 Rahul Mittal
Rahul Mittal Nicole Bencie
Nicole Bencie James M. Parrish
James M. Parrish George Liu1
George Liu1 Jeenu Mittal
Jeenu Mittal Denise Yan
Denise Yan Xue Zhong Liu
Xue Zhong Liu- 1Department of Otolaryngology, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Department of Human Genetics, University of Miami Miller School of Medicine, Miami, FL, United States
Cyclic nucleotide phosphodiesterases (PDEs) refer to a class of enzymes that degrade the phosphodiester bonds in cAMP and cGMP secondary messenger molecules (Tetsi et al., 2017; Weber et al., 2017). Due to their ability to regulate signal transduction, PDEs are vital in the modulation of biological signaling pathways in sensory systems including auditory and visual systems. The importance of PDEs in maintaining homeostatic environments in both the inner ear and the eye is highlighted by the fact that mutations in PDEs have been associated with various phenotypic abnormalities, including sensorineural hearing loss (SNHL), vision disorders, and other related syndromes (Bayés et al., 1995; Saga et al., 1998; Tetsi et al., 2017).
PDEs affect many biological systems (Fusco and Paldino, 2017), and PDE genetic mutations have been associated with the pathogenesis of several conditions, including sudden sensorineural hearing loss (SSNHL), retinitis pigmentosa (RP), and Usher Syndrome. While more research must be conducted and it is likely that the mechanisms surrounding SSNHL are complex, current data and several studies strongly supports the notion that PDE specifically PDE4D plays a role in causing SSNHL. PDE4D is expressed in the inner ear (Degerman et al., 2017) potentially playing a role in maintaining cochlear homeostasis and hearing function. In addition, PDE4Ds have been associated with atherosclerosis, which is a risk factor for SSNHL (Fischer et al., 2015; Rajati et al., 2016). While PDE4Ds are proposed as key players in SSNHL, PDE6 genes most likely play a role in the development of RP. PDE6 is an important component of visual phototransduction cascade (Majumder et al., 2015; Lagman et al., 2016) and therefore, low levels of PDE6 due to mutations can lead to rod-cone degeneration. This article will discuss the association of SSNHL, RP and Usher syndrome with PDE mutations and seek to elucidate the potential for common treatment modalities in such syndromes due to their shared etiologies.
PDE Mutations and Hearing Loss
The SNHL is defined as hearing loss resulting from damage to the inner ear or the auditory nerves that aid in the sound transducing mechanism to the brain (Luo et al., 2017). SSNHL is a form of SNHL that characterizes the sudden loss of hearing, usually in one ear and affects an estimated 5-27/100,000 persons per year (Alexander and Harris, 2013; Lin et al., 2017).
Until 2010, PDEs were known to be present in the cochlea and inner ear, but remained unidentified. Observation of the nitric oxide (NO) signaling pathway elucidated the existence and role that PDEs play in inner ear homeostasis. Most recently, PDE receptors were specified in the cochlea via reference to the guinea pig genome, which is up to 95% similar to the human genome. One study used the previously published full genome sequence of guinea pigs on the University of California, Santa Cruz (UCSC) genome bioinformatics browser to find PDEs present in humans, mice, and guinea pigs (Huang et al., 2010). The 22 PDE genes present in all three species were then located in guinea pig cochlear tissues using PCR. As a result, six PDE cDNA genes were identified in the guinea pig inner ear: CpPde3a, CpPde4d, CpPde8a, CpPde8b, CpPde9a, and CpPde11a. These genes were thus assumed to be present in the human ear based on the existence of cross-species gene superfamilies (Huang et al., 2010).
The identification of these PDEs in the cochlea led to studies that analyzed the correlations between genetic polymorphisms in these loci and SSNHL. In one Taiwanese study, mutations in the PDE4D gene were examined for their effects on the cochlea and possible pathogenic outcomes (Chien et al., 2016). As PDE4D mutations are known to cause ischemic stroke and cardiac abnormalities (Dichgans, 2007; Jørgensen et al., 2015; Shao et al., 2015) with associated hearing impairment, it was hypothesized that such mutations might play a role in the development of SSNHL. Results revealed a sex-differential effect of the PDE4D gene on SSNHL where only females had phenotypic disease symptoms (Chien et al., 2016). A rare T allele, coming from the rs702553 (SNP56) polymorphism of the PDE4D gene, increased prevalence of the condition in the female population. While studies have uncovered some correlations between the PDE4D gene and hearing function, the current lack of information warrants further study to better understand this relationship. A better knowledge about how PDE mutations affects cochlear function will help in developing novel treatment modalities for hearing loss.
PDE Mutations, Retinitis Pigmentosa and Usher Syndrome
Besides SNHL, retinitis pigmentosa (RP) demonstrates a heterogeneous genetic etiology. RP is a disorder characterized by gradual devolution of the retina and it affects between 1 in 3,000 and 8,000 people worldwide (Ali et al., 2017). The disorder can display X-linked, autosomal dominant or autosomal recessive inheritance patterns and has been associated with about 70 genes (Dias et al., 2017). The majority of such genes are expressed in photoreceptors of the pigment epithelium. The PDE6 complex includes α, β, and γ subunits, which together, play an integral role in rod photoreceptor phototransduction (Majumder et al., 2015; Gopalakrishna et al., 2017). It has been demonstrated that PDE6 maintains intracellular levels of cGMP by hydrolyzing cGMP when activated by light (Lagman et al., 2016). While the mechanism in RP that leads to rod photoreceptor death remains unknown, it is hypothesized to be a result of a defect in PDE6 (Ali et al., 2017; Table 1).
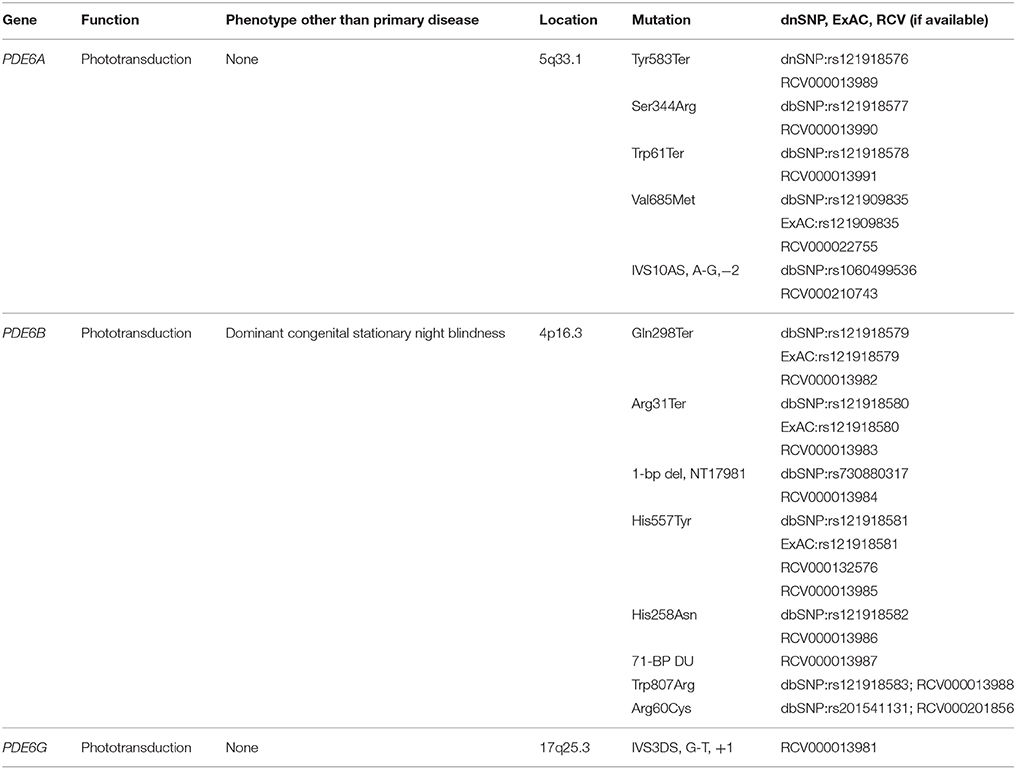
Table 1. Phosphodiesterase (PDE) gene variants associated with retinitis pigmentosa.
For autosomal recessive RP (ARRP), over 40 genes have been mapped, including PDE6A, PDE6B, and PDE6G (RetNet, 2014). PDE6A and PDE6B are the second most common cause of ARRP, accounting for up to 2–5% of all such cases (Ali et al., 2017). While ARRP can be non-syndromic, it also presents as part of a constellation of signs in the form of a syndrome. Approximately 20–30% of ARRP patients have reported associations with non-ocular disorders. Usher syndrome is one such disease that includes both ARRP and hearing impairment. While Usher syndrome etiology is classically regarded as stemming from genes that do not contain PDE6A, PDE6B, or PDE6G, researchers have yet to identify whether the mechanisms that underlie vision loss in Usher syndrome are identical to those present in non-syndromic RP (Hmani-Aifa et al., 2009). Some patients diagnosed with Usher syndrome have been found to possess a so-called “double hit” of both classically regarded Usher mutations (e.g., MYO7A, USH1C, CDH23) in combination with PDE6 mutations. This “double hit” not only appears to contribute to a greater severity of retinal devolution, but it also demonstrates the coexistence of SNHL, ARRP, and a mutation in PDE. The aforementioned genetic syndromes, along with a growing number of disorders that are associated with PDE, all make the case that a successful PDE-treatment modality might not only be applied to multiple disorders, but is certain to have vast interdisciplinary effects.
Conclusion and Future Directions
While disorders such as SNHL, RP, and Usher syndrome encompass heterogeneous genetic etiologies, there is lack of information regarding PDE-related molecular mechanisms underlying each disease. There is a need to explore the tissue specificity of different PDEs and how PDE mutations lead to phenotypic consequences. In addition, the careful analysis of genetic etiology is imperative in diagnosis and development of new treatment modalities. The advent of new genome editing technologies including clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 holds a great potential as a therapeutic strategy for genetic diseases including SNHL and RP (Hsu et al., 2014; Zou et al., 2015). As a proof of this concept, CRISPR/Cas9 genome editing has been demonstrated to produce substantial improvement in mice with regard to retinal cone function and rod survival via the NRL gene (Peng et al., 2017; Yu et al., 2017). In the future, combining CRISPR/Cas9 with other powerful tools including adeno-associated virus (AAV) and iPSCs will open up avenues to develop effective therapeutic strategies to treat SNHL and RP having an underlying genetic etiology.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The research work in Dr. Liu's Laboratory is supported by grants R01 DC05575, R01 DC01246, and R01 DC012115 from the National Institutes of Health/National Institute on Deafness and Other Communication Disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RM, XL, and DY is a recipient of a grant from Action on Hearing Loss. NB is a recipient of the 2017 LINK/SEW Award from Brown University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. We are thankful to April Mann for the critical reading of the manuscript.
References
Alexander, T. H., and Harris, J. P. (2013). Incidence of sudden sensorineural hearing loss. Otol. Neurotol. 34, 1586–1589. doi: 10.1097/MAO.0000000000000222
Ali, M. U., Rahman, M. S. U., Cao, J., and Yuan, P. X. (2017). Genetic characterization and disease mechanism of retinitis pigmentosa; current scenario. 3 Biotech. 7, 251. doi: 10.1007/s13205-017-0878-3
Bayés, M., Giordano, M., Balcells, S., Grinberg, D., Vilageliu, L., Martínez, I., et al. (1995). Homozygous tandem duplication within the gene encoding the beta-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum. Mutat. 5, 228–234. doi: 10.1002/humu.1380050307
Chien, C. Y., Tai, S. Y., Wang, L. F., Hsi, E., Chang, N. C., Wang, H. M., et al. (2016). Phosphodiesterase 4D gene polymorphisms in sudden sensorineural hearing loss. Eur. Arch. Otorhinolaryngol. 273, 2403–2409. doi: 10.1007/s00405-015-3804-5
Degerman, E., In 't Zandt, R., Pålbrink, A., Eliasson, L., Cayé-Thomasen, P., and Magnusson, M. (2017). Inhibition of phosphodiesterase 3, 4, and 5 induces endolymphatic hydrops in mouse inner ear, as evaluated with repeated 9.4T MRI. Acta Otolaryngol. 137, 8–15. doi: 10.1080/00016489.2016.1211320
Dias, M. F., Joo, K., Kemp, J. A., Fialho, S. L., da Silva Cunha, A. Jr., Woo, S. J., et al. (2017). Molecular genetics and emerging therapies for retinitis pigmentosa: basic research and clinical perspectives. Prog. Retin. Eye Res. doi: 10.1016/j.preteyeres.2017.10.004. [Epub ahead of print].
Dichgans, M. (2007). Genetics of ischaemic stroke. Lancet Neurol. 6, 149–161. doi: 10.1016/S1474-4422(07)70028-5
Fischer, M. E., Schubert, C. R., Nondahl, D. M., Dalton, D. S., Huang, G. H., Keating, B. J., et al. (2015). Subclinical atherosclerosis and increased risk of hearing impairment. Atherosclerosis 238, 344–349. doi: 10.1016/j.atherosclerosis.2014.12.031
Fusco, F. R., and Paldino, E. (2017). Role of Phosphodiesterases in Huntington's disease. Adv Neurobiol. 17, 285–304. doi: 10.1007/978-3-319-58811-7_11
Gopalakrishna, K. N., Boyd, K., and Artemyev, N. O. (2017). Mechanisms of mutant PDE6 proteins underlying retinal diseases. Cell. Signal. 37, 74–80. doi: 10.1016/j.cellsig.2017.06.002
Hmani-Aifa, M., Benzina, Z., Zulfiqar, F., Dhouib, H., Shahzadi, A., Ghorbel, A., et al. (2009). Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur. J. Hum. Genet. 17, 474–482. doi: 10.1038/ejhg.2008.167
Hsu, P. D., Lander, E. S., and Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. doi: 10.1016/j.cell.2014.05.010
Huang, M. Q., Zhang, J. G., and Long, Y. S. (2010). Identification of novel cyclic nucleotide phosphodiesterase gene cDNAs in the cochlea of guinea pig (Cavia porcellus) through conserved homologous sequences. Mol. Biotechnol. 44, 66–70. doi: 10.1007/s12033-009-9209-1
Jørgensen, C., Yasmeen, S., Iversen, H. K., and Kruuse, C. (2015). Phosphodiesterase4D (PDE4D)- A risk factor for atrial fibrillation and stroke? J. Neurol. Sci. 359, 266–274. doi: 10.1016/j.jns.2015.11.010
Lagman, D., Franzén, I. E., Eggert, J., Larhammar, D., and Abalo, X. M. (2016). Evolution and expression of the phosphodiesterase 6 genes unveils vertebrate novelty to control photosensitivity. BMC Evol. Biol. 16:124. doi: 10.1186/s12862-016-0695-z
Lin, H. C., Chou, Y. C., Wang, C. H., Hung, L. W., Shih, C. P., Kang, B. H., et al. (2017). Correlation between auditory brainstem response and hearing prognosis in idiopathic sudden sensorineural hearing loss patients. Auris Nasus Larynx. 44, 678–684. doi: 10.1016/j.anl.2017.01.004
Luo, J., Bai, X., Zhang, F., Xiao, Y., Gu, L., Han, Y., et al. (2017). Prevalence of mutations in deafness-causing genes in cochlear implanted patients with profound non-syndromic sensorineural hearing loss in Shandong Province, China. Ann. Hum. Genet. 81, 258–266. doi: 10.1111/ahg.12207
Majumder, A., Pahlberg, J., Muradov, H., Boyd, K. K., Sampath, A. P., and Artemyev, N. O. (2015). Exchange of cone for rod phosphodiesterase 6 catalytic subunits in rod photoreceptors mimics in part features of light adaptation. J. Neurosci. 35, 9225–9235. doi: 10.1523/JNEUROSCI.3563-14.2015
Peng, Y. Q., Tang, L. S., Yoshida, S., and Zhou, Y. D. (2017). Applications of CRISPR/Cas9 in retinal degenerative diseases. Int. J. Ophthalmol. 10, 646–651. doi: 10.18240/ijo.2017.04.23
Rajati, M., Azarpajooh, M. R., Mouhebati, M., Nasrollahi, M., Salehi, M., Khadivi, E., et al. (2016). Is sudden hearing loss associated with atherosclerosis? Iran. J. Otorhinolaryngol. 28, 189–195. doi: 10.4172/lpma.1000186
RetNet (2014). Daiger, S. P. PhD, Administrator. The University of Texas Health Science Center. Houston: The Retinal Information Network. Available online at: http://www.sph.uth.tmc.edu/RetNet/
Saga, M., Mashima, Y., Akeo, K., Kudoh, J., Oguchi, Y., and Shimizu, N. (1998). A novel homozygous Ile535Asn mutation in the rod cGMP phosphodiesterase beta-subunit gene in two brothers of a Japanese family with autosomal recessive retinitis pigmentosa. Curr. Eye Res. 17, 332–335. doi: 10.1076/ceyr.17.3.332.5214
Shao, M., Yi, X., Chi, L., Lin, J., Zhou, Q., and Huang, R. (2015). Ischemic stroke risk in a southeastern Chinese population: Insights from 5-lipoxygenase activating protein and phosphodiesterase 4D single-nucleotide polymorphisms. J. Formos. Med. Assoc. 114, 422–429. doi: 10.1016/j.jfma.2013.12.004
Tetsi, L., Charles, A. L., Paradis, S., Lejay, A., Talha, S., Geny, B., et al. (2017). Effects of cyclic nucleotide phosphodiesterases (PDEs) on mitochondrial skeletal muscle functions. Cell. Mol. Life Sci. 74, 1883–1893. doi: 10.1007/s00018-016-2446-0
Weber, S., Zeller, M., Guan, K., Wunder, F., Wagner, M., and El-Armouche, A. (2017). PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell. Signal. 38, 76–84. doi: 10.1016/j.cellsig.2017.06.020
Yu, W., Mookherjee, S., Chaitankar, V., Hiriyanna, S., Kim, J. W., Brooks, M., et al. (2017). Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 8:14716. doi: 10.1038/ncomms14716
Keywords: phosphodiesterase, CRISPR/Cas9, hearing loss, genetics, retinitis pigmentosa, usher syndrome
Citation: Mittal R, Bencie N, Parrish JM, Liu G, Mittal J, Yan D and Liu XZ (2018) An Update on Phosphodiesterase Mutations Underlying Genetic Etiology of Hearing Loss and Retinitis Pigmentosa. Front. Genet. 9:9. doi: 10.3389/fgene.2018.00009
Received: 05 November 2017; Accepted: 09 January 2018;
Published: 08 February 2018.
Edited by:
Musharraf Jelani, King Abdulaziz University, Saudi ArabiaReviewed by:
Muhammad Tariq, University of Tabuk, Saudi ArabiaMuzammil Ahmad Khan, Gomal University, Pakistan
Copyright © 2018 Mittal, Bencie, Parrish, Liu, Mittal, Yan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rahul Mittal, ci5taXR0YWwxMUBtZWQubWlhbWkuZWR1
Xue Zhong Liu, eGxpdUBtZWQubWlhbWkuZWR1