 Blanka Rogina
Blanka Rogina- Department of Genetics and Genome Sciences, Institute for Systems Genomics, School of Medicine, University of Connecticut Health Center, Farmington, CT, United States
The Indy (I’m Not Dead Yet) gene encodes the fly homolog of the mammalian SLC13A5 citrate transporter. Reduced expression of the Indy gene in flies and worms extends their longevity. INDY is expressed in the plasma membrane of metabolically active tissues. Decreased expression of Indy in worms, flies, mice, and rats alters metabolism in a manner similar to calorie restriction. Reducing INDY activity prevents weight gain in flies, worms, and mice, and counteracts the negative effects of age or a high fat diet on metabolism and insulin sensitivity. The metabolic effects of reducing INDY activity are the result of reduced cytoplasmic citrate. Citrate is a key metabolite and has a central role in energy status of the cell by effecting lipid and carbohydrate metabolism and energy production. Thereby newly described drugs that reduce INDY transporting activity increase insulin sensitivity and reduce hepatic lipid levels via its effect on hepatic citrate uptake. A recent report presented the first direct link between increased hepatic levels of human INDY, insulin resistance, and non-alcoholic fatty liver disease in obese humans. Similarly increased hepatic mIndy levels were observed in non-human primates fed on a high fat diet for 2 years. This effect is mediated via the stimulatory effect of the interleukin-6/Stat3 pathway on mINDY hepatic expression. These findings make INDY a potential and very promising target for the treatment of metabolic disorders in humans.
INDY Reduction Affects Metabolism, Health, and Longevity
The Indy (I’m Not Dead Yet) gene encodes the fly homolog of the mammalian SLC13A5 transporter of the tricarboxylic acid (TCA) cycle intermediates (Rogina et al., 2000; Knauf et al., 2002, 2006). INDY is a member of the SLC13 protein family of Na+-coupled di- and tri-carboxylate/sulfate transporters in prokaryotes and eukaryotes (Bhutia et al., 2017). In flies, INDY mediates a cation independent and electroneutral high affinity bidirectional transport of the TCA intermediate across the plasma membrane (Knauf et al., 2002, 2006). Fly INDY has the highest substrate affinity for transporting citrate and lower affinities to other Krebs cycle intermediates such as succinate, malate, and fumarate (Knauf et al., 2002, 2006). INDY homologs in bacteria and mammals have the highest affinity for transporting citrate, and lower for other Krebs cycle intermediates but this transport is Na-dependent. The Na-citrate stoichiometry is 1:1 in bacteria and 4:1 for mINDY (Inoue et al., 2002a,b; Bhutia et al., 2017; von Loeffelholz et al., 2017).
Although all mammalian INDY transporters are Na+-dependent, the human mINDY is a high-capacity and low affinity transporter, while the rodent mINDY are low-capacity and high affinity transporters (Bhutia et al., 2017; von Loeffelholz et al., 2017).
Reduced expression of the Indy gene in flies and worms extends longevity in all but one study (Rogina et al., 2000; Fei et al., 2003, 2004; Toivonen et al., 2007; Wang et al., 2009; Rogina and Helfand, 2013; Rogers and Rogina, 2014; Schwarz et al., 2015). INDY is expressed on the plasma membrane of metabolically active tissues. In flies INDY is predominantly expressed in the midgut, fat body, and oenocytes (fly liver) (Rogina et al., 2000; Knauf et al., 2002). In humans, Indy mRNA is mainly expressed in the liver, less in the brain and testis, while small levels of Indy mRNA expression were found in the kidneys, thymus, ovaries, adipose tissue, stomach, and colon (Inoue et al., 2002b; von Loeffelholz et al., 2017). Decreased expression of Indy in worms, flies, mice, and rats alters metabolism in a manner similar to calorie restriction (CR; Rogina et al., 2000; Fei et al., 2003; Bross et al., 2005; Neretti et al., 2009; Wang et al., 2009; Birkenfeld et al., 2011; Schwarz et al., 2015). This is supported by similar phenotypes found in CR wild type flies and in Indy flies that were kept on a high calorie diet. These Indy flies have lower lipid levels, increased mitochondrial biogenesis, increased spontaneous physical activity and a reduction in components of the insulin-signaling pathway activity (Figure 1; Neretti et al., 2009; Wang et al., 2009; Rogers and Rogina, 2014). Indy flies are protected from weight gain when aged on a high calorie diet (Wang et al., 2009). Under standard condition, heterozygous Indy flies do not experience any negative effects on health and have the same negative geotaxis, metabolic rate and maximal flight velocity (Marden et al., 2003). Furthermore, Indy heterozygous flies laid more eggs during their life compared to controls (Marden et al., 2003). However, under CR condition, Indy heterozygous flies have reduced fecundity due to lower energy resource caused by the effect of reduced Indy on metabolism (Marden et al., 2003). Consistently, CR does not further extend longevity of long-lived Indy heterozygous flies and shortens longevity of Indy homozygous flies (Wang et al., 2009).
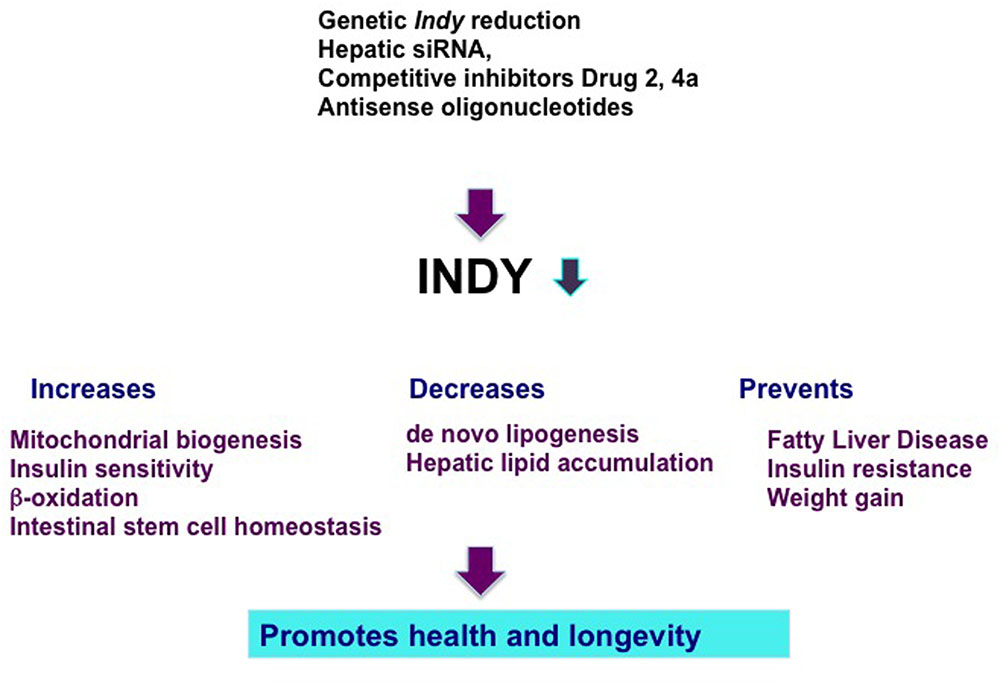
FIGURE 1. Genetics and pharmacological manipulations that result in hepatic Indy reduction prevent negative effects of a high fat diet and promote health and longevity. INDY reduction increases mitochondrial biogenesis, insulin sensitivity, β-oxidation, and preserves intestinal stem cell homeostasis. Reduced INDY decreases de novo lipogenesis and hepatic lipid accumulation. Decrease in INDY transporting activity prevents fatty liver disease, insulin resistance, and weight gain.
Preservation of intestinal stem cell (ISC) homeostasis has a key role in maintaining normal midgut function and contributes to extended health and longevity in flies (Biteau et al., 2010). Changes in mitochondrial biogenesis found in the midgut of Indy flies, combined with increased antioxidant activity and reduced production of reactive oxygen species preserve ISC homeostasis and intestinal integrity in Indy flies. These changes maintain midgut function and mediate extended health and longevity of Indy flies (Rogers and Rogina, 2014).
Reduced activity of the Indy homologs in other organisms is associated with similar metabolic effects that mimic CR. siRNA mediated knockdown of Indy/CeNac2, the worm Indy homolog, results in worms that are smaller, have reduced lipid levels, and have extended longevity (Fei et al., 2004; Schwarz et al., 2015). mIndy-/- knockout mice are protected from the negative effects of aging or a high-fat diet on metabolism, which include hepatic fat accumulation, obesity, and insulin insensitivity (Birkenfeld et al., 2011). These mice have increased energy expenditure, reduced hepatic lipogenesis, increased mitochondrial biogenesis, and enhanced hepatic fatty acid (FA) oxidation (Birkenfeld et al., 2011). Increased liver accumulation of diacylglycerols (DAG) and ceramides have been linked to insulin resistance and development of type 2-diabetes (T2D) (Jornayvaz and Shulman, 2012). mIndy-/- mice have reduced DAG levels, which most likely contributes to their protection against insulin resistance. Whole-genome microarray studies comparing mIndy-/- and mIndy-/+ revealed that transcriptional changes found in the liver of mIndy-/- mice are 80% identical to changes found in the liver of CR mice (Birkenfeld et al., 2011). All of these findings confer that INDY reduction creates a state similar to CR.
The metabolic effects of reduced INDY activity are a result of decreased cytoplasmic citrate levels. Citrate is converted to oxaloacetate and acetyl-CoA by ATP-citrate lyase. Acetyl-CoA is precursor for biosynthesis of triglycerides, FAs, low-density lipoproteins, and cholesterol. Citrate inhibits catabolism of glucose by inhibiting phosphofructokinase through allosteric modulation. Citrate also activates acetyl-CoA carboxylase, thereby affecting de novo lipogenesis. Thus, cytoplasmic citrate levels affect lipids and glucose metabolism, as well as energy production in mitochondria. Reduced Indy levels are associated with reduced ATP levels in mIndy-/- mice and in worms in which INDY levels are reduced by siIndy (Birkenfeld et al., 2011; Schwarz et al., 2015). The low ATP/ADP ratio activates AMPK, an energy sensor in the cells, which increases mitochondrial biogenesis by activating mitochondrial transcriptional co-activator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (Guarente, 2008; Neretti et al., 2009; Birkenfeld et al., 2011; Rogers and Rogina, 2014; Schwarz et al., 2015). AMPK also increases insulin sensitivity, contributing to the beneficial effects of Indy reduction on glucose metabolism. When cytoplasmic citrate levels are high, FA β-oxidation is down regulated, while FA synthesis is upregulated. The opposite is found in mIndy-/- mice, in which low cytoplasmic citrate levels result in reduced FA synthesis, while FA β-oxidation and insulin sensitivity is increased (Birkenfeld et al., 2011).
Regulation of mIndy Transcription
The levels of fly INDY are affected by age and by caloric content of the food. Aging flies on a standard diet, young flies on a high calories diet, or young flies treated with paraquat (creating oxidative stress), have increased Indy mRNA and protein levels in the midgut (Rogers and Rogina, 2014). In contrast, flies aged on a CR diet have Indy mRNA reduced to 50% of the levels found in controls (Wang et al., 2009; Rogers and Rogina, 2014). Recent work showed that mIndy levels in primary rat hepatocytes are regulated by glucagon released during early starvation (Neuschafer-Rube et al., 2015). Glucagon binds to the CREB (cAMP-dependent and cAMP-responsive element protein)-dependent binding site in the promoter region of mIndy and transiently increases mIndy expression (Figure 2) (Neuschafer-Rube et al., 2014). In vivo studies shown increased hepatic mIndy levels in high-fat-diet-streptozotocin diabetic rats, in which CREB is constitutively active.
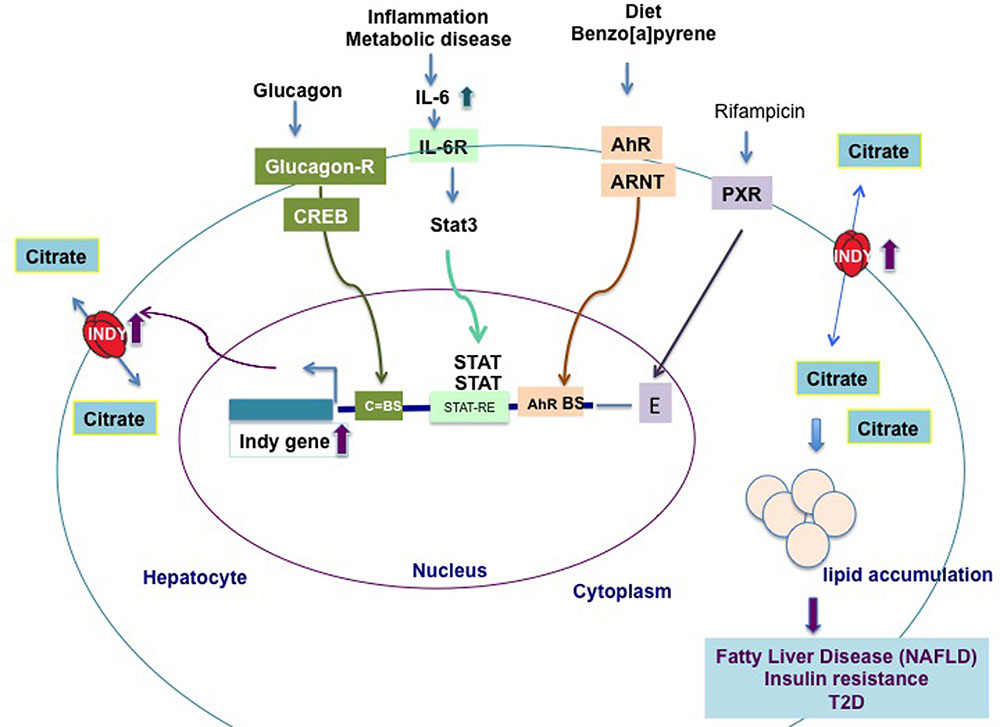
FIGURE 2. Transcriptional regulation of hepatic mIndy mRNA levels in model organisms and humans. Hepatic mIndy expression levels are upregulated by glucagon released during early starvation via a cAMP and cAMP-responsive-element binding-protein (CREB)-dependent mechanism. A CREB binding site (C-BS) was identified in the promoter region of mINDY. Increased levels of interleukin-6 (IL-6) associated with inflammation and metabolic disease, activate transcription of hepatic mIndy via the signal transducer and activator of transcription 3 (STAT-3) signaling pathway. Diet and benzo[a]pyrene activate aryl hydrocarbon receptor (AhR) and its heterodimerization to AhR nuclear translocator (ARNT). In nucleus AhR activates transcription of mIndy via binding to potential binding site (BS) in the mIndy promoter (Neuschafer-Rube et al., 2015). Rifampicin activates the pregnane X receptor (PXR), which increases mIndy transcription by activation of two enhancer modules located upstream of the mIndy (SLC13A5) transcriptional start site (Li et al., 2015). Increased INDY levels result in higher citrate uptake and its incorporation in lipids, leading to non-alcoholic fatty liver disease (NAFLD), insulin resistance and type 2-diabetes (T2D).
Additional findings link increased INDY to lipid metabolism. SLC13A5 has been identified as a novel transcriptional target of the pregnane X receptor (PXR) (Li et al., 2015). The PXR has been linked to lipid metabolism and energy homeostasis. Rifampicin activates the PXR, which increases mIndy transcription by activation of two enhancer modules located upstream of the mIndy (SLC13A5) transcriptional start site (Li et al., 2015). Rifampicin-induced PXR activation induces SLC13A5 mRNA and protein levels, and the lipid content in human primary hepatocytes, while SLC13A5 knockdown expression significantly reduces the lipid content in HepG2 cells (Li et al., 2015). These data suggest a role of increased INDY levels in drug-induced steatosis in human liver.
Energy-rich diet and benzo[a]pyrene activate aryl hydrocarbon receptor (AhR). AhR heterodimerization to AhR nuclear translocator (ARNT) allows its translocation to nucleus and activation of mIndy transcription via binding to potential binding site in the mIndy promoter. AhR putative binding site has been identified in the promoter region of both rat and human mIndy (Neuschafer-Rube et al., 2015). Activation of the AhR leads to fatty liver disease.
Patients undergoing Li+ treatment have dyslipidemia and gain body weight. The stimulatory effects of Li+ on the transporting activity of mINDY have been described, suggesting possible clinically relevant connection between increased INDY activity and obesity in humans (Inoue et al., 2003). Taken together, increased hINDY levels may contribute to clinically relevant hepatic metabolic dysfunction associated with T2D.
Increased INDY Levels are Linked to Non-Alcoholic Fatty Liver Disease in Humans
Two recent reports linked increased mIndy levels to non-alcoholic fatty liver disease (NAFLD) in an experimental mice model of NAFLD and human patients with NAFLD (Brachs et al., 2016; von Loeffelholz et al., 2017). NAFLD is associated with development of insulin resistance, T2D and liver cirrhosis (Farrell and Larter, 2006; Postic and Girard, 2008). NAFLD results from sedentary life style, increased energy uptake, reduced FA oxidation, and possible altered gut microbiota (Postic and Girard, 2008). Diet-induced NAFLD in adult C57BL/6J mice fed a western diet was prevented by weekly injection of liver specific siRNA against mINDY, which suppressed 60% of liver mIndy levels after 8 weeks (Brachs et al., 2016). These mice have improved hepatic insulin sensitivity, reduced hepatic lipid accumulation, and are protected against diet-induced lipid-steatosis. Notably, activation of the AhR leads to fatty liver disease and induces mIndy expression in rat hepatocytes, Figure 2 (Neuschafer-Rube et al., 2015).
The first direct link between increased hepatic mIndy levels and lipid steatosis in human NAFLD patients was reported by von Loeffelholz et al. (2017). The authors showed that, in lean individuals, low levels of hepatic fat are associated with decreased mIndy expression, while in individuals with an increased body mass index and a high liver fat content mINDY levels are increased. Similarly, mINDY levels in the liver are increased in non-human primates aged on a high-fat or high-sucrose diet and in mice on a high-fat or a high fat-nonalcoholic steatohepatitis (NASH) inducing (low methionine, choline-deficient) diet. Liver microarray studies performed in samples from NAFLD patients and controls revealed an association between changes in whole-genome transcriptional levels in NAFLD liver samples and an increase in the expression of genes involved in lipid synthesis, metabolism, and immunological processes (von Loeffelholz et al., 2017). An increase in interleukin-6 (IL-6) levels activates mIndy transcription via binding to the IL-6-receptor, which is associated with phosphorylation, activation of the transcription factor Stat3, and transfer of the latter to the nucleus, where it binds to the putative STAT-responsive element (STAT-RE) binding site in the mIndy promoter. The in vivo activation of the IL-6/Stat3 pathway increases mIndy expression and thereby citrate uptake and hepatic lipogenesis (Figure 2). In other in vivo studies, the administration of IL-6 for 14 days increased citrate uptake and FA synthesis in the livers of mIndy+/+ mice, by increasing mIndy mRNA levels (von Loeffelholz et al., 2017). This result confirmed the link between IL-6 and increased mIndy. The IL-6 mediated increase in mIndy citrate activity was confirmed by the lack of citrate uptake in hepatocytes isolated from mIndy-/- knockout mice, consistent with the link between mIndy and IL-6 (von Loeffelholz et al., 2017).
mIndy is a Potential Therapeutic Target for Treating Hepatic Steatosis and Insulin Resistance
Considering the beneficial effects of the reduced transporting activity of INDY on metabolism and health, we and others have suggested mIndy (SLC13A5) as a target for the treatment of metabolic disorders (Figure 1; Rogina et al., 2000; Birkenfeld et al., 2011; Frankel and Rogina, 2012; Willmes and Birkenfeld, 2013; Rogers and Rogina, 2015). Several recent reports have described successful efforts to target the transporting activity of INDY and novel compounds have been identified that demonstrate the therapeutic potential of mINDY inhibitors in the treatment of NAFLD and insulin resistance. Temporal administrations of inducible hepatic 2′-O-methoxyethyl chimeric antisense oligonucleotides to knockdown mIndy levels in rats alleviated the negative effects of a high fat diet (HFD) and prevented diet-induced hepatic steatosis and hepatic insulin resistance without affecting rat body weight (Figure 1; Pesta et al., 2015). These rats had lower serum triglyceride levels and improved hepatic insulin sensitivity. Several competitive stereo-specific inhibitors of mIndy transporting activity have been identified (Huard et al., 2015, 2016). Small dicarboxylate molecules selectively inhibit mIndy citrate uptake in vitro in human hepatocytes and hepatic citrate uptake in vivo in mice. Compound 2 is a competitive inhibitor of mIndy that results in lower hepatic lipid levels, reduced plasma glucose levels, and a reversal of glucose intolerance in HFD-fed mice (Huard et al., 2015). Compound 4a is a potent mIndy inhibitor (Huard et al., 2016). Rodents treated with compound 4a had reduced citrate uptake in the liver, kidneys, and testis, which resulted in modest improvements in glucose metabolism (Huard et al., 2016).
Mutations in mIndy (SLC13A5) Lead to Autosomal-Recessive Epileptic Encephalopathy with Neonatal Seizures
Mutations in human mIndy cause autosomal-recessive epileptic encephalopathy with seizures during the first days of life and lead to developmental delays (Thevenon et al., 2014). mIndy encodes the only known neuronal plasma membrane Na-dependent citrate transporter, and gene mutations typically involve the sodium-binding domains of the respective protein. The key steps in mINDY-mediated citrate transport across the plasma membrane depend on conformational changes associated with Na-binding to critical residues within two mINDY domains. Thus, it has been speculated that the severe phenotype induced by human mIndy mutations is most likely due to the inability of mutated INDY to bind the sodium required for transporting citrate across the plasma membrane (Thevenon et al., 2014). In vitro studies showed that COS-7 cells transiently transfected with mIndy mutant genes lack citrate transport capacity (Klotz et al., 2016). Recently, the European Consortium identified additional mIndy mutations that give rise to premature stop codons or amino acid substitutions in mINDY and result in the absence of citrate transport (Hardies et al., 2015; Weeke et al., 2017). These mIndy mutations are also associated with neonatal epilepsy, developmental delays, and variable cognitive impairment. Several patients have dental hypoplasia or hypodontia (Hardies et al., 2015). Neuroimaging findings of eight full-term infants with mIndy mutations showed punctate white-matter lesions (Weeke et al., 2017). Once in the cytoplasm, citrate is used for lipid, cholesterol, glucose, and glutamate synthesis. Malate derived from citrate is transported into mitochondria, where it is used for energy production. Thus, epilepsy may be due to disrupted energy production and altered metabolism in brain cells, an imbalance in GABA and glutamate production, or a reduced inhibition of excitatory N-methyl-D-aspartate (NMDA) receptors (Hardies et al., 2015). However, the pathophysiology of mIndy mutations and the mechanism underlying severe phenotypes are unknown. Notably, mIndy-/- mice or mice treated with small mIndy competitive inhibitors did not develop seizures.
Summary and Concluding Thoughts
Reduction of Indy gene activity in flies and worms extends their health and longevity (Rogers and Rogina, 2015). Genetically reduced INDY expression has beneficial effects on metabolism and prevents diet-induced obesity in flies and mice, suggesting INDY as a target in the treatment of metabolic disorders in humans (Wang et al., 2009; Birkenfeld et al., 2011; Rogers and Rogina, 2015). This potential is supported by several findings. For example, reduced plasma insulin and plasma triglyceride levels as well as an improvement of hepatic steatosis were observed in HFD-fed mice and rats in which liver-specific knockdown of Indy was accomplished using a low-dose of a chimeric anti-sense oligonucleotide (Pesta et al., 2015). Additional pharmacological treatments were recently described, such as competitive inhibitors of INDY transport activity that improve insulin sensitivity in rats and mice in vivo and in cultured human cells (Huard et al., 2015, 2016).
By contrast, high levels of INDY are associated with negative effects on metabolism and health, while mIndy gene mutations cause autosomal-recessive epileptic encephalopathy in newborns as well as developmental delays (Thevenon et al., 2014; von Loeffelholz et al., 2017). Further studies are needed to reveal the mechanisms that lead to this severe phenotype in patients. Recently, increased hepatic levels of mINDY were linked to insulin resistance and NAFLD in obese humans (von Loeffelholz et al., 2017). These findings illustrate both the relevance of the mIndy gene to human health and a highly conserved role for INDY in the metabolism of a broad range of species. Thus, mIndy has emerged as a novel target for the treatment of age- and diet-associated metabolic syndrome, NAFLD, and T2D. The further development of mIndy inhibitors may additionally provide effective interventions targeting the debilitating health effects that are often associated with aging and will thereby allow a healthier life.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Funding
This work was supported by grant from the National Institute on Health RO1AG 023088 to BR. BR is a recipient of a Glenn Award for Research in Biological Mechanisms of Aging.
Conflict of Interest Statement
The author declares that she is a co-owner of the INDY US Patent (#7,118,873).
Acknowledgments
I thank Suzanne Kowalski, Robert Pijewski, and Stewart Frankel for critical reading of the manuscript.
References
Bhutia, Y. D., Kopel, J. J., Lawrence, J. J., Neugebauer, V., and Gabapathy, V. (2017). Plasma membrane Na+-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules 22:378. doi: 10.3390/molecules22030378
Birkenfeld, A. L., Lee, H.-Y., Guebre-Egziabher, F., Alves, T. C., Jurczak, M. J., Jornayvaz, F. R., et al. (2011). Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14, 184–195. doi: 10.1016/j.cmet.2011.06.009
Biteau, B., Karpac, J., Supoyo, S., DeGennaro, M., Lehmann, R., and Jasper, H. (2010). Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 6:e1001159. doi: 10.1371/journal.pgen.1001159
Brachs, S., Winkel, A. F., Tang, H., Birkenfeld, A. L., Brunner, B., Jahn-Hofmann, K., et al. (2016). Inhibition of citrate transporter Slc13A5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 5, 1072–1082. doi: 10.1016/j.molmet.2016.08.004
Bross, T. G., Rogina, B., and Helfand, S. L. (2005). Behavioral, physical, and demographic changes in Drosophila populations through dietary restriction. Aging Cell 4, 309–317. doi: 10.1111/j.1474-9726.2005.00181.x
Farrell, G. C., and Larter, C. Z. (2006). Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43, S99–S112. doi: 10.1002/hep.20973
Fei, Y. J., Inoue, K., and Ganapathy, V. (2003). Structural and functional characteristics of two sodium-coupled dicarboxylate transporters (ceNaDC1 and ceNaDC2) from Caenorhabditis elegans and their relevance to life span. J. Biol. Chem. 278, 6136–6144. doi: 10.1074/jbc.M208763200
Fei, Y. J., Liu, J. C., Inoue, K., Zhuang, L., Miyake, K., Miyauchi, S., et al. (2004). Relevance of NAC-2, and Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem. J. 379, 191–198. doi: 10.1042/bj20031807
Frankel, S., and Rogina, B. (2012). Indy mutants: live long and prosper. Front. Genet. 3:13. doi: 10.3389/fgene.2012.00013
Guarente, L. (2008). Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell 132, 171–176. doi: 10.1016/j.cell.2008.01.007
Hardies, K., de Kovel, C. G., Weckhuysen, S., Asselbergh, B., Geuens, T., Deconinck, T., et al. (2015). Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 138, 3238–3250. doi: 10.1093/brain/awv263
Huard, K., Brown, J., Jones, J. C., Cabral, S., Futatsugi, K., Gorgoglione, M., et al. (2015). Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep. 5, 17391. doi: 10.1038/srep17391
Huard, K., Gosset, J. R., Montgomery, J. I., Gilbert, A., Hayward, M. M., Magee, T. V., et al. (2016). Optimization of a dicarboxylic series for in vivo inhibition of citrate transport by the solute carrier 13 (SLC13) Family. J. Med. Chem. 59, 1165–1175. doi: 10.1021/acs.jmedchem.5b01752
Inoue, K., Fei, Y. J., Huang, W., Zhuang, L., Chen, Z., and Ganapathy, V. (2002a). Functional identity of Drosophila melanogaster Indy as a cation-independent, electroneutral transporter for tricarboxylic acid-cycle intermediates. Biochem. J. 367, 313–319.
Inoue, K., Zhuang, L., Maddox, D. M., Smith, S. B., and Ganapathy, V. (2002b). Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 277,39469–39476.
Inoue, K., Zhuang, L., Maddox, D. M., Smith, S. B., and Ganapathy, V. (2003). Human sodium-coupled citrate transporter, the orthologue of Drosophila indy, as a novel target for lithium action. Biochem. J. 374, 21–26. doi: 10.1042/bj20030827
Jornayvaz, F. R., and Shulman, G. I. (2012). Diacylglycerol activation of protein kinase Cε and hepatic insulin resistance. Cell. Metabol. 15, 574–584. doi: 10.1016/j.cmet.2012.03.005
Klotz, J., Porter, B. E., Colas, C., Schlessinger, A., and Pajor, A. M. (2016). Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 26, 22. doi: 10.2119/molmed.2016.00077
Knauf, F., Mohebbi, N., Teichert, C., Herold, D., Rogina, B., Helfand, S., et al. (2006). The life-extending gene indy encodes an exchanger for Krebs-cycle intermediates. Biochem. J. 397, 25–29. doi: 10.1042/BJ20060409
Knauf, F., Rogina, B., Jiang, Z., Aronson, P. S., and Helfand, S. L. (2002). Functional characterization and immunolocalization of the transporter encoded by the life-extending gene Indy. Proc. Natl. Acad. Sci. U.S.A. 99, 14315–14319. doi: 10.1073/pnas.222531899
Li, L., Li, H., Garzel, B., Yang, H., Sueyoshi, T., Li, G., et al. (2015). SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 87, 674–682. doi: 10.1124/mol.114.097287
Marden, J., Rogina, B., Montooth, K. L., and Helfand, S. L. (2003). Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc. Natl. Acad. Sci. U.S.A. 100, 3369–3373. doi: 10.1073/pnas.0634985100
Neretti, N., Wang, P.-Y., Brodsky, A. S., Nyguyen, H. H., White, K. P., Rogina, B., et al. (2009). Long-lived Indy induces reduced mitochondrial ROS production and oxidative damage. Proc. Natl. Acad. Sci. U.S.A. 106, 2277–2282. doi: 10.1073/pnas.0812484106
Neuschafer-Rube, F., Lieske, S., Kuna, M., Henkel, J., Perry, R. J., Erion, D. M., et al. (2014). The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes Metab. Res. Rev. 63, 1048–1057. doi: 10.2337/db13-0749
Neuschafer-Rube, F., Schraplau, A., Schewe, B., Lieske, S., Krutzfeldt, J.-M., Ringel, S., et al. (2015). Arylhydrocarbon receptor-dependent mIndy (Slc13a5) induction as possible contributor to benzo[a]pyrene-induced lipid accumulation in hepatocytes. Toxicology 337, 1–9. doi: 10.1016/j.tox.2015.08.007
Pesta, D. H., Perry, R. J., Guebre-Egziabher, F., Zhang, D., Jurczak, M., Fischer-Rosinsky, A., et al. (2015). Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging 7, 1086–1093. doi: 10.18632/aging.100854
Postic, C., and Girard, J. (2008). Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Investig. 118, 829–838. doi: 10.1172/JCI34275
Rogers, R. P., and Rogina, B. (2014). Increased mitochondrial biogenesis preserves intestinal stem cell homeostasis and contributes to longevity in Indy mutant flies. Aging 6, 335–350. doi: 10.186332/aging.100658
Rogers, R. P., and Rogina, B. (2015). The role of INDY in metabolism, health and longevity. Front. Genet. 6:204. doi: 10.3389/fgene.2015.00204
Rogina, B., and Helfand, S. L. (2013). Indy mutations and Drosophila longevity. Front. Genet. 4:47. doi: 10.3389/fgene.2013.00047
Rogina, B., Reenan, R. A., Nielsen, S. P., and Helfand, S. L. (2000). Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 290, 2137–2140. doi: 10.1126/science.290.5499.2137
Schwarz, F., Karadeniz, Z., Fischer-Rosinsky, A., Willmes, D. M., Spranger, J., and Birkenfeld, A. L. (2015). Knockdown of Indy/CeNac2 extends Caenorhbditis elegans life span by inducing AMPK/aak-2. Aging 7, 553–567. doi: 10.18632/aging.100791
Thevenon, J., Milh, M., Feillet, F., St-Onge, J., Duffourd, Y., Jugé, C., et al. (2014). Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 95, 113–120. doi: 10.1016/j.ajhg.2014.06.006
Toivonen, J. M., Walker, G. A., Martinez-Diaz, P., Bjedov, I., Driege, Y., Jacobs, H. T., et al. (2007). No influence of Indy on lifespan in Drosophila after correction for genetic and cytoplasmic background effects. PLoS Genet. 3:e95. doi: 10.1371/journal.pgen.0030095
von Loeffelholz, C., Lieske, S., Neuschäfer-Rube, F., Willmes, D. M., Raschzok, N., Sauer, I. M., et al. (2017). The human longevity gene homolog Indy and Interleukin 6 interact in hepatic lipid metabolism. Hepatology doi: 10.1002/hep.29089 [Epub ahead of print].
Wang, P. Y., Neretti, N., Whitaker, R., Hosier, S., Chang, C., Lu, D., et al. (2009). Long-lived Indy and calorie restriction interact to extend life span. Proc. Natl. Acad. Sci. U.S.A. 106, 9262–9267. doi: 10.1073/pnas.0904115106
Weeke, L. C., Brilstra, E., Braun, K. P., Zonneveld-Huijssoon, E., Salomons, G. S., Koeleman, B. P., et al. (2017). Punctate white matter lesions in full-term infants with neonatal seizures associated with SLC13A5 mutations. Eur. J. Paediatr. Neurol. 2, 396–403. doi: 10.1016/j/ejpn.2016.11.002
Keywords: mIndy, SLC13A5, aging, metabolism, longevity gene, calorie restriction, non-alcoholic fatty liver disease
Citation: Rogina B (2017) INDY—A New Link to Metabolic Regulation in Animals and Humans. Front. Genet. 8:66. doi: 10.3389/fgene.2017.00066
Received: 25 March 2017; Accepted: 09 May 2017;
Published: 24 May 2017.
Edited by:
Elena G. Pasyukova, Institute of Molecular Genetics of Russian Academy of Sciences, RussiaReviewed by:
Svetlana Radyuk, Southern Methodist University, United StatesAdam Salmon, University of Texas Health Science Center at San Antonio, United States
Copyright © 2017 Rogina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Blanka Rogina, cm9naW5hQHVjaGMuZWR1