 Laura M. Senatus
Laura M. Senatus Ann Marie Schmidt
Ann Marie Schmidt- Diabetes Research Program, Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, New York University School of Medicine, New York, NY, United States
The process of advanced glycation leads to the generation and accumulation of an heterogeneous class of molecules called advanced glycation endproducts, or AGEs. AGEs are produced to accelerated degrees in disorders such as diabetes, renal failure, inflammation, neurodegeneration, and in aging. Further, AGEs are present in foods and in tobacco products. Hence, through both endogenous production and exogenous consumption, AGEs perturb vascular homeostasis by a number of means; in the first case, AGEs can cause cross-linking of long-lived molecules in the basement membranes such as collagens, thereby leading to “vascular stiffening” and processes that lead to hyperpermeability and loss of structural integrity. Second, AGEs interaction with their major cell surface signal transduction receptor for AGE or RAGE sets off a cascade of events leading to modulation of gene expression and loss of vascular and tissue homeostasis, processes that contribute to cardiovascular disease. In addition, it has been shown that an enzyme, which plays key roles in the detoxification of pre-AGE species, glyoxalase 1 (GLO1), is reduced in aged and diabetic tissues. In the diabetic kidney devoid of Ager (gene encoding RAGE), higher levels of Glo1 mRNA and GLO1 protein and activity were observed, suggesting that in conditions of high AGE accumulation, natural defenses may be mitigated, at least in part through RAGE. AGEs are a marker of arterial aging and may be detected by both biochemical means, as well as measurement of “skin autofluorescence.” In this review, we will detail the pathobiology of the AGE-RAGE axis and the consequences of its activation in the vasculature and conclude with potential avenues for therapeutic interruption of the AGE-RAGE ligand-RAGE pathways as means to forestall the deleterious consequences of AGE accumulation and signaling via RAGE.
Introduction
Our population is aging and living longer. Although the average life expectancy of the population is increasing in the United States, there remain significant consequences of the aging process. An average 1 death every 40 s is due to cardiovascular disease (Mozaffarian et al., 2016). The prevalence of heart failure is highest among the adult population 65 years or older (Lakatta and Levy, 2003). As one in four, or 25% of individuals, will be over 65 years of age in the United States by the year 2035, these statistics underscore the fact that significant increases in heart failure are to be expected over this time frame (Lakatta and Sollott, 2002). Clinical and experimental evidence suggests that natural aging imbues inherent risk for the development of cardiovascular complications, such as arterial stiffness, atherosclerosis, and hypertension, which eventually may lead to myocardial infarction, stroke, and heart failure (Safar et al., 2003; Izzo, 2004; Sethi et al., 2014). Hence, understanding the underlying mechanisms may aid in the discovery of new therapeutic approaches to deter pathological vascular aging.
Changes in the components of large arteries due to advancing age have been described in humans and animals (Spinetti et al., 2004; Pepe and Lakatta, 2005). Age-associated blood vessel remodeling includes such features as dilation of the lumina, intimal and medial thickening, changes in the extracellular matrix (ECM), and augmented stiffness (Gaballa et al., 1998). In addition to these structural changes, other mechanisms contribute to the overall consequences of aging to the arterial wall, including such phenomena as inflammation, endothelial dysfunction, and oxidative stress (Xu et al., 2003). Fibroblasts and smooth muscle cells (SMC) contribute to aging in the vasculature, in part by increasing ECM; macrophages contribute by increasing inflammatory factors that have a wide range of possible consequences, such as vascular hyperpermeability and an increase in the procoagulant state (Sprague and Khalil, 2009; Strait and Lakatta, 2012). These pathobiological events adversely affect the vessel wall and all of its components (Najjar et al., 2005; Greenwald, 2007), potentially contributing to arterial aging.
It has been shown that the aged human arterial wall exhibits a more proinflammatory signature, with increased expression and activity of matrix metalloproteinases (MMPs) and chemokines (Wang et al., 2007). Atop these considerations is the effect of co-morbid conditions in aging, which may augment production of inflammatory mediators and exacerbate the impact of arterial aging, examples of which include diabetes mellitus (types 1 or 2 or the rarer forms of diabetes); chronic renal disease; and chronic immune/inflammatory disorders.
AGEs: Production and Functions in Arterial Aging
Advanced glycation endproducts (AGEs) are a diverse group of macromolecules and at least 20 different specific AGEs have been described to date. Among the major groups of AGEs are carboxymethyl lysine (CML), carboxyethyl lysine (CEL), pentosidine, glucosepane, methylglyoxal lysine dimer, glyoxal lysine dimer, and glycolic acid lysine amide (Henning and Glomb, 2016). AGEs form throughout life via the process of non-enzymatic glycation of proteins and lipids, and this process is accelerated during hyperglycemia, oxidative stress, aging, advanced renal disease, and inflammation (Daffu et al., 2013; Singh et al., 2014; Baig et al., 2017). Humans and animals are also exposed to exogenous sources of AGEs ingested through food-derived AGEs and tobacco products (Luevano-Contreras and Chapman-Novakofski, 2010; Uribarri et al., 2015). It has been shown that restriction in dietary AGE intake may increase the lifespan in animals (Cai et al., 2002; Luevano-Contreras and Chapman-Novakofski, 2010).
AGEs accumulate in aging tissues and on vulnerable plasma proteins. Higher levels of circulating AGEs have been linked to chronic diseases in aging subjects (Semba et al., 2015). The accumulation of AGEs is increased and accelerated in hypertensive subjects (McNulty et al., 2007) and is also associated with diabetes (Soulis et al., 1997; Yan et al., 2003). In fact, aged subjects, even though healthy, may have higher AGE accumulation compared to younger subjects with diabetes and its complications, thus underscoring that AGE production and accumulation accompanies the normal aging process (Hadi and Suwaidi, 2007). Therefore, multiple factors such as the rate of accumulation of AGE ligand, the absolute concentration of the ligand, and individual susceptibility to AGE formation may be important in determining an individual's AGE burden.
AGEs modify collagen and elastin in the vascular wall (Meerwaldt et al., 2004); because of reduced turnover of such proteins, they become more susceptible to glycation during the aging process (Schleicher et al., 1997; Sell and Monnier, 2012). Elevated levels of plasma CML-AGEs are associated with diastolic dysfunction in aging (Campbell et al., 2012). In experiments in diabetic rats, higher AGE crosslinking of collagen was associated with increased stiffness of the aorta (Reddy, 2004). This may change the beneficial functions of several important molecules of the ECM, which can mediate vascular dysfunction. Numerous studies have confirmed the correlation between AGE accumulation and increased artery stiffness (Goldin et al., 2006; Campbell et al., 2011). Arterial stiffness is associated with greater risk for aging-associated cardio- and cerebrovascular diseases and mortality (Laurent et al., 2001; Mattace-Raso et al., 2006; Kaess et al., 2012). AGE accumulation causes upregulation of inflammation and destruction of collagen and elastin, along with other proteins of the ECM (Sims et al., 1996; Greenwald, 2007; Peppa and Raptis, 2008; Baulmann et al., 2010).
Measuring AGEs
AGEs can affect virtually every tissue in the body, either through mediation of cellular damage via protein cross-linking and/or through their binding to cell surface receptors. Since various diseases have been linked to AGEs, it is plausible that AGEs can be utilized as biomarkers, such as for predilection to disease, state of disease activity, and/or response to therapeutic interventions.
Measurment of skin autofluorescence (SAF) estimates the skin tissue AGE content and may predict cardiovascular complications, at least in certain subjects (Lutgers et al., 2009; Noordzij et al., 2012; Tanaka et al., 2012). Some AGEs are fluorescent and can be non-invasively measured in skin by autofluorescence, as a representative marker of the total AGE modifications of other long-lived tissues in association with vascular disease. In a recent study, SAF was measured using an autofluorescent spectrometer followed by measurement of endothelial function and arterial stiffness. The results indicated that SAF was associated with increased arterial stiffness in the older individuals and that arterial function was blunted by the advancing age of the subject (Sturmer et al., 2015). It is important to note, however, that there are limitations to the use of SAF; first, it only measures fluorescence and not all AGEs are fluorescent; second, skin fluorophores exist that are not related to AGEs, and therefore, such measurement is not reflective of the AGE pool; third, in subjects with darker skin pigmentation, SAF measurements have been found to be less reliable, thereby possibly reducing the applicability of this technique across diverse groups of subjects; and fourth, certain skin creams, such as agents used to “tan” or “brown” the skin may cause direct interference with SAF measurements. Hence, although the technique does not require biopsies or invasive approaches, there are notable limitations that must be considered in its use (Da Moura Semedo et al., 2017).
Traditionally, the precise detection of AGE measurement includes HPLC (High-Performance liquid chromatography) (Ashraf et al., 2015) with fluorescence-based detection. Using HPLC methods, high levels of serum AGE, such as CML-AGE and pentosidine were shown to increase with age and in patients with diabetes (Aso et al., 2000). The use of ELISA (Enzyme-linked immunosorbent assay), an immunological technique for the determination of AGEs, proved to be an alternative for detection of AGEs in samples such as serum and plasma (Munch et al., 1997; Takeuchi et al., 1999). Tissue AGE concentrations using immunohistochemical methods can also be measured using antibodies to detect CML-AGE (Soulis et al., 1997). Using these methods, AGE have been identified in arterial disease and have been localized to early atherosclerotic plaques and cellular constituents, including macrophages and SMCs (Stitt et al., 1997). LC-MS (liquid chromatography- mass spectroscopy) is another technique for the detection of AGEs and the early glycation products. LC-MS allows for AGEs such as pyrraline to be detected in human skin and plasma in very low concentrations (picomolar range) (Pitt, 2009). The possibility of using AGE measurements to gauge the state of AGE-related disease activity and the effectiveness of therapeutic intervention underscores the importance of using reliable methods for the detection of AGEs.
AGEs and their Pathobiological Actions: Interactions with Cellular Receptor RAGE
RAGE (Receptor for Advanced glycation end products) was identified in 1992 from bovine lung as a protein that bound AGEs in a dose-dependent manner (Wautier et al., 1994). RAGE has many ligands that increase in aging, even beyond AGEs, and it is a cell surface macromolecule. RAGE contains extracellular domains composed of one V (variable)-type domain and two C (constant)-type immunoglobulin—like domains (C1 And C2); these are followed by a single transmembrane spanning helix (Koch et al., 2010), and the cytoplasmic domain, which is essential for signal transduction (Xue et al., 2014). RAGE binds a diverse group of ligands, including AGEs, at least certain members of the S100/calgranulins, high mobility group Box-1 (HMGB1), Mac-1, and amyloid- β peptide, particularly its oligomeric forms (Herold et al., 2007).
Mechanisms by which AGEs could alter the vasculature and increase arterial stiffness include generation of inflammation (Chavakis et al., 2004) and oxidative stress (Tan et al., 2007). Further, AGEs binding to endothelial cell surface RAGE can lead to stimulation of NADPH oxidase, thereby increasing the production of reactive oxygen species (ROS) (Wautier et al., 2001). Additional mechanisms such as mitochondrial stress may further increase the production of ROS (Rubattu et al., 2013; Li et al., 2014; Montezano and Touyz, 2014). Previous studies proposed that one of the mechanisms by which AGE/RAGE contributes to endothelial dysfunction is through regulation of the production and expression of tumor necrosis factor (TNF)-α. The transcription factor nuclear factor-κB (NF-κB), triggered by inflammation and by ROS, plays a key role in cytokine and inflammatory mediator expression, thereby exacerbating microvasculopathy and mediating pathological changes in gene expression, at least in part through RAGE ligand-RAGE interactions and activation of cellular signal transduction (Gao et al., 2008; Kay et al., 2016).
Evidence of RAGE-mediated perturbation in vivo has also been demonstrated. Diabetic apolipoprotein E (ApoE) deficient mice that are also devoid of Ager (gene encoding RAGE) display reduced atherosclerosis and lower expression of vascular cell adhesion molecule (VCAM)-1 and tissue factor (Kislinger et al., 2001). AGEs also induce vascular endothelial growth factor (VEGF) expression in microvascular endothelial cells (Yamagishi et al., 1997), which may have implications for the diabetic retina, as an example. In addition to the chronic conditions of AGE formation discussed above, such as aging, diabetes, and chronic inflammatory conditions, research has illustrated that AGEs may form rapidly in settings of acute stress as well. For example, endothelial cells subjected to in vitro-applied hypoxia release AGEs within minutes of exposure to reduced levels of oxygen (Chang et al., 2008). These considerations indicate that it was important to identify means to block AGE-RAGE interactions in the vasculature.
In animal studies, treatment with soluble RAGE (sRAGE), the soluble extracellular domains of RAGE, which sequester AGEs and RAGE ligands, thereby blocking their interaction with RAGE demonstrated significant reductions in atherosclerotic lesion area (Park et al., 1998), in a manner independent of changes in lipid or glucose levels. In other studies, sRAGE treatment in rodents significantly mitigated diabetic vascular hyperpermeability (Schmidt et al., 1999).
Hallam and colleagues demonstrated that aged 24 month-old Fischer 344 rats displayed higher vascular RAGE expression in the aorta, and higher expression of the polyol pathway enzyme, aldose reductase (AR), which stimulates metabolic pathways that increase AGE formation. Aging-related vascular dysfunction was evident in these rats on account of impaired endothelial relaxation in response to acetylcholine (Hallam et al., 2010). Treatment of aged Fischer 344 rats with sRAGE improved endothelial dependent relaxation in response to acetylcholine (Hallam et al., 2010). Taken together, these studies illustrate that increased AGE burden and RAGE expression mediate vascular dysfunction and that these perturbations may be suppressed by administration of antagonists of ligand-RAGE interactions in vivo.
Glyoxalase 1 (GLO1) and Amplification of AGE Accumulation
In addition to increased direct mediators of damage in aging, defense mechanisms may also be attenuated in aging. (GLO1) contributes to the regulation of the levels of the pre-AGE methylglyoxal (MG) and MG-derived AGEs (Giacco et al., 2014). MG, formed mainly inside cells, is a potent glycating agent (Rabbani and Thornalley, 2014).
Published work has suggested a link between RAGE and Glo1. Exposure of cultured endothelial cells to high glucose increases expression of RAGE and various RAGE ligands, such as S100B, AGEs, and HMGB1; this was prevented by overexpression of Glo1 (Brouwers et al., 2011, 2014). Reiniger and colleagues showed that renal accumulation of AGEs promotes kidney dysfunction and that when Ager is deleted in OVE26 diabetic mice, reduced pathological, and functional derangements in the kidney ensued, in parallel with reduced MG levels and higher levels of GLO1 in the kidney (Reiniger et al., 2010). These authors showed that in Ager null diabetic OVE26 kidney, levels of MG were lower than those of wild-type diabetic OVE26 controls, despite equal levels of high glucose. Reiniger and colleagues traced the mechanism to RAGE-dependent downregulation of Glo1 mRNA and activity in diabetes (Reiniger et al., 2010). Thus, RAGE activation may perpetuate AGE accumulation and deletion of Ager may exert its protection, at least in part by downregulation of Glo1.
It is possible that increasing GLO1 expression and/or activity may slow down age-related damage, as acceleration in glycation in aged rats was attenuated by transgenic (Tg) expression of Glo1 in these animals (Jo-Watanabe et al., 2014). Interestingly, exercise training in aged rats resulted in activation of GLO1, with consequent reduction in the formation of MG and CML, along with lower RAGE expression in the aorta (Gu et al., 2014). Overall, agents that augment GLO1 to block formation of AGEs may serve as therapeutic strategies for averting complications in vascular disorders in which AGEs accumulate.
RAGE/DIAPH1 Signal Transduction Axis: Link to Vascular Dysfunction
RAGE requires its cytoplasmic domain for signal transduction. Hudson and colleagues demonstrated the interaction of the cytoplasmic domain of RAGE tail with mammalian diaphanous 1 or DIAPH1 (Hudson et al., 2008). The cytoplasmic domain or tail of RAGE (ctRAGE) binds specifically to the formin homology 1 (FH1) domain of DIAPH1 (Hudson et al., 2008). Formins are actin-binding molecules that contribute to Rho GTPase down-stream signals (Hudson et al., 2008) in cells such as vascular cells, monocytes/macrophages, and transformed cells. DIAPH1 has also been shown to be an effector of serum response factors (SRFs), which are linked to gene regulation mechanisms, and cellular signaling mechanisms such as AKT and GSK-3beta (Toure et al., 2012).
In SMCs, DIAPH1 was required for RAGE ligand (S100B)-induced c-Src translocation to the plasma membrane, RAC1 activation, generation of ROS and cellular migration. RAC1 modulates the actin cytoskeleton, the arrangement of which governs cell motility and regulates signal transduction pathways (Toure et al., 2012). To verify the RAGE-DIAPH1 interaction, Shekhtman and colleagues used NMR spectroscopy to identify the four key amino acids in the RAGE cytoplasmic domain (Q3, R4, R5, and Q6 corresponding to Q364, R365, R366, and Q367 of the full-length RAGE) that are essential for the interaction of ctRAGE with the FH1 domain of DIAPH1. When R5/Q6 were mutated to alanine residues and expressed in murine SMCs, AKT signaling and cellular migration and proliferation in response to RAGE ligand S100B, but not to non-RAGE ligands, were significantly reduced (Rai et al., 2012).
A role for DIAPH1 in RAGE signaling in macrophages has also been demonstrated. In macrophages devoid of Diaph1, hypoxia-mediated upregulation of the transcription factor Egr1, which upregulates inflammatory and prothrombotic mediators, was prevented (Xu et al., 2010). To test these points in vivo and the role of DIAPH1 in mediating the effects of RAGE ligands, studies are underway in animals of diabetes, aging, and vascular perturbation to probe the potential impact of DIAPH1 in vascular dysfunction.
Taken together, extensive evidence is building to implicate AGEs and RAGE in the pathogenesis of vascular perturbation, which stimulate processes that lead to the development of arterial stiffness, an established harbinger of cardiovascular disease and aging. AGEs, via RAGE stimulate endothelial cells to generate ROS and to activate cellular signaling pathways, at least in part through the cytoplasmic domain of RAGE binding to DIAPH1; processes which lead to activation of seminal transcription factors such as NF-κB (Figure 1). In addition to AGE-RAGE activation of endothelial cells and mediation of endothelial dysfunction, AGEs, via RAGE, may also stimulate macrophages and other immune cells, to induce migration and recruitment of inflammatory cells into AGE-laden foci in the tissues. Further, research has shown that a natural anti-AGE mechanism, the enzyme GLO1, which detoxifies pre-AGE species, is downregulated by the actions of RAGE, such as in the diabetic kidney. Hence, AGE-RAGE activation stimulates a feed forward loop, in which AGE-RAGE interaction causes vascular perturbation and, in parallel, a mechanism to perpetuate AGE production and accumulation.
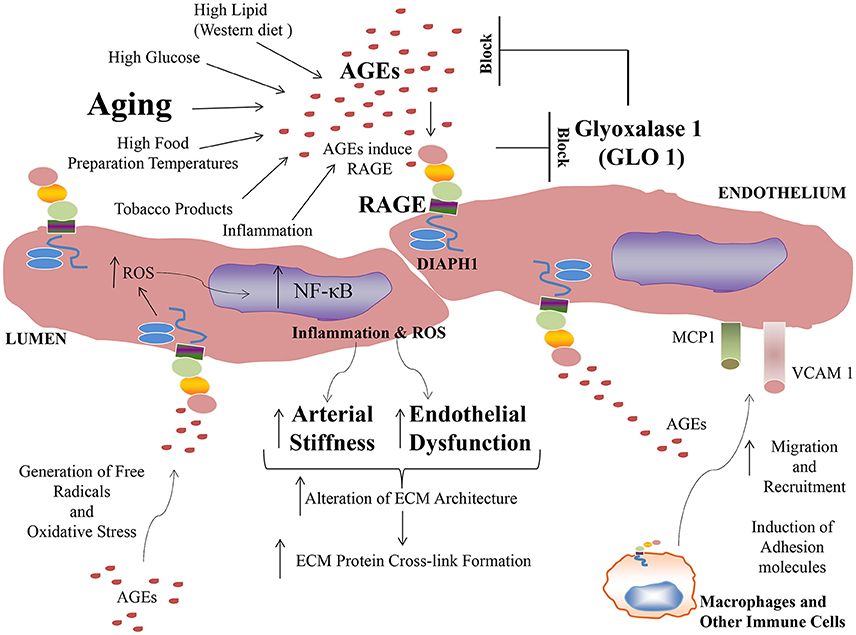
Figure 1. Formation of AGEs and mechanisms of their accumulation and pathobiological actions via the signal transduction receptor RAGE.
In the section to follow, we consider therapeutic opportunities in halting the detrimental actions of this AGE-RAGE pathway.
Therapeutic Strategies: Targeting AGE and RAGE
Propelled by the epidemiological and experimental evidence linking AGE and RAGE to the pathogenesis of arterial stiffness and vascular perturbation, AGEs and RAGE have been identified as targets for therapeutic intervention in these settings. In the sections to follow, we detail examples of some of the strategies to block AGEs and RAGEs for their possible benefits in cardiovascular diseases (Figure 2).
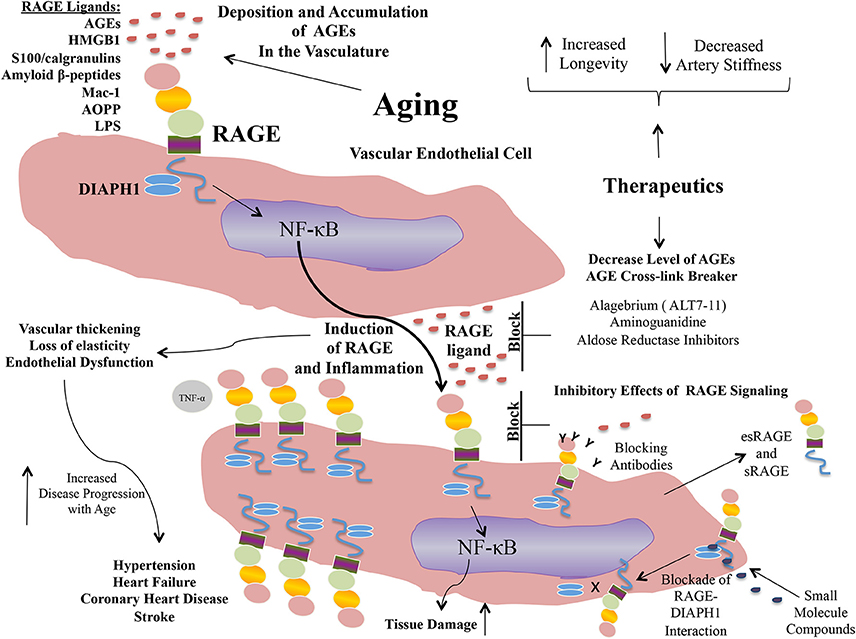
Figure 2. Targeting AGEs and RAGE: examples of putative therapeutic strategies.
Anti-AGE Strategies
Alagebrium, or ALT7-11, is an AGE cross-link breaker. In animals and humans, this agent improved ventricular function and arterial compliance (Kass et al., 2001; Vaitkevicius et al., 2001); reduced expression of RAGE and collagen accumulation in vascular tissues; and in patients with systolic hypertension, it improved endothelial function (Zieman et al., 2007). Although Alagebrium is no longer available, its use served as an important test of the AGE hypothesis in vascular stiffness and function.
Other anti-AGE strategies, such as aminoguanidine, which blocks AGE production, also had beneficial effects in increasing vascular elasticity and in augmenting left ventricular arterial coupling, as well as decreasing vascular permeability in diabetic rats (Wu et al., 2008). Atherosclerosis was attenuated in streptozotocin-induced diabetic Apoe-deficient mice treated with aminoguanidine (Forbes et al., 2004). Aminoguanidine also reduced AGE accumulation from food sources (He et al., 1999). Finally, other strategies to reduce AGEs are being investigated, such as aldose reductase inhibitors (ARI). ARI have been shown to suppress AGE accumulation in the atherosclerotic plaques and, in parallel, to reduce atherosclerotic plaque lesions (Vikramadithyan et al., 2005).
In summary, it is noteworthy that despite testing of multiple classes of anti-AGE agents, none have obtained, at least to date, approval for anti-AGE indications. Although there are many possible reasons for this, we propose that one reason is that solely targeting AGEs fails to capture the pathobiological effects of distinct RAGE ligands. Therefore, it is not surprising that attempts are underway to directly target RAGE as a therapeutic strategy.
Anti-RAGE Strategies
Approaches to limit RAGE ligand AGEs have been accompanied by efforts to block RAGE itself and these have been tested in vitro and in vivo; in addition, human clinical trial testing is also underway. In vitro, pre-treatment of AGE-stimulated endothelial cells with anti-RAGE antibodies or anti-oxidants blocked cellular perturbation (Yan et al., 1994). Another RAGE blocking agent currently in Phase III clinical trials in Alzheimer's disease is the small molecule Azeliragon, which inhibits the receptor for advanced glycation endproducts through its first extracellular V-type domain, which prevents RAGE ligands from interacting with RAGE. It is orally bioavailable (Sabbagh et al., 2011).
The AGER gene may be alternatively spliced to result in the generation of several RAGE isoforms (Yonekura et al., 2003; Kalea et al., 2009). The C- terminally truncated RAGE (known as endogenous secretory RAGE (esRAGE) or RAGEv1, does not contain the transmembrane domain, and is secreted. Other forms of soluble RAGE also exist, as the cell surface RAGE can be proteolytically cleaved by MMPs or ADAM10, thereby resulting in the release of soluble RAGE (sRAGE) (Hanford et al., 2004).
In mouse models, sRAGE treatment suppressed acceleration and blocked the progression of established atherosclerosis in diabetic Apoe null mice (Park et al., 1998; Bucciarelli et al., 2002). Various studies in human subjects have sought to determine whether the plasma sRAGE or esRAGE level is associated with cardiometabolic diseases (Choi et al., 2009). Generally, plasma sRAGE/esRAGE levels are lower in subjects with these disorders vs. healthy controls. Thus, recombinant sRAGE might be of benefit in arterial aging and metabolic diseases.
Beyond targeting RAGE and the extracellular domains, distinct therapeutic opportunities have arisen regarding RAGE signaling via blockade of RAGE-DIAPH1 interaction. Manigrasso and colleagues developed a high throughput RAGE tail-DIAPH1 binding assay and screened a library of >58,000 small molecule compounds to identify molecules that blocked this interaction. A series of 13 compounds was identified that exhibited high affinity binding to ctRAGE domain. In vitro and in vivo studies illustrated that these compounds displayed inhibitory effects on RAGE signal transduction in SMCs in vitro, and in vivo, on RAGE ligand-stimulated inflammatory gene expression in liver and kidney tissue (Manigrasso et al., 2016). Therefore, the discovery that the cytoplasmic domain of RAGE bound DIAPH1 may aid in the identification of a distinct class of RAGE signaling intracellular antagonists.
Conclusions and Perspectives
Evidence is accruing that exposure to AGEs contributes to detrimental aging-related outcomes and to reduced health and life span. In vitro and in vivo animal model studies have shown that AGEs affect and disrupt cellular and tissue homeostasis. AGEs can cause alteration of ECM architecture, thereby affecting cellular permeability and signaling; mediate ECM and circulating protein cross-linking; and they can activate cellular signaling and modulate transcription factor activities and subsequent gene expression via receptors such as RAGE. AGE accumulation may result in the increased expression of RAGE in a ligand-enriched environment and exacerbate proinflammatory mechanisms, thereby accelerating aging-associated arterial diseases.
RAGE is expressed on a number of important cell types implicated in arterial aging and vascular pathology. Once AGEs are formed, albeit by diverse intrinsic and environmentally-triggered mechanisms, their interaction with RAGE on endothelial cells, SMCs, and immune cells such as macrophages, results in upregulation of inflammatory and oxidative stress-provoking factors, thereby providing a mechanism to link AGE-RAGE to arterial aging and its consequences, such as stroke, hypertension, atherosclerosis, myocardial infarction, and heart failure. Of note, as hyperglycemia accelerates AGE formation, these AGE-RAGE processes are amplified in diabetes. Epidemiological studies assuredly support the exacerbation of cardiovascular disease in subjects with diabetes.
Certainly, more research is required to understand the entire scope of RAGE signaling and the extent to which blocking AGEs/RAGE/DIAPH1 interaction may intercept the full pathobiology of RAGE activation. Key remaining questions include whether interventions to reduce AGEs and/or to block RAGE extracellular and/or intracellular domains, might provide the greatest protection in attenuation of arterial aging and vascular dysfunction (See Table 1 for a summary of some of the key anti-AGE and anti-RAGE therapeutic approaches). Clinical studies to address these concepts are required to optimize strategies to protect the vasculature from the adverse effects of AGEing.
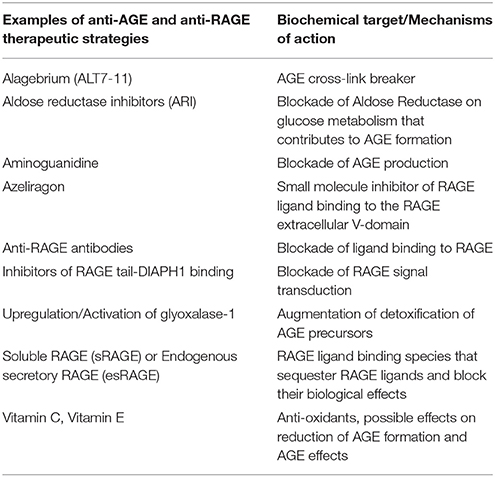
Table 1. Key anti-AGE and anti-RAGE therapeutic approaches.
Materials and Methods
Search Strategies
Arterial Aging:
11,140 refs
https://www.ncbi.nlm.nih.gov/pubmed/?term=arterial+aging
Arterial aging and glycation:
116 refs:
https://www.ncbi.nlm.nih.gov/pubmed/?term=arterial+aging+and+glycation
Arterial aging and advanced glycation end product:
95 refs:
https://www.ncbi.nlm.nih.gov/pubmed/?term=arterial+aging+and+advanced+glycation+end+product
Arterial aging and receptor for advanced glycation end products
22 refs:
Author Contributions
LS and AS: Wrote and edited the manuscript.
Funding
This work was funded by grants from the U.S. Public Health Service (Grant no. HL60901).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AGEs, advanced glycation endproducts; ALT7-11, Alagebrium; AR, aldose reductase; ARI, aldose reductase inhibitors; CML, carboxymethyl lysine; CEL, carboxyethyl lysine; ctRAGE, cytoplasmic domain of RAGE; DIAPH1, diaphanous-1; ELISA, enzyme-linked immunosorbent assay; esRAGE, endogenous secretory RAGE; ECM, extracellular matrix; FH1, formin homology 1; Glo1, Glyoxalase 1; HMGB1, high mobility group Box-1; HPLC, high performance liquid chromatography; LC-MS, liquid chromatography tandem mass spectrometry; MG, methylglyoxal; ROS, reactive oxygen species; VCAM-1, vascular cell adhesion molecule−1; VEGF, vascular endothelial growth factor; RAGE, receptor for advanced glycation end products; SAF, Skin autofluorescence; SMC, smooth muscle cells; sRAGE, soluble RAGE; SRFs, serum response factors; Tg, transgenic.
References
Ashraf, J. M., Ahmad, S., Choi, I., Ahmad, N., Farhan, M., Tatyana, G., et al. (2015). Recent advances in detection of AGEs: immunochemical, bioanalytical and biochemical approaches. IUBMB Life 67, 897–913. doi: 10.1002/iub.1450
Aso, Y., Inukai, T., Tayama, K., and Takemura, Y. (2000). Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol. 37, 87–92. doi: 10.1007/s005920070025
Baig, M. H., Jan, A. T., Rabbani, G., Ahmad, K., Ashraf, J. M., Kim, T., et al. (2017). Methylglyoxal and advanced glycation end products: insight of the regulatory machinery affecting the myogenic program and of its modulation by natural compounds. Sci. Rep. 7:5916. doi: 10.1038/s41598-017-06067-5
Baulmann, J., Nurnberger, J., Slany, J., Schmieder, R., Schmidt-Trucksass, A., Baumgart, D., et al. (2010). [Arterial stiffness and pulse wave analysis]. Dtsch. Med. Wochenschr. 135(Suppl. 1), S4–S14. doi: 10.1055/s-0030-1249183
Brouwers, O., Niessen, P. M., Ferreira, I., Miyata, T., Scheffer, P. G., Teerlink, T., et al. (2011). Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 286, 1374–1380. doi: 10.1074/jbc.M110.144097
Brouwers, O., Niessen, P. M., Miyata, T., Ostergaard, J. A., Flyvbjerg, A., Peutz-Kootstra, C. J., et al. (2014). Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 57, 224–235. doi: 10.1007/s00125-013-3088-5
Bucciarelli, L. G., Wendt, T., Qu, W., Lu, Y., Lalla, E., Rong, L. L., et al. (2002). RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 106, 2827–2835. doi: 10.1161/01.CIR.0000039325.03698.36
Cai, W., Gao, Q.-D., Zhu, L., Peppa, M., He, C., and Vlassara, H. (2002). Oxidative stress-inducing carbonyl compounds from common foods: novel mediators of cellular dysfunction. Mol. Med. 8, 337–346.
Campbell, D. J., Somaratne, J. B., Jenkins, A. J., Prior, D. L., Yii, M., Kenny, J. F., et al. (2011). Impact of type 2 diabetes and the metabolic syndrome on myocardial structure and microvasculature of men with coronary artery disease. Cardiovasc. Diabetol. 10, 80–80. doi: 10.1186/1475-2840-10-80
Campbell, D. J., Somaratne, J. B., Jenkins, A. J., Prior, D. L., Yii, M., Kenny, J. F., et al. (2012). Diastolic dysfunction of aging is independent of myocardial structure but associated with plasma advanced glycation end-product levels. PLoS ONE 7:e49813. doi: 10.1371/journal.pone.0049813
Chang, J. S., Wendt, T., Qu, W., Kong, L., Zou, Y. S., Schmidt, A. M., et al. (2008). Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ. Res. 102, 905–913. doi: 10.1161/CIRCRESAHA.107.165308
Chavakis, T., Bierhaus, A., and Nawroth, P. P. (2004). RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microb. Infect. 6, 1219–1225. doi: 10.1016/j.micinf.2004.08.004
Choi, K. M., Yoo, H. J., Kim, H. Y., Lee, K. W., Seo, J. A., Kim, S. G., et al. (2009). Association between endogenous secretory RAGE, inflammatory markers and arterial stiffness. Int. J. Cardiol. 132, 96–101. doi: 10.1016/j.ijcard.2007.10.047
Daffu, G., del Pozo, C. H., O'Shea, K. M., Ananthakrishnan, R., Ramasamy, R., and Schmidt, A. M. (2013). Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int. J. Mol. Sci. 14, 19891–19910. doi: 10.3390/ijms141019891
Da Moura Semedo, C., Webb, M., Waller, H., Khunti, K., and Davies, M. (2017). Skin autofluorescence, a non-invasive marker of advanced glycation end products: clinical relevance and limitations. Postgrad. Med. J. 93, 289–294. doi: 10.1136/postgradmedj-2016-134579
Forbes, J. M., Yee, L. T., Thallas, V., Lassila, M., Candido, R., Jandeleit-Dahm, K. A., et al. (2004). Advanced glycation end product interventions reduce diabetes-accelerated atherosclerosis. Diabetes 53, 1813–1823. doi: 10.2337/diabetes.53.7.1813
Gaballa, M. A., Jacob, C. T., Raya, T. E., Liu, J., Simon, B., and Goldman, S. (1998). Large artery remodeling during aging: biaxial passive and active stiffness. Hypertension 32, 437–443. doi: 10.1161/01.HYP.32.3.437
Gao, X., Zhang, H., Schmidt, A. M., and Zhang, C. (2008). AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 295, H491–H498. doi: 10.1152/ajpheart.00464.2008
Giacco, F., Du, X., D'Agati, V. D., Milne, R., Sui, G., Geoffrion, M., et al. (2014). Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 63, 291–299. doi: 10.2337/db13-0316
Goldin, A., Beckman, J. A., Schmidt, A. M., and Creager, M. A. (2006). Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114, 597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
Greenwald, S. E. (2007). Ageing of the conduit arteries. J. Pathol. 211, 157–172. doi: 10.1002/path.2101
Gu, Q., Wang, B., Zhang, X. F., Ma, Y. P., Liu, J. D., and Wang, X. Z. (2014). Contribution of receptor for advanced glycation end products to vasculature-protecting effects of exercise training in aged rats. Eur. J. Pharmacol. 741, 186–194. doi: 10.1016/j.ejphar.2014.08.017
Hadi, H. A. R., and Suwaidi, J. A. (2007). Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 3, 853–876.
Hallam, K. M., Li, Q., Ananthakrishnan, R., Kalea, A., Zou, Y. S., Vedantham, S., et al. (2010). Aldose reductase and AGE–RAGE pathways: central roles in the pathogenesis of vascular dysfunction in aging rats. Aging Cell 9, 776–784. doi: 10.1111/j.1474-9726.2010.00606.x
Hanford, L. E., Enghild, J. J., Valnickova, Z., Petersen, S. V., Schaefer, L. M., Schaefer, T. M., et al. (2004). Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J. Biol. Chem. 279, 50019–50024. doi: 10.1074/jbc.M409782200
He, C., Sabol, J., Mitsuhashi, T., and Vlassara, H. (1999). Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 48, 1308–1315. doi: 10.2337/diabetes.48.6.1308
Henning, C., and Glomb, M. A. (2016). Pathways of the Maillard reaction under physiological conditions. Glycoconj. J. 33, 499–512. doi: 10.1007/s10719-016-9694-y
Herold, K., Moser, B., Chen, Y., Zeng, S., Yan, S. F., Ramasamy, R., et al. (2007). Receptor for advanced glycation end products (RAGE) in a dash to the rescue: inflammatory signals gone awry in the primal response to stress. J. Leukoc. Biol. 82, 204–212. doi: 10.1189/jlb.1206751
Hudson, B. I., Kalea, A. Z., Del Mar Arriero, M., Harja, E., Boulanger, E., D'Agati, V., et al. (2008). Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 283, 34457–34468. doi: 10.1074/jbc.M801465200
Izzo, J. L. Jr. (2004). Arterial stiffness and the systolic hypertension syndrome. Curr. Opin. Cardiol. 19, 341–352. doi: 10.1097/01.hco.0000126581.89648.10
Jo-Watanabe, A., Ohse, T., Nishimatsu, H., Takahashi, M., Ikeda, Y., Wada, T., et al. (2014). Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell 13, 519–528. doi: 10.1111/acel.12204
Kaess, B. M., Rong, J., Larson, M. G., Hamburg, N. M., Vita, J. A., Levy, D., et al. (2012). Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 308, 875–881. doi: 10.1001/2012.jama.10503
Kalea, A. Z., Reiniger, N., Yang, H., Arriero, M., Schmidt, A. M., and Hudson, B. I. (2009). Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 23, 1766–1774. doi: 10.1096/fj.08-117739
Kass, D. A., Shapiro, E. P., Kawaguchi, M., Capriotti, A. R., Scuteri, A., deGroof, R. C., et al. (2001). Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation 104, 1464–1470. doi: 10.1161/hc3801.097806
Kay, A. M., Simpson, C. L., and Stewart, J. A. (2016). The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J. Diabetes Res. 2016:6809703. doi: 10.1155/2016/6809703
Kislinger, T., Tanji, N., Wendt, T., Qu, W., Lu, Y., Ferran, L. J. Jr., et al. (2001). Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 21, 905–910. doi: 10.1161/01.ATV.21.6.905
Koch, M., Chitayat, S., Dattilo, B. M., Schiefner, A., Diez, J., Chazin, W. J., et al. (2010). Structural basis for ligand recognition and activation of RAGE. Structure 18, 1342–1352. doi: 10.1016/j.str.2010.05.017
Lakatta, E. G., and Levy, D. (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation 107, 139–146. doi: 10.1161/01.CIR.0000048892.83521.58
Lakatta, E. G., and Sollott, S. J. (2002). Perspectives on mammalian cardiovascular aging: humans to molecules. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 132, 699–721. doi: 10.1016/S1095-6433(02)00124-1
Laurent, S., Boutouyrie, P., Asmar, R., Gautier, I., Laloux, B., Guize, L., et al. (2001). Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37, 1236–1241. doi: 10.1161/01.HYP.37.5.1236
Li, H., Horke, S., and Förstermann, U. (2014). Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 237, 208–219. doi: 10.1016/j.atherosclerosis.2014.09.001
Luevano-Contreras, C., and Chapman-Novakofski, K. (2010). Dietary advanced glycation end products and aging. Nutrients 2, 1247–1265. doi: 10.3390/nu2121247
Lutgers, H. L., Gerrits, E. G., Graaff, R., Links, T. P., Sluiter, W. J., Gans, R. O., et al. (2009). Skin autofluorescence provides additional information to the UK Prospective Diabetes Study (UKPDS) risk score for the estimation of cardiovascular prognosis in type 2 diabetes mellitus. Diabetologia 52, 789–797. doi: 10.1007/s00125-009-1308-9
Manigrasso, M. B., Pan, J., Rai, V., Zhang, J., Reverdatto, S., Quadri, N., et al. (2016). Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci. Rep. 6:22450. doi: 10.1038/srep22450
Mattace-Raso, F. U., van der Cammen, T. J., Hofman, A., van Popele, N. M., Bos, M. L., Schalekamp, M. A., et al. (2006). Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation 113, 657–663. doi: 10.1161/CIRCULATIONAHA.105.555235
McNulty, M., Mahmud, A., and Feely, J. (2007). Advanced glycation end-products and arterial stiffness in hypertension. Am. J. Hypertens. 20, 242–247. doi: 10.1016/j.amjhyper.2006.08.009
Meerwaldt, R., Graaff, R., Oomen, P. H. N., Links, T. P., Jager, J. J., Alderson, N. L., et al. (2004). Simple non-invasive assessment of advanced glycation endproduct accumulation. Diabetologia 47, 1324–1330. doi: 10.1007/s00125-004-1451-2
Montezano, A. C., and Touyz, R. M. (2014). Reactive oxygen species, vascular noxs, and hypertension: focus on translational and clinical research. Antioxid. Redox Signal. 20, 164–182. doi: 10.1089/ars.2013.5302
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2016). Executive summary: heart disease and stroke statistics−2016 update: a report from the american heart association. Circulation 133, 447–454. doi: 10.1161/CIR.0000000000000366
Münch, G., Keis, R., Wessels, A., Riederer, P., Bahner, U., Heidland, A., et al. (1997). Determination of advanced glycation end products in serum by fluorescence spectroscopy and competitive ELISA. Eur. J. Clin. Chem. Clin. Biochem. 35, 669–677. doi: 10.1515/cclm.1997.35.9.669
Najjar, S. S., Scuteri, A., and Lakatta, E. G. (2005). Arterial aging: is it an immutable cardiovascular risk factor? Hypertension 46, 454–462. doi: 10.1161/01.HYP.0000177474.06749.98
Noordzij, M. J., Lefrandt, J. D., Loeffen, E. A., Saleem, B. R., Meerwaldt, R., Lutgers, H. L., et al. (2012). Skin autofluorescence is increased in patients with carotid artery stenosis and peripheral artery disease. Int. J. Cardiovasc. Imaging 28, 431–438. doi: 10.1007/s10554-011-9805-6
Park, L., Raman, K. G., Lee, K. J., Lu, Y., Ferran, L. J. Jr., Chow, W. S., et al. (1998). Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 4, 1025–1031. doi: 10.1038/2012
Pepe, S., and Lakatta, E. G. (2005). Aging hearts and vessels: masters of adaptation and survival. Cardiovasc. Res. 66, 190–193. doi: 10.1016/j.cardiores.2005.03.004
Peppa, M., and Raptis, S. A. (2008). Advanced glycation end products and cardiovascular disease. Curr. Diabetes Rev. 4, 92–100. doi: 10.2174/157339908784220732
Pitt, J. J. (2009). Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. Clin. Biochem. Rev. 30, 19–34.
Rabbani, N., and Thornalley, P. J. (2014). The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 63, 50–52. doi: 10.2337/db13-1606
Rai, V., Maldonado, A. Y., Burz, D. S., Reverdatto, S., Schmidt, A. M., and Shekhtman, A. (2012). Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of c-terminal rage (ctRAGE) and its binding to mDia1. J. Biol. Chem. 287, 5133–5144. doi: 10.1074/jbc.M111.277731
Reddy, G. K. (2004). AGE-related cross-linking of collagen is associated with aortic wall matrix stiffness in the pathogenesis of drug-induced diabetes in rats. Microvasc. Res. 68, 132–142. doi: 10.1016/j.mvr.2004.04.002
Reiniger, N., Lau, K., McCalla, D., Eby, B., Cheng, B., Lu, Y., et al. (2010). Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 59, 2043–2054. doi: 10.2337/db09-1766
Rubattu, S., Mennuni, S., Testa, M., Mennuni, M., Pierelli, G., Pagliaro, B., et al. (2013). Pathogenesis of chronic cardiorenal syndrome: is there a role for oxidative stress? Int. J. Mol. Sci. 14, 23011–23032. doi: 10.3390/ijms141123011
Sabbagh, M. N., Agro, A., Bell, J., Aisen, P. S., Schweizer, E., and Galasko, D. (2011). PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 25, 206–212. doi: 10.1097/WAD.0b013e318204b550
Safar, M. E., Levy, B. I., and Struijker-Boudier, H. (2003). Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation 107, 2864–2869. doi: 10.1161/01.CIR.0000069826.36125.B4
Schleicher, E. D., Wagner, E., and Nerlich, A. G. (1997). Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J. Clin. Invest. 99, 457–468. doi: 10.1172/JCI119180
Schmidt, A. M., Yan, S. D., Wautier, J. L., and Stern, D. (1999). Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 84, 489–497. doi: 10.1161/01.RES.84.5.489
Sell, D. R., and Monnier, V. M. (2012). Molecular basis of arterial stiffening: role of glycation - a mini-review. Gerontology 58, 227–237. doi: 10.1159/000334668
Semba, R. D., Sun, K., Schwartz, A. V., Varadhan, R., Harris, T. B., Satterfield, S., et al. (2015). Serum carboxymethyl-lysine, an advanced glycation end product, is associated with arterial stiffness in older adults. J. Hypertens. 33, 797–803; discussion: 803. doi: 10.1097/HJH.0000000000000460
Sethi, S., Rivera, O., Oliveros, R., and Chilton, R. (2014). Aortic stiffness: pathophysiology, clinical implications, and approach to treatment. Integr. Blood Press. Control 7, 29–34. doi: 10.2147/IBPC.S59535
Sims, T. J., Rasmussen, L. M., Oxlund, H., and Bailey, A. J. (1996). The role of glycation cross-links in diabetic vascular stiffening. Diabetologia 39, 946–951. doi: 10.1007/BF00403914
Singh, V. P., Bali, A., Singh, N., and Jaggi, A. S. (2014). Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 18, 1–14. doi: 10.4196/kjpp.2014.18.1.1
Soulis, T., Thallas, V., Youssef, S., Gilbert, R. E., McWilliam, B. G., Murray-McIntosh, R. P., et al. (1997). Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 40, 619–628. doi: 10.1007/s001250050725
Spinetti, G., Wang, M., Monticone, R., Zhang, J., Zhao, D., and Lakatta, E. G. (2004). Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler. Thromb. Vasc. Biol. 24, 1397–1402. doi: 10.1161/01.ATV.0000134529.65173.08
Sprague, A. H., and Khalil, R. A. (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 78, 539–552. doi: 10.1016/j.bcp.2009.04.029
Stitt, A. W., He, C., Friedman, S., Scher, L., Rossi, P., Ong, L., et al. (1997). Elevated AGE-modified ApoB in sera of euglycemic, normolipidemic patients with atherosclerosis: relationship to tissue AGEs. Mol. Med. 3, 617–627.
Strait, J. B., and Lakatta, E. G. (2012). Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 8, 143–164. doi: 10.1016/j.hfc.2011.08.011
Stürmer, M., Šebeková, K., Fazeli, G., Bahner, U., Stäb, F., and Heidland, A. (2015). 25-hydroxyvitamin d and advanced glycation endproducts in healthy and hypertensive subjects: are there interactions? J. Ren. Nutr. 25, 209–216. doi: 10.1053/j.jrn.2014.10.027
Takeuchi, M., Makita, Z., Yanagisawa, K., Kameda, Y., and Koike, T. (1999). Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol. Med. 5, 393–405.
Tan, A. L., Forbes, J. M., and Cooper, M. E. (2007). AGE, RAGE, and ROS in diabetic nephropathy. Semin. Nephrol. 27, 130–143. doi: 10.1016/j.semnephrol.2007.01.006
Tanaka, K., Tani, Y., Asai, J., Nemoto, F., Kusano, Y., Suzuki, H., et al. (2012). Skin autofluorescence is associated with severity of vascular complications in Japanese patients with type 2 diabetes. Diabet. Med. 29, 492–500. doi: 10.1111/j.1464-5491.2011.03448.x
Touré, F., Fritz, G., Li, Q., Rai, V., Daffu, G., Zou, Y. S., et al. (2012). Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ. Res. 110, 1279–1293. doi: 10.1161/CIRCRESAHA.111.262519
Uribarri, J., del Castillo, M. D., de la Maza, M. P., Filip, R., Gugliucci, A., Luevano-Contreras, C., et al. (2015). Dietary advanced glycation end products and their role in health and disease. Adv. Nutr. 6, 461–473. doi: 10.3945/an.115.008433
Vaitkevicius, P. V., Lane, M., Spurgeon, H., Ingram, D. K., Roth, G. S., Egan, J. J., et al. (2001). A cross-link breaker has sustained effects on arterial and ventricular properties in older rhesus monkeys. Proc. Natl. Acad. Sci. U.S.A. 98, 1171–1175. doi: 10.1073/pnas.98.3.1171
Vikramadithyan, R. K., Hu, Y., Noh, H. L., Liang, C. P., Hallam, K., Tall, A. R., et al. (2005). Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J. Clin. Invest. 115, 2434–2443. doi: 10.1172/JCI24819
Wang, M., Zhang, J., Jiang, L. Q., Spinetti, G., Pintus, G., Monticone, R., et al. (2007). Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 50, 219–227. doi: 10.1161/HYPERTENSIONAHA.107.089409
Wautier, J. L., Wautier, M. P., Schmidt, A. M., Anderson, G. M., Hori, O., Zoukourian, C., et al. (1994). Advanced Glycation End Products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: a link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. U.S.A. 91, 7742–7746. doi: 10.1073/pnas.91.16.7742
Wautier, M. P., Chappey, O., Corda, S., Stern, D. M., Schmidt, A. M., and Wautier, J. L. (2001). Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 280, E685–E694.
Wu, M. S., Liang, J. T., Lin, Y. D., Wu, E. T., Tseng, Y. Z., and Chang, K. C. (2008). Aminoguanidine prevents the impairment of cardiac pumping mechanics in rats with streptozotocin and nicotinamide-induced type 2 diabetes. Br. J. Pharmacol. 154, 758–764. doi: 10.1038/bjp.2008.119
Xu, B., Chibber, R., Ruggiero, D., Kohner, E., Ritter, J., and Ferro, A. (2003). Impairment of vascular endothelial nitric oxide synthase activity by advanced glycation end products. FASEB J. 17, 1289–1291. doi: 10.1096/fj.02-0490fje
Xu, Y., Toure, F., Qu, W., Lin, L., Song, F., Shen, X., et al. (2010). Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 285, 23233–23240. doi: 10.1074/jbc.M110.117457
Xue, J., Ray, R., Singer, D., Böhme, D., Burz, D. S., Rai, V., et al. (2014). The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 53, 3327–3335. doi: 10.1021/bi500046t
Yamagishi, S., Yonekura, H., Yamamoto, Y., Katsuno, K., Sato, F., Mita, I., et al. (1997). Advanced glycation end products-driven angiogenesis in vitro. Induction of the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. J. Biol. Chem. 272, 8723–8730. doi: 10.1074/jbc.272.13.8723
Yan, S. D., Schmidt, A. M., Anderson, G. M., Zhang, J., Brett, J., Zou, Y. S., et al. (1994). Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 269, 9889–9897.
Yan, S. F., Ramasamy, R., Naka, Y., and Schmidt, A. M. (2003). Glycation, inflammation, and RAGE: a scaffold for the macrovascular complications of diabetes and beyond. Circ. Res. 93, 1159–1169. doi: 10.1161/01.RES.0000103862.26506.3D
Yonekura, H., Yamamoto, Y., Sakurai, S., Petrova, R. G., Abedin, M. J., Li, H., et al. (2003). Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 370, 1097–1109. doi: 10.1042/bj20021371
Zieman, S. J., Melenovsky, V., Clattenburg, L., Corretti, M. C., Capriotti, A., Gerstenblith, G., et al. (2007). Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J. Hypertens. 25, 577–583. doi: 10.1097/HJH.0b013e328013e7dd
Keywords: RAGE, glycation, arterial diseases, inflammation, aging
Citation: Senatus LM and Schmidt AM (2017) The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 8:187. doi: 10.3389/fgene.2017.00187
Received: 06 September 2017; Accepted: 10 November 2017;
Published: 05 December 2017.
Edited by:
Mingyi Wang, National Institutes of Health (NIH), United StatesReviewed by:
Alun Hughes, University College London, United KingdomBeatrice Charreau, University of Nantes, France
Copyright © 2017 Senatus and Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ann Marie Schmidt, YW5ubWFyaWUuc2NobWlkdEBueXVtYy5vcmc=