 Marianna De Cinque1,2
Marianna De Cinque1,2 Orazio Palumbo3
Orazio Palumbo3 Ermelinda Mazzucco1
Ermelinda Mazzucco1 Antonella Simone1,2
Antonella Simone1,2 Pietro Palumbo3
Pietro Palumbo3 Renata Ciavatta4Giuliana Maria4Rosangela Ferese5Stefano Gambardella5
Renata Ciavatta4Giuliana Maria4Rosangela Ferese5Stefano Gambardella5 Antonella Angiolillo1Massimo Carella3
Antonella Angiolillo1Massimo Carella3 Silvio Garofalo1*
Silvio Garofalo1*- 1Dipartimento di Medicina e Scienze della Salute “V. Tiberio”, Università degli Studi del Molise, Campobasso, Italy
- 2Unità Operativa di Medicina Trasfusionale, Azienda Sanitaria Regionale del Molise, Presidio Ospedaliero San Timoteo, Termoli, Italy
- 3Unità Operativa Complessa di Genetica Medica, Poliambulatorio “Giovanni Paolo II”, IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy
- 4Ufficio per la Tutela della Salute Neurologica e Psichica dell’Età Evolutiva, Azienda Sanitaria Regionale del Molise, Termoli, Italy
- 5IRCCS Neuromed, Pozzilli, Italy
Terminal deletion of chromosome 6q is a rare chromosomal abnormality associated with variable phenotype spectrum. Although intellectual disability, facial dysmorphism, seizures and brain abnormalities are typical features of this syndrome, genotype–phenotype correlation needs to be better understood. We report the case of a 6-year-old Caucasian boy with a clinical diagnosis of intellectual disability, delayed language development and dyspraxia who carries an approximately 8 Mb de novo heterozygous microdeletion in the 6q26-q27 locus identified by karyotype and defined by high-resolution SNP-array analysis. This patient has no significant structural brain or other organ malformation, and he shows a very mild phenotype compared to similar 6q26-qter deletion. The patient phenotype also suggests that a dyspraxia susceptibility gene is located among the deleted genes.
Case Presentation
Children with intellectual disability in the Molise (Italy) school district were enrolled in genetic screening for chromosomal defects and copy number variations (CNVs). This study was carried out following the recommendations of the Declaration of Helsinki and the Italian law for biomedical experimentation with written informed consent from all subjects. The protocol was approved and authorized by Italian National Health Service Azienda Sanitaria Regionale del Molise (ASREM) bio-ethical Committee with protocol no. 68 – November 5th, 2015 under the denomination “Epidemiologia delle malattie genetiche rare associate a sindromi dismorfiche e disabilità psichica nella Regione Molise.” All subjects or their legal guardians gave written informed consent under the Declaration of Helsinki and the approved protocol, including full consent to the publication of the case and related picture. Patients were carefully selected to undergo a preliminary cytogenetic screening followed by chromosome microarray analysis (CMA).
The patient was born from a 38-year old mother and a 40-year old father. He is the third son of non-consanguineous Caucasian parents and was born at 37 weeks of gestation by cesarean section. Birth weight was 2400 g (3rd centile), length 48 cm (21st centile) and head circumference 31.5 cm (3rd centile). The Apgar score was eight at 1 min and nine at 5 min.
The patient did not experience seizures, but his motor and language development was delayed. He started walking when he was 18 months old, and he pronounced the first words only around 3 years old. When he was 4 years old, the neuropsychological assessment showed developmental disorders of speech and language, hyperkinetic disorder, and global developmental delay.
At the age of 6 years, the patient was referred to a clinical geneticist for evaluation of developmental delay and dysmorphic features. His physical examination showed microcephaly (head circumference, 48.5 cm, 1st centile), hypertelorism, depressed nasal root, bulbous tip, winged scapula, and toe walking (Figure 1A). He was clumsy in movements. Weight was 23 kg (72nd centile) and height was 117 cm (63rd centile).
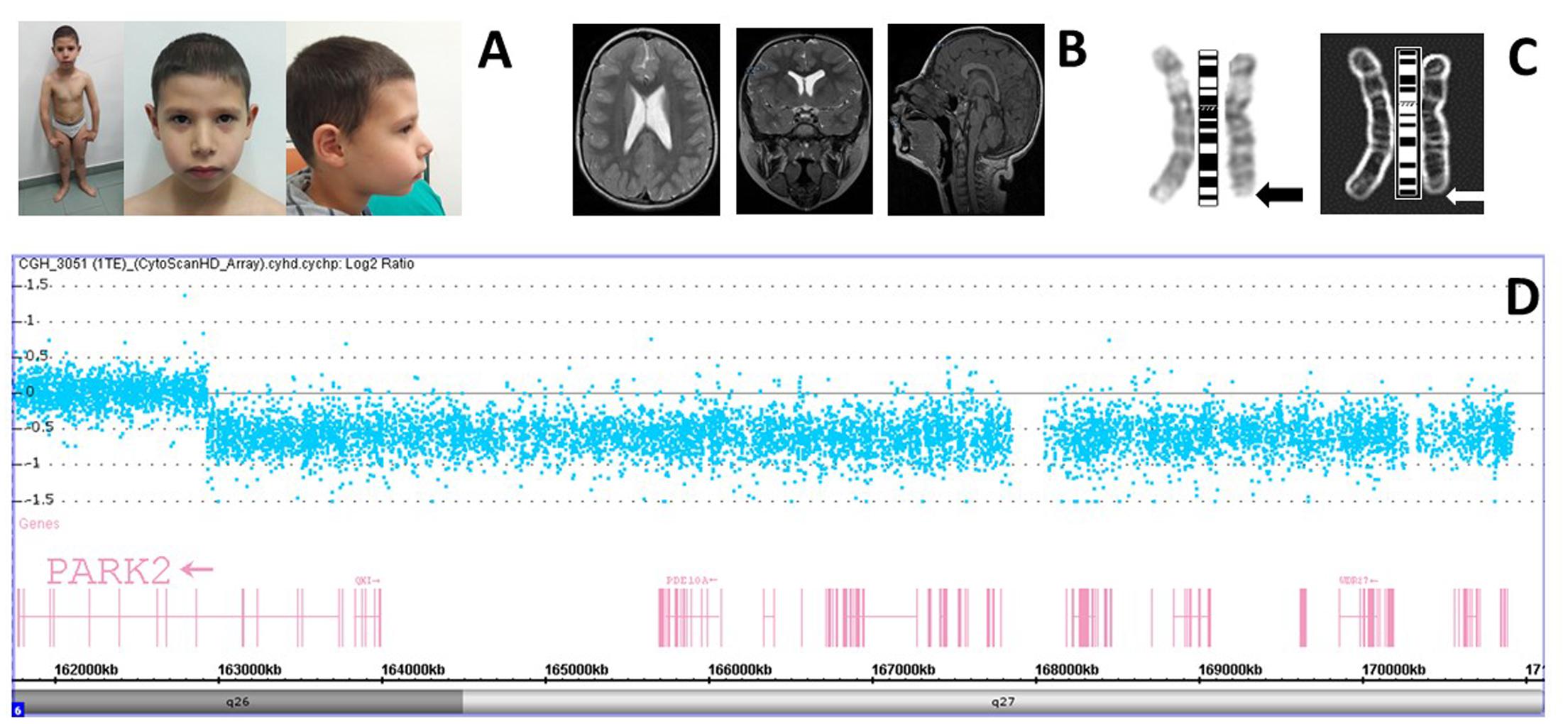
FIGURE 1. (A) The patient and his facies showing mild dysmorphological features. (B) Patient brain MRI is showing slight enlargement of the occipital horns with normal morphology. (C) Positive and negative views of the patient GTC-banded chromosomes six at 400 bands resolution with the arrows pointing to the deleted region. (D) High-resolution chromosome microarray analysis (CMA) in the patient. Deviations of probe Log2 ratios from zero of the SNP probes on chromosome 6q26-q27 cytoband indicate the deleted region identified in the genomic profile of the patient.
Infant neuropsychiatric evaluation revealed mild cognitive delay (IQ of 67 using WPPSI-III), speech delay and learning difficulties, and developmental coordination disorder (DCD) (dyspraxia). The EEG examination showed the presence of minor electrical abnormalities (potential 4-5 c/s, 30–50 μV) prevailing on the left posterior regions. MRI of the brain detected slight enlargement of the ventricular system mainly in occipital horns with normal morphology (Figure 1B). Cardiac examination with echocardiogram was normal as well-ophthalmological and audiological evaluation. Ultrasound of kidneys and urinary tract shows the absence of anomalies. His medical history also included surgery to remove a thyroglossal duct cyst and frequent episodes of wetting and encopresis.
Chromosomes were obtained from cultured peripheral lymphocytes using Synchroset kit (Euroclone, Milano, Italy). The conventional technique of G banding analysis was used. DNA was extracted from whole blood using kit QIAamp DNA Mini (Qiagen, Hilden, Germany). DNA concentration and purity were determined with a ND-1000 Spectrophotometer (NanoDrop Technologies, Berlin, Germany).
The CytoScan HD array contains more than 2.6 million markers for the copy number analysis. Of these markers, 1,950,000 are unique, non-polymorphic oligonucleotide probes, and 750,000 are SNP probes used for genotyping. The average marker spacing is one probe per 1.1 kb, with a mean spacing of one probe per 1.7 kb on non-gene backbones and one probe per 880 bp in intragenic regions. The CytoScanHD assay was performed according to the manufacturer’s protocol, starting with 250 ng DNA. Briefly: total genomic DNA was digested with a restriction enzyme (NspI), ligated to an appropriate adapter for the enzyme, and subjected to PCR amplification using a single primer. After digestion, with DNase I, the PCR products were labeled with a biotinylated nucleotide analog, using terminal deoxynucleotidyl transferase and hybridized to the microarray. Hybridization was carried out in the Hybridization Oven 645 while subsequent washing and staining were performed using the Fluidics Station 450. The array was then scanned with the Scanner 3000 7G and both quality control step and copy number analysis were performed using the Chromosome Analysis Suite Software version 3.1. The clinical significance of each CNV detected was assessed by comparison with an internal database of 3,000 clinical samples and public database of CNVs (ISCA, ClinVar, DECIPHER, DGV). All nucleotide positions were based on the February 2009 human reference sequence, assembly GRCh37/hg19, produced by the Genome Reference Consortium.
Twenty metaphases of the patient and the parents were analyzed after GTG banding at 400 chromosome band resolution. Karyotype showed a small deletion in the terminal region of the long arm of chromosome 6 in all the analyzed metaphases. The karyotype of the child was 46, XY, del(6)(q26→qter) (Figure 1C). This rearrangement was not detected in the parents. To map deletion breakpoint more accurately high-resolution CMA was made.
Results of SNP array analysis of the patient displayed a copy number loss in chromosome bands 6q26-q27 of about 7,988 Mb ranging from nucleotide 162,931,432 to 170,919,482 (Figure 1D). In this interval there are 36 protein-coding genes: PARK2, PACRG, QKI, C6orf118, PDE10A, SDIM1, TFT, PRR18, SFT2D1, MPC1, RPS6KA2, RNASET2, FGFR10P, CCR6, GPR31, TCP10L2, UNC93A, TTLL2, TCP10, C6orf123, MLLT4, KIF25, FRMD1, DACT2, SMOC2, THBS2, WDR27, C6orf120, PHF10, TCTE3, ERMARD, DLL1, FAM120B, PSMB1, TBP, PDCD2 (according to DECIPHER v9.15 https://decipher.sanger.ac.uk/). The haploinsufficiency of some genes must be implicated in the clinical phenotype of this patient with the telomeric deletion of 6q26-q27.
According to the International System for Human Cytogenetic Nomenclature [ISCN, 2016], the molecular karyotype of the patient (showing both the minimum and maximum extent of the deletion) was defined as follows: arr[GRCh37] 6q26-q27(162,931,432-170,919,482)x1 dn. No other pathological CNVs were identified, excluding well-known benign CNVs variants.
The PARK2 gene maps in 6q26 deletion breakpoint. Consequently, PARK2 coding sequence (MIM number: 602544; NM_004562), exon/intron boundaries and flanking intronic regions, were analyzed by PCR and direct sequencing (Kitada et al., 1998) using ABI BigDye Terminator Sequencing Kit v.3.3 (Applied Biosystems, Foster City, CA, United States) and an ABI 310 Genetic Analyzer (Applied Biosystems). Sequencing did not show any other mutation.
Background
About 1/2000 patients affected by mental retardation with developmental delay shows terminal deletion of the long arm of chromosome 6 (Lee et al., 2011). This condition is a rare cytogenetic disorder characterized by extended variation in the size of the deleted region that can go from cytogenetically visible to small submicroscopic deletions, ranging from 0.4 to 12 Mb (Lee et al., 2011). Only molecular cytogenetic techniques as fluorescent in situ hybridization (FISH) and comparative genomic hybridization (CGH) can precisely recognize and define the smaller terminal deletions (Erdel et al., 1997; Birnbacher et al., 2001; Sherr et al., 2005).
Patients with distal deletion of chromosome 6q show also variable phenotypic features with only a few common symptoms that include, besides intellectual disability and developmental delay (Bertini et al., 2006; Striano et al., 2006; Mosca et al., 2010), also hypotonic muscle, seizure (Elia et al., 2006; Striano et al., 2006), dysmorphic features with spinal cord and brain anomalies (Dupé et al., 2011; Nair et al., 2012). Agenesis/hypoplasia of the corpus callosum (Sherr et al., 2005), hydrocephalus (Li et al., 2015) or non-specific ventricle enlargement, lissencephaly, pachygyria, polymicrogyria, neuronal migration abnormality with periventricular nodular heterotopia (PNH) (Conti et al., 2013), olfactory bulb aplasia and anosmia (Gerber et al., 2011) are the most frequently described brain defects in these patients. Craniofacial anomalies (Hopkin et al., 1997) are also common, and they include hypertelorism with broad nasal bridge, midface hypoplasia with cleft palate, long philtrum, thin upper lip and ear anomalies. Also, heart defects (Nair et al., 2012), retinal abnormalities (Rivas et al., 1986; Abu-Amero et al., 2010), vertebral column defects (Li et al., 2015), joint laxity with elbow and knee anomalies, and attention deficit hyperactivity disorder (ADHD) are described in numerous patients.
Several authors have tried to correlate the distal deletion of chromosome 6q to a distinct clinical phenotype (Stevenson et al., 2004; Bertini et al., 2006; Rigon et al., 2011; Zhou et al., 2014). Others have sought to identify the minimum deleted interval containing the critical genes responsible for the major clinical problems (Eash et al., 2005; Rooms et al., 2006; Peddibhotla et al., 2015). However, it is still very problematic to identify clinically patients carrying such deletion and to correlate deleted genes with a clinical phenotype (Rigon et al., 2011).
Discussion
Here, we report the clinical and genomic characterization of a patient with an intellectual disability, delayed language developmental and dyspraxia and minor dysmorphic features due to a non-recurrent 6q26-qter deletion identified using high-resolution CMA in a screening of patients with non-syndromic intellectual disability. In this patient haplodeficiency of chromosome 6q subtelomeric region is the result of a de novo event and it is not due to familiar unbalanced translocation. The breakpoint is mapped in 6q26 where is located the common fragile site FRA6E (Figure 1D). It is well-known that FRA6E is a genomic hotspot that predisposes to chromosomal deletion and it is considered the third most mutation-prone fragile site of the human genome (Palumbo et al., 2010; Ambroziak et al., 2015). In the center of this genomic site is located the PARK2 gene that is a huge gene implicated in the autosomal recessive early-onset type of Parkinson disease. The PARK2 gene encodes the ubiquitin ligase Parkin that has high expression level in human neurons. We believe that PARK2 haploinsufficiency may have a role in the etiology of intellectual disability of this patient since we can exclude mutations in the other PARK2 allele. Micro-deletions or duplications and CNV in PARK2 are found in patients with Autism Spectrum Disorder (Scheuerle and Wilson, 2011). In mouse, Park2 knock out does not reproduce any sign of the Parkinson disease (Perez and Palmiter, 2005). Instead, Parkin null mouse and the quaking mutant mouse, who carries a 1,8 Mb deletion of mouse chromosome 17 including Park2, show mainly neurological and behavioral problems (Lorenzetti et al., 2004; Wilson et al., 2010).
The patient displays a cytogenetically visible deletion of about 8 Mb in size that causes haploinsufficiency of other 36 proteins coding genes. RNASET2, CCR6, DACT2, SMOC2, DLL1, and TBP are other brain-specific genes located in the deletion interval. They are all excellent candidate genes to affect brain functions and to generate with Parkin the clinical spectrum of the 6qter syndrome. However, the patient shows a mild phenotype. He has microcephaly, minimal enlargement of the cerebral ventricular system mainly in occipital horns and minor electrical abnormalities prevailing on the left posterior regions of the brain. Cerebral malformations are absent as eye and heart anomalies.
As described in previous studies (Stevenson et al., 2004; Bertini et al., 2006; Zhou et al., 2014), the 6q distal deletions are characterized by highly variable clinical symptoms that do not depend on the extension of the chromosomal abnormality. This condition can also happen in other contiguous gene deletion syndromes because any CNV can also affect the expression level of genes situated nearby the deleted or duplicated region, modulating the clinical consequences of haploinsufficiency (Stranger et al., 2007). This “position effect” can explain the complexity and heterogeneity of the phenotype associated with terminal 6q deletion (Stranger et al., 2007).
Peddibhotla et al. (2015) have reported a very similar 6q terminal deletion in a female newborn. She had a 7.6 Mb heterozygous deletion at 6q26-qter between nucleotides 162,784,565–170,899,992 which determined multiple organ anomalies (right-sided subependymal nodular gray matter heterotopia and agenesis of the corpus callosum, bicuspid aortic valve, imperforate anus with perineal fistula, segmentation anomalies of the sacrum, dextroconvex scoliosis of the lumbosacral spine and tethered spinal cord). None of these symptoms are present in this patient. However, both patients showed hypertelorism, broad nasal bridge, and global developmental delay. Correlation between the two patients indicates that terminal 6q deletions that are genotypically almost overlapping can be remarkably dissimilar in phenotype and clinical severity.
Concluding Remarks
To our knowledge, this is the first case of association between terminal 6q deletion and DCD. However, there are several genes in 6q26-27 region. Studies may narrow down the DCD candidates in the near future.
Author Contributions
MDC, SiG, RC, and GM were responsible for clinical data and sample collection. MDC, AS, EM, PP, and OP made DNA extraction, karyotype, and CMA. RF and StG Park2 sequencing. MDC, AA, MC, and SiG analyzed the data and wrote the manuscript.
Funding
This work was supported by Regione Molise Research Projects “Obiettivi di carattere prioritario 2012” no. 17 – “Un ambulatorio mobile itinerante per le malattie genetiche rare nei piccoli Comuni della Regione Molise” and no. 7 – “Cariotipo Molecolare nella diagnosi di autismo, schizofrenia e ritardo mentale ed interventi per la diagnosi precoce delle demenze” to SiG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would also like to show our gratitude to the patient and his family for the participation to this study. They also thank Maria Grazia Mastromonaco for nursing assistance and Dr. Roberto Fratangelo for help to recruit patients for this project.
References
Abu-Amero, K. K., Hellani, A., Salih, M. A., Al Hussain, A., Al Obailan, M., Zidan, G., et al. (2010). Ophthalmologic abnormalities in a de novo terminal 6q deletion. Ophthalmic Genet. 31, 1–11. doi: 10.3109/13816810903312535
Ambroziak, W., Koziorowski, D., Duszyc, K., Górka-Skoczylas, P., Potulska-Chromik, A., Sławek, J., et al. (2015). Genomic instability in the PARK2 locus is associated with Parkinson’s disease. J. Appl. Genet. 56, 451–461. doi: 10.1007/s13353-015-0282-9
Bertini, V., De Vito, G., Costa, R., Simi, P., and Valetto, A. (2006). Isolated 6q terminal deletions: an emerging new syndrome. Am. J. Med. Genet. A. 140, 74–81. doi: 10.1002/ajmg.a.31020
Birnbacher, R., Chudoba, I., Pirc-Danoewinata, H., Konig, M., Kohlhauser, C., Schnedl, W., et al. (2001). Microdissection and reverse painting reveals a microdeletion 6(q26qter) in a de novo r(6) chromosome. Ann. Genet. 44, 13–18. doi: 10.1016/S0003-3995(00)01033-9
Conti, V., Carabalona, A., Pallesi-Pocachard, E., Parrini, E., Leventer, R. J., Buhler, E., et al. (2013). Periventricular heterotopia in 6q terminal deletion syndrome: role of the C6orf70 gene. Brain 136(Pt 11), 3378–3394. doi: 10.1093/brain/awt249
Dupé, V., Rochard, L., Mercier, S., Le Pétillon, Y., Gicquel, I., Bendavid, C., et al. (2011). NOTCH, a new signaling pathway implicated in holoprosencephaly. Hum. Mol. Genet. 20, 1122–1131. doi: 10.1093/hmg/ddq556
Eash, D., Waggoner, D., Chung, J., Stevenson, D., and Martin, C. L. (2005). Calibration of 6q subtelomere deletions to define genotype/phenotype correlations. Clin. Genet. 67, 396–403. doi: 10.1111/j.1399-0004.2005.00424.x
Elia, M., Striano, P., Fichera, M., Gaggero, R., Castiglia, L., Galesi, O., et al. (2006). 6q terminal deletion syndrome associated with a distinctive EEG and clinical pattern: a report of five cases. Epilepsia 47, 830–838. doi: 10.1111/j.1528-1167.2006.00522.x
Erdel, M., Duba, H. C., Verdorfer, I., Lingenhel, A., Geiger, R., Gutenberger, K. H., et al. (1997). Comparative genomic hybridization reveals a partial de novo trisomy 6q23-qter in an infant with congenital malformations: delineation of the phenotype. Hum. Genet. 99, 596–601. doi: 10.1007/s004390050412
Gerber, J. C., Neuhann, T. M., Tyshchenko, N., Smitka, M., and Hackmann, K. (2011). Expanding the clinical and neuroradiological phenotype of 6q27 microdeletion: olfactory bulb aplasia and anosmia. Am. J. Med. Genet. A. 155A, 1981–1986. doi: 10.1002/ajmg.a.34079
Hopkin, R. J., Schorry, E., Bofinger, M., Milatovich, A., Stern, H. J., Jayne, C., et al. (1997). New insights into the phenotypes of 6q deletions. Am. J. Med. Genet. 70, 377–386. doi: 10.1002/(SICI)1096-8628(19970627)70:4<377::AID-AJMG9>3.0.CO;2-Q
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Lee, J. Y., Cho, Y. H., and Hallford, G. (2011). Delineation of subtelomeric deletion of the long arm of chromosome 6. Ann. Hum. Genet. 75, 755–764. doi: 10.1111/j.1469-1809.2011.00675.x
Li, Y., Choy, K. W., Xie, H. N., Chen, M., He, W. Y., Gong, Y. F., et al. (2015). Congenital hydrocephalus and hemivertebrae associated with de novo partial monosomy 6q (6q25.3→qter). Balkan J. Med. Genet. 18, 77–84. doi: 10.1515/bjmg-2015-0009
Lorenzetti, D., Antalffy, B., Vogel, H., Noveroske, J., Armstrong, D., and Justice, M. (2004). The neurological mutant quaking(viable) is Parkin deficient. Mamm. Genome 15, 210–217. doi: 10.1007/s00335-003-2333-5
Mosca, A. L., Callier, P., Masurel-Paulet, A., Thauvin-Robinet, C., Marle, N., Nouchy, M., et al. (2010). Cytogenetic and array-CGH characterization of a 6q27 deletion in a patient with developmental delay and features of Ehlers-Danlos syndrome. Am. J. Med. Genet. A 152A, 1314–1317. doi: 10.1002/ajmg.a.33254
Nair, S., Varghese, R., Hashim, S., and Scariah, P. (2012). Dysmorphic features and congenital heart disease in chromosome 6q deletion: a short report. Indian J. Hum. Genet. 18, 127–129. doi: 10.4103/0971-6866.96682
Palumbo, E., Matricardi, L., Tosoni, E., Bensimon, A., and Russo, A. (2010). Replication dynamics at common fragile site FRA6E. Chromosoma 119, 575–587. doi: 10.1007/s00412-010-0279-4
Peddibhotla, S., Nagamani, S. C., Erez, A., Hunter, J. V., Holder, J. L., Carlin, M. E., et al. (2015). Delineation of candidate genes responsible for structural brain abnormalities in patients with terminal deletions of chromosome 6q27. Eur. J. Hum. Genet. 23, 54–60. doi: 10.1038/ejhg.2014.51
Perez, F. A., and Palmiter, R. D. (2005). Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 102, 2174–2179. doi: 10.1073/pnas.0409598102
Rigon, C., Salviati, L., Mandarano, R., Donà, M., and Clementi, M. (2011). 6q27 subtelomeric deletions: Is there a specific phenotype? Am. J. Med. Genet. A 155A, 1213–1214. doi: 10.1002/ajmg.a.33877
Rivas, F., Ruiz, C., Rivera, H., Moller, M., Serrano-Lucas, J. I., and Cantu, J. M. (1986). De novo del(6)(q25) associated with macular degeneration. Ann. Genet. 29, 42–44.
Rooms, L., Reyniers, E., Scheers, S., van Luijk, R., Wauters, J., Van Aerschot, L., et al. (2006). TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. Eur. J. Hum. Genet. 14, 1090–1096. doi: 10.1038/sj.ejhg.5201674
Scheuerle, A., and Wilson, K. (2011). PARK2 copy number aberrations in two children presenting with autism spectrum disorder: further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 413–420. doi: 10.1002/ajmg.b.31176
Sherr, E. H., Owen, R., Albertson, D. G., Pinkel, D., Cotter, P. D., Slavotinek, A. M., et al. (2005). Genomic microarray analysis identifies candidate loci in patients with corpus callosum anomalies. Neurology 65, 1496–1498. doi: 10.1212/01.wnl.0000183066.09239.b6
Stevenson, D. A., Brothman, A. R., Carey, J. C., Chen, Z., Dent, K. M., Bale, J. F., et al. (2004). 6q subtelomeric deletion: Is there a recognizable syndrome? Clin. Dysmorphol. 13, 103–106. doi: 10.1097/00019605-200404000-00010
Stranger, B. E., Forrest, M. S., Dunning, M., Ingle, C. E., Beazley, C., Thorne, N., et al. (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853. doi: 10.1126/science.1136678
Striano, P., Malacarne, M., Cavani, S., Pierluigi, M., Rinaldi, R., Cavaliere, M. L., et al. (2006). Clinical phenotype and molecular characterization of 6q terminal deletion syndrome: five new cases. Am. J. Med. Genet. A 140, 1944–1949. doi: 10.1002/ajmg.a.31435
Wilson, G. R., Wang, H. X., Egan, G. F., Robinson, P. J., Delatycki, M. B., O’Bryan, M. K., et al. (2010). Deletion of the Parkin co-regulated gene causes defects in ependymal ciliary motility and hydrocephalus in the quakingviable mutant mouse. Hum. Mol. Genet. 19, 1593–1602. doi: 10.1093/hmg/ddq031
Keywords: terminal 6q deletion, mental retardation, dyspraxia, Parkin, FRA6E
Citation: De Cinque M, Palumbo O, Mazzucco E, Simone A, Palumbo P, Ciavatta R, Maria G, Ferese R, Gambardella S, Angiolillo A, Carella M and Garofalo S (2017) Developmental Coordination Disorder in a Patient with Mental Disability and a Mild Phenotype Carrying Terminal 6q26-qter Deletion. Front. Genet. 8:206. doi: 10.3389/fgene.2017.00206
Received: 04 August 2017; Accepted: 24 November 2017;
Published: 06 December 2017.
Edited by:
Musharraf Jelani, King Abdulaziz University, Saudi ArabiaReviewed by:
Changsoo Kang, Sungshin Women’s University, South KoreaNaveed Wasif, University of Ulm, Germany
Copyright © 2017 De Cinque, Palumbo, Mazzucco, Simone, Palumbo, Ciavatta, Maria, Ferese, Gambardella, Angiolillo, Carella and Garofalo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvio Garofalo, c2lsdmlvLmdhcm9mYWxvQHVuaW1vbC5pdA==