbored in the additional CNVs, whereas 11 miRNA, all related to biological pathways present in the 22q11.2DS, have been detected in 19/21 patients. CNVs and miRNAs are new entities that have changed the order of complexity at the level of gene expression and regulation, thus CNV-miRNAs (miRNA harbored in the CNVs) are potential functional variants that should be considered high priority candidate variants in genotype-phenotype association studies. Deletion of DGCR8, the main actor in miRNA biogenesis, amplifies this variability. To our knowledge, this is the first report that focus on the miRNA-CNVs in 22q11.2DS, with the aim of trying to better understand their role in the variable expressivity and incomplete penetrance.
Introduction
In humans, the most common genomic disorder is the hemizygous deletion of the chromosome 22q11.2 region that results in the “22q11.2 deletion syndrome” (22q11.2DS) (MIM #188400/#192430), also referred to as Di George, Velocardiofacial or Shprintzen syndrome.
A peculiarity of 22q11.2DS is its great phenotypic variability that makes this pathology a classic example of a syndrome with variable expressivity and incomplete penetrance.
22q11.2DS phenotype is characterized by a constellation of clinical signs, whose characterization has expanded considerably within the last decade. The phenotype includes many associated findings such as congenital heart disease (CHD; 50–75%), hypocalcemia (17–60%), immunological disorders (35–40%), palatal (69–100%), craniofacial (90%), skeletal (17–19%), gastroenterological (35%) anomalies and a range of neurocognitive, and psychiatric disorders (85%; McDonald-McGinn and Sullivan, 2011). The severity of symptoms is also variable, ranging from quite severe to near-normal life conditions (Bassett et al., 2011). 22q11.2 deletion can be transmitted by a normal parent with no clinically obvious phenotype, to an affected child (Leana-Cox et al., 1996) and this provides additional evidence that genetic modifiers, beyond the deleted genes themselves, play a key role in determining the 22q11.2DS phenotypic severity.
This variability can be due to stochastic and environmental processes as well as to genetic effects, with all these factors potentially acting alone or in combination. Currently, no mechanistic explanation exists, and the reasons underlying this phenotypic variability are still debated.
Extension of the deleted region could be a genetic cause underlying the phenotypic variability. Deletions in 22q11.2 region are a consequence of non-allelic homologous recombination (NAHR) due to misalignment of low copy repeats (LCRs) during meiosis (Edelmann et al., 1999; Kumar et al., 2008). Eight LCRs (named LRC22-A to H) have been identified, but only the four centromeric ones (LCR22-A to D) are implicated in this syndrome. Deletions mediated by different specific LCRs will result in different sets of genes being deleted and this is a cause of phenotypic variability. In any case, this does not play a major role, since over 90% of patients share a deletion between LCR22-A and LCR22-D.
Deletions that appear to have roughly the same size, since mediated by the same couple of LCRs, may exhibit variability at the sites of the NAHR event. A fine mapping of the breakpoints within the LCRs has been performed only in a few studies (Mantripragada et al., 2004; Urban et al., 2006; Bittel et al., 2009). These small differences of the breakpoint localization within the same LCR may influence the expression of genes that could have a major role in the clinical phenotype of 22q11.2DS.
The presence of additional CNVs in the rest of the genome could be another factor responsible for this variability. Human genome is much more genetically variable than previously appreciated. To date, nearly 10–15% of it has been annotated as copy-number variations (CNVs), namely microdeletions and microduplications, ranging from 1 kb to several Mb (Lee et al., 2007). Their presence may contribute significantly toward human phenotypic variability, complex behavioral traits, and disease susceptibility.
Epistatic interactions between genes in the 22q11.2DS region and other genes harbored in CNVs elsewhere in the genome could explain the variable expressivity and reduced penetrance of this syndrome.
According to the recent literature data, CNVs are a substantial risk factor for a range of neurodevelopment and psychiatric disorders such as autism, ADHD, schizophrenia, epilepsy, and intellectual disability (Kirov et al., 2009; Cooper et al., 2011; Vaishnavi et al., 2013).
In 22q11.2DS cases, a correlation between CNVs and schizophrenia has been explored (Williams et al., 2013). However, little is known about to the role of CNVs in modulating the outcome of other phenotypic aspects of this syndrome. Two recent articles focused on the role of CNVs as genetic modifiers involved in the variable cardiac phenotype, studying a very large cohort of 22q11.2 patients. Except for a common CNV, a SLC2A3 duplication, that was significantly associated with CHDs (cardiac heart diseases), no differences were found in the number or in the size of common CNVs between 22q11DS cases with or without CHDs.
When rare CNVs were carefully examined for their gene content and function, specific cardiac networks appeared to be overrepresented in 22q11DS CHD cases but not in 22q11DS controls with no cardiac phenotype (Mlynarski et al., 2015, 2016).
In this view, we focused our attention on these two genetic factors as potential cause of variability, and, in particular (1) on the localization of the deletion breakpoints within LCRs, (2) and on the additional CNVs elsewhere in the genome, by analyzing their gene content. Both protein-coding genes and microRNAs (miRNAs) were evaluated, in order to discover possible epistatic interactions between genes of the 22q11.2 region and the rest of the genome.
Materials and Methods
Subjects
Our study group consisted of 21 subjects with known 22q11.2 deletion who had been referred to our Institution for molecular-cytogenetic investigations (9 males, 12 females), with age ranging from to 3 months to 37 years. This study was approved by the Pediatric Committee of the Tuscany Region, Meyer Hospital, Firenze, Italy. An informed consent was signed prior to the genetic analyses. Main clinical aspects are reported in Table 1.
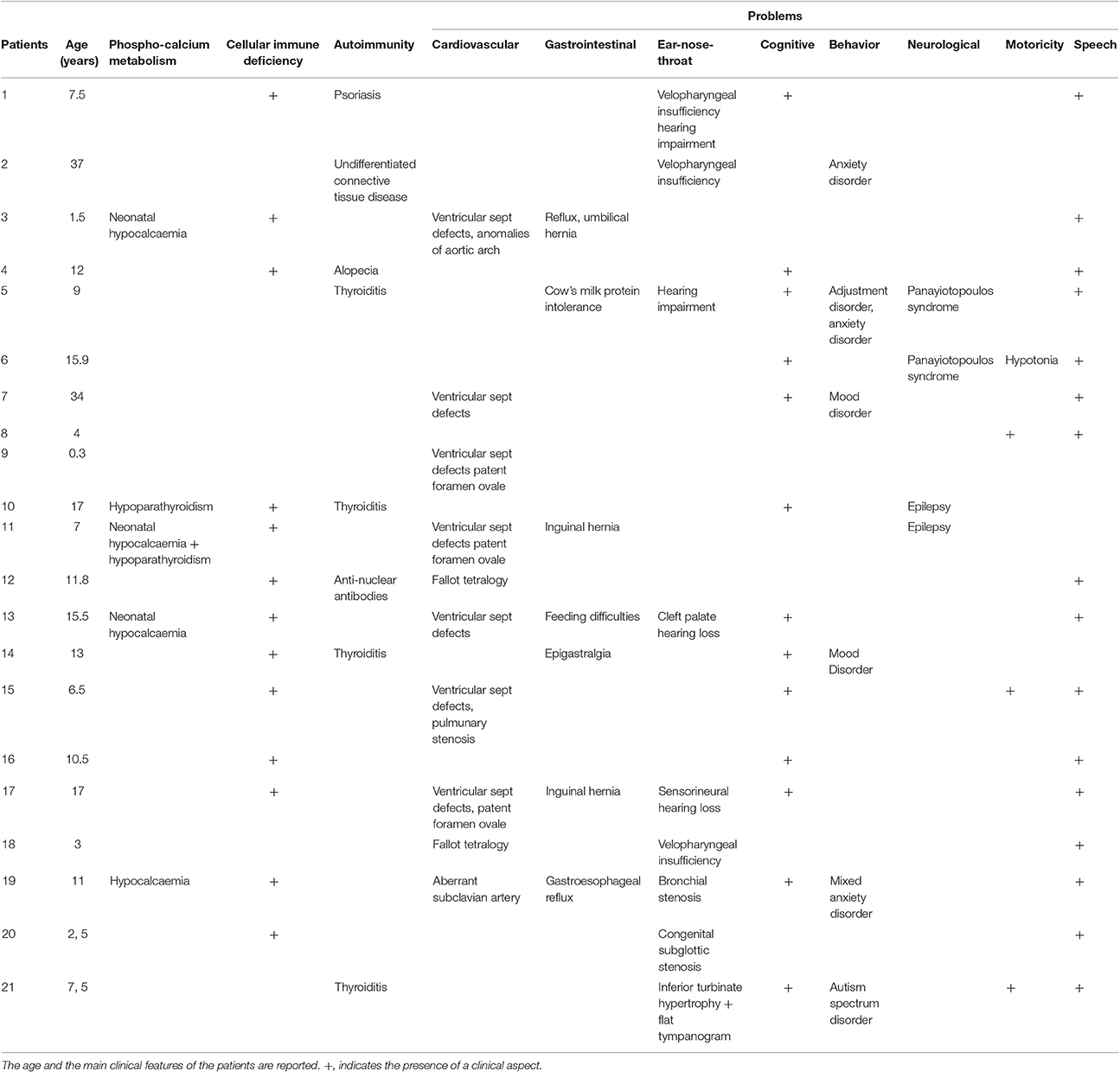
Table 1. Clinical Findings in the 21 patients studied.
Array-CGH
Genomic DNA of the patients was isolated from peripheral blood by standard methods; DNA from healthy subjects (a male and a female) was used as controls (Agilent Technologies, Santa Clara, California, USA). Five-hundred nanograms of genomic DNA both from the patient (test sample) and the control (reference sample) were differentially labeled with Cy5-dCTP or with Cy3-dCTP using random primer labeling according to manufacturer's protocol (Agilent). The labeling reactions were applied to the oligo-arrays and incubated for 24 h at 67°C in an oven. Slides were washed and scanned using the Agilent scanner, and the identification of individual spots on scanned arrays and quality slide evaluation was performed using the Agilent dedicated software (Feature Extraction, Agilent).
The array CGH was performed on 180K SurePrint G3 Human CGH Microarray (Agilent), that have a 13 KB overall median probe spacing (11 KB in Refseq genes). In order to better define the deletion breakpoints, we have also used 60K SurePrint G3 Unrestricted CGH ISCA v2, that has a 60 KB overall median probe spacing but higher in pathological or gene rich regions.
Given that the quality of the experiment may greatly influence the CNVs analysis (number and type of calls, probes included/excluded from a call), in order to have more powerful and clean data, we elaborated only those experiments that met the “excellent” criteria as determined by the QC report (Cytogenomic software, Agilent). In particular, the Derivative Log Ratio Spread (DLRS) was the main value considered for further analysis of the data: when >0.16, the experiment was discarded and repeated. CNVs were identified with Cytogenomics 3.0.6.6. (Agilent), using the Aberration Detection Method-2 (ADM-2) algorithm. This algorithm identifies aberrant intervals in a sample that has consistently high or low log ratios based on their statistical score. The score represents the deviation of the weighted average of the normalized log ratios from its expected value of zero and incorporates quality information about each probe measurement. ADM-2 uses an iterative procedure to find all genomic intervals with a score above a user/specified statistical threshold value. The threshold was set to a minimum of six with the minimum number of three probes required in a region and a minimum absolute log ratio of 0.25. In particular, we have examined the 22q11.2 region, focusing on probes delimiting the breakpoints, together with all the CNVs meeting the statistical parameter described above. We analyzed all the CNVs >3 contiguous probes for deletions and >4 probes for duplications, independently of their absolute size; they were compared to those reported in the http://dgv.tcag.ca/dgv/app/home. The gene content was established by UCSC Genome Browser (http://genome.ucsc.edu/) (NCBI37/hg19 assembly) and the gene function by RefSeq (https://www.ncbi.nlm.nih.gov/refseq/rsg/).
Retrieval of CNV-microRNAs and Target Prediction
The miRNA content in CNVs was identified by analyzing the chromosomal coordinates in UCSC Genome Browser.
The function of these miRNAs and their validated and putative targets were identified from the miRWalk database (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/). miRWalk (Dweep et al., 2011) is a comprehensive database that provides information on predicted as well as validated binding sites of miRNAs on their target genes and the functional patterns they are part of. For the prediction of putative targets by miRWalk, filters were set and applied to identify the 3′UTR that has a minimum seed match of seven nucleotides and retained only the targets predicted by multiple databases (consistently predicted by miRanda, miRDB, miRWalk, and TargetScan). This integration between multiple databases improves the accuracy or coverage of predictions by balancing out the precision and recall.
The biological pathways of genes targeted by these miRNA were analyzed with Gene Ontology (GO; http://www.geneontology.org/). GO has developed three structured controlled vocabularies to describe gene products in terms of their associated biological processes, cellular components and molecular functions (Ashburner et al., 2000). All the related functions associated with their enrichment scores and p-values derived with the multiple test adjustment set were done by Benjamini and Hochberg. Only the results with a corrected p < 0.05 were considered to be significant. For each miRNA, significative data on the biological process (GOBP) were imported into a Microsoft Excel Table.
Results
We analyzed 21 subjects with known 22q11.2 deletion using the Agilent 180K whole genome and the 60K ISCA microarrays. A scheme of the deletion extent is reported in Figure 1A. Eighteen out of twenty-one patients had a 22q11.2 deletion of ~3 Mb mediated by LCR22-A and D, whereas 3/21 had smaller deletions, of 1.3 Mb (pt. 19), 2.4 Mb (pt. 20), and 745 kb (pt. 21), respectively.
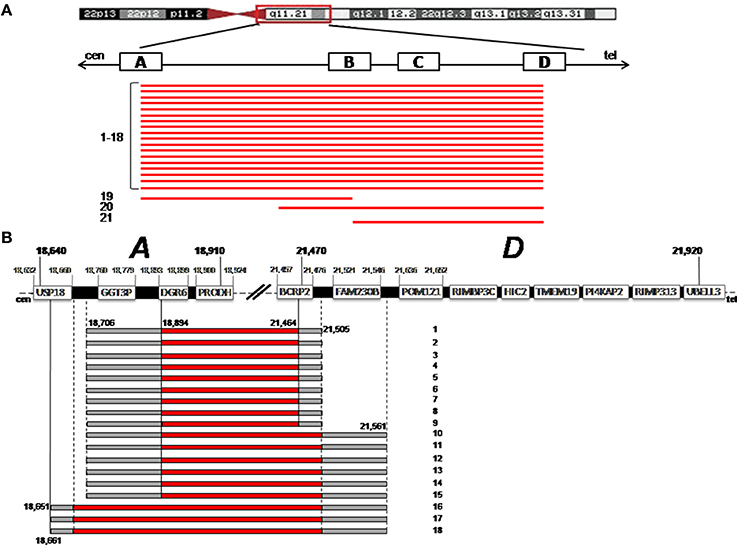
Figure 1. (A) The deletion extent is shown for the 21 patients (bottom, black bars). On the top, a schematic representation of chromosome 22 is given. The four Low Copy Repeats A to D (LCRA-D) involved in the deletion are indicated with white blocks. Cen is centromere; Tel is telomere. (B) Precise definition of the breakpoints is shown for patients 1–18. On the top, a detailed structure of LCR A–D is shown, with the genes harbored and their positions (hg19 Genome Build). The dark bars indicates the region deleted, the gray bars indicate the region that could or could not be deleted, with the probe positions.
In the 18 patients with the common ~3 Mb deletion, using the above sets of arrays, we were able to accurately define the deletion boundaries. LCR22-A and D extend for 270 and 450 Kb, respectively, and harbor many genes (Figure 1B); the “gray zone” (i.e., the region comprised between the last non-deleted oligo and the first deleted one) was restricted to a small interval. As far as the LCR22-A breakpoint is concerned, in three cases (pt n.16,17,18) USP18 (Ubiquitin specific peptidase 18) was the most proximal deleted gene with breakpoint boundaries limited to a 10 Kb interval. In all the other patients, DGCR6 is the first functional gene involved in the deletion since GGT3P (Gamma-glutamyltransferase 3 pseudogene) is a pseudogene.
As far as the LCR22-D breakpoint is concerned, FAM230B (Homo sapiens family with sequence similarity 230 member B) is not deleted in patients 1–9, whereas it is in the “gray zone” in the other nine cases (pts. 10–18). FAM230B is a long non-protein coding RNA, with unknown functions. BCRP2 (breakpoint cluster region pseudogene 2) that may represent a difference in patients 1–9, is a pseudogene.
In the above 18 patients, the deleted region encompasses about 50 RefSeq genes, whose function has been well-established only in a small proportion. There is an abundance of non-coding RNA (a total of 13, 6 of which are miRNA) and nine genes (MRPL40, PRODH, SLC25A1, TXNRD2, T10, ZDHHC, COMT, UFD1L, DGCR8) that code for proteins, either localized to mitochondria or connected to mithocondrial functions. DGCR8 (Di George Critical Region Gene 8), in particular, is a gene that also has a primary role in miRNA biogenesis.
CNVs
In our cohort, besides the 22q11.2 deletion, array CGH showed the presence of additional CNVs. All the CNVs detected are reported in Table 2, independently of their size, position, or gene content; CNVs spanning segmental duplications were included, since they are often gene rich. Both “rare” (when not reported in normal population studies or with a frequency of <1% in a previously published control populations) and “common” (reported in several studies or with a frequence equal or higher than 1% in the control populations) were considered (Pinto et al., 2007; Vogler et al., 2010; Cooper and Mefford, 2011; Cooper et al., 2011; Coe et al., 2014).
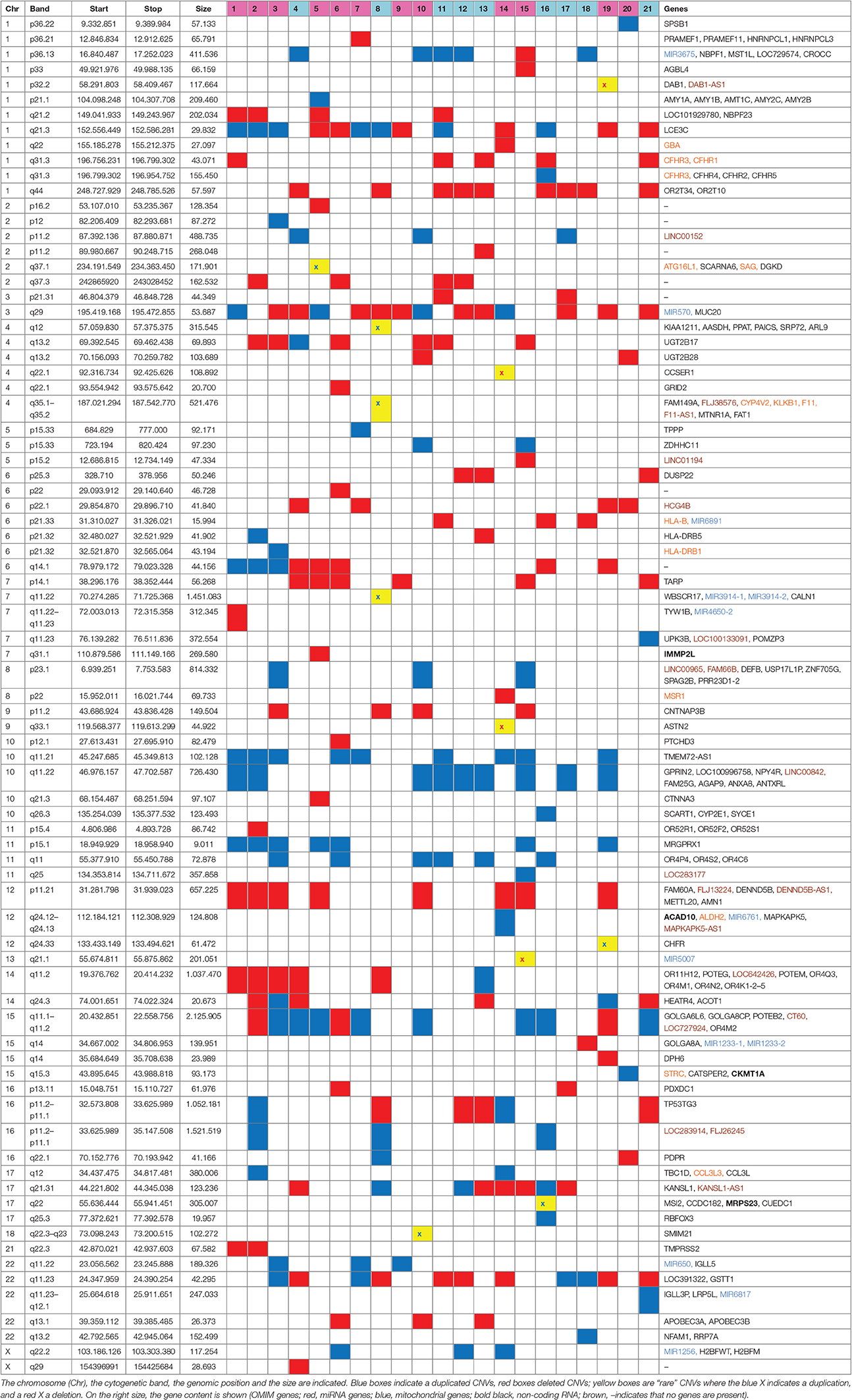
Table 2. Additional CNVs detected in the 21 patients are shown.
The average number of CNVs per patient was 11; these CNVs are present on all the chromosomes (except chromosome 19 and 20), with a size ranging from 9 kb to 2.1 Mb (average size 249 kb), and include roughly the same number of deletions and duplications.
Among the 81 CNVs, 11 were rare, detected in 7 patients (Table 2).
As far as their gene content is concerned, at first glance it emerges that among the total amount of 177 RefSeq, olfactory receptor, immunoglobulin family, HLA, and amylase genes are the most represented, as expected. Fourteen genes, both in common than in rare CNVs, are reported in the OMIM database (https://www.omim.org/); four genes related to mitochondrial function (IMMP2L, ACAD10, MRPS23, CKMT1A) were detected. Interestingly, among the other genes, there is a preponderance of miRNA (11). Nineteen out of 21 patients present at least one CNV-miRNA; some miRNAs are present only in deleted CNVs (i.e., miRNA6891, miRNA4650-2, miRNA5007, miRNA12331-1, miRNA1233-2), some only in duplicated CNVs (miRNA3914-1, miRNA3914-2, miRNA6761, miRNA650, miRNA6817, miRNA1256) and some in both (miRNA3675, miRNA570; Tables 2, 3).
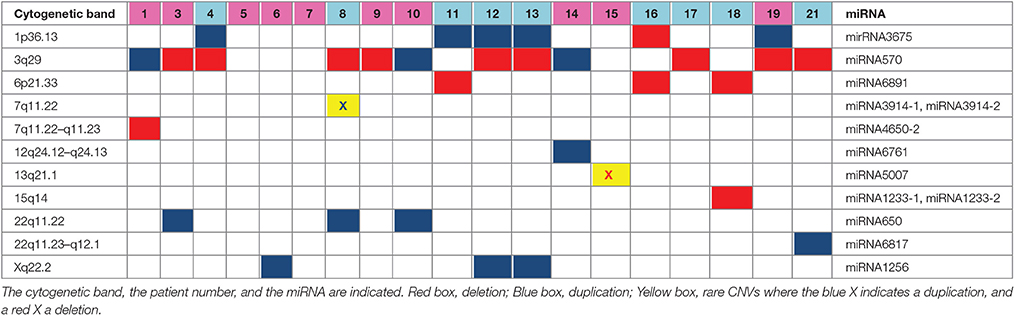
Table 3. The MiRNAs contained in the CNVs are shown in details.
Functional Pathways of miRNA
These miRNAs regulate a large number of target genes, as indicated by the miRWalk database (Dweep et al., 2011; more than 400 entries, data not shown). Analyzing the biological pathways with GO, only those significant (p < 0.05) were considered. Many entries are related to clinical features of 22q11.2DS and we have grouped them into nine macro-areas, including: phospho-calcium metabolism; neurological, cardiovascular, immunity, cognitive-behavior, and gastrointestinal pathways; ear-nose-throat, skeletal development. As can be seen in Figure 2 and in Table 4, each miRNA has a regulatory role on the genes that are involved in the vast majority of these macro-areas.
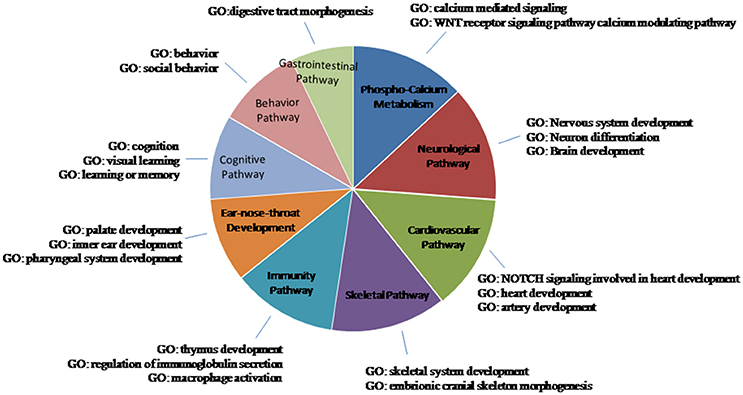
Figure 2. Schematic representation of the biological pathways of the miRNA-CNVs. GO is the Gene ontology process. Only the results with a p < 0.05 were considered to be significant. Many entries were related to clinical features of 22q11.2DS; they were grouped them into nine macro-areas.
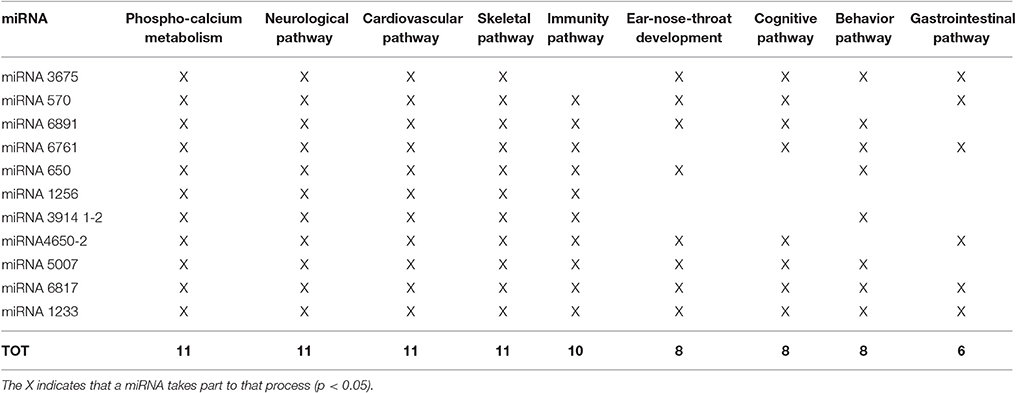
Table 4. Gene Ontology Biological Process (GOBP) of CNV-miRNA, with relation to the 22q11.2 deletion syndrome clinical signs.
Discussion
As known, the clinical phenotype of 22q211.2DS is very variable and it can be considered the prototype of those syndromes with variable expressivity and reduced penetrance. The reasons for this variability have not been elucidated yet, and the molecular substrates underlying the different clinical features of 22q11.2DS are still debated.
In consideration of this, we collected a cohort of 21 patients and analyzed them by applying very homogeneous and strict criteria, in order to detect some of the genetic differences that may influence this variability. Two aspects were investigated: (1) the precise breakpoint localization and (2) the additional CNVs elsewhere in the genome in order to elucidate the possible interactions between their genes and those in the 22q11.2 region.
Breakpoint Localization
According to the literature, it is known that the vast majority of 22q11.2 deletions happen between LCR22-A and D, giving rise to the “common” ~3 Mb deletion. Also in our cohort, 18/21 pts have the deletion between A and D, whereas three cases have smaller deletions (Figure 1B).
In the 18 patients sharing the ~3 Mb deletion, the fine mapping of the breakpoints within LCR22-A and D allowed us to evaluate whether small differences of the gene content at the boundaries may play a role in the clinical variability of 22q.11.2 DS. Since LCR22-A and D are quite large and harbor many genes, it is expected that a certain variability could be present at the deletion sites. However, the combination of two array CGH platforms surprisingly showed that in our cohort the breakpoints localization is rather clustered. The only difference concerns the USP18 and FAM230B genes, at the proximal and at the distal breakpoint, respectively.
USP18 (NM_017414) codes for a protein that belongs to the ubiquitin-specific proteases (UBP) family of enzymes that cleave ubiquitin from ubiquitinated protein substrates. Mice lacking this gene are hypersensitive to interferon; in humans, no data are available about its loss-of-function. FAM230B (Homo sapiens family with sequence similarity 230 member B) is a long non-protein coding RNA, the function of which is unknown.
Even if our cohort is limited, it emerges from these data that the breakpoints within the LCR22-A and D are clustered and the small differences concern genes that are not directly related to clinical signs of 22q11.2DS, thus it can be inferred that the breakpoint localization does not have a major role in 22q11.2DS phenotypic variability.
Additional CNVs and Their Gene Content
22q11.2DS patients show a wide spectrum of clinical signs, but single aspects are recurrent (i.e., cardiological 50–75%, immunological 35–40%, neurobehavioral 85%), and cannot be related only to the presence of rare CNVs, which are defined as having a frequency equal or <1% in control populations. For this reason, in the present study, analysis of the gene content was not limited to the rare CNVs, but was extended also to the common ones. In Table 2, all the 81 CNVs detected in our cohort are reported, regardless of their size or frequency, and their genes content is shown.
Before discussing these results, it is worth noting that additional CNVs represent a common background in all the microdeletion syndromes, but most of them do not show the phenotypic variability seen in the 22q11.2DS. If we assume that CNVs could have a major impact on clinical variability, we should address the reason why CNVs affect the 22q11.2DS phenotype more severely than other microdeletion syndromes.
To find the genetic peculiarities that could justify such a preponderant role of CNVs only in 22q11.2DS, first of all we focused on the functional role of genes harbored in the 22q11.2 deleted region.
Peculiarity of the Genes in the 22q11.2 Region
The ~3 Mb region encompasses about 50 RefSeq genes; many of them have not been functionally well-characterized and little is known about their contribution to single clinical aspects of 22q11DS. However, at first glance, the region contains a large number of genes related to mitochondrial functions, and an abundance of non-coding RNA (a total of 13, 6 of which are miRNA) together with the presence of DGCR8 (Di George Critical Region Gene 8), a gene with a primary role in miRNA biogenesis.
Mitochondria and Embryonic development
During the embryo differentiation and early postnatal development, cells actively divide, and these divisions require a great amount of energy, with intense mitochondrial activity; it is expected that deficits in the bioenergetic machinery could set the basis for pleiotropic effects in the majority of developing organs and tissues.
Nine genes in 22q11.2 region code for proteins which are either localized in the mitochondria or are connected to mitochondrial functions (MRPL40, PRODH, SLC25A1, TXNRD2, T10, ZDHHC, COMT, UFD1L, DGCR8) (Meechan et al., 2011). 22q11.2DS is clearly due to an altered embryonal development that compromises early morphogenesis in the pharyngeal arches, heart, skeleton, and brain and thus it could be the consequence of metabolic and energetic problems, rather than of a decreased dosage of gene with a morphogenetic role.
Among the additional CNVs, four genes with mitochondrial function (IMMP2L, ACAD10, MRPS23, and CKMT1A) have been found. Even if our cohort is limited and it is not possible to demonstrate a correlation between clinical variability and mitochondrial pathways, a search for genes involved in mitochondria has not been performed yet, and it should deserve further attention in the future.
DGCR8 and CNV-miRNAs
DGCR8 has a primary role in miRNA biogenesis (Han et al., 2004). MiRNAs exert a precise control over the spatiotemporal expression of individual genes as well as large gene networks. The way these tiny post-transcriptional gene regulators function is not yet fully known. There is a functional redundancy, since a single miRNA may have multiple mRNA targets, and a target may be also regulated by multiple miRNAs. DGCR8 is a main actor in miRNA biogenesis, but alternative pathways are known (Berezikov et al., 2007). Variations in miRNA genes can severely affect downstream-regulated genes and their pathways.
As expected, haploinsufficency of DGCR8 interferes with the processing of many miRNAs, and it is clearly emerging that some of phenotypic features of the 22q11.2DS could be ascribed not only to haploinsufficiency of coding genes but to an altered dosage of miRNAs.
In a knock-out mouse model, Dgcr8 hemizygosity causes a 20–70% reduction in a subset of miRNAs in prefrontal cortex and hippocampus with a consequent dysregulation of the mRNA targets. Dgcr8+/− mice show behavioral and cognitive dysfunction (Stark et al., 2008; Fénelon et al., 2011), present also in the human 22q11.2DS (Schofield et al., 2011; Ouchi et al., 2013). Dgcr8 is essential for miRNA biogenesis in embryonic stem (ES) cells and regulates efficient ES-cell differentiation (Wang et al., 2007). Cardiomyocyte-specific deletion of Dgcr8+/− demonstrated downregulation of a subset of mature cardiac enriched miRNAs; these mice experienced premature lethality due to cardiac failure, pointing out a critical role for miRNAs in maintaining cardiac function in mature cardiomyocytes (Rao et al., 2009).
More recently, the human miRNA expression pattern in the peripheral blood of 22q11.2DS has revealed a peculiar profile that differs from that of the controls; the deregulated miRNAs show a connection with the immunological, cardiac and hypocalcemic pathways (de la Morena et al., 2013; Sellier et al., 2014), suggesting that they may contribute to these phenotypic aspects in 22q11.2DS. Interestingly, processing of miRNAs is not globally altered in 22q11DS, but limited to the DGCR8 targets, providing compelling evidence of specific miRNA DGCR8-mediated dysregulation (Sellier et al., 2014).
In view of this, the role of the CNVs on the phenotypic variability of 22q11.2DS could be correlated non just to protein coding genes, but to miRNA gene content. Most of the studies that have dealt with CNVs and 22q11.2DS variability have been focused only on the protein coding genes, and the impact of altered dosage of non-coding regulatory miRNAs is largely unexplored.
Recently, some articles have highlighted a consistent co-localization of miRNA loci with CNV regions (CNV-miRNAs) (Marcinkowska et al., 2011; Veerappa et al., 2014). In a study on 1,715 subjects, about 5% of all miRNAs are harbored in the CNVs, ~9% of CNVs were shown to harbor miRNA genes and these CNV-miRNAs were identified in 83.8% individuals (Veerappa et al., 2014).
In our cohort, 11 CNV-miRNAs were identified (Table 3): as can be seen, some miRNAs are deleted, some duplicated, some others are present both in deletions and in duplications.
It seems obvious that the expression of miRNAs correlates with the CNV dose, as the expression of protein-coding genes depends on the copy number. CNV-miRNA with one allele duplicated (copy number = 3) or deleted (copy number = 1) is expected to have a level of expression roughly 1.5 or 0.5 times, respectively, compared to the wild type (copy number = 2). A change in a miRNA dosage will affect the expression of all of its target genes and may have a pleiotropic effect.
In 22q11.2DS patients, where DGCR8 activity is reduced by about 50% (Sellier et al., 2014), the expression pattern of CNV-miRNAs is completely dysregulated. The dosage of a miRNA in a deleted CNV is further decreased, whereas the dosage of miRNA in a duplicated CNV could be compensated by DGCR8 hemizygosity. This dysregulation take place simultaneously on all miRNAs of an individual according to his CNV background.
It is worth noting that all the 11 miRNAs detected in the present cohort of patients have a biological role in pathways related to some phenotypic characteristics of 22q11.2DS (Table 4).
These changes in miRNAs expression are likely to contribute significantly toward 22q11.2DS phenotypic variability. Therefore, DGCR8 could represent the genetic particularity according to which the additional CNVs affect the 22q11.2DS phenotype more greatly than other microdeletion syndromes.
In view of this, 22q11.2DS could be mainly related to a dysregulation of post-transcriptional modifications that lead to an altered dosage of proteins crucial for phenotype development.
Concluding Remarks
In this paper some of the genetic factors that can influence 22q11.2DS variability have been analyzed, and, even if the cohort of patients is limited, interesting observations can be made.
22q11.2 breakpoints are rather clustered, and it is unlikely that they play a major role in the variability.
A detailed analysis of the gene content of 22q11.2 deleted region indicates that this syndrome could be a metabolic disorder or consequence of altered post-transcriptional gene modifications.
In the additional CNVs, four genes with mitochondrial functions and 11 miRNAs, all related to the biological pathways present in the 22q11.2DS, have been detected in 4/21 and in19/21 patients, respectively.
CNVs and miRNAs are new entities that have changed the order of complexity at the level of gene expression and regulation, thus CNV-miRNAs are potential functional variants that should be considered high priority candidate variants in genotype-phenotype association studies. Deletion of DGCR8, the main actor in miRNA biogenesis, amplifies this variability.
To our knowledge, this is the first report that focuses on the CNV-microRNAs in 22q11.2DS, with the aim of trying to better understand their role in the variable expressivity and reduced penetrance.
In the near future, it will be important to characterize the pathways of these CNV-miRNAs in a much more comprehensive manner in order to improve the genetic networks that can contribute to the pathophysiology of 22q11.2.
Author Contributions
AV, VB, and RC participated in the design of this study. VB, AA, and AV performed the array CGH and the CNV gene content analyses; and contributed to writing the manuscript. AL and RC performed the immunological and the clinical characterization of the patients. RM and RB performed the clinical and neuropsychiatric investigations. All authors have read and approve the final version of this manuscript.
Funding
This work has been supported by a fund (PRA_2015_0104) Progetto di Ricerca di Ateneo 2015 (PRA), Università di Pisa.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Prof. Wendy Doherty (Assistant Professor of English Literature, University of Pisa) for her critical revision of the manuscript and English editing. We also thank Alessandra Consani, for technical help in the array CGH experiments.
References
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bassett, A. S., McDonald-McGinn, D. M., Devriendt, K., Digilio, M. C., Goldenberg, P., Habel, A., et al. (2011). Practical guidelines for managing patients with 22q11.2 deletion syndrome. J. Pediatr. 159, 332.e1–339.e1. doi: 10.1016/j.jpeds.2011.02.039
Berezikov, E., Chung, W. J., Willis, J., Cuppen, E., and Lai, E. C. (2007). Mammalian mirtron genes. Mol. Cell 28, 328–336. doi: 10.1016/j.molcel.2007.09.028
Bittel, D. C., Yu, S., Newkirk, H., Kibiryeva, N., Holt, A., Butler, M. G., et al. (2009). Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH. Cytogenet. Genome Res. 124, 113–120. doi: 10.1159/000207515
Coe, B. P., Witherspoon, K., Rosenfeld, J. A., van Bon, B. W., Vulto-van Silfhout, A. T., Bosco, P., et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 46, 1063–1071. doi: 10.1038/ng.3092
Cooper, G. M., and Mefford, H. C. (2011). Detection of copy number variation using SNP genetyping. Methods Mol. Biol. 767, 243–252. doi: 10.1007/978-1-61779-201-4_18
Cooper, G. M., Coe, B. P., Girirajan, S., Rosenfeld, J. A., Vu, T. H., Baker, C., et al. (2011). A copy number variation morbidity map of developmental delay. Nat. Genet. 14, 839–846. doi: 10.1038/ng.909
de la Morena, M. T., Eitson, J. L., Dozmorov, I. M., Belkaya, S., Hoover, A. R., Anguiano, E., et al. (2013). Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. Clin. Immunol. 147, 11–22. doi: 10.1016/j.clim.2013.01.011
Dweep, H., Sticht, C., Pandey, P., and Gretz, N. (2011). miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 44, 839–847. doi: 10.1016/j.jbi.2011.05.002
Edelmann, L., Pandita, R. K., and Morrow, B. E. (1999). Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am. J. Hum. Genet. 64, 1076–1086. doi: 10.1086/302343
Fénelon, K., Mukai, J., Xu, B., Hsu, P. K., Drew, L. J., Karayiorgou, M., et al. (2011). Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 108, 4447–4452. doi: 10.1073/pnas.1101219108
Han, J., Lee, Y., Yeom, K. H., Kim, Y. K., Jin, H., and Kim, V. N. (2004). The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18, 3016–3027. doi: 10.1101/gad.1262504
Kirov, G., Grozeva, D., Norton, N., Ivanov, D., Mantripragada, K. K., Holmans, P., et al. (2009). Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 18, 1497–1503. doi: 10.1093/hmg/ddp043
Kumar, R. A., KaraMohamed, S., Sudi, J., Conrad, D. F., Brune, C., Badner, J. A., et al. (2008). Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 17, 628–638. doi: 10.1093/hmg/ddm376
Leana-Cox, J., Pangkanon, S., Eanet, K. R., Curtin, M. S., and Wulfsberg, E. A. (1996). Familial DiGeorge/velocardiofacial syndrome with deletions of chromosome area 22q11.2: report of five families with a review of the literature. Am. J. Med. Genet. 65, 309–316. doi: 10.1002/(SICI)1096-8628(19961111)65:4<309::AID-AJMG12>3.0.CO;2-Y
Lee, C., Iafrate, A. J., and Brothman, A. R. (2007). Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet. 39, S48–S54. doi: 10.1038/ng2092
Mantripragada, K. K., Tapia-Páez, I., Blennow, E., Nilsson, P., Wedell, A., and Dumanski, J. P. (2004). DNA copy-number analysis of the 22q11 deletion syndrome region using array-CGH with genomic and PCR-based targets. Int. J. Mol. Med. 13, 273–279. doi: 10.3892/ijmm.13.2.273
Marcinkowska, M., Szymanski, M., Krzyzosiak, W. J., and Kozlowski, P. (2011). Copy number variation of microRNA genes in the human genome. BMC Genomics 12:183. doi: 10.1186/1471-2164-12-183
McDonald-McGinn, D. M., and Sullivan, K. E. (2011). Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore). 90, 1–18. doi: 10.1097/MD.0b013e3182060469
Meechan, D. W., Maynard, T. M., Tucker, E. S., and LaMantia, A. S. (2011). Three phases of DiGeorge/22q11 deletion syndrome pathogenesis during brain development: patterning, proliferation, and mitochondrial functions of 22q11 genes. Int. J. Dev. Neurosci. 29, 283–294. doi: 10.1016/j.ijdevneu.2010.08.005
Mlynarski, E. E., Sheridan, M. B., Xie, M., Guo, T., Racedo, S. E., McDonald-McGinn, D. M., et al. (2015). Copy-number variation of the glucose transporter gene SLC2A3 and congenital heart defects in the 22q11.2 deletion syndrome. Am. J. Hum. Genet. 96, 753–764. doi: 10.1016/j.ajhg.2015.03.007
Mlynarski, E. E., Xie, M., Taylor, D., Sheridan, M. B., Guo, T., Racedo, S. E., et al. (2016). Rare copy number variants and congenital heart defects in the 22q11.2 deletion syndrome. Hum. Genet. 135, 273–285. doi: 10.1007/s00439-015-1623-9
Ouchi, Y., Banno, Y., Shimizu, Y., Ando, S., Hasegawa, H., Adachi, K., et al. (2013). Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J. Neurosci. 33, 9408–9419. doi: 10.1523/JNEUROSCI.2700-12.2013
Pinto, D., Marshall, C., Feuk, L., and Scherere, S. W. (2007). Copy-number variation in control population cohorts. Hum. Mol. Genet. 16, R168–R173. doi: 10.1093/hmg/ddm241
Rao, P. K., Toyama, Y., Chiang, H. R., Gupta, S., Bauer, M., Medvid, R., et al. (2009). Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 105, 585–594. doi: 10.1161/CIRCRESAHA.109.200451
Schofield, C. M., Hsu, R., Barker, A. J., Gertz, C. C., Blelloch, R., and Ullian, E. M. (2011). Monoallelic deletion of the microRNA biogenesis gene Dgcr8 produces deficits in the development of excitatory synaptic transmission in the prefrontal cortex. Neural Dev. 6:11. doi: 10.1186/1749-8104-6-11
Sellier, C., Hwang, V. J., Dandekar, R., Durbin-Johnson, B., Charlet-Berguerand, N., Ander, B. P., et al. (2014). Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLoS ONE 9:e103884. doi: 10.1371/journal.pone.0103884
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760. doi: 10.1038/ng.138
Urban, A. E., Korbel, J. O., Selzer, R., Richmond, T., Hacker, A., Popescu, G. V., et al. (2006). High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 103, 4534–4539. doi: 10.1073/pnas.0511340103
Vaishnavi, V., Manikandan, M., Tiwary, B. K., and Munirajan, A. K. (2013). Insights on the functional impact of microRNAs present in autism-associated copy number variants. PLoS ONE 8:e56781. doi: 10.1371/journal.pone.0056781
Veerappa, A. M., Murthy, M. N., Vishweswaraiah, S., Lingaiah, K., Suresh, R. V., Nachappa, S. A., et al. (2014). Copy number variations burden on miRNA genes reveals layers of complexities involved in the regulation of pathways and phenotypic expression. PLoS ONE 9:e90391. doi: 10.1371/journal.pone.0090391
Vogler, C., Gschwind, L., Röthlisberger, B., Huber, A., Filges, I., Miny, P., et al. (2010). Microarray-based maps of copy-number variant regions in European and sub-Saharan populations. PLoS ONE 5:e15246. doi: 10.1371/journal.pone.0015246
Wang, Y., Medvid, R., Melton, C., Jaenisch, R., and Blelloch, R. (2007). DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 39, 380–385. doi: 10.1038/ng1969
Keywords: 22q11 deletion syndrome, array CGH, copy number variations, miRNA, CNV-miRNA, DGCR8
Citation: Bertini V, Azzarà A, Legitimo A, Milone R, Battini R, Consolini R and Valetto A (2017) Deletion Extents Are Not the Cause of Clinical Variability in 22q11.2 Deletion Syndrome: Does the Interaction between DGCR8 and miRNA-CNVs Play a Major Role? Front. Genet. 8:47. doi: 10.3389/fgene.2017.00047
Received: 30 December 2016; Accepted: 30 March 2017;
Published: 01 May 2017.
Edited by:
Babajan Banganapalli, King Abdulaziz University, Saudi ArabiaReviewed by:
Tommaso Pizzorusso, Consiglio Nazionale Delle Ricerche, ItalyMaria Paola Lombardi, University of Amsterdam, Netherlands
Alex Vincent Postma, University of Amsterdam, Netherlands
Copyright © 2017 Bertini, Azzarà, Legitimo, Milone, Battini, Consolini and Valetto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelo Valetto, YS52YWxldHRvQGFvLXBpc2EudG9zY2FuYS5pdA==